Retinal Cyclic Nucleotide-Gated Channels: From Pathophysiology to Therapy



Center for Integrated Protein Science Munich (CIPSM), Department of Pharmacy—Center for Drug Research, Ludwig-Maximilians-Universität München, Butenandtstr, 5-13, 81377 Munich, Germany
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(3), 749; https://doi.org/10.3390/ijms19030749
Submission received: 1 February 2018
/
Revised: 28 February 2018
/
Accepted: 6 March 2018
/
Published: 7 March 2018
(This article belongs to the Special Issue cGMP-Signalling in Cells: Molecular and Functional Features)
Abstract
:The first step in vision is the absorption of photons by the photopigments in cone and rod photoreceptors. After initial amplification within the phototransduction cascade the signal is translated into an electrical signal by the action of cyclic nucleotide-gated (CNG) channels. CNG channels are ligand-gated ion channels that are activated by the binding of cyclic guanosine monophosphate (cGMP) or cyclic adenosine monophosphate (cAMP). Retinal CNG channels transduce changes in intracellular concentrations of cGMP into changes of the membrane potential and the Ca2+ concentration. Structurally, the CNG channels belong to the superfamily of pore-loop cation channels and share a common gross structure with hyperpolarization-activated cyclic nucleotide-gated (HCN) channels and voltage-gated potassium channels (KCN). In this review, we provide an overview on the molecular properties of CNG channels and describe their physiological role in the phototransduction pathways. We also discuss insights into the pathophysiological role of CNG channel proteins that have emerged from the analysis of CNG channel-deficient animal models and human CNG channelopathies. Finally, we summarize recent gene therapy activities and provide an outlook for future clinical application.
Keywords:
CNG; cyclic nucleotide-gated channel; vision; channelopathies; Ca2+; knockout; photoreceptor; cGMP; gene therapy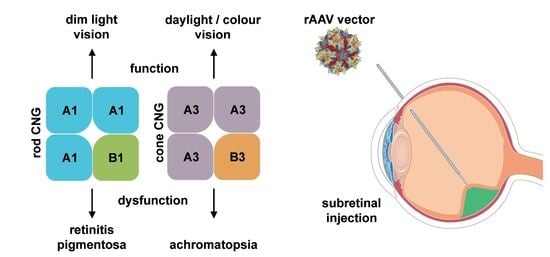
1. Introduction
Cyclic nucleotides, such as cAMP and cGMP, regulate the activity of several classes of proteins including cyclic nucleotide-binding domain (CNBD)-containing cation channels. Structurally, cyclic nucleotide-regulated channels belong to the superfamily of pore-loop cation channels [1]. In vertebrates, three subfamilies of CNBD-containing ion channels exist: the CNG, the HCN and the KCN channels [2,3]. These channels share a similar principal architecture but differ from each other in terms of their activation mode and ion selectivity. Cyclic nucleotide-gated (CNG) channels are strictly ligand-gated because their opening requires the binding of cGMP or cAMP. CNG channels have been also found in several non-vertebrate species including insect species and C. elegans. In mammals, the CNG channel family comprises six homologous members (CNGA1-4, CNGB1 and CNGB3) encoding for structurally similar A and B subunits that assemble in distinct combinations into cell type-specific heterotetrameric complexes [4] (Figure 1). The A subunits confer the principal channel properties, whereas the B subunits are essential for proper outer segment localization and contribute specific biophysical properties (e.g., fast gating kinetics) to the native channel complex [2].
2. Structure, Basic Properties and Activation of CNG Channels
CNG channels form a distinct branch within the superfamily of voltage-gated-like (VGL) channels [1,5]. In mammals, the CNG channel family comprises six homologous members, which are classified as A subunits (CNGA1-4) and B subunits (CNGB1 and CNGB3). A subunits and B subunits share the same principal membrane topology. Each of these subunits possesses a transmembrane channel core consisting of six α-helical segments (S1–S6), a reentrant pore (P) loop between S5 and S6, and cytosolic N- and C-termini. The CNBD is located in the C-terminus and is connected to the S6 by the C-linker. Native CNG channels in photoreceptor outer segments are heterotetramers assembled by three A subunits (CNGA1 in rods and CNGA3 in cones) and one B subunit (CNGB1 in rods and CNGB3 in cones) [6,7,8,9]. So far, no structure of mammalian CNG channels is available. However, a structure of the CNG channel from C. elegans TAX-4 has been recently determined by single-particle electron cryo-microscopy [10]. Like other CNBD-containing channels [3], CNG channels are tetramers with the four subunits arranged around the centrally located pore [10]. The ion conducting pore is lined by the P-loops and the S6 segments of the four subunits. The TAX-4 voltage-sensor-like (VSL) domain is unique among known VSL structures as it appears segmented. This segmentation might preclude voltage-dependent movements within the membrane and thus help explain the lack of voltage-dependent gating of CNG channels [10]. However, the TAX-4 structure only reflects the cGMP-bound open state and a closed state structure is missing. Based on comparisons of available open and closed state structures from related channels and previous mutagenesis studies, activation of CNG channels is thought to involve coordinated movements of at least three basic elements, the CNBD, the C-linker and the channel gate [10,11,12,13,14,15]. Binding of cyclic nucleotides to the CNBD leads to a rotational change of the entire C-terminus relative to the pore. The C-linker, a domain that allosterically couples cyclic nucleotide-binding to the channel gate also follows this rotation and partially moves upwards. The channel gate located in the intracellular portion of the S6 segment is constricted and maintained in the closed state presumably by steady forces from the C-linker [16,17]. Upon binding of the ligand, the movements described above relieve the inhibitory forces of the C-linker and the channel pore becomes delated, thereby allowing for ion permeation.
While all six mammalian CNG channel subunits share significant sequence homology, only CNGA1, CNGA2 and CNGA3 can form functional homotetrameric channels in heterologous expression systems. The remaining subunits (CNGA4, CNGB1 and CNGB3) do not assemble to functional homotetramers. However, when present in heterotetrameric CNG channel complexes, they confer key functional properties specific for native CNG channels (e.g., single channel flickering, altered affinity for cyclic nucleotides, distinct permeation properties, regulation by Ca2+) (for detailed review see [2,18]).
CNG channels are non-selective for several monovalent or divalent cations, such as Na+ and K+, Ca2+ and Mg2+. However, Ca2+ and Mg2+ can also act as a voltage-dependent blocker of monovalent cation permeability [19,20]. Another characteristic feature of CNG channels is the lack of desensitization or inactivation upon exposure to cyclic nucleotides. In addition, CNG channels show Ca2+-dependent feedback inhibition, which is thought to be calmodulin (CaM)-mediated [21,22]. In rods, Ca2+-inhibition is conferred by binding of CaM to an IQ-type binding site located at the N-terminus of the CNGB1 subunit [23,24]. In cone photoreceptors this inhibitory effect is more pronounced and most likely mediated by another Ca2+-binding protein termed CNG-modulin [25].
3. Signal Transduction in Photoreceptors
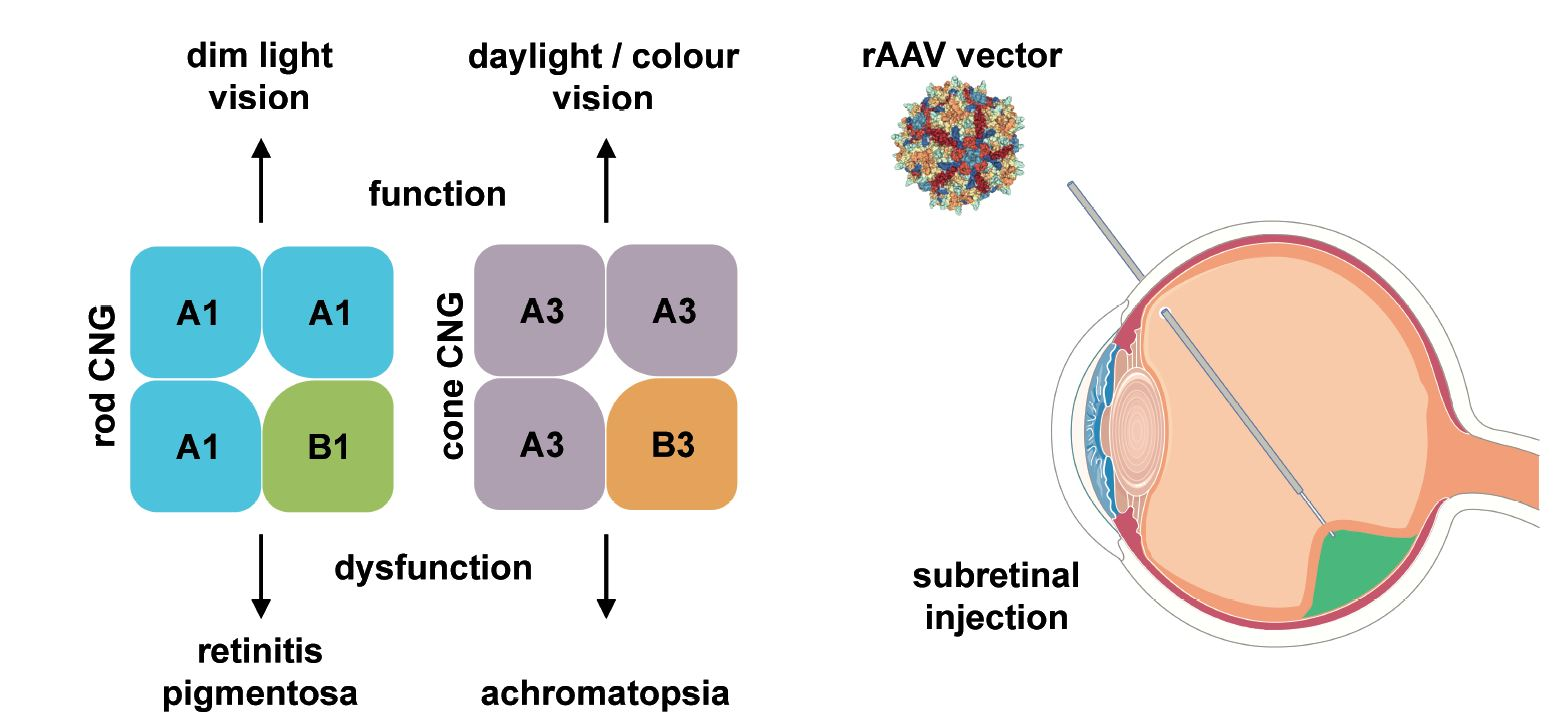
Rod and cone photoreceptors possess similar, but distinct phototransduction pathways which fulfill the required initial processing of visual stimuli at different ambient light conditions (Figure 2). Rods mediate vision at dim light levels, whereas vision at higher light levels (e.g., daylight) is provided by cones with minor contribution from rods. The cone visual system also enables color vision as it can discriminate between wavelengths by comparing inputs from two (in most vertebrates) or three (in humans and some non-human primates) types of cones equipped with different cone opsin variants (short-, medium- and long-wave sensitive) with distinct spectral sensitivities [26,27]. In both rods and cones, signal transduction follows the same principle and is facilitated by enzymes controlling the levels of the second messenger cGMP. cGMP in turn controls the activation of the CNG channel present in the plasma membrane of the photoreceptor outer segment (Figure 1). In the dark, constant activity of transmembrane guanylyl cyclases [28,29] results in high levels of cGMP, which maintains the CNG channel in an opened conformation. As mentioned in the previous section, CNG channels carry a steady non-inactivating Na+ and Ca2+ current (“dark current”) which depolarizes the photoreceptor promoting glutamate release at the photoreceptor synaptic terminal. Following light stimulation photopigments (opsins) initiate a G protein (transducin) mediated signaling cascade leading to activation of a cGMP phosphodiesterase (PDE6). PDE6 hydrolyses cGMP and thereby leads to closure of the CNG channel. Consequently, the photoreceptor hyperpolarizes and the synaptic glutamate release decreases. CNG channels provide the only source for Ca2+ influx into rod and cone outer segments [2,30,31]. Ca2+ entry is balanced by Ca2+ extrusion through the activity of a Na+/Ca2+-K+ exchanger [31,32,33,34,35]. Thus, light stimulation of photoreceptors results in a decrease of the intracellular Ca2+ concentration because it shuts down Ca2+ entry via the CNG channels while the Na+/Ca2+-K+ exchanger continues to clear Ca2+ from the cytosol. The reduced Ca2+ level upon light absorption contributes to the recovery from light response by several mechanisms including phosphorylation of visual pigments and PDE6, as well as restoration of cGMP levels by guanylyl cyclase activating proteins (GCAPs; [36])-mediated activation of the guanylyl cyclase.
