The In Vitro Effects of Enzymatic Digested Gliadin on the Functionality of the Autophagy Process

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
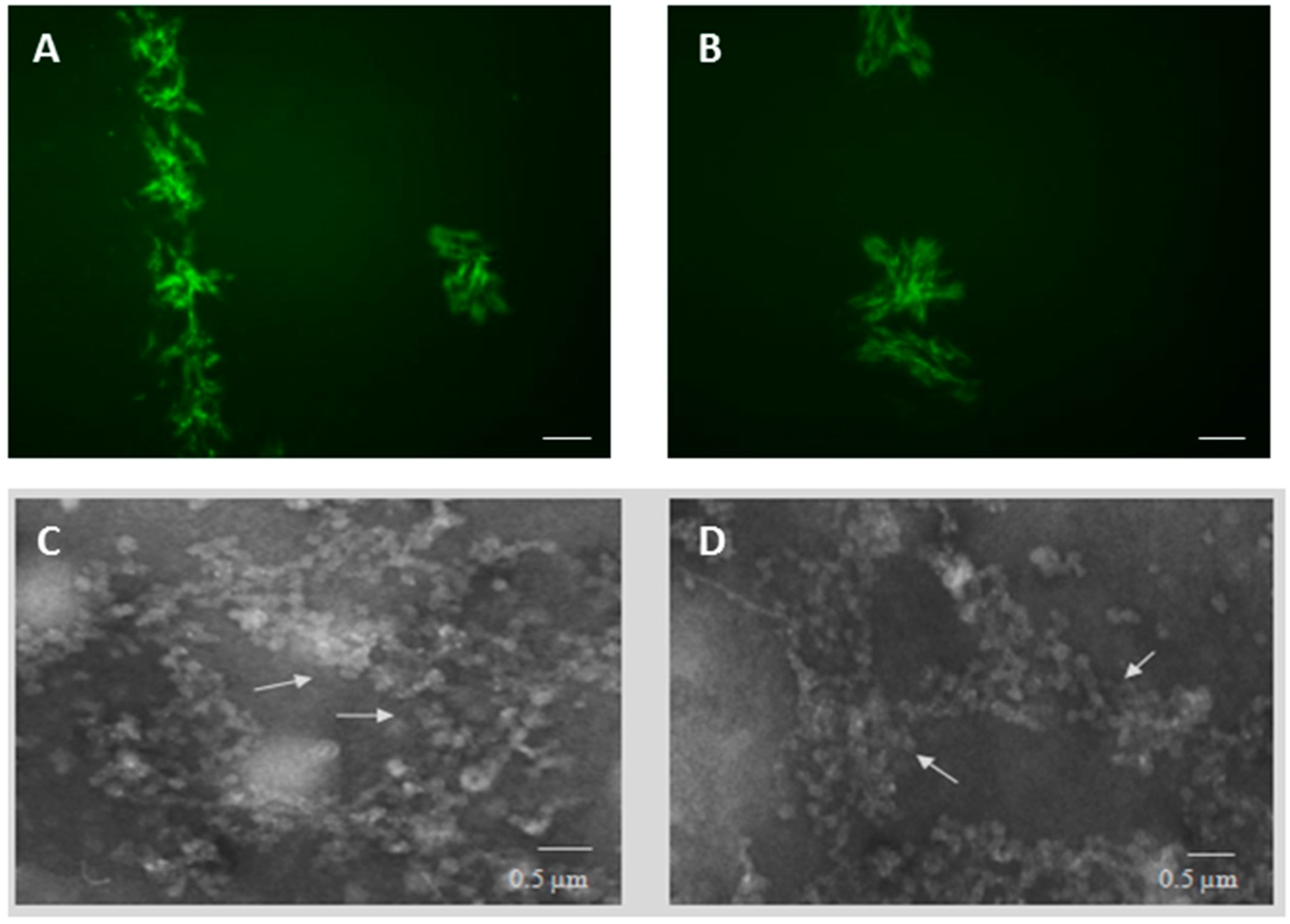
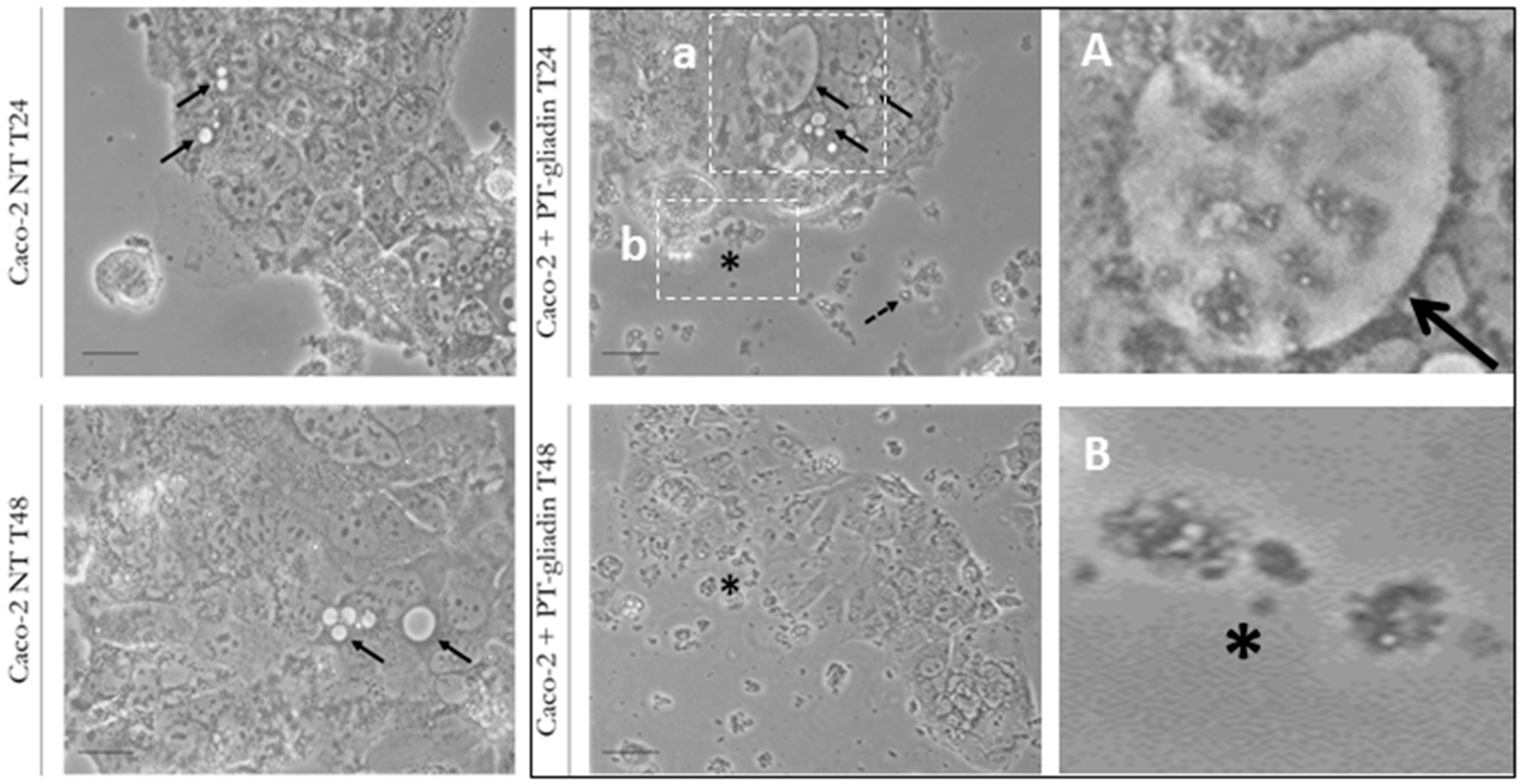
