Transcriptomic Profiles of Brain Provide Insights into Molecular Mechanism of Feed Conversion Efficiency in Crucian Carp (Carassius auratus)


Abstract
:1. Introduction
2. Results
2.1. Transcriptome Sequencing and Statistics of Unigenes
2.2. Functional Annotation and Classification of Unigenes
2.3. Identification of Differentially Expressed Genes (DEGs)
2.4. Enrichment for Functional Analysis of DEGs
2.5. Critical DEGs Involved in Feed Conversion Efficiency of Crucian Carp
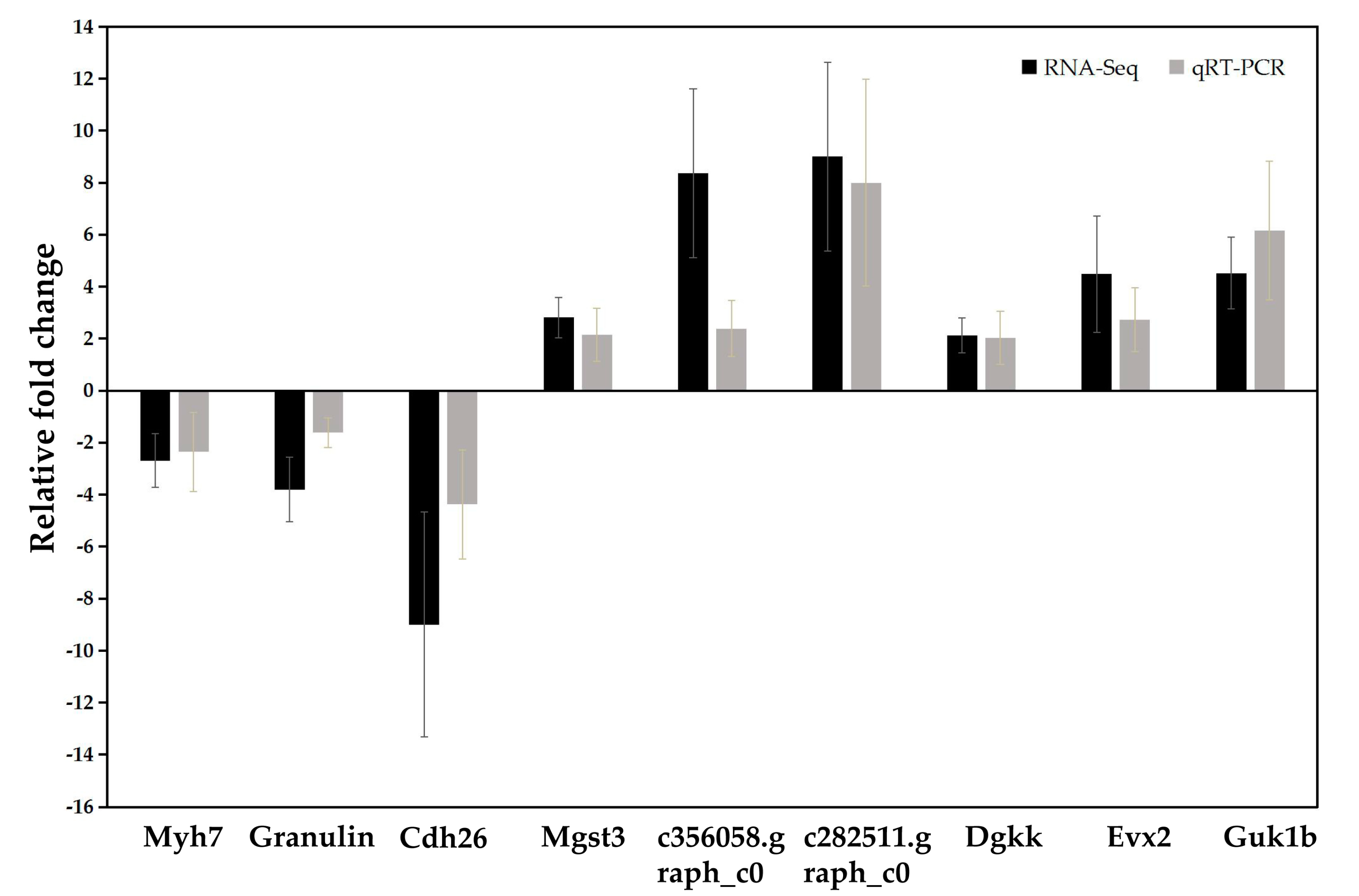
2.6. Validation of RNA-Seq Results by Quantitative Real-Time RT-PCR (qRT-PCR)
3. Discussion
4. Materials and Methods
4.1. Sample Collection and RNA Preparation
4.2. RNA Extraction, Library Preparation and Transcriptome Sequencing
4.3. Transcriptome Assembly and Functional Annotation
4.4. Analysis of Differentially Expressed Genes (DEGs)
4.5. Validation of RNA-Seq Results by qRT-PCR
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Merino, G.; Barange, M.; Blanchard, J.L.; Harle, J.; Holmes, R.; Allen, I.; Allison, E.H.; Badjeck, M.B.; Dulvy, N.K.; Holt, J.; et al. Can marine fisheries and aquaculture meet fish demand from a growing human population in a changing climate? Glob. Environ. Chang. 2012, 22, 795–806. [Google Scholar] [CrossRef]
- Easterling, W.E. Climate change and the adequacy of food and timber in the 21st century. Proc. Natl. Acad. Sci. USA 2007, 104, 19679. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.C.; Garcia, S.M. Fisheries, food security, climate change, and biodiversity: Characteristics of the sector and perspectives on emerging issues. ICES J. Mar. Sci. 2011, 68, 1343–1353. [Google Scholar] [CrossRef]
- Alexandratos, N. How to Feed the World in 2050. Available online: http://www.indiaenvironmentportal.org.in/files/How_to_Feed_the_World_in_2050.pdf. (accessed on 1 Octobor 2009).
- Wringe, B.F.; Devlin, R.H.; Ferguson, M.M.; Moghadam, H.K.; Sakhrani, D.; Danzmann, R.G. Growth-related quantitative trait loci in domestic and wild rainbow trout (Oncorhynchus mykiss). BMC Genet. 2010, 11, 63. [Google Scholar] [CrossRef] [PubMed]
- Laghari, M.Y.; Zhang, Y.; Lasgari, P.; Zhang, X.; Xu, P.; Xin, B.; Sun, X. Quantitative trait loci (QTL) associated with growth rate trait in common carp (Cyprinus carpio). Aquac. Int. 2013, 21, 1373–1379. [Google Scholar] [CrossRef]
- Gjedrem, T.; Baranski, M. Selective Breeding in Aquaculture: An Introduction. Rev. Methods Technol. Fish Biol. Fish. 2009, 10, 570–572. [Google Scholar]
- Sherman, E.L.; Nkrumah, J.D.; Li, C.; Bartusiak, R.; Murdoch, B.; Moore, S.S. Fine mapping quantitative trait loci for feed intake and feed efficiency in beef cattle. J. Anim. Sci. 2009, 87, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Barendse, W.; Reverter, A.; Bunch, R.J.; Harrison, B.E.; Barris, W.; Thomas, M.B. A validated whole-genome association study of efficient food conversion in cattle. Genetics 2007, 176, 1893–1905. [Google Scholar] [CrossRef] [PubMed]