Photoresponses measured from isolated rods and cones differ in sensitivity and kinetics [2,37]. Rod and cone CNG channels share the same principle functional properties and differ only in some specific features. Notable examples are the higher Ca2+-permeability of the cone CNG channel and the stronger Ca2+-dependent inhibition of the ligand-sensitivity in the rod CNG channel [2,38]. However, these differences cannot explain the differences in rod and cone photoresponses, which most likely result from multiple distinct signaling cascade molecules and distinct morphological characteristics (e.g., outer segment compartmentalization).
4. In Vivo Analysis of CNG Channel Function: Analysis of Genetic Mouse Models and Human Channelopathies
Genetic studies have dramatically increased our knowledge on the significance of retinal CNG channels. The major findings of these studies will be summarized in the following sections.
4.1. CNGA1 and CNGB1
Mutations in the rod-specific CNGA1 or CNGB1 genes cause autosomal recessive retinitis pigmentosa (RP) [39,40] (Table 1). RP comprises a genetically diverse group of progressive degenerative retinal diseases affecting the photoreceptors of the retina [41,42]. The most common symptoms of RP include night blindness, progressive concentric reduction of the visual field, and abnormal accumulation of pigmentation in the retina [43]. Fast progressing RP can lead to legal blindness in advanced stages. So far, RP has been mapped to >60 genes (http://www.sph.uth.tmc.edu/Retnet, 22 February 2018) encoding proteins involved in the visual transduction pathway or required for the maintenance of the rod architecture. Depending on the genetic background, mutations in CNGA1 account for 1–8% of cases of autosomal recessive RP (arRP) [40,41,44,45,46]. Most of the identified CNGA1 mutations cause deletions of important functional domains or result in defective membrane trafficking [2,40,47,48]. As a result, the affected patients lack rod-mediated light responses. Mutations in CNGB1 account for approx. 4% of arRP cases [39,41,49,50]. Although the known CNGB1 mutations cause only minor deletions or single amino acid substitutions, the phenotype is comparable to the severe RP phenotype in CNGA1 patients. So far, only a few CNGB1 mutations have been functionally characterized. These mutations lead to protein instability, impaired channel targeting or functionally inactive CNG channels [39,49,51,52].
Mice lacking Cnga1 have not been described so far. However, transgenic mice overexpressing Cnga1 antisense mRNA were generated [53]. Although no functional characterization of these mice has been reported, histological analysis revealed some morphological features of RP (e.g., reduced number of photoreceptors, apoptotic death of retinal cells). A naturally occurring Cnga1 mutation has been recently identified in a Shetland sheepdog breed with progressive retinal atrophy [54]. However, functional characterization of the retinal phenotype is still missing.
Genetic deletion of Cngb1 in mice results in a phenotype that recapitulates the principal pathology of RP patients. In particular, Cngb1 knockout (KO) mice have strongly diminished rod photoreceptor function and impaired rod-mediated vision [55,56,57]. The early functional defects are followed by a progressive degeneration of rods and secondary degeneration of primarily non-affected cones. The degeneration process is rather slow and results in loss of about 50% of rods at 6 months and 90% at 12 months of age [55,57,58]. At the cellular level, the loss of Cngb1 induces down-regulation of several proteins of the phototransduction cascade and degradation of the CNGA1 subunit. A naturally occurring Cngb1 mutation was also identified in a Papillon dog breed with markedly reduced or absent rod function, and accompanied by progressive retinal degeneration [59]. Comparative analysis suggested that CNGB1-deficient RP patients and mouse and dog models have a similar phenotype characterized by early loss of rod function and slow rod photoreceptor degeneration along with a secondary decline in cone function [58].
4.2. CNGA3 and CNGB3
Mutations in either CNGA3 [60] or CNGB3 [61,62] cause achromatopsia (ACHM), also known as rod monochromatism or total color blindness (Table 1). ACHM is an inherited eye disease associated with congenital absence of cone photoreceptor function. In contrast to color blindness, in which changes in expression of opsin genes merely affect spectral sensitivity but not the physiology of photoreceptors [63,64], the complete unresponsiveness of cones in ACHM has grave consequences for vision, particularly with respect to the densely cone-packed human fovea. Affected individuals suffer from strongly impaired daylight vision, photophobia, nystagmus, and lack of color discrimination [65,66,67]. The prevalence of ACHM is approximately 1:30,000 [68,69,70]. Currently, six genes have been linked to ACHM: ATF6, CNGA3, CNGB3, GNAT2, PDE6C and PDE6H. Up to 80% of the patients carry mutations in the genes CNGA3 and CNGB3 encoding the two subunits of the cone CNG channel. To date, more than 100 mutations in CNGA3 and more than 50 mutations in CNGB3 were found to cause inherited ACHM in humans [71]. Mutations in CNGB3 (ACHM1) are more common in the general western population and account for almost 50% of ACHM cases [72]. Most CNGB3 mutations are nonsense, frameshift or splice mutations resulting in truncated or strongly impaired channel proteins [68,69]. A missense mutation in the CNGB3 gene (S435F) was identified in colorblind individuals originating from the Pingelap atoll of Micronesia [62]. In this small island ACHM is very frequent and affects nearly 10% of the native population [73]. Approximately 28% of patients in the western population carry mutations in CNGA3 (ACHM2). In Middle East and Arabic populations, mutations in CNGA3 account for up to 60% of ACHM cases. The majority of CNGA3 mutations are missense mutations affecting only single amino acid residues of the protein [68,69].
Loss of function mutations in CNGA3 result in non-functional cone CNG channels because CNGB3 cannot form functional CNG channels in the absence of CNGA3 [2,47]. Genetic inactivation of Cnga3 in mice leads to selective loss of cone-mediated light responses [74] accompanied by progressive degeneration and cell death of cones [75]. An early hallmark of cone degeneration is the strong accumulation of the second messenger cGMP, suggesting its involvement in the process of degeneration [76,77]. Cone degeneration affects M- and S-cones differentially and cell death proceeds significantly faster in ventral and nasal (S-cone-rich) than in dorsal and temporal (M-cone-rich) parts of the retina. Ventral cones are almost completely missing after the third postnatal month whereas residual dorsal cones are present even in aged knockout mice [75]. In addition, a naturally occurring mouse model of achromatopsia—the cpfl5 mouse with a Cnga3 point mutation—was described with a phenotype similar to the Cnga3 KO mouse [78]. Moreover, a sheep model of ACHM2 was identified with diminished cone, but normal rod function [79,80]. Affected lambs were homozygous for a Cnga3 mutation which results in a premature stop codon [79]. Recently, two spontaneous canine models of ACHM2 have been described [81]: a German shepherd, carrying a CNGA3/p.R424W mutation, and a Labrador retriever with a CNGA3/p.V644del mutation.