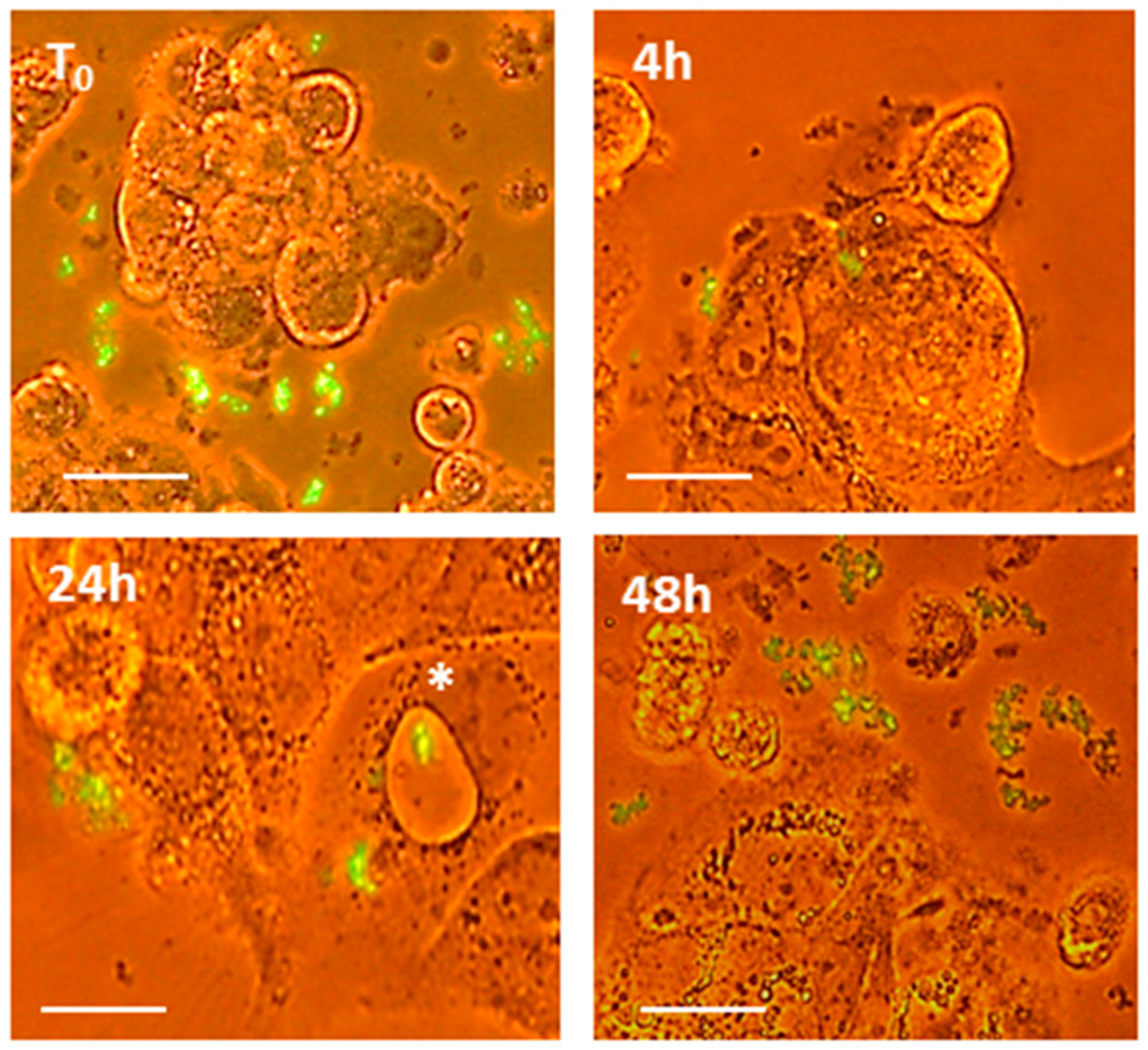
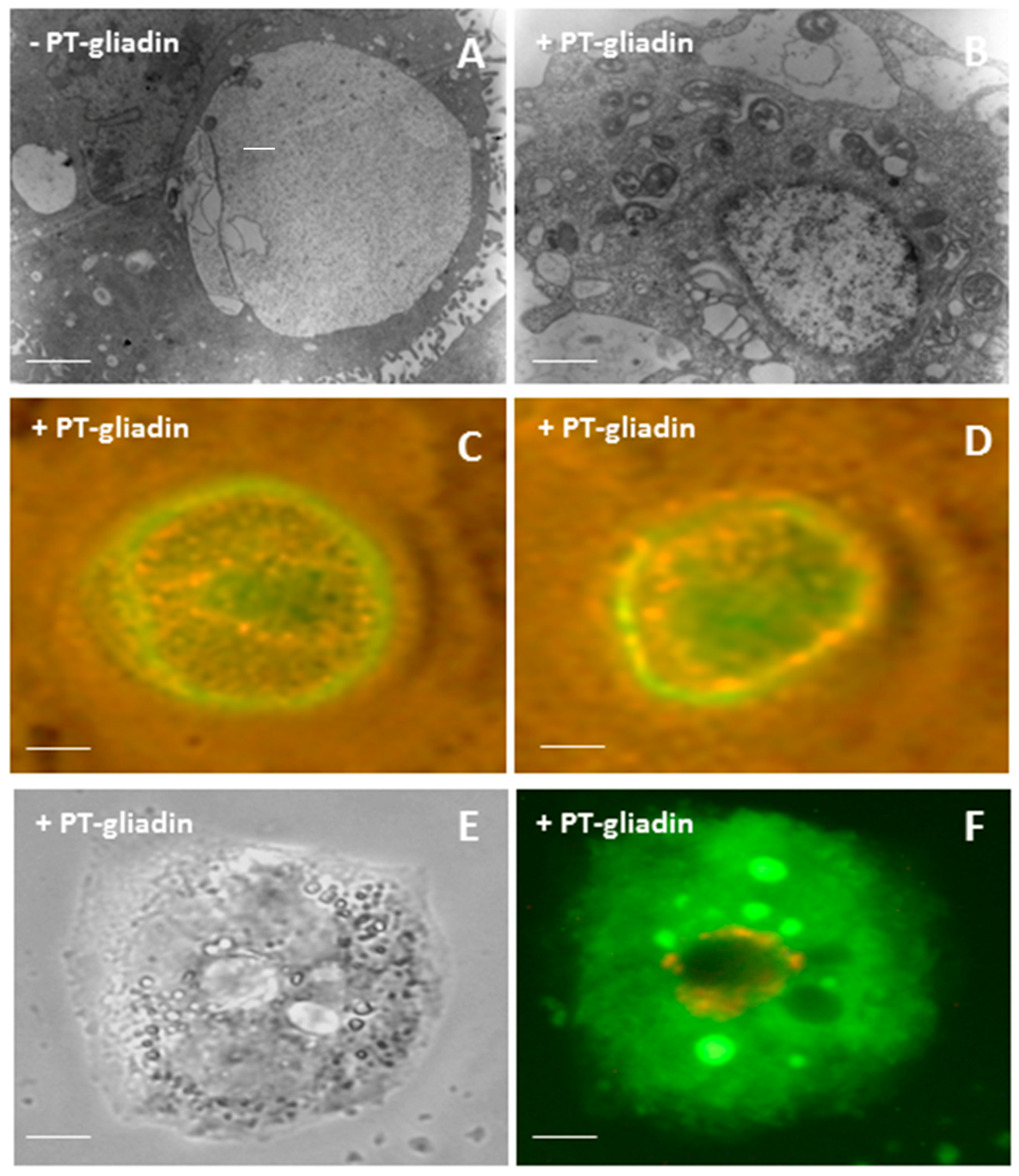
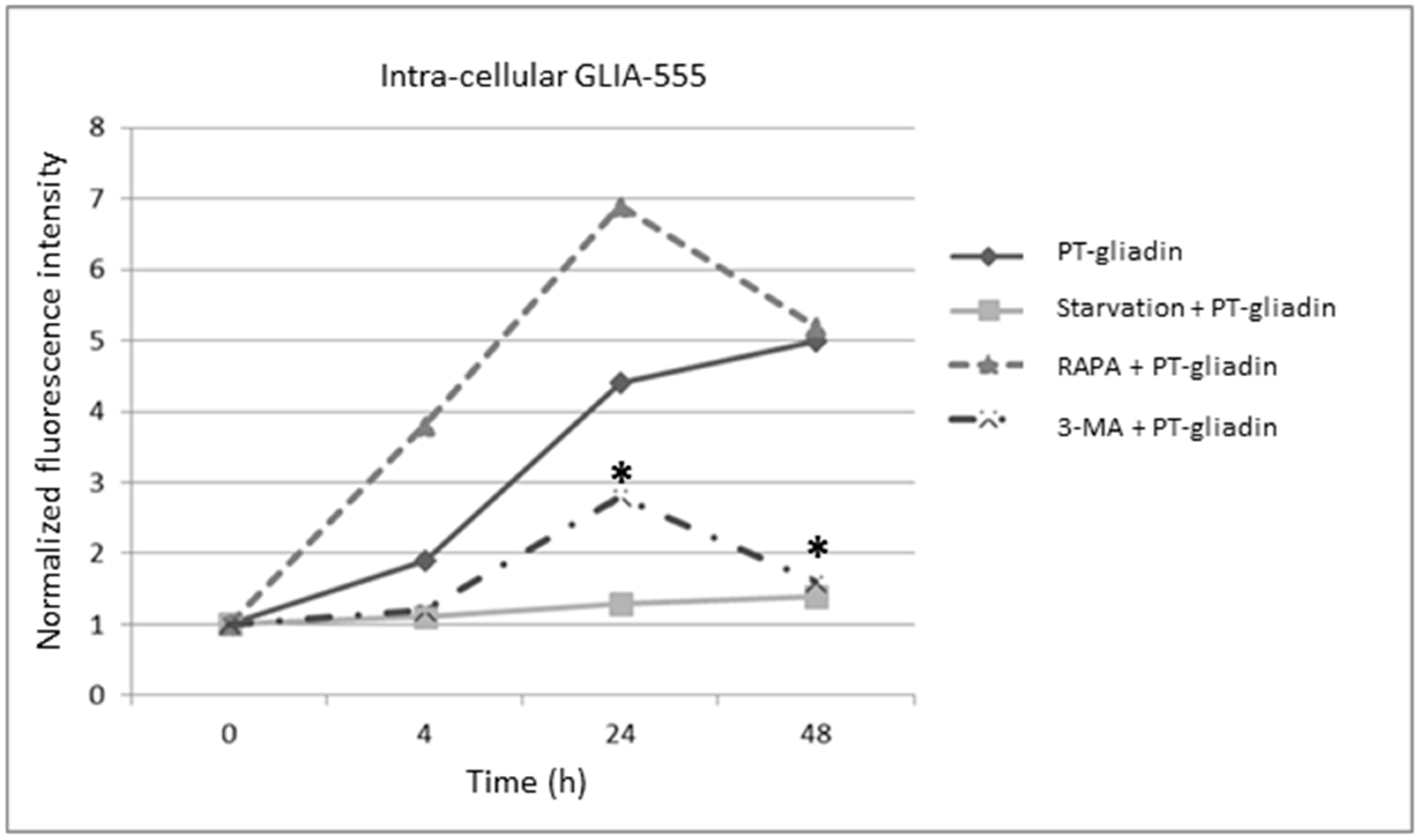