- Do, D.N.; Strathe, A.B.; Ostersen, T.; Pant, S.D.; Kadarmideen, H.N. Genome-wide association and pathway analysis of feed efficiency in pigs reveal candidate genes and pathways for residual feed intake. Front. Genet. 2014, 5, 307. [Google Scholar] [CrossRef] [PubMed]
- Nkrumah, J.D.; Sherman, E.L.; Li, C.; Marques, E.; Crews, D.H.J.; Bartusiak, R.; Murdoch, B.; Wang, Z.; Basarab, J.A.; Moore, S.S. Primary genome scan to identify putative quantitative trait loci for feedlot growth rate, feed intake, and feed efficiency of beef cattle. J. Anim. Sci. 2007, 85, 3170–3181. [Google Scholar] [CrossRef] [PubMed]
- Mignon-Grasteau, S.; Rideau, N.; Gabriel, I.; Chantry-Darmon, C.; Boscher, M.; Sellier, N.; Chabault, M.; Bihan-Duval, E.L.; Narcy, A. Detection of QTL controlling feed efficiency and excretion in chickens fed a wheat-based diet. Genet. Sel. Evol. 2015, 47, 74. [Google Scholar] [CrossRef] [PubMed]
- De Koning, D.J.; Windsor, D.; Hocking, P.M.; Burt, D.W.; Law, A.; Haley, C.S.; Morris, A.; Vincent, J.; Griffin, H. Quantitative trait locus detection in commercial broiler lines using candidate regions. J. Anim. Sci. 2003, 81, 1158–1165. [Google Scholar] [CrossRef] [PubMed]
- De Koning, D.J.; Haley, C.S.; Windsor, D.; Hocking, P.M.; Griffin, H.; Morris, A.; Vincent, J.; Burt, D.W. Segregation of QTL for production traits in commercial meat-type chickens. Genet. Res. 2004, 83, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Wolc, A.; Arango, J.; Jankowski, T.; Settar, P.; Fulton, J.E.; O’Sullivan, N.P.; Fernando, R.; Garrick, D.J.; Dekker, J.C. Pedigree and genomic analyses of feed consumption and residual feed intake in laying hens. Poult. Sci. 2013, 92, 2270–2275. [Google Scholar] [CrossRef] [PubMed]
- Do, D.N.; Ostersen, T.; Strathe, A.B.; Mark, T.; Jensen, J.; Kadarmideen, H.N. Genome-wide association and systems genetic analyses of residual feed intake, daily feed consumption, backfat and weight gain in pigs. BMC Genet. 2014, 15, 27. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Peng, L.; Ruan, X.; Chen, X.; Ji, H.; Ma, J.; Ni, H.; Jiang, S.; Guo, D. Transcriptome profile analysis reveals cardiotoxicity of maduramicin in primary chicken myocardial cells. Arch. Toxicol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Liu, G.; Liu, Y.; Xu, Q.; Zhang, M.; Fang, M. Transcriptome profile at different physiological stages reveals potential mode for curly fleece in Chinese tan sheep. PLoS ONE 2013, 8, e71763. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Liu, B.; Fan, S.; Li, H.; Chen, M.; Zhang, B.; Su, J.; Meng, Z.; Yu, D. Differentially expressed immune-related genes in hemocytes of the pearl oyster Pinctada fucata against allograft identified by transcriptome analysis. Fish Shellfish Immunol. 2017, 62, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Cao, X.; Xu, X.; Huang, S.; Liu, C.; Tomljanovic, T. Developmental transcriptome analysis and identification of genes involved in formation of intestinal air-breathing function of Dojo loach, Misgurnus anguillicaudatus. Sci. Rep. 2016, 6, 31845. [Google Scholar] [CrossRef] [PubMed]