Various animal models also exist for CNGB3-linked ACHM. Knockout of Cngb3 in mice strongly impairs cone function and leads to progressive cone degeneration [82,83]. A residual cone function can be observed in this model, most likely conferred by irregular homomeric CNGA3 channels. In addition to the Cngb3 knockout (KO) mouse, multiple naturally occurring canine models exist that carry recessive mutations in exon 6 or a genomic deletion of the entire Cngb3 gene [84]. The latter results in a disease sharing the same clinical phenotype as human patients characterized by day blindness and absence of cone function [85,86,87,88].
5. Gene Therapy for the Treatment of CNG Channelopathies
No curative treatment for CNG channelopathies (ACHM or RP) exists thus far and clinical management is currently limited to specialized genetic counseling, the use of low vision aids, tinted contact lenses or glasses to reduce symptoms of photophobia [70,107]. Our improved knowledge on CNG channel biology and the availability of suitable animal models together with the advent of recombinant adeno-associated virus (rAAV) vectors as efficient and safe retinal gene delivery tools has led to the initiation of several promising gene therapy programs (Table 1).
ACHM and RP caused by mutations in CNG channels are both inherited in an autosomal recessive manner. Therefore, most treatment approaches for CNG channelopathies focus on so-called gene supplementation. Gene supplementation aims at complementing affected photoreceptor cells with a healthy copy of the disease-causing gene. In the following paragraphs, we will provide an overview on rAAV vectors, review current preclinical activities and finally summarize ongoing clinical trials.
rAAV vectors are based on AAVs, which are small (25 nm diameter), non-enveloped and non-pathogenic parvoviruses with a genome packaging capacity of approx. 4.7 kb [108]. rAAV vectors are devoid of most AAV genomic sequences and contain only short palindromic sequences, so called inverted terminal repeats (ITRs), which flank the therapeutic gene expression cassette [108]. In recent years, rAAV vectors have evolved as the gold standard gene delivery vector for retinal photoreceptors. Their success is based on the fact that they are easy to produce at large scale and their small genome can be easily manipulated [109,110]. Moreover, a large and growing number of naturally occurring or engineered AAV capsids are available allowing for packaging of pseudotyped rAAV of different serotypes with distinct properties, e.g., specificity for different cell types [111,112]. Finally, rAAVs are generally considered as very safe gene therapy vectors with low immunogenicity and toxicity [113,114]. After transduction, in contrast to the native unmodified AAV that integrates into a preferred genome site, the rAAV vector genome stays predominantly in an episomal state and no considerable integration into the host genome has been reported [115,116,117].
6. Gene Therapy for CNG Channel-Linked RP
So far, no gene therapy approaches have been reported for CNGA1-linked RP (RP49). This is most likely due to the lack of well-characterized animal models for this form of RP. For CNGB1-linked RP (RP45) successful proof-of-concept studies for rAAV-based gene supplementation have been reported in both the Cngb1 KO mouse model [90] and the Cngb1 mutant dog model [58]. To enable efficient packaging and rod-specific expression of the relatively large full-length Cngb1 cDNA (~4 kb), the two studies used an rAAV expression cassette with a short rod- [90] or photoreceptor-specific [58] promoter combined with short regulatory elements (e.g., SV40 polyA). In both species, subretinal injection of therapeutic rAAV gene supplementation vectors (serotype 5 or 8) led to (i) restoration of CNG channel expression and localization, (ii) gain of retinal function and preservation of retinal morphology and (iii) improvement of vision-guided behavior. Apart from the expression of full-length CNGB1, rAAV treatment also restored normal levels of the otherwise degraded endogenous CNGA1 subunit of the rod CNG channel in this mouse model. Both proteins co-localized in rod outer segments and formed regular CNG channel complexes within the treated area of the Cngb1 KO or mutant retina, leading to significant morphological preservation and a delay of retinal degeneration. In the electroretinographic analysis, substantial restoration of rod-driven light responses was observed. Finally, treated Cngb1 KO mice as well as Cngb1 mutant dogs performed significantly better than untreated controls in rod-dependent vision-guided behavior tests [58,90]. These promising results facilitated the initiation of translational studies, which might culminate in a first in man clinical trial.
7. Gene Therapy for CNG Channel-Linked ACHM
Successful preclinical proof-of-concept animal studies for rAAV-based gene supplementation therapy exist for CNGA3 and CNGB3 [76,78,92,93,101,102,103]. In these studies wildtype or tyrosine mutant AAV2, AAV5 or AAV8 vectors expressing the corresponding wildtype cDNAs under control of cone-specific or ubiquitous promoters were used to evaluate rAAV-mediated gene supplementation as a potential treatment of ACHM. The studies utilized engineered or naturally occurring mouse, dog or sheep models of ACHM and reported beneficial effects of the treatment on cone-specific visual function. Treated animals showed cone-driven light responses and showed structural preservation of cone photoreceptors. Our group studied in detail the effect of rAAV-mediated gene supplementation in Cnga3 KO mice [76,93]. In addition to the gain of retinal function, we could show that the treatment also had beneficial effects on retinal morphology and degenerative cellular processes. For instance, the therapy normalized pathologically elevated cellular cGMP levels in cones, delayed cone cell death and diminished the inflammatory response of Müller glia cells that is a hallmark of retinal degenerations [75]. Follow-up studies suggested that the therapeutic effect was long-lasting and could be applied at more advanced disease stages (e.g., at three months of age) [93].
rAAV-based gene supplementation has proven equally effective in preclinical models of both CNGA3- [76,78,92] and CNGB3-linked ACHM [101,103] with the only exception being the reduced treatment efficiency of the treatment in advanced stage Cngb3 mutant dogs [101]. However, the efficiency in this dog model could be improved when rAAV gene supplementation was combined with the administration of ciliary neurotrophic factor (CNTF) which is known to cause a temporal deconstruction of photoreceptor outer segments [102].
The very encouraging preclinical studies have led to the initiation of various translational projects to bring gene supplementation therapies for CNGA3- and CNGB3-linked ACHM to the clinics (Table 1). The first study to recruit patients was a phase I/II exploratory, dose-escalation study, which tests safety and efficacy of a single dose subretinal injection of rAAV8.hCNGA3 in patients with ACHM caused by CNGA3 mutations (clinicaltrials.gov identifier: NCT02610582) [118]. This clinical trial was conducted by the RD-CURE consortium encompassing clinicians and researchers from the University Eye Hospital Tübingen and the Ludwig-Maximilians-University Munich. Nine patients were treated in this trial and publication of study results is expected in 2018. Three additional interventional phase I/II clinical trials with ACHM patients are currently in progress. Two studies concern CNGB3-linked ACHM (NCT02599922 and NCT03001310) and are sponsored by Applied Genetic Technologies Corp (AGTC) and MeiraGTx UK II Ltd., respectively. These studies also have a non-randomized, open-label and dose-escalation design and test safety and efficacy of rAAV2tYF-PR1.7-hCNGB3 (AGTC study) or AAV2/8-hCARp.hCNGB3 (MeiraGTx study) in patients with ACHM caused by mutations in CNGB3. Both studies started enrolling patients in 2016. Another phase I/II clinical trial testing safety and efficacy of rAAV2tYF-PR1.7-hCNGA3 in patients with CNGA3-linked ACHM sponsored by AGTC (NCT02935517) enrolled the first patient in May 2017. These studies together bring hope to patients with CNG channel-linked eye diseases and suggest that gene therapy products targeting CNG channelopathies could follow the path of Luxturna® (voretigene neparvovec-rzyl), the first in class rAAV gene therapy product for the treatment of biallelic RPE65 mutation-linked retinal dystrophy, which recently received marketing authorization from the U.S. Food and Drug Administration. Pricing negotiations for Luxturna® are ongoing and we will have to wait and see how affordable such treatments will be.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (EXC 114).