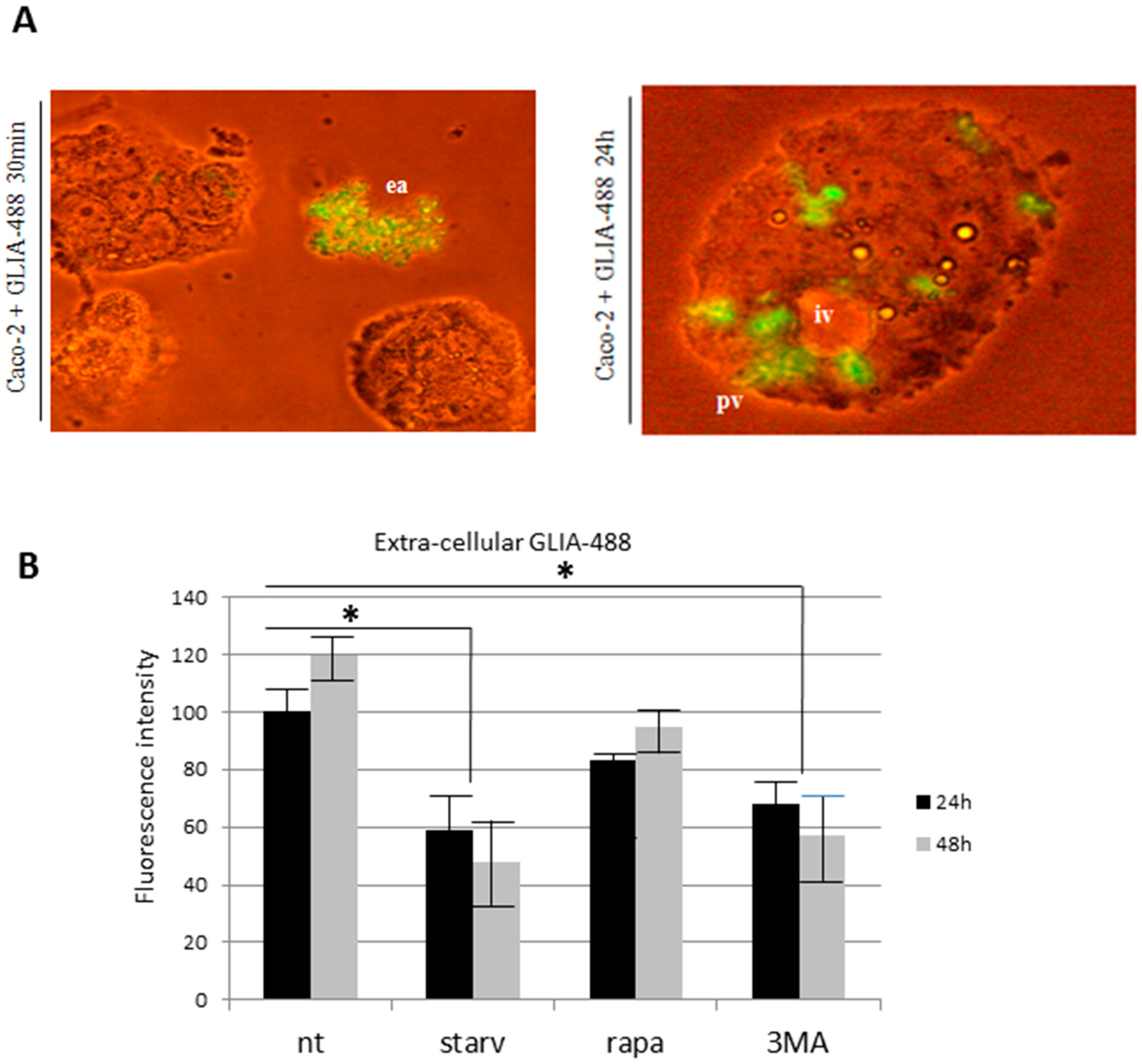
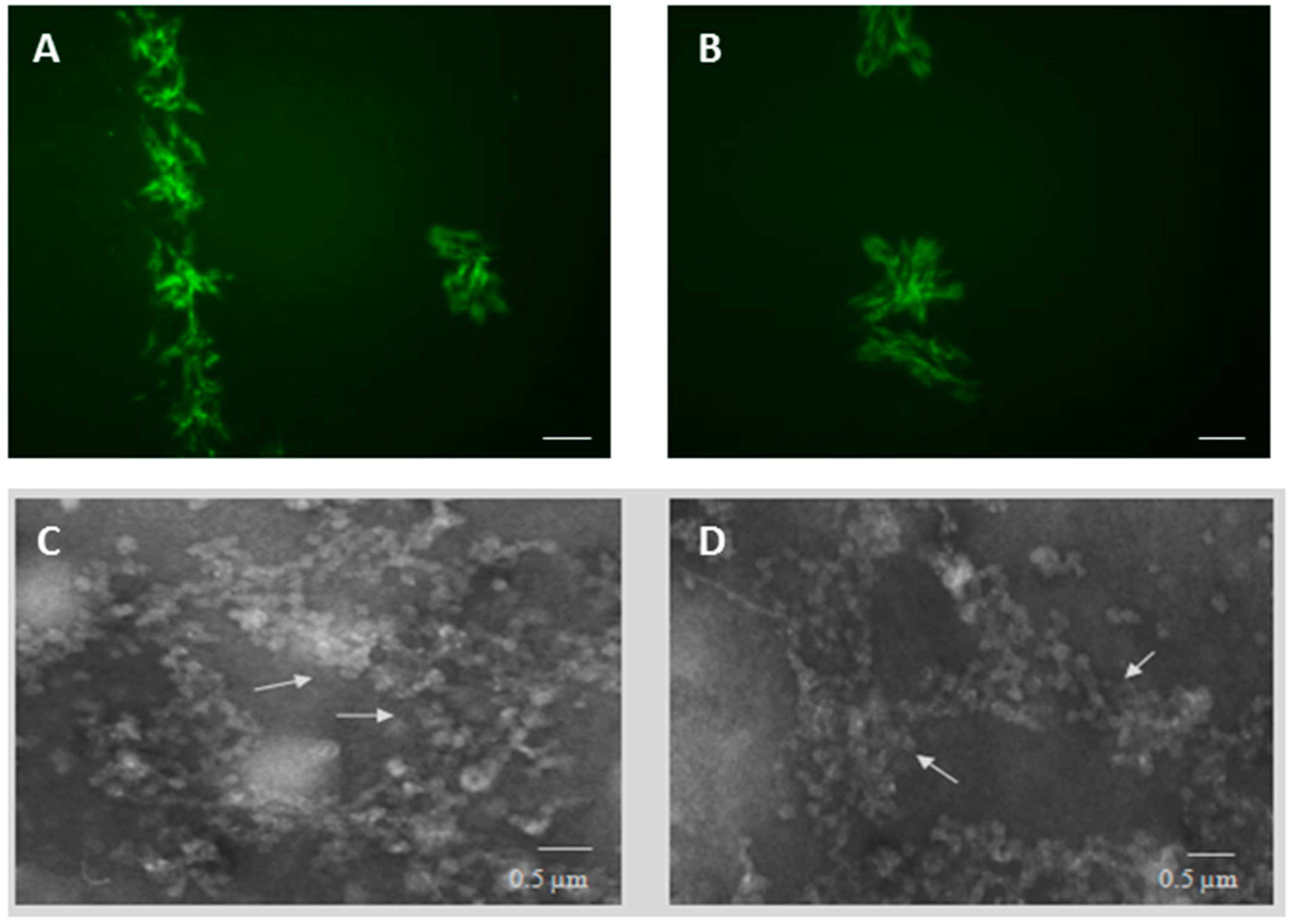
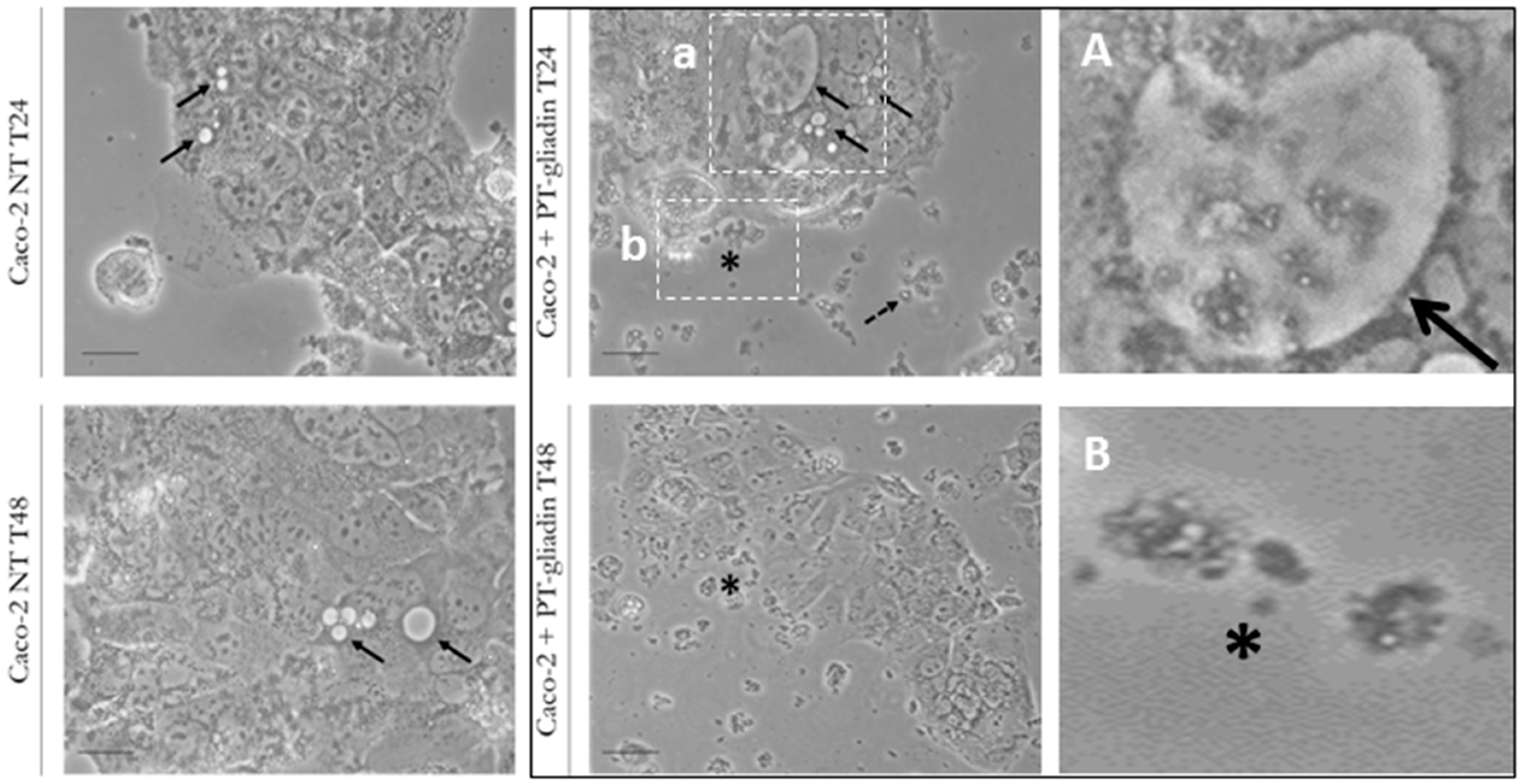