- Yi, G.; Yuan, J.; Bi, H.; Yan, W.; Yang, N.; Qu, L. In-Depth Duodenal Transcriptome Survey in Chickens with Divergent Feed Efficiency Using RNA-Seq. PLoS ONE 2015, 10, e0136765. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Hou, Y.; Liu, F.; Liu, A.; Jing, L.; Zhao, C.; Luan, Y.; Miao, Y.; Zhao, S.; Li, X. Transcriptome Analysis Reveals that Vitamin A Metabolism in the Liver Affects Feed Efficiency in Pigs. G3 Genes Genomes Genet. 2016, 6, 3615–3624. [Google Scholar] [CrossRef] [PubMed]
- Paradis, F.; Yue, S.; Grant, J.R.; Stothard, P.; Basarab, J.A.; Fitzsimmons, C. Transcriptomic analysis by RNA sequencing reveals that hepatic interferon-induced genes may be associated with feed efficiency in beef heifers. J. Anim. Sci. 2015, 93, 3331–3341. [Google Scholar] [CrossRef] [PubMed]
- Jégou, M.; Gondret, F.; Vincent, A.; Tréfeu, C.; Gilbert, H.; Louveau, I. Whole Blood Transcriptomics Is Relevant to Identify Molecular Changes in Response to Genetic Selection for Feed Efficiency and Nutritional Status in the Pig. PLoS ONE 2016, 11, e0146550. [Google Scholar] [CrossRef] [PubMed]
- Gondret, F.; Vincent, A.; Houée-Bigot, M.; Siegel, A.; Lagarrigue, S.; Causeur, D.; Gilbert, H. A transcriptome multi-tissue analysis identifies biological pathways and genes associated with variations in feed efficiency of growing pigs. BMC Genom. 2017, 18, 244. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Laghari, M.Y.; Zheng, X.; Cao, D.; Zhang, X.; Kuang, Y.; Li, C.; Cheng, L.; Mahboob, S.; Al-Ghanim, K.A.; et al. Mapping quantitative trait loci and identifying candidate genes affecting feed conversion ratio based onto two linkage maps in common carp (Cyprinus carpio L.). Aquaculture 2017, 468, 585–596. [Google Scholar] [CrossRef]
- Laghari, M.Y.; Lashari, P.; Zhang, Y.; Sun, X. Identification of Quantitative Trait Loci (QTLs) in Aquaculture Species. Rev. Fish. Sci. Aquac. 2014, 22, 221–238. [Google Scholar] [CrossRef]
- Rhee, J.S.; Jeong, C.B.; Kim, I.C.; Lee, Y.S.; Lee, C.; Lee, J.S. Immune gene discovery in the crucian carp Carassius auratus. Fish Shellfish Immunol. 2013, 36, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Lü, Y.P.; Wang, X.F.; Zhang, X.P. Experiment on using the extract of perilla seed as feed additives for Allogynogenetic crucian carp. China Feed 2007, 8, 32–33. [Google Scholar]
- Luo, W.; Wang, G.; Gong, W.; Xie, J.; Yu, D.; Li, Z. Effects of bioflocs technology (BFT) on growth and water quality of crucian carp (Carassius auratus var. pengze). J. South. Agric. 2014, 45, 318–322. [Google Scholar]