Author Contributions
Stylianos Michalakis, Elvir Becirovic and Martin Biel wrote the manuscript and compiled the artwork.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Yu, F.H.; Yarov-Yarovoy, V.; Gutman, G.A.; Catterall, W.A. Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacol. Rev. 2005, 57, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Kaupp, U.B.; Seifert, R. Cyclic nucleotide-gated ion channels. Physiol. Rev. 2002, 82, 769–824. [Google Scholar] [CrossRef] [PubMed]
- James, Z.M.; Zagotta, W.N. Structural insights into the mechanisms of cnbd channel function. J. Gen. Physiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Biel, M.; Michalakis, S. Cyclic nucleotide-gated channels. Handb. Exp. Pharmacol. 2009, 111–136. [Google Scholar] [CrossRef]
- Hofmann, F.; Biel, M.; Kaupp, U.B. International union of pharmacology. Li. Nomenclature and structure-function relationships of cyclic nucleotide-regulated channels. Pharmacol. Rev. 2005, 57, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Weitz, D.; Ficek, N.; Kremmer, E.; Bauer, P.J.; Kaupp, U.B. Subunit stoichiometry of the cng channel of rod photoreceptors. Neuron 2002, 36, 881–889. [Google Scholar] [CrossRef]
- Zheng, J.; Trudeau, M.C.; Zagotta, W.N. Rod cyclic nucleotide-gated channels have a stoichiometry of three cnga1 subunits and one cngb1 subunit. Neuron 2002, 36, 891–896. [Google Scholar] [CrossRef]
- Zhong, H.; Molday, L.L.; Molday, R.S.; Yau, K.W. The heteromeric cyclic nucleotide-gated channel adopts a 3a:1b stoichiometry. Nature 2002, 420, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Shuart, N.G.; Haitin, Y.; Camp, S.S.; Black, K.D.; Zagotta, W.N. Molecular mechanism for 3:1 subunit stoichiometry of rod cyclic nucleotide-gated ion channels. Nat. Commun. 2011, 2, 457. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhou, X.; Wang, S.; Michailidis, I.; Gong, Y.; Su, D.; Li, H.; Li, X.; Yang, J. Structure of a eukaryotic cyclic-nucleotide-gated channel. Nature 2017, 542, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Zagotta, W.N.; Olivier, N.B.; Black, K.D.; Young, E.C.; Olson, R.; Gouaux, E. Structural basis for modulation and agonist specificity of hcn pacemaker channels. Nature 2003, 425, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Siegelbaum, S.A. Gating of hcn channels by cyclic nucleotides: Residue contacts that underlie ligand binding, selectivity, and efficacy. Structure 2007, 15, 655–670. [Google Scholar] [CrossRef] [PubMed]
- Taraska, J.W.; Zagotta, W.N. Cyclic nucleotide-regulated ion channels: Spotlight on symmetry. Structure 2007, 15, 1023–1024. [Google Scholar] [CrossRef] [PubMed]
- Flynn, G.E.; Black, K.D.; Islas, L.D.; Sankaran, B.; Zagotta, W.N. Structure and rearrangements in the carboxy-terminal region of spih channels. Structure 2007, 15, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Mazzolini, M.; Arcangeletti, M.; Marchesi, A.; Napolitano, L.M.R.; Grosa, D.; Maity, S.; Anselmi, C.; Torre, V. The gating mechanism in cyclic nucleotide-gated ion channels. Sci. Rep. 2018, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Lee, A.; Chen, J.; Cadene, M.; Chait, B.T.; MacKinnon, R. Crystal structure and mechanism of a calcium-gated potassium channel. Nature 2002, 417, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Giorgetti, A.; Nair, A.V.; Codega, P.; Torre, V.; Carloni, P. Structural basis of gating of cng channels. FEBS Lett. 2005, 579, 1968–1972. [Google Scholar] [CrossRef] [PubMed]
- Matulef, K.; Zagotta, W.N. Cyclic nucleotide-gated ion channels. Annu. Rev. Cell Dev. Biol. 2003, 19, 23–44. [Google Scholar] [CrossRef] [PubMed]
- Frings, S.; Seifert, R.; Godde, M.; Kaupp, U.B. Profoundly different calcium permeation and blockage determine the specific function of distinct cyclic nucleotide-gated channels. Neuron 1995, 15, 169–179. [Google Scholar] [CrossRef]
- Dzeja, C.; Hagen, V.; Kaupp, U.B.; Frings, S. Ca2+ permeation in cyclic nucleotide-gated channels. EMBO J. 1999, 18, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.; Reisert, J.; Frings, S. Regulation of cyclic nucleotide-gated channels. Curr. Opin. Neurobiol. 2005, 15, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Pifferi, S.; Boccaccio, A.; Menini, A. Cyclic nucleotide-gated ion channels in sensory transduction. FEBS Lett. 2006, 580, 2853–2859. [Google Scholar] [CrossRef] [PubMed]
- Weitz, D.; Zoche, M.; Muller, F.; Beyermann, M.; Korschen, H.G.; Kaupp, U.B.; Koch, K.W. Calmodulin controls the rod photoreceptor cng channel through an unconventional binding site in the n-terminus of the beta-subunit. EMBO J. 1998, 17, 2273–2284. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.; Bonigk, W.; Yau, K.W.; Frings, S. Calmodulin permanently associates with rat olfactory cng channels under native conditions. Nat. Neurosci. 2004, 7, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Rebrik, T.I.; Botchkina, I.; Arshavsky, V.Y.; Craft, C.M.; Korenbrot, J.I. Cng-modulin: A novel ca-dependent modulator of ligand sensitivity in cone photoreceptor cgmp-gated ion channels. J. Neurosci. 2012, 32, 3142–3153. [Google Scholar] [CrossRef] [PubMed]
- Ahnelt, P.K.; Kolb, H. The mammalian photoreceptor mosaic-adaptive design. Prog. Retin. Eye Res. 2000, 19, 711–777. [Google Scholar] [CrossRef]
- Yokoyama, S. Molecular evolution of vertebrate visual pigments. Prog. Retin. Eye Res. 2000, 19, 385–419. [Google Scholar] [CrossRef]
- Yang, R.B.; Foster, D.C.; Garbers, D.L.; Fulle, H.J. Two membrane forms of guanylyl cyclase found in the eye. Proc. Natl. Acad. Sci. USA 1995, 92, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Pugh, E.N., Jr.; Duda, T.; Sitaramayya, A.; Sharma, R.K. Photoreceptor guanylate cyclases: A review. Biosci. Rep. 1997, 17, 429–473. [Google Scholar] [CrossRef] [PubMed]
- Yau, K.W.; Nakatani, K. Cation selectivity of light-sensitive conductance in retinal rods. Nature 1984, 309, 352–354. [Google Scholar] [CrossRef] [PubMed]
- Hodgkin, A.L.; McNaughton, P.A.; Nunn, B.J. The ionic selectivity and calcium dependence of the light-sensitive pathway in toad rods. J. Physiol. 1985, 358, 447–468. [Google Scholar] [CrossRef] [PubMed]
- Yau, K.W.; Nakatani, K. Light-induced reduction of cytoplasmic free calcium in retinal rod outer segment. Nature 1985, 313, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Schnetkamp, P.P. The slc24 Na+/Ca2+-K+ exchanger family: Vision and beyond. Pflugers Arch. 2004, 447, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Cervetto, L.; Lagnado, L.; Perry, R.J.; Robinson, D.W.; McNaughton, P.A. Extrusion of calcium from rod outer segments is driven by both sodium and potassium gradients. Nature 1989, 337, 740–743. [Google Scholar] [CrossRef] [PubMed]
- Vinberg, F.; Wang, T.; De Maria, A.; Zhao, H.; Bassnett, S.; Chen, J.; Kefalov, V.J. The Na+/Ca2+, K+ exchanger nckx4 is required for efficient cone-mediated vision. eLife 2017, 6, e24550. [Google Scholar] [CrossRef] [PubMed]