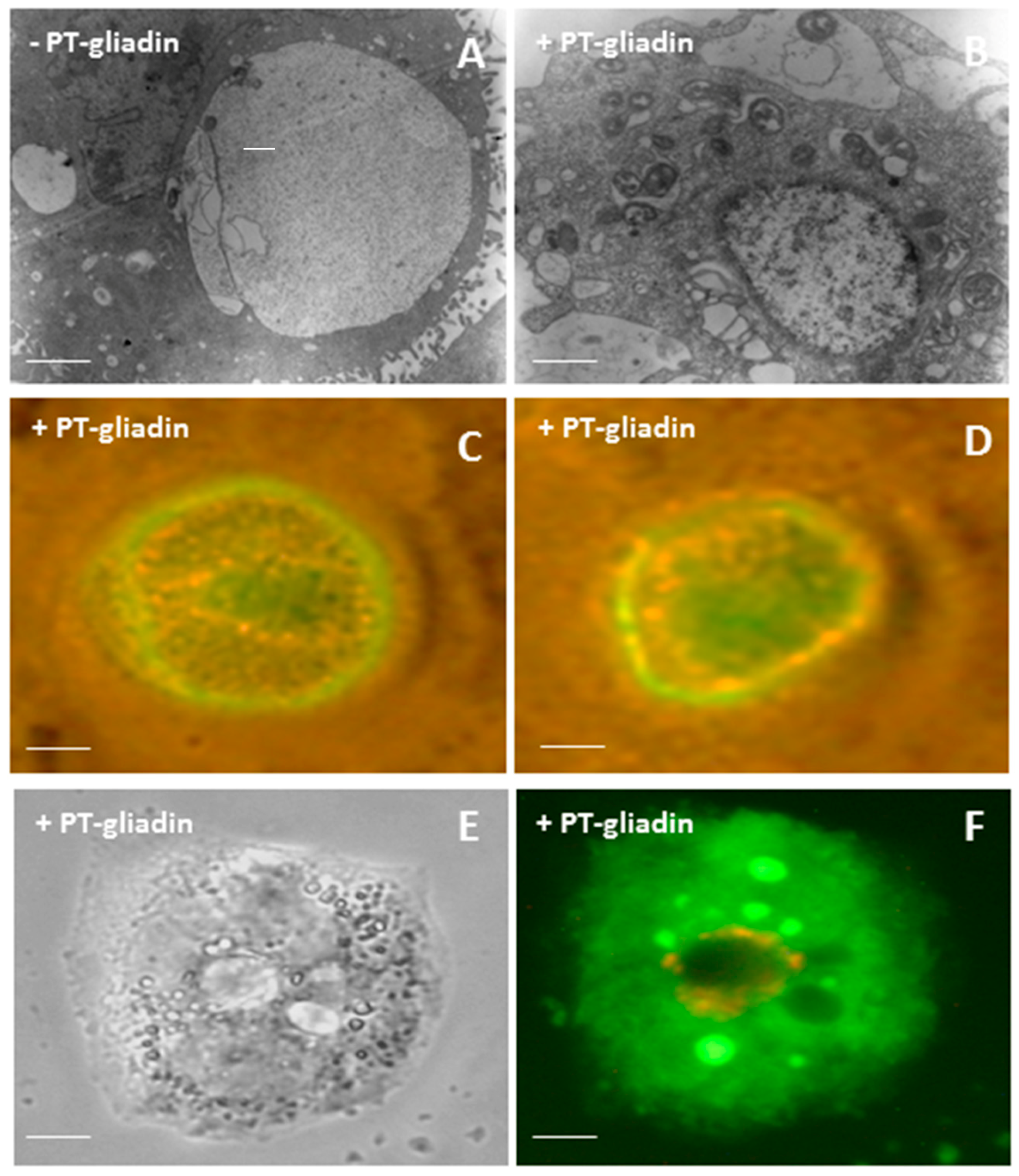
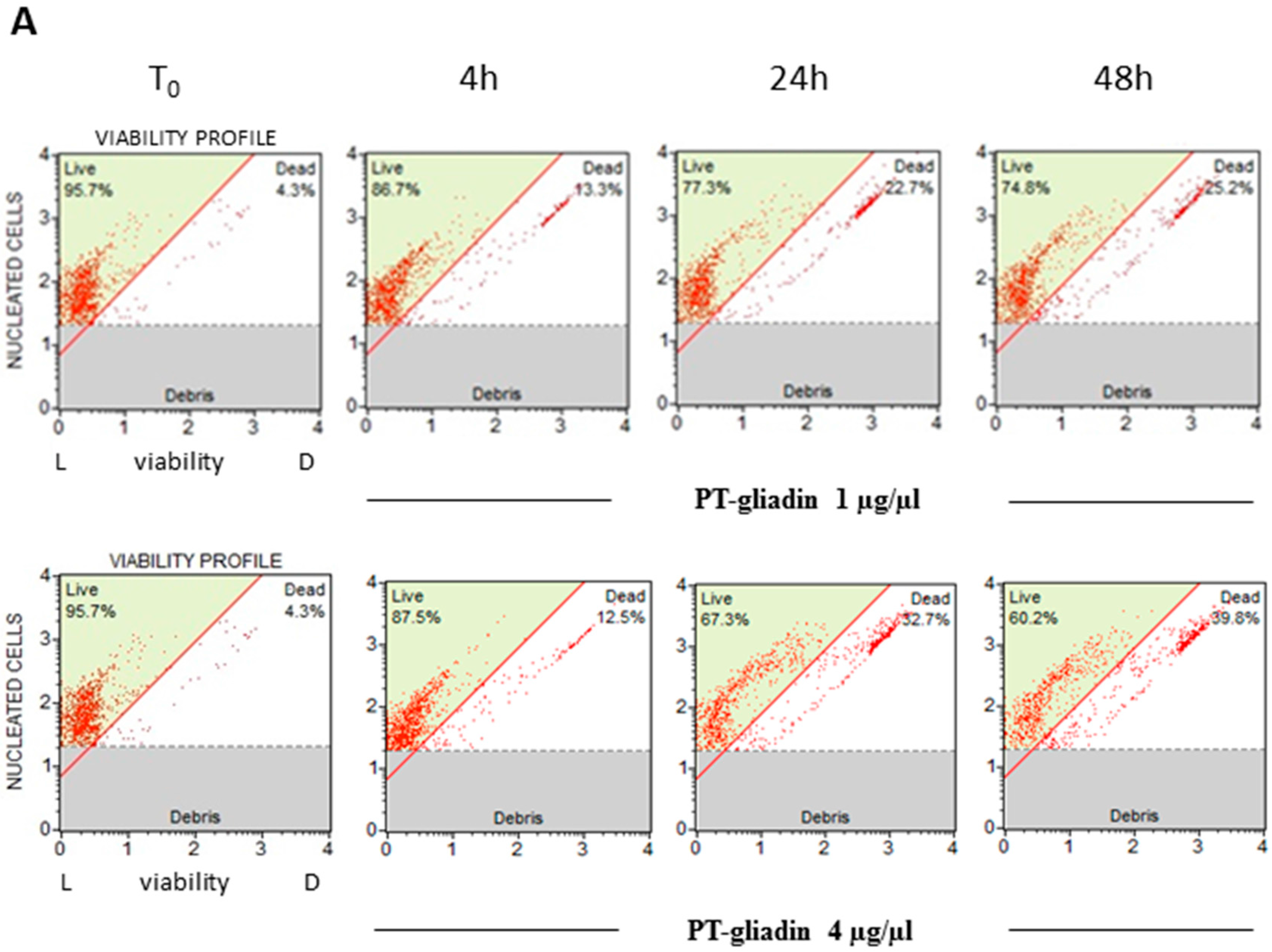
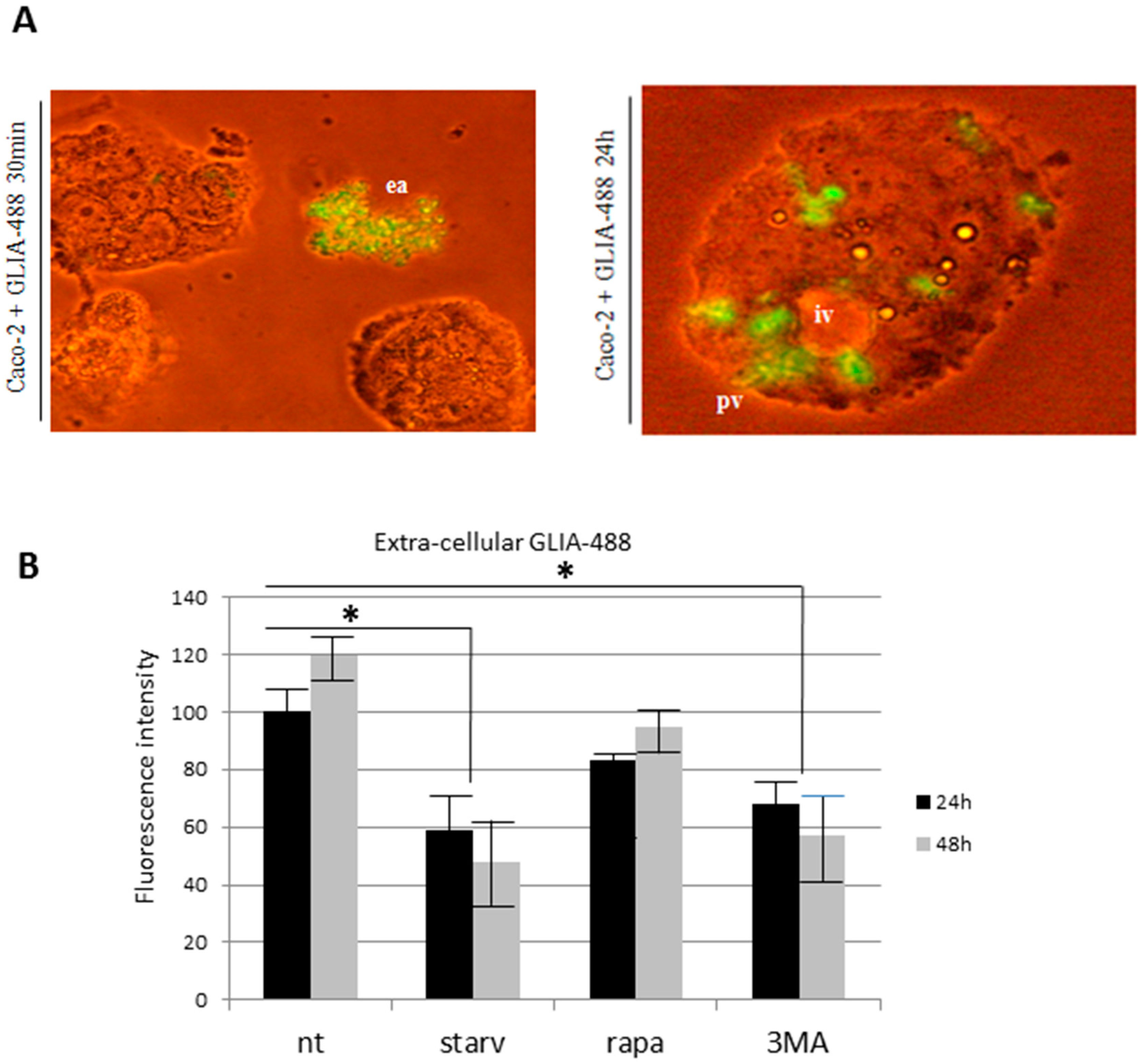