- Pang, M.; Fu, B.; Yu, X.; Liu, H.; Wang, X.; Yin, Z.; Xie, S.; Tong, J. Quantitative trait loci mapping for feed conversion efficiency in crucian carp (Carassius auratus). Sci. Rep. 2017, 7, 16971. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Cheng, L.; Xu, P.; Lu, G.; Wachholtz, M.; Sun, X.; Chen, S. Transcriptome analysis of crucian carp (Carassius auratus), an important aquaculture and hypoxia-tolerant species. PLoS ONE 2013, 8, e62308. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Li, J.T.; Kuang, Y.Y.; Xu, R.; Zhao, Z.X.; Hou, G.Y.; Liang, H.W.; Sun, X.W. The transcriptomes of the crucian carp complex (Carassius auratus) provide insights into the distinction between unisexual triploids and sexual diploids. Int. J. Mol. Sci. 2014, 15, 9386–9406. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.Y.; Li, J.Q.; Wu, S.F.; Zhu, Y.P.; Chen, Y.W.; He, F.C. Integrated nr Database in Protein Annotation System and Its Localization. Comput. Eng. 2006, 32, 71–74. [Google Scholar]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The Universal Protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Niemann, H.; Kuhla, B.; Flachowsky, G. Perspectives for feed-efficient animal production. J. Anim. Sci. 2011, 89, 4344–4363. [Google Scholar] [CrossRef] [PubMed]
- Doupé, R.G.; Lymbery, A.J. Toward the Genetic Improvement of Feed Conversion Efficiency in Fish. J. World Aquac. Soc. 2003, 34, 245–254. [Google Scholar] [CrossRef]
- Dickerson, G.E. Animal size and efficiency: Basic concepts. Anim. Prod. 1978, 27, 367–379. [Google Scholar] [CrossRef]
- Kushwaha, R.; Singh, A.K.; Kumar, M. Residual feed intake-An emerging system for selection of efficient animals: A review. Agric. Rev. 2016, 37, 66–71. [Google Scholar] [CrossRef]
- Ogata, H.Y.; Oku, H.; Murai, T. Growth, feed efficiency and feed intake of offspring from selected and wild Japanese flounder (Paralichthys olivaceus). Aquaculture 2002, 211, 183–193. [Google Scholar] [CrossRef]
- Imai, S.; Kai, M.; Yasuda, S.; Kanoh, H.; Sakane, F. Identification and characterization of a novel human type II diacylglycerol kinase, DGK kappa. J. Biol. Chem. 2005, 280, 39870–39881. [Google Scholar] [CrossRef] [PubMed]
- Thameem, F.; Yang, X.; Permana, P.A.; Wolford, J.K.; Bogardus, C.; Prochazka, M. Evaluation of the microsomal glutathione S-transferase 3 (MGST3) locus on 1q23 as a Type 2 diabetes susceptibility gene in Pima Indians. Hum. Genet. 2003, 113, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Shimma, Y.; Nishikawa, A.; bin Kassim, B.; Eto, A.; Jigami, Y. A defect in GTP synthesis affects mannose outer chain elongation in Saccharomyces cerevisiae. Mol. Gen. Genet. 1997, 256, 469–480. [Google Scholar] [PubMed]
- Weber, K.L.; Rincon, G.; Eenennaam, A.V. Genome-Wide Association Study and Differential Expression Analysis of Feed Efficiency using HD SNP Genotyping and Multiple Tissue RNAseq. In Proceedings of the International Plant and Animal Genome Conference XXII, San Diego, CA, USA, 10–15 January 2014. [Google Scholar]
- Serão, N.V.L.; Gonzálezpeña-Peña, D.; Beever, J.E.; Faulkner, D.B.; Southey, B.R.; Rodriguez-Zas, S.L. Single nucleotide polymorphisms and haplotypes associated with feed efficiency in beef cattle. BMC Genet. 2013, 14, 94. [Google Scholar] [CrossRef] [PubMed]