- Palczewski, K.; Sokal, I.; Baehr, W. Guanylate cyclase-activating proteins: Structure, function, and diversity. Biochem. Biophys. Res. Commun. 2004, 322, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Korenbrot, J.I. Speed, sensitivity, and stability of the light response in rod and cone photoreceptors: Facts and models. Prog. Retin. Eye Res. 2012, 31, 442–466. [Google Scholar] [CrossRef] [PubMed]
- Ohyama, T.; Hackos, D.H.; Frings, S.; Hagen, V.; Kaupp, U.B.; Korenbrot, J.I. Fraction of the dark current carried by Ca2+ through cgmp-gated ion channels of intact rod and cone photoreceptors. J. Gen. Physiol. 2000, 116, 735–754. [Google Scholar] [CrossRef] [PubMed]
- Bareil, C.; Hamel, C.P.; Delague, V.; Arnaud, B.; Demaille, J.; Claustres, M. Segregation of a mutation in cngb1 encoding the beta-subunit of the rod cgmp-gated channel in a family with autosomal recessive retinitis pigmentosa. Hum. Genet. 2001, 108, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Dryja, T.P.; Finn, J.T.; Peng, Y.W.; McGee, T.L.; Berson, E.L.; Yau, K.W. Mutations in the gene encoding the alpha subunit of the rod cgmp-gated channel in autosomal recessive retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 1995, 92, 10177–10181. [Google Scholar] [CrossRef] [PubMed]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Kennan, A.; Aherne, A.; Humphries, P. Light in retinitis pigmentosa. Trends Genet. 2005, 21, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Kalloniatis, M.; Fletcher, E.L. Retinitis pigmentosa: Understanding the clinical presentation, mechanisms and treatment options. Clin. Exp. Optom. 2004, 87, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhao, K.; Sheng, X.; Li, Y.; Gao, X.; Zhang, X.; Kang, X.; Pan, X.; Liu, Y.; Jiang, C.; et al. Targeted sequencing of 179 genes associated with hereditary retinal dystrophies and 10 candidate genes identifies novel and known mutations in patients with various retinal diseases. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2186–2197. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, S.; Akahori, M.; Sergeev, Y.; Yoshitake, K.; Ikeo, K.; Furuno, M.; Hayashi, T.; Kondo, M.; Ueno, S.; Tsunoda, K.; et al. Whole exome analysis identifies frequent cnga1 mutations in japanese population with autosomal recessive retinitis pigmentosa. PLoS ONE 2014, 9, e108721. [Google Scholar] [CrossRef] [PubMed]
- Paloma, E.; Martinez-Mir, A.; Garcia-Sandoval, B.; Ayuso, C.; Vilageliu, L.; Gonzalez-Duarte, R.; Balcells, S. Novel homozygous mutation in the alpha subunit of the rod cgmp gated channel (cnga1) in two spanish sibs affected with autosomal recessive retinitis pigmentosa. J. Med. Genet. 2002, 39, E66. [Google Scholar] [CrossRef] [PubMed]
- Biel, M.; Michalakis, S. Function and dysfunction of cng channels: Insights from channelopathies and mouse models. Mol. Neurobiol. 2007, 35, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Mallouk, N.; Ildefonse, M.; Pages, F.; Ragno, M.; Bennett, N. Basis for intracellular retention of a human mutant of the retinal rod channel alpha subunit. J. Membr. Biol. 2002, 185, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Kondo, H.; Qin, M.; Mizota, A.; Kondo, M.; Hayashi, H.; Hayashi, K.; Oshima, K.; Tahira, T.; Hayashi, K. A homozygosity-based search for mutations in patients with autosomal recessive retinitis pigmentosa, using microsatellite markers. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4433–4439. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.A.; Clark, G.R.; Alexander, S.; Silvestri, G.; Willoughby, C.E. Molecular diagnosis for heterogeneous genetic diseases with targeted high-throughput DNA sequencing applied to retinitis pigmentosa. J. Med. Genet. 2011, 48, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Becirovic, E.; Nakova, K.; Hammelmann, V.; Hennel, R.; Biel, M.; Michalakis, S. The retinitis pigmentosa mutation c.3444+1g>a in cngb1 results in skipping of exon 32. PLoS ONE 2010, 5, e8969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalakis, S.; Zong, X.; Becirovic, E.; Hammelmann, V.; Wein, T.; Wanner, K.T.; Biel, M. The glutamic acid-rich protein is a gating inhibitor of cyclic nucleotide-gated channels. J. Neurosci. 2011, 31, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Leconte, L.; Barnstable, C.J. Impairment of rod cgmp-gated channel alpha-subunit expression leads to photoreceptor and bipolar cell degeneration. Investig. Ophthalmol. Vis. Sci. 2000, 41, 917–926. [Google Scholar]
- Wiik, A.C.; Ropstad, E.O.; Ekesten, B.; Karlstam, L.; Wade, C.M.; Lingaas, F. Progressive retinal atrophy in shetland sheepdog is associated with a mutation in the cnga1 gene. Anim. Genet. 2015, 46, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Hüttl, S.; Michalakis, S.; Seeliger, M.; Luo, D.G.; Acar, N.; Geiger, H.; Hudl, K.; Mader, R.; Haverkamp, S.; Moser, M.; et al. Impaired channel targeting and retinal degeneration in mice lacking the cyclic nucleotide-gated channel subunit cngb1. J. Neurosci. 2005, 25, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Molday, L.L.; Molday, R.S.; Sarfare, S.S.; Woodruff, M.L.; Fain, G.L.; Kraft, T.W.; Pittler, S.J. Knockout of garps and the beta-subunit of the rod cgmp-gated channel disrupts disk morphogenesis and rod outer segment structural integrity. J. Cell Sci. 2009, 122, 1192–1200. [Google Scholar] [CrossRef] [PubMed]
- Blank, T.; Goldmann, T.; Koch, M.; Amann, L.; Schon, C.; Bonin, M.; Pang, S.; Prinz, M.; Burnet, M.; Wagner, J.E.; et al. Early microglia activation precedes photoreceptor degeneration in a mouse model of cngb1-linked retinitis pigmentosa. Front. Immunol. 2017, 8, 1930. [Google Scholar] [CrossRef] [PubMed]
- Petersen-Jones, S.M.; Occelli, L.M.; Winkler, P.A.; Lee, W.; Sparrow, J.R.; Tsukikawa, M.; Boye, S.L.; Chiodo, V.; Capasso, J.E.; Becirovic, E.; et al. Patients and animal models of cngbeta1-deficient retinitis pigmentosa support gene augmentation approach. J. Clin. Investig. 2018, 128, 190–206. [Google Scholar] [CrossRef] [PubMed]
- Petersen-Jones, S.M.; Winkler, P.A.; Bartoe, J.T.; Venta, P.J.; Ekenstedt, K. Large animal model of autosomal recessive rp due to a cngb1 gene mutation. Investig. Ophthalmol. Vis. Sci. 2013, 2013, 684. [Google Scholar]
- Kohl, S.; Marx, T.; Giddings, I.; Jagle, H.; Jacobson, S.G.; Apfelstedt-Sylla, E.; Zrenner, E.; Sharpe, L.T.; Wissinger, B. Total colourblindness is caused by mutations in the gene encoding the alpha-subunit of the cone photoreceptor cgmp-gated cation channel. Nat. Genet. 1998, 19, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Kohl, S.; Baumann, B.; Broghammer, M.; Jagle, H.; Sieving, P.; Kellner, U.; Spegal, R.; Anastasi, M.; Zrenner, E.; Sharpe, L.T.; et al. Mutations in the cngb3 gene encoding the beta-subunit of the cone photoreceptor cgmp-gated channel are responsible for achromatopsia (achm3) linked to chromosome 8q21. Hum. Mol. Genet. 2000, 9, 2107–2116. [Google Scholar] [CrossRef] [PubMed]