2.1. PT-Gliadin Aggregation and Internalization in Caco-2 Cells
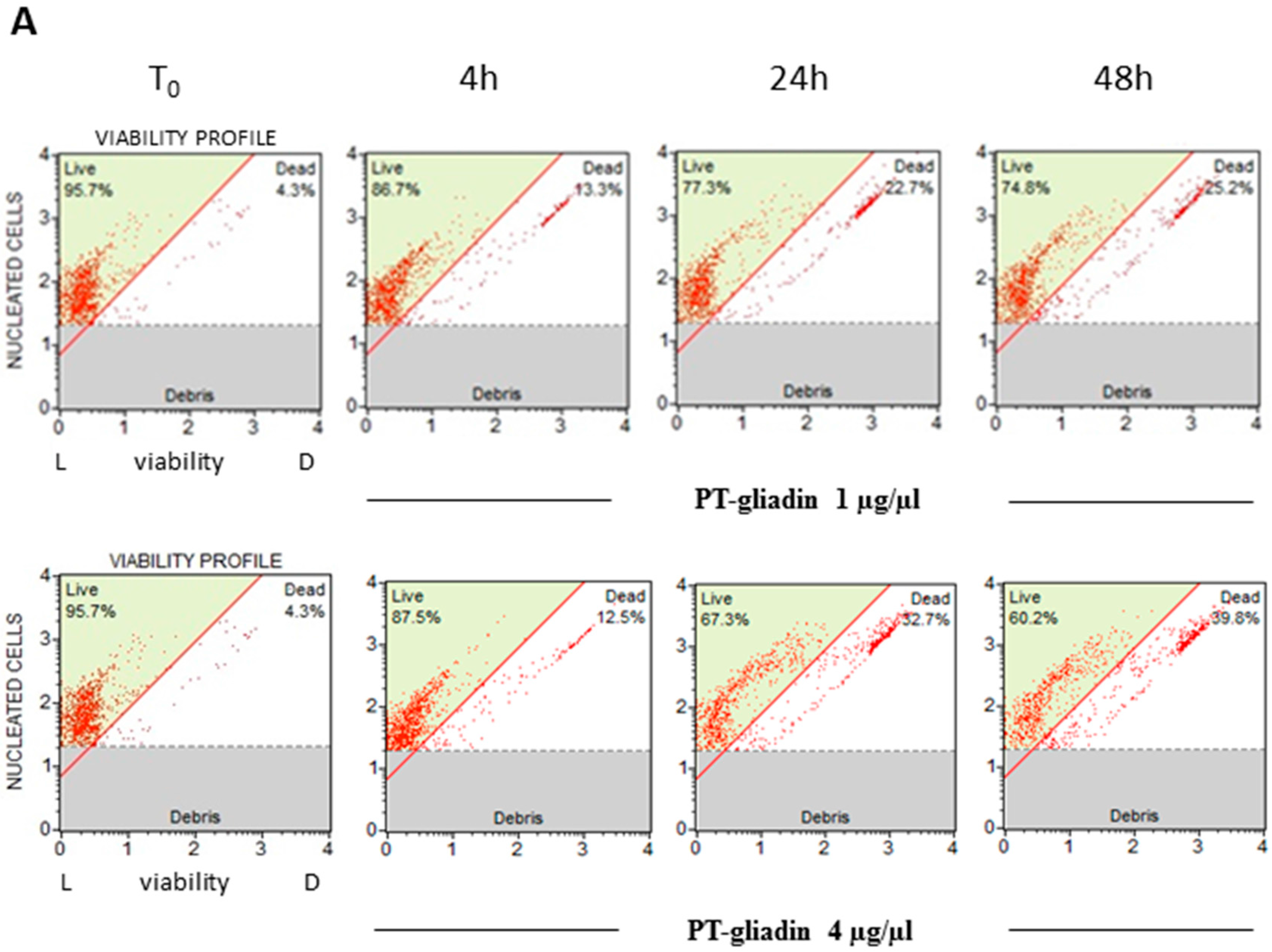