- Rasal, K.D.; Shah, T.M.; Vaidya, M.; Jakhesara, S.J.; Joshi, C.G. Analysis of consequences of non-synonymous SNP in feed conversion ratio associated TGF-β receptor type 3 gene in chicken. Meta Gene 2015, 4, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Oksbjerg, N.; Gondret, F.; Vestergaard, M. Basic principles of muscle development and growth in meat-producing mammals as affected by the insulin-like growth factor (IGF) system. Domest. Anim. Endocrinol. 2004, 27, 219–240. [Google Scholar] [CrossRef] [PubMed]
- Kuemmerle, J.F. Insulin-like growth factors in the gastrointestinal tract and liver. Endocrinol. Metab. Clin. N. Am. 2012, 41, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huang, X.L.; Cong, L.; Peng, Z.J.; Zhang, D.X. The study on the relations of chicken IGF-1 gene and feed conversion traits. Heilongjiang Anim. Sci. Vet. Med. 2013, 7, 37–39. [Google Scholar]
- Kinoshita, S.; Siddique, B.S.; Ceyhun, S.B.; Asaduzzaman, M.; Asakawa, S.; Watabe, S. Species-specific expression variation of fish MYH14, an ancient vertebrate myosin heavy chain gene orthologue. Fish. Sci. 2011, 77, 847–853. [Google Scholar] [CrossRef]
- Cui, X.; Hou, Y.; Yang, S.; Xie, Y.; Zhang, S.; Zhang, Y.; Zhang, Q.; Lu, X.; Liu, G.E.; Sun, D. Transcriptional profiling of mammary gland in Holstein cows with extremely different milk protein and fat percentage using RNA sequencing. BMC Genom. 2014, 15, 226. [Google Scholar] [CrossRef] [PubMed]
- Coble, D.J.; Fleming, D.; Persia, M.E.; Ashwell, C.M.; Rothschild, M.F.; Schmidt, C.J. RNA-seq analysis of broiler liver transcriptome reveals novel responses to high ambient temperature. BMC Genom. 2014, 15, 1084. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, Y.; Liang, X.; Cao, X.; Huang, L.; Yan, J.; Wei, Y.; Gao, J. Hepatic transcriptome analysis and identification of differentially expressed genes response to dietary oxidized fish oil in loach Misgurnus anguillicaudatus. PLoS ONE 2017, 12, e0172386. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2012, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Samples | H1 | H2 | H3 | L1 | L2 | L3 |
|---|---|---|---|---|---|---|
| Raw reads | 51,467,792 | 50,558,463 | 54,265,382 | 53,444,150 | 56,248,579 | 55,784,163 |
| Clean reads | 50,530,680 | 49,250,952 | 53,769,034 | 52,491,591 | 55,011,113 | 54,460,392 |
| Q30 | 93.20% | 93.76% | 93.65% | 93.37% | 93.84% | 93.79% |
| GC-content | 48.04% | 46.68% | 48.84% | 49.44% | 47.37% | 45.33% |
| Mapped reads | 29,173,543 | 31,871,361 | 31,789,018 | 30,732,102 | 33,802,037 | 35,592,419 |
| Mapped ratio | 57.73% | 64.71% | 59.12% | 58.55% | 61.45% | 65.35% |
| Length Range | Transcript | Unigene |
|---|---|---|
| 200–300 | 283,078 (18.32%) | 209,625 (38.49%) |
| 300–500 | 264,160 (17.09%) | 151,108 (27.75%) |
| 500–1000 | 316,468 (20.48%) | 102,120 (18.75%) |
| 1000–2000 | 360,982 (23.36%) | 50,873 (9.34%) |
| 2000+ | 320,919 (20.76%) | 30,886 (5.67%) |