- Sundin, O.H.; Yang, J.M.; Li, Y.; Zhu, D.; Hurd, J.N.; Mitchell, T.N.; Silva, E.D.; Maumenee, I.H. Genetic basis of total colourblindness among the pingelapese islanders. Nat. Genet. 2000, 25, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Deeb, S.S. The molecular basis of variation in human color vision. Clin. Genet. 2005, 67, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Jagla, W.M.; Jagle, H.; Hayashi, T.; Sharpe, L.T.; Deeb, S.S. The molecular basis of dichromatic color vision in males with multiple red and green visual pigment genes. Hum. Mol. Genet. 2002, 11, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Eksandh, L.; Kohl, S.; Wissinger, B. Clinical features of achromatopsia in swedish patients with defined genotypes. Ophthalmic Genet. 2002, 23, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Pokorny, J.; Smith, V.C.; Pinckers, A.J.; Cozijnsen, M. Classification of complete and incomplete autosomal recessive achromatopsia. Graefes Arch. Clin. Exp. Ophthalmol. 1982, 219, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Michalakis, S.; Schön, C.; Becirovic, E.; Biel, M. Gene therapy for achromatopsia. J. Gene Med. 2017, 19. [Google Scholar] [CrossRef] [PubMed]
- Aboshiha, J.; Dubis, A.M.; Carroll, J.; Hardcastle, A.J.; Michaelides, M. The cone dysfunction syndromes. Br. J. Ophthalmol. 2016, 100, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Poloschek, C.M.; Kohl, S. [achromatopsia]. Ophthalmologe 2010, 107, 571–580; quiz 581–572. [Google Scholar] [CrossRef] [PubMed]
- Zobor, D.; Zobor, G.; Kohl, S. Achromatopsia: On the doorstep of a possible therapy. Ophthalmic Res. 2015, 54, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Remmer, M.H.; Rastogi, N.; Ranka, M.P.; Ceisler, E.J. Achromatopsia: A review. Curr. Opin. Ophthalmol. 2015, 26, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Kohl, S.; Varsanyi, B.; Antunes, G.A.; Baumann, B.; Hoyng, C.B.; Jagle, H.; Rosenberg, T.; Kellner, U.; Lorenz, B.; Salati, R.; et al. Cngb3 mutations account for 50% of all cases with autosomal recessive achromatopsia. Eur. J. Hum. Genet. 2005, 13, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Sacks, O.W. The Island of the Colourblind; Alfred A. Knopf: New York, NY, USA, 1997. [Google Scholar]
- Biel, M.; Seeliger, M.; Pfeifer, A.; Kohler, K.; Gerstner, A.; Ludwig, A.; Jaissle, G.; Fauser, S.; Zrenner, E.; Hofmann, F. Selective loss of cone function in mice lacking the cyclic nucleotide-gated channel cng3. Proc. Natl. Acad. Sci. USA 1999, 96, 7553–7557. [Google Scholar] [CrossRef] [PubMed]
- Michalakis, S.; Geiger, H.; Haverkamp, S.; Hofmann, F.; Gerstner, A.; Biel, M. Impaired opsin targeting and cone photoreceptor migration in the retina of mice lacking the cyclic nucleotide-gated channel cnga3. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1516–1524. [Google Scholar] [CrossRef] [PubMed]
- Michalakis, S.; Mühlfriedel, R.; Tanimoto, N.; Krishnamoorthy, V.; Koch, S.; Fischer, M.D.; Becirovic, E.; Bai, L.; Huber, G.; Beck, S.C.; et al. Restoration of cone vision in the cnga3−/− mouse model of congenital complete lack of cone photoreceptor function. Mol. Ther. 2010, 18, 2057–2063. [Google Scholar] [CrossRef] [PubMed]
- Michalakis, S.; Xu, J.; Biel, M.; Ding, X.Q. Detection of cgmp in the degenerating retina. Methods Mol. Biol. 2013, 1020, 235–245. [Google Scholar] [PubMed]
- Pang, J.J.; Deng, W.T.; Dai, X.; Lei, B.; Everhart, D.; Umino, Y.; Li, J.; Zhang, K.; Mao, S.; Boye, S.L.; et al. Aav-mediated cone rescue in a naturally occurring mouse model of cnga3-achromatopsia. PLoS ONE 2012, 7, e35250. [Google Scholar] [CrossRef] [PubMed]
- Reicher, S.; Seroussi, E.; Gootwine, E. A mutation in gene cnga3 is associated with day blindness in sheep. Genomics 2010, 95, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Shamir, M.H.; Ofri, R.; Bor, A.; Brenner, O.; Reicher, S.; Obolensky, A.; Averbukh, E.; Banin, E.; Gootwine, E. A novel day blindness in sheep: Epidemiological, behavioural, electrophysiological and histopathological studies. Vet. J. 2010, 185, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Dutrow, E.V.; Miyadera, K.; Delemotte, L.; MacDermaid, C.M.; Reinstein, S.L.; Crumley, W.R.; Dixon, C.J.; Casal, M.L.; Klein, M.L.; et al. Canine cnga3 gene mutations provide novel insights into human achromatopsia-associated channelopathies and treatment. PLoS ONE 2015, 10, e0138943. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.Q.; Harry, C.S.; Umino, Y.; Matveev, A.V.; Fliesler, S.J.; Barlow, R.B. Impaired cone function and cone degeneration resulting from cngb3 deficiency: Down-regulation of cnga3 biosynthesis as a potential mechanism. Hum. Mol. Genet. 2009, 18, 4770–4780. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Morris, L.; Fliesler, S.J.; Sherry, D.M.; Ding, X.Q. Early-onset, slow progression of cone photoreceptor dysfunction and degeneration in cng channel subunit cngb3 deficiency. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3557–3566. [Google Scholar] [CrossRef] [PubMed]
- Sidjanin, D.J.; Lowe, J.K.; McElwee, J.L.; Milne, B.S.; Phippen, T.M.; Sargan, D.R.; Aguirre, G.D.; Acland, G.M.; Ostrander, E.A. Canine cngb3 mutations establish cone degeneration as orthologous to the human achromatopsia locus achm3. Hum. Mol. Genet. 2002, 11, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, G.D.; Rubin, L.F. Pathology of hemeralopia in the alaskan malamute dog. Investig. Ophthalmol. 1974, 13, 231–235. [Google Scholar]
- Aguirre, G.D.; Rubin, L.F. The electroretinogram in dogs with inherited cone degeneration. Investig. Ophthalmol. 1975, 14, 840–847. [Google Scholar]
- Rubin, L.F. Hemeralopia in alaskan malamute pups. J. Am. Vet. Med. Assoc. 1971, 158, 1699–1701. [Google Scholar] [PubMed]
- Rubin, L.F. Clinical features of hemeralopia in the adult alaskan malamute. J. Am. Vet. Med. Assoc. 1971, 158, 1696–1698. [Google Scholar] [PubMed]
- Winkler, P.A.; Ekenstedt, K.J.; Occelli, L.M.; Frattaroli, A.V.; Bartoe, J.T.; Venta, P.J.; Petersen-Jones, S.M. A large animal model for cngb1 autosomal recessive retinitis pigmentosa. PLoS ONE 2013, 8, e72229. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Sothilingam, V.; Garcia Garrido, M.; Tanimoto, N.; Becirovic, E.; Koch, F.; Seide, C.; Beck, S.C.; Seeliger, M.W.; Biel, M.; et al. Gene therapy restores vision and delays degeneration in the cngb1(−/−) mouse model of retinitis pigmentosa. Hum. Mol. Genet. 2012, 21, 4486–4496. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.J.; Alexander, J.; Lei, B.; Deng, W.; Zhang, K.; Li, Q.; Chang, B.; Hauswirth, W.W. Achromatopsia as a potential candidate for gene therapy. Adv. Exp. Med. Biol. 2010, 664, 639–646. [Google Scholar] [PubMed]