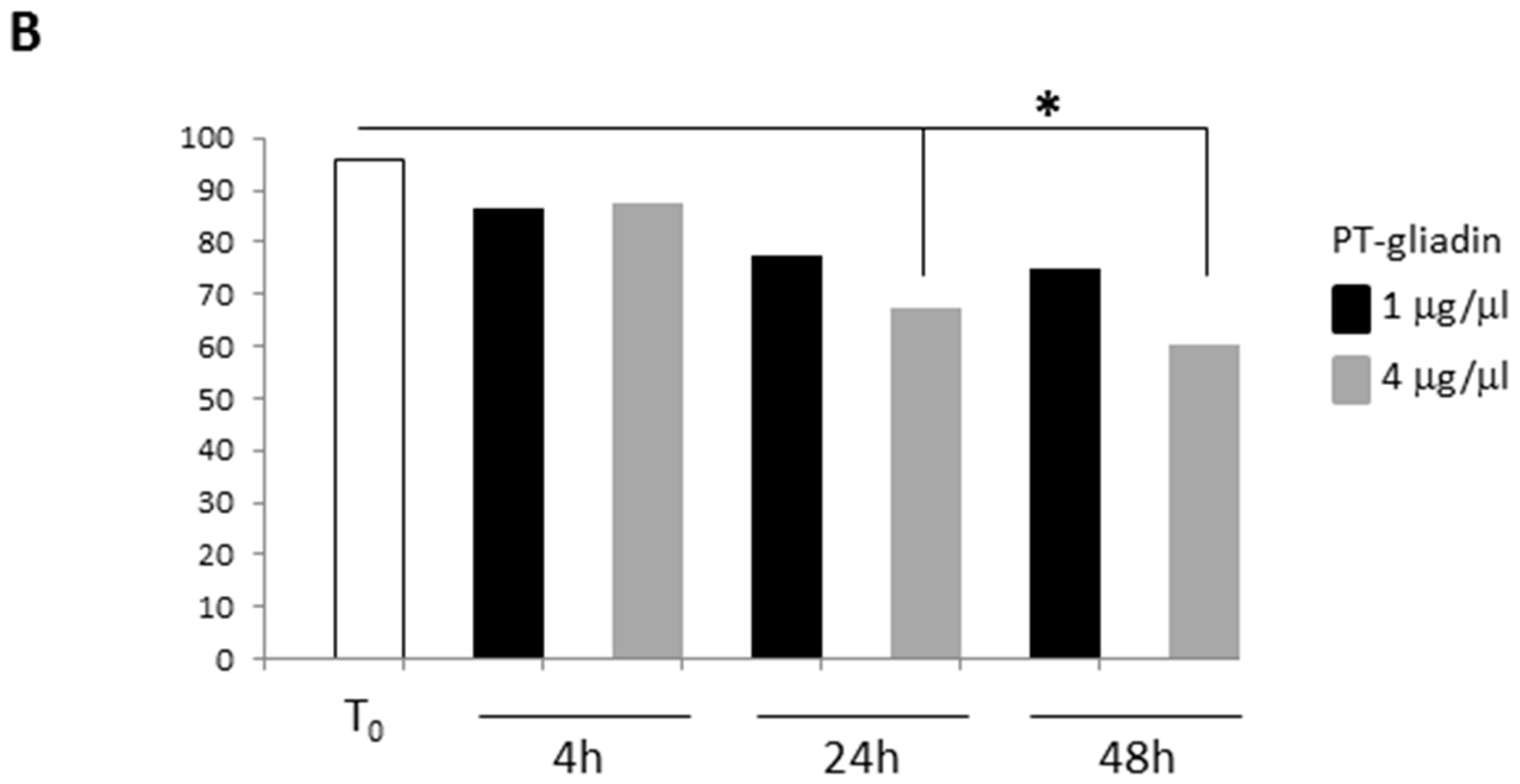
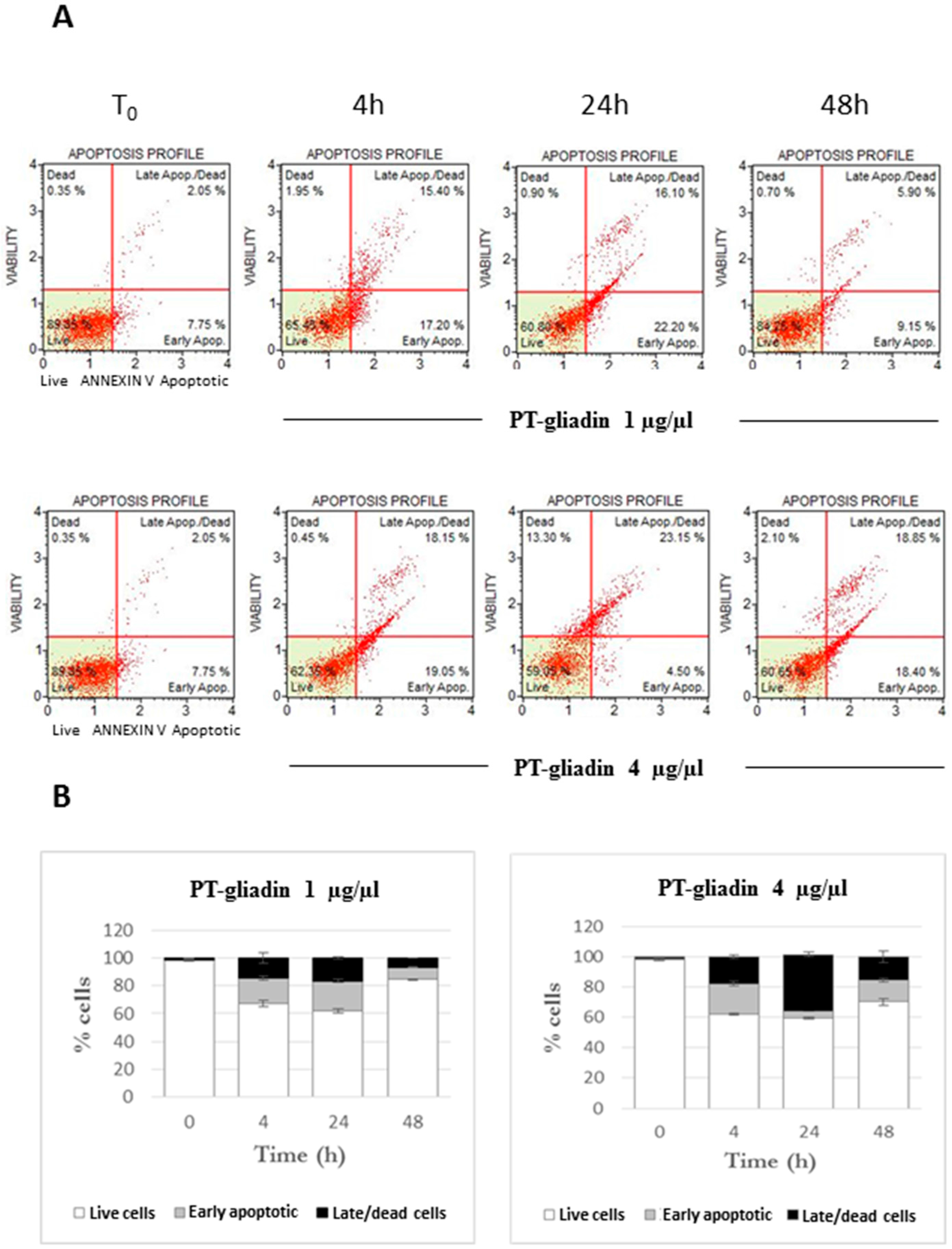
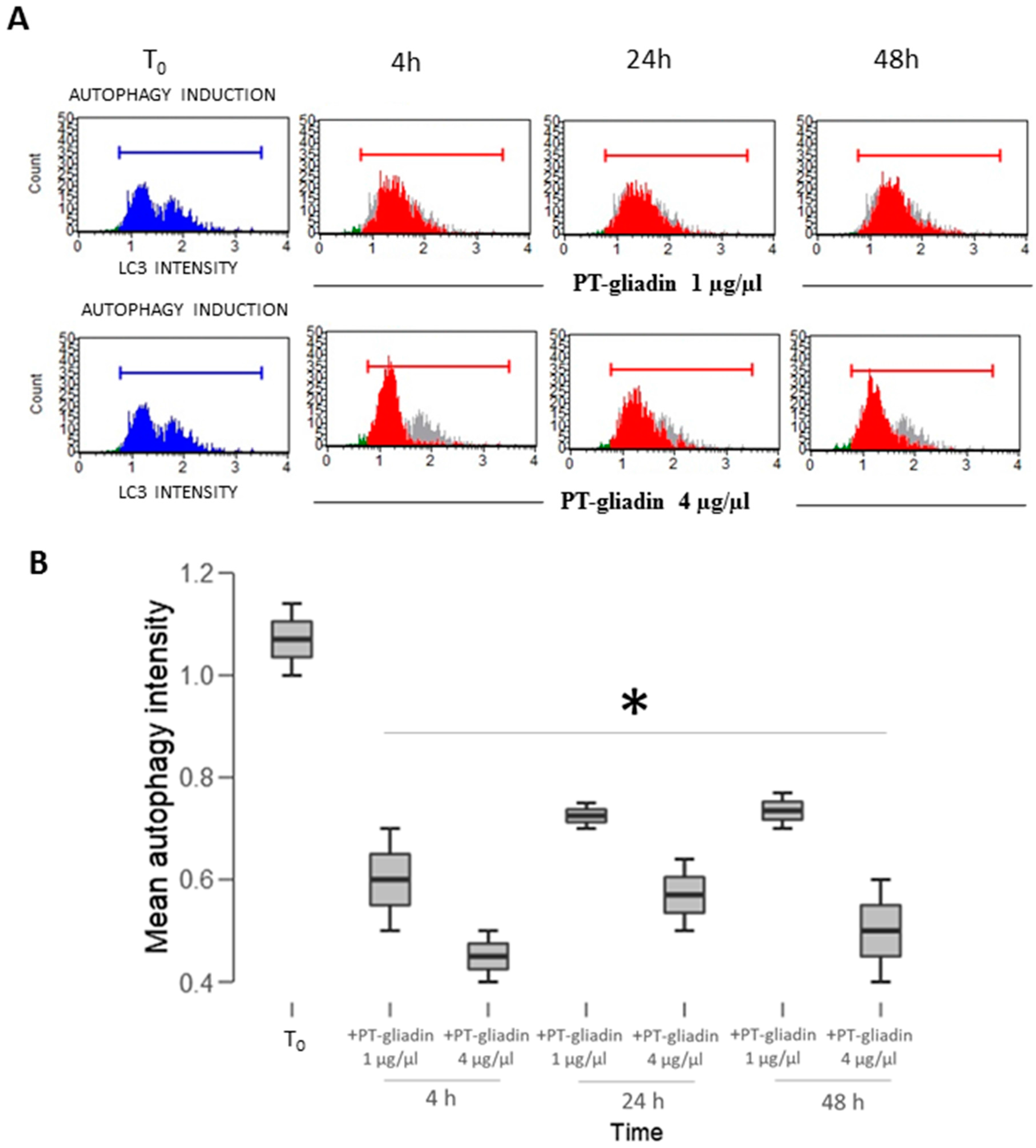