| Total Number | 1,545,607 | 544,612 |
| Total Length | 1,940,292,597 | 350,936,744 |
| N50 Length | 2091 | 965 |
| Mean Length | 1,255.36 | 644.38 |
| KEGG Category | Pathway Name | Pathway ID | DEGs | |
|---|---|---|---|---|
| Cellular Processes | Cell growth and death | Apoptosis | ko04210 | Endog |
| Cell cycle | ko04110 | Crebrtc2 | ||
| Cell motility | Regulation of actin cytoskeleton | ko04810 | Nckap1 | |
| Cellular community | Tight junction | ko04530 | Myh7, Myh | |
| Environmental Information Processing | Membrane transport | ABC transporters | ko02010 | Abcb11 |
| Signal transduction | Calcium signaling pathway | ko04020 | Vdnccsa1b, Tacr2, Htr7 | |
| ErbB signaling pathway | ko04012 | Tgfα | ||
| MAPK signaling pathway | ko04010 | Vdnccsa1b, Nr4a1 | ||
| Phosphatidylinositol signaling system | ko04070 | Dgkk | ||
| Signaling molecules and interaction | Cytokine-cytokine receptor interaction | ko04060 | Xcr1 | |
| Neuroactive ligand-receptor interaction | ko04080 | Tacr2, Htr7 | ||
| Genetic Information Processing | Replication and repair | Base excision repair | ko03410 | Cdcpcec1 |
| Transcription | Basal transcription factors | ko03022 | Qtf2f2b | |
| Translation | RNA transport | ko03013 | Tef1 | |
| Metabolism | Lipid metabolism | Glycerolipid metabolism | ko00561 | Dgkk |
| Glycerophospholipid metabolism | ko00564 | Dgkk | ||
| Metabolism of other amino acids | Glutathione metabolism | ko00480 | Mgst 3 | |
| Nucleotide metabolism | Purine metabolism | ko00230 | Guk1b | |
| Xenobiotics biodegradation and metabolism | Drug metabolism—cytochrome P450 | ko00982 | Mgst 3 | |
| Metabolism of xenobiotics by cytochrome P450 | ko00980 | Mgst 3 | ||
| Organismal Systems | Circulatory system | Adrenergic signaling in cardiomyocytes | ko04261 | Myh7 |
| Cardiac muscle contraction | ko04260 | Myh7 | ||
| Trait | Low Group | High Group |
|---|---|---|
| BWI (g) | 0.69 ± 0.12 | 0.78 ± 0.35 |
| BWF (g) | 1.93 ± 0.37 | 4.25 ± 1.87 |
| FI (g) | 3.32 ± 0.59 | 4.63 ± 2.20 |
| FCE | 37.0 ± 4.1% | 76.0 ± 3.0% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pang, M.; Luo, W.; Fu, B.; Yu, X.; Zhou, Y.; Tong, J. Transcriptomic Profiles of Brain Provide Insights into Molecular Mechanism of Feed Conversion Efficiency in Crucian Carp (Carassius auratus). Int. J. Mol. Sci. 2018, 19, 858. https://doi.org/10.3390/ijms19030858
Pang M, Luo W, Fu B, Yu X, Zhou Y, Tong J. Transcriptomic Profiles of Brain Provide Insights into Molecular Mechanism of Feed Conversion Efficiency in Crucian Carp (Carassius auratus). International Journal of Molecular Sciences. 2018; 19(3):858. https://doi.org/10.3390/ijms19030858
Chicago/Turabian StylePang, Meixia, Weiwei Luo, Beide Fu, Xiaomu Yu, Ying Zhou, and Jingou Tong. 2018. "Transcriptomic Profiles of Brain Provide Insights into Molecular Mechanism of Feed Conversion Efficiency in Crucian Carp (Carassius auratus)" International Journal of Molecular Sciences 19, no. 3: 858. https://doi.org/10.3390/ijms19030858