- Banin, E.; Gootwine, E.; Obolensky, A.; Ezra-Elia, R.; Ejzenberg, A.; Zelinger, L.; Honig, H.; Rosov, A.; Yamin, E.; Sharon, D.; et al. Gene augmentation therapy restores retinal function and visual behavior in a sheep model of cnga3 achromatopsia. Mol. Ther. 2015, 23, 1423–1433. [Google Scholar] [CrossRef] [PubMed]
- Mühlfriedel, R.; Tanimoto, N.; Schon, C.; Sothilingam, V.; Garcia Garrido, M.; Beck, S.C.; Huber, G.; Biel, M.; Seeliger, M.W.; Michalakis, S. Aav-mediated gene supplementation therapy in achromatopsia type 2: Preclinical data on therapeutic time window and long-term effects. Front. Neurosci. 2017, 11, 292. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Tao, Y.; Deng, W.T.; Zhu, P.; Li, J.; Dai, X.; Zhang, Y.; Shi, W.; Liu, X.; Chiodo, V.A.; et al. Vitreal delivery of aav vectored cnga3 restores cone function in cnga3−/−/nrl−/− mice, an all-cone model of cnga3 achromatopsia. Hum. Mol. Genet. 2015, 24, 3699–3707. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; He, Y.; Zhang, H.; Zhang, Y.; Liu, Y.; Wang, M.; Chen, H.; Pang, J.J. Long-term retinal cone rescue using a capsid mutant aav8 vector in a mouse model of cnga3-achromatopsia. PLoS ONE 2017, 12, e0188032. [Google Scholar] [CrossRef] [PubMed]
- Gootwine, E.; Abu-Siam, M.; Obolensky, A.; Rosov, A.; Honig, H.; Nitzan, T.; Shirak, A.; Ezra-Elia, R.; Yamin, E.; Banin, E.; et al. Gene augmentation therapy for a missense substitution in the cgmp-binding domain of ovine cnga3 gene restores vision in day-blind sheep. Investig. Ophthalmol. Vis. Sci. 2017, 58, 1577–1584. [Google Scholar] [CrossRef] [PubMed]
- Reichel, F.F.; Dauletbekov, D.L.; Klein, R.; Peters, T.; Ochakovski, G.A.; Seitz, I.P.; Wilhelm, B.; Ueffing, M.; Biel, M.; Wissinger, B.; et al. Aav8 can induce innate and adaptive immune response in the primate eye. Mol. Ther. 2017, 25, 2648–2660. [Google Scholar] [CrossRef] [PubMed]
- Gootwine, E.; Ofri, R.; Banin, E.; Obolensky, A.; Averbukh, E.; Ezra-Elia, R.; Ross, M.; Honig, H.; Rosov, A.; Yamin, E.; et al. Safety and efficacy evaluation of raav2tyf-pr1.7-hcnga3 vector delivered by subretinal injection in cnga3 mutant achromatopsia sheep. Hum. Gene Ther. Clin. Dev. 2017, 28, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Ochakovski, G.A.; Peters, T.; Michalakis, S.; Wilhelm, B.; Wissinger, B.; Biel, M.; Bartz-Schmidt, K.U.; Fischer, M.D.; Consortium, R.-C. Subretinal injection for gene therapy does not cause clinically significant outer nuclear layer thinning in normal primate foveae. Investig. Ophthalmol. Vis. Sci. 2017, 58, 4155–4160. [Google Scholar] [CrossRef] [PubMed]
- Seitz, I.P.; Michalakis, S.; Wilhelm, B.; Reichel, F.F.; Ochakovski, G.A.; Zrenner, E.; Ueffing, M.; Biel, M.; Wissinger, B.; Bartz-Schmidt, K.U.; et al. Superior retinal gene transfer and biodistribution profile of subretinal versus intravitreal delivery of aav8 in nonhuman primates. Investig. Ophthalmol. Vis. Sci. 2017, 58, 5792–5801. [Google Scholar] [CrossRef] [PubMed]
- Komaromy, A.M.; Alexander, J.J.; Rowlan, J.S.; Garcia, M.M.; Chiodo, V.A.; Kaya, A.; Tanaka, J.C.; Acland, G.M.; Hauswirth, W.W.; Aguirre, G.D. Gene therapy rescues cone function in congenital achromatopsia. Hum. Mol. Genet. 2010, 19, 2581–2593. [Google Scholar] [CrossRef] [PubMed]
- Komaromy, A.M.; Rowlan, J.S.; Corr, A.T.; Reinstein, S.L.; Boye, S.L.; Cooper, A.E.; Gonzalez, A.; Levy, B.; Wen, R.; Hauswirth, W.W.; et al. Transient photoreceptor deconstruction by cntf enhances raav-mediated cone functional rescue in late stage cngb3-achromatopsia. Mol. Ther. 2013, 21, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, L.S.; Xu, J.; Pearson, R.A.; Smith, A.J.; Bainbridge, J.W.; Morris, L.M.; Fliesler, S.J.; Ding, X.Q.; Ali, R.R. Long-term and age-dependent restoration of visual function in a mouse model of cngb3-associated achromatopsia following gene therapy. Hum. Mol. Genet. 2011, 20, 3161–3175. [Google Scholar] [CrossRef] [PubMed]
- Ye, G.J.; Komaromy, A.M.; Zeiss, C.; Calcedo, R.; Harman, C.D.; Koehl, K.L.; Stewart, G.A.; Iwabe, S.; Chiodo, V.A.; Hauswirth, W.W.; et al. Safety and efficacy of aav5 vectors expressing human or canine cngb3 in cngb3-mutant dogs. Hum. Gene Ther. Clin. Dev. 2017, 28, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Ye, G.J.; Budzynski, E.; Sonnentag, P.; Nork, T.M.; Miller, P.E.; Sharma, A.K.; Ver Hoeve, J.N.; Smith, L.; Arndt, T.; Calcedo, R.; et al. Safety and biodistribution evaluation in cynomolgus macaques of raav2tyf-pr1.7-hcngb3, a recombinant aav vector for treatment of achromatopsia. Hum. Gene Ther. Clin. Dev. 2016, 27, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Ye, G.J.; Budzynski, E.; Sonnentag, P.; Nork, T.M.; Miller, P.E.; McPherson, L.; Ver Hoeve, J.N.; Smith, L.; Arndt, T.; Mandapati, S.; et al. Safety and biodistribution evaluation in cngb3-deficient mice of raav2tyf-pr1.7-hcngb3, a recombinant aav vector for treatment of achromatopsia. Hum. Gene Ther. Clin. Dev. 2016, 27, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Kohl, S.; Hamel, C. Clinical utility gene card for: Achromatopsia—Update 2013. Eur. J. Hum. Genet. 2013, 21. [Google Scholar] [CrossRef] [PubMed]
- Daya, S.; Berns, K.I. Gene therapy using adeno-associated virus vectors. Clin. Microbiol. Rev. 2008, 21, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Schön, C.; Becirovic, E.; Biel, M.; Michalakis, S. Design and development of aav-based gene supplementation therapies for achromatopsia and retinitis pigmentosa. Methods Mol. Biol. 2018, 1715, 33–46. [Google Scholar] [PubMed]
- Schön, C.; Biel, M.; Michalakis, S. Retinal gene delivery by adeno-associated virus (aav) vectors: Strategies and applications. Eur. J. Pharm. Biopharm. 2015, 95, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Lisowski, L.; Tay, S.S.; Alexander, I.E. Adeno-associated virus serotypes for gene therapeutics. Curr. Opin. Pharmacol. 2015, 24, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Trapani, I.; Puppo, A.; Auricchio, A. Vector platforms for gene therapy of inherited retinopathies. Prog. Retin. Eye Res. 2014, 43, 108–128. [Google Scholar] [CrossRef] [PubMed]
- Willett, K.; Bennett, J. Immunology of aav-mediated gene transfer in the eye. Front. Immunol. 2013, 4, 261. [Google Scholar] [CrossRef] [PubMed]
- Dismuke, D.J.; Tenenbaum, L.; Samulski, R.J. Biosafety of recombinant adeno-associated virus vectors. Curr. Gene Ther. 2013, 13, 434–452. [Google Scholar] [CrossRef] [PubMed]
- Mingozzi, F.; High, K.A. Therapeutic in vivo gene transfer for genetic disease using aav: Progress and challenges. Nat. Rev. Genet. 2011, 12, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Kaeppel, C.; Beattie, S.G.; Fronza, R.; van Logtenstein, R.; Salmon, F.; Schmidt, S.; Wolf, S.; Nowrouzi, A.; Glimm, H.; von Kalle, C.; et al. A largely random aav integration profile after lpld gene therapy. Nat. Med. 2013, 19, 889–891. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Mehta, A.; Wang, G.; Hauswirth, W.W.; Chiodo, V.; Boye, S.L.; Guy, J. Next-generation sequencing of mitochondrial targeted aav transfer of human nd4 in mice. Mol. Vis. 2013, 19, 1482–1491. [Google Scholar] [PubMed]