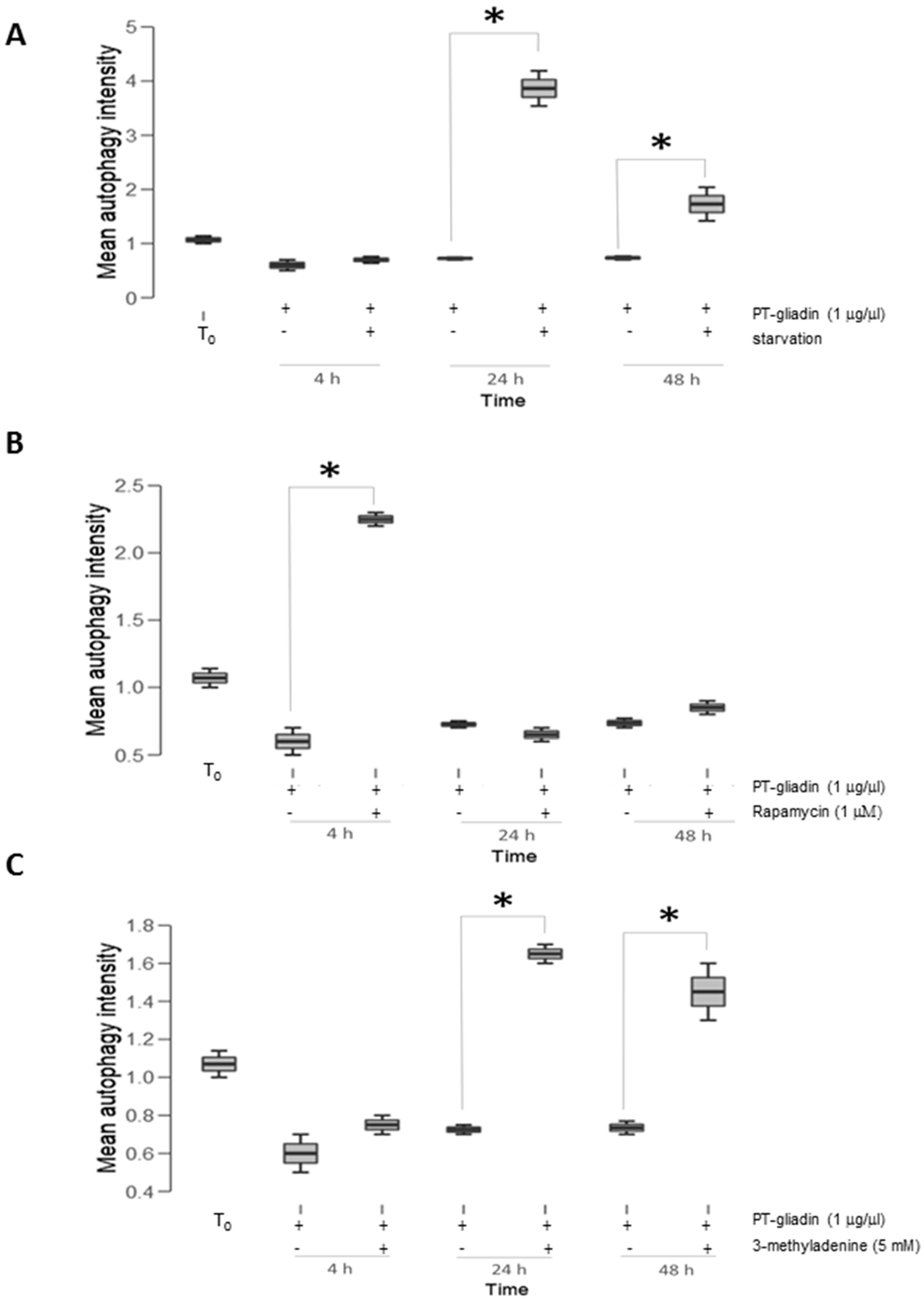
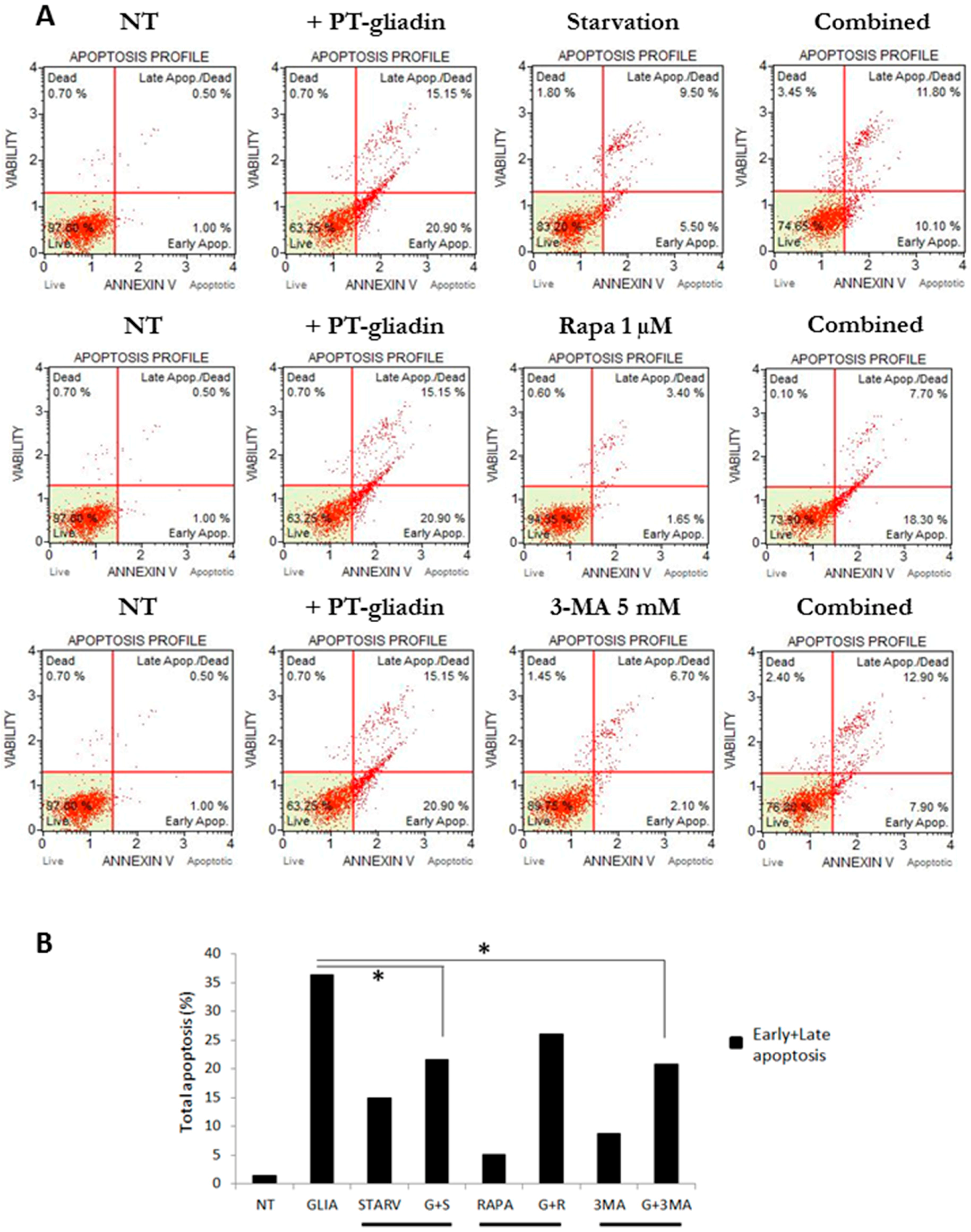
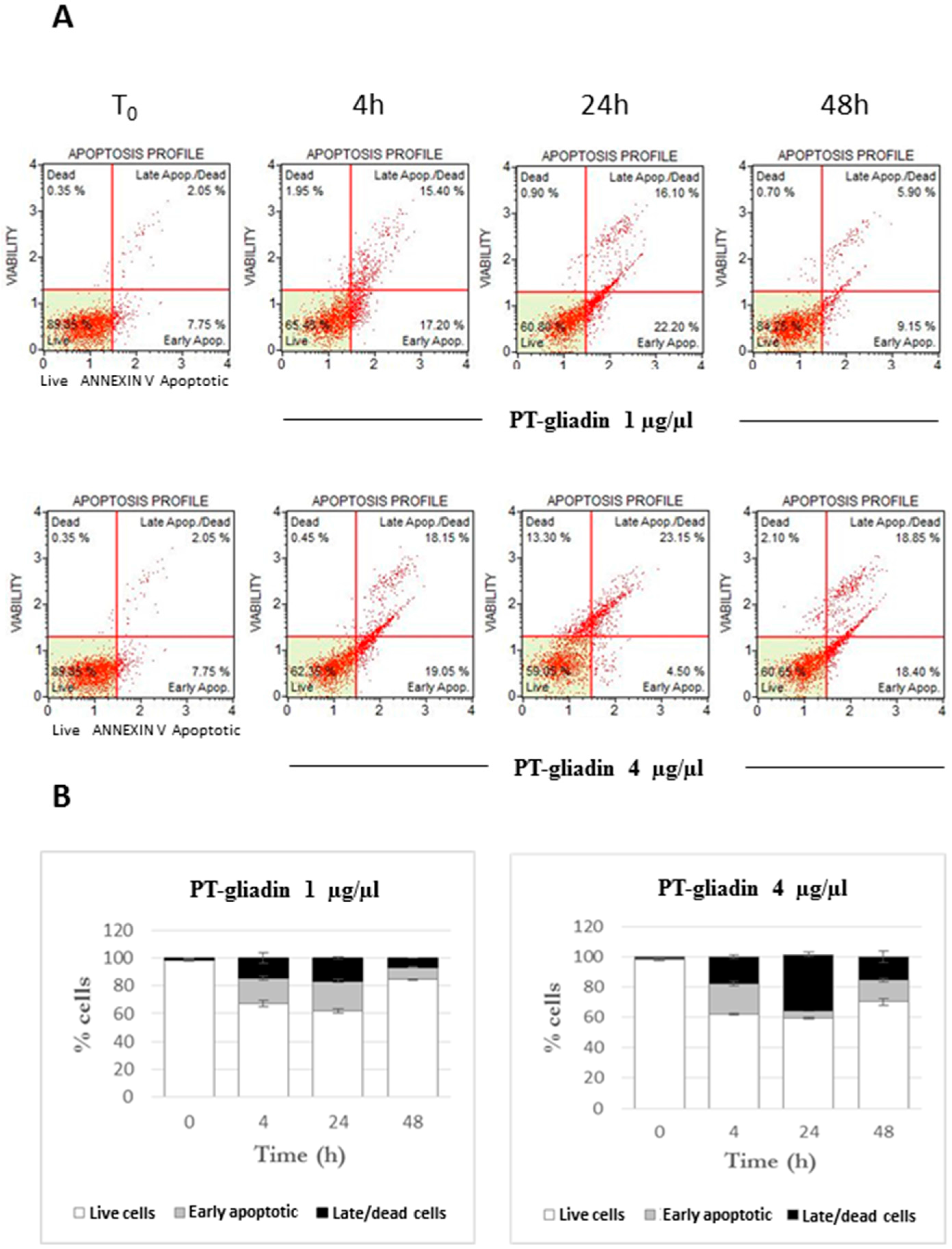