- Fischer, M.D.; Wilhelm, B.; Michalakis, S.; Zobor, D.; Kohl, S.; Seeliger, M.; Zrenner, E.; Ueffing, M.; Wissinger, B.; Biel, M.; et al. Successful delivery of raav8.Cnga3 in a patient with cnga3 achromatopsia. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5207. [Google Scholar]
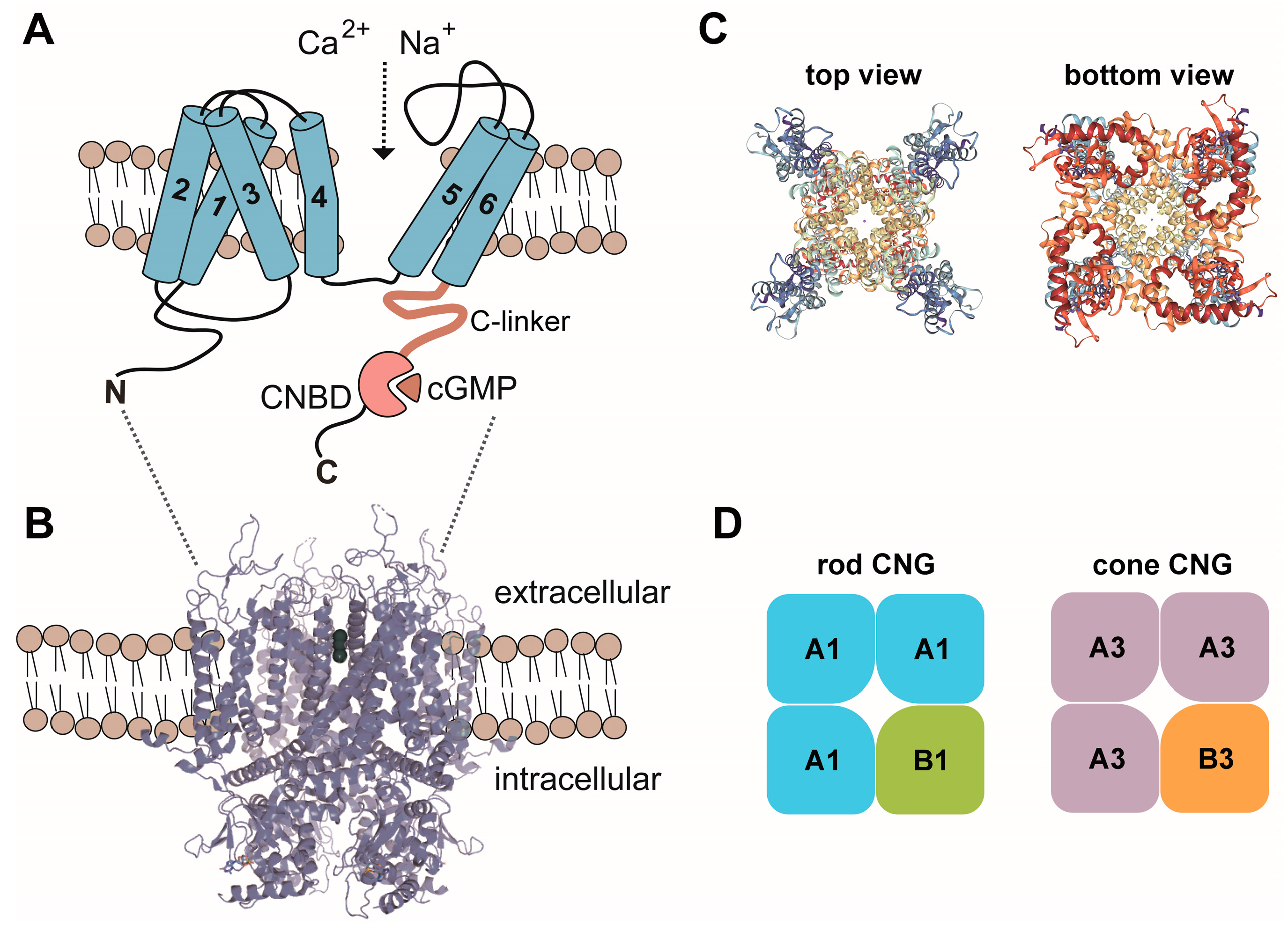
Figure 1.
(A) Membrane topology of CNG channel subunits. 1–6, transmembrane segment 1–6; C, carboxy-terminus; CNBD, cyclic nucleotide binding domain; N, amino-terminus. (B) Model of the CNG channel complex embedded in the plasma membrane based on the TAX-4 structure. (C) Top and bottom views of the tetrameric TAX-4 C. elegans CNG channel complex. (D) Subunit composition of the CNG channels from rods and cones. A1, CNGA1; A3, CNGA3; B1, CNGB1; B3, CNGB3. Structures in this figure were generated with the RSCB PDB 3D View tool (www.rcsb.org/3d-view/) based on PDB 5H3O.
Figure 1.
(A) Membrane topology of CNG channel subunits. 1–6, transmembrane segment 1–6; C, carboxy-terminus; CNBD, cyclic nucleotide binding domain; N, amino-terminus. (B) Model of the CNG channel complex embedded in the plasma membrane based on the TAX-4 structure. (C) Top and bottom views of the tetrameric TAX-4 C. elegans CNG channel complex. (D) Subunit composition of the CNG channels from rods and cones. A1, CNGA1; A3, CNGA3; B1, CNGB1; B3, CNGB3. Structures in this figure were generated with the RSCB PDB 3D View tool (www.rcsb.org/3d-view/) based on PDB 5H3O.

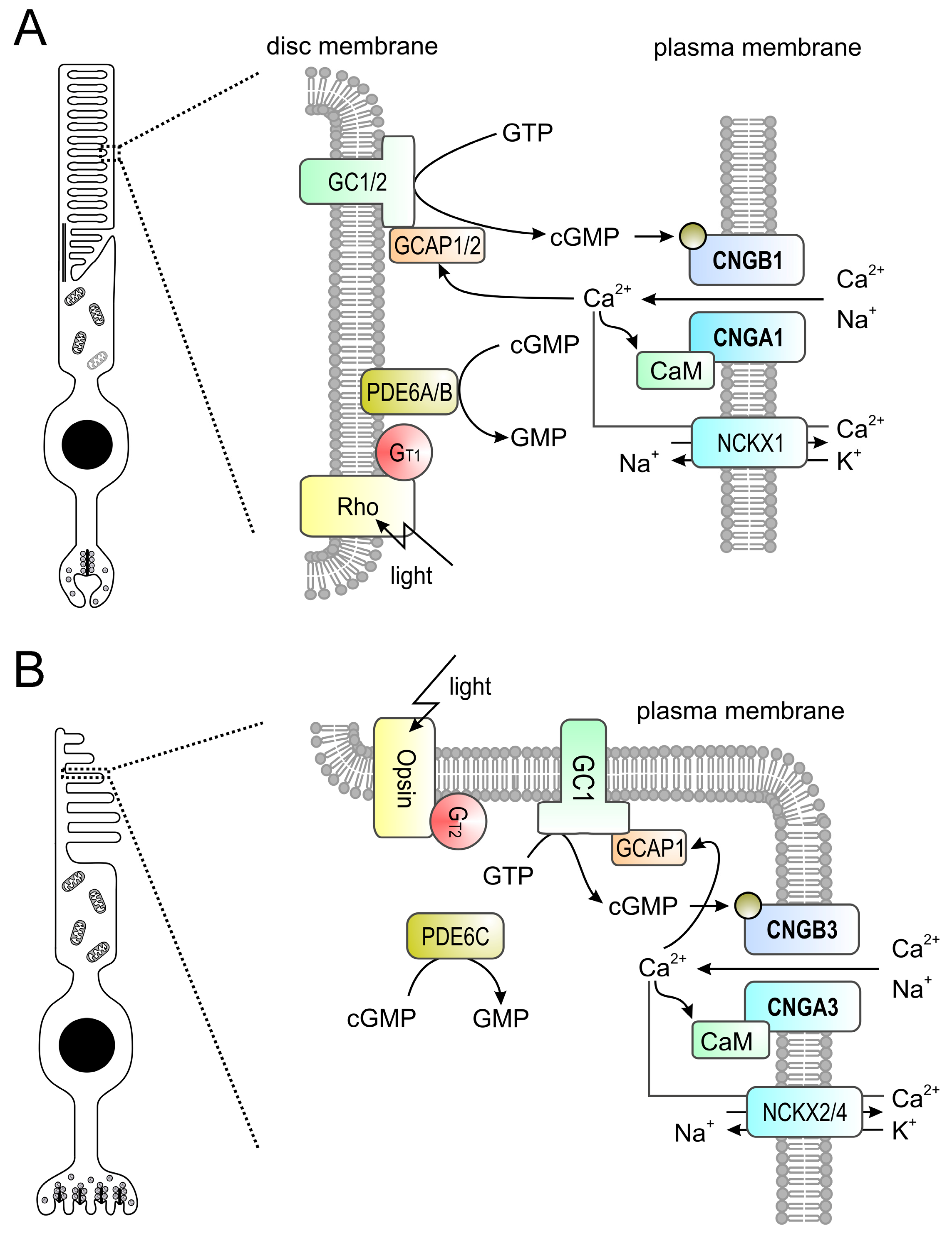
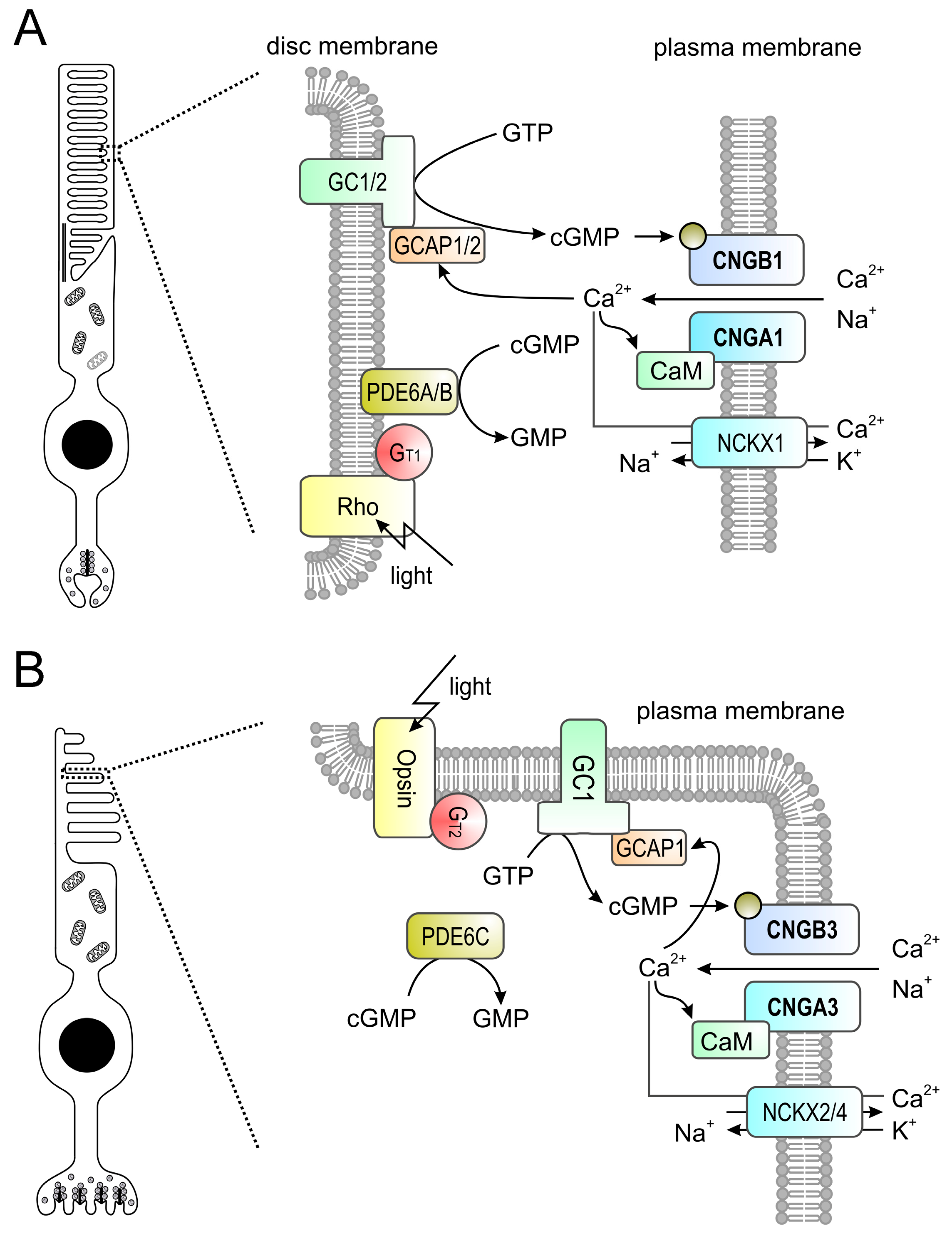
Figure 2.
(A,B) Phototransduction in mouse rod (A) and cone (B) outer segments. The principle of the phototransduction is similar in both cell types. In the dark, the cyclic nucleotide-gated (CNG) channel (CNGA1/B1 in rods and CNGA3/B3 in cones) of the outer membrane is kept open by high concentrations of cyclic guanosine monophosphate (cGMP) produced by retinal guanylyl cyclases (GCs) 1 and 2 (GC1/2 in rods, GC1 only in cones) present in the disc membrane. The resulting influx of Na+ and Ca2+ depolarizes the plasma membrane. Light activates rhodopsin (Rh) which in turn activates transducin (Gt1 in rods, Gt2 in cones) whose alpha subunit activates a phosphodiesterase (PDE6A/B in rods, PDE6C in cones) leading to hydrolysis of cGMP. The drop in the cGMP concentration leads to the closure of the CNG channel yielding to membrane hyperpolarization. Ca2+ is an important regulator of phototransduction. At high concentrations Ca2+ binds to guanylyl cyclase-activating proteins (GCAPs; GCAP1/2 in rods, GCAP1 only in cones) leading to an inhibition of guanylyl cyclases. High levels of Ca2+ also lead to a slight reduction of the cGMP affinity of the CNG channel via CaM-mediated feedback inhibition. Ca2+ is cleared from the outer segment via a Na+-Ca2+-K+-exchanger (NCKX1 in rods, NCKX2/4 in cones). At low Ca2+ levels GCAPs switch to the Ca2+-free form that is an activator of GCs.
Figure 2.
(A,B) Phototransduction in mouse rod (A) and cone (B) outer segments. The principle of the phototransduction is similar in both cell types. In the dark, the cyclic nucleotide-gated (CNG) channel (CNGA1/B1 in rods and CNGA3/B3 in cones) of the outer membrane is kept open by high concentrations of cyclic guanosine monophosphate (cGMP) produced by retinal guanylyl cyclases (GCs) 1 and 2 (GC1/2 in rods, GC1 only in cones) present in the disc membrane. The resulting influx of Na+ and Ca2+ depolarizes the plasma membrane. Light activates rhodopsin (Rh) which in turn activates transducin (Gt1 in rods, Gt2 in cones) whose alpha subunit activates a phosphodiesterase (PDE6A/B in rods, PDE6C in cones) leading to hydrolysis of cGMP. The drop in the cGMP concentration leads to the closure of the CNG channel yielding to membrane hyperpolarization. Ca2+ is an important regulator of phototransduction. At high concentrations Ca2+ binds to guanylyl cyclase-activating proteins (GCAPs; GCAP1/2 in rods, GCAP1 only in cones) leading to an inhibition of guanylyl cyclases. High levels of Ca2+ also lead to a slight reduction of the cGMP affinity of the CNG channel via CaM-mediated feedback inhibition. Ca2+ is cleared from the outer segment via a Na+-Ca2+-K+-exchanger (NCKX1 in rods, NCKX2/4 in cones). At low Ca2+ levels GCAPs switch to the Ca2+-free form that is an activator of GCs.

{kind=link}
{kind=link}
{kind=link}
Table 1.
Overview of retinal CNG genes, associated human diseases, animal models, preclinical and clinical gene therapy studies. ID, www.clinicaltrials.gov identifier. POC, proof-of-concept.
Table 1.
Overview of retinal CNG genes, associated human diseases, animal models, preclinical and clinical gene therapy studies. ID, www.clinicaltrials.gov identifier. POC, proof-of-concept.
| Gene (Cell Type) | Associated Human Disease | Animal Models | POC Studies | Preclinical Safety Studies | Clinical Trials (ID) |
|---|---|---|---|---|---|
| CNGA1 (rods) | Retinitis pigmentosa, RP49 | Cnga1 antisense expressing mice: retinal degeneration [53] Canine model (missense mutation): Progressive retinal atrophy [54] | - | - | - |
| CNGB1 (rods) | Retinitis pigmentosa, RP45 | Cngb1-deficient mice: impaired rod function and retinal degeneration [55,56] Canine model (missense mutation): impaired rod function and retinal degeneration [89] | [58,90] | - | - |
| CNGA3 (cones) | Achromatopsia, ACHM2 | Cnga3-deficient mice: loss of cone function and cone degeneration [74] cpfl5 mouse: loss of cone function and cone cell degeneration [91] Sheep model (missense mutation): loss of cone function and day blindness [80] | [76,78,92,93,94,95,96] | [97,98,99,100] | NCT02610582 NCT02935517 |
| CNGB3 (cones) | Achromatopsia, ACHM1 | Canine models (null-deletion or missense mutation): cone degeneration [84] Cngb3-deficient mice: Impaired cone function and cone degeneration [82] | [101,102,103] | [98,104,105,106] | NCT02599922 NCT03001310 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Michalakis, S.; Becirovic, E.; Biel, M. Retinal Cyclic Nucleotide-Gated Channels: From Pathophysiology to Therapy. Int. J. Mol. Sci. 2018, 19, 749. https://doi.org/10.3390/ijms19030749
AMA Style
Michalakis S, Becirovic E, Biel M. Retinal Cyclic Nucleotide-Gated Channels: From Pathophysiology to Therapy. International Journal of Molecular Sciences. 2018; 19(3):749. https://doi.org/10.3390/ijms19030749
Chicago/Turabian StyleMichalakis, Stylianos, Elvir Becirovic, and Martin Biel. 2018. "Retinal Cyclic Nucleotide-Gated Channels: From Pathophysiology to Therapy" International Journal of Molecular Sciences 19, no. 3: 749. https://doi.org/10.3390/ijms19030749
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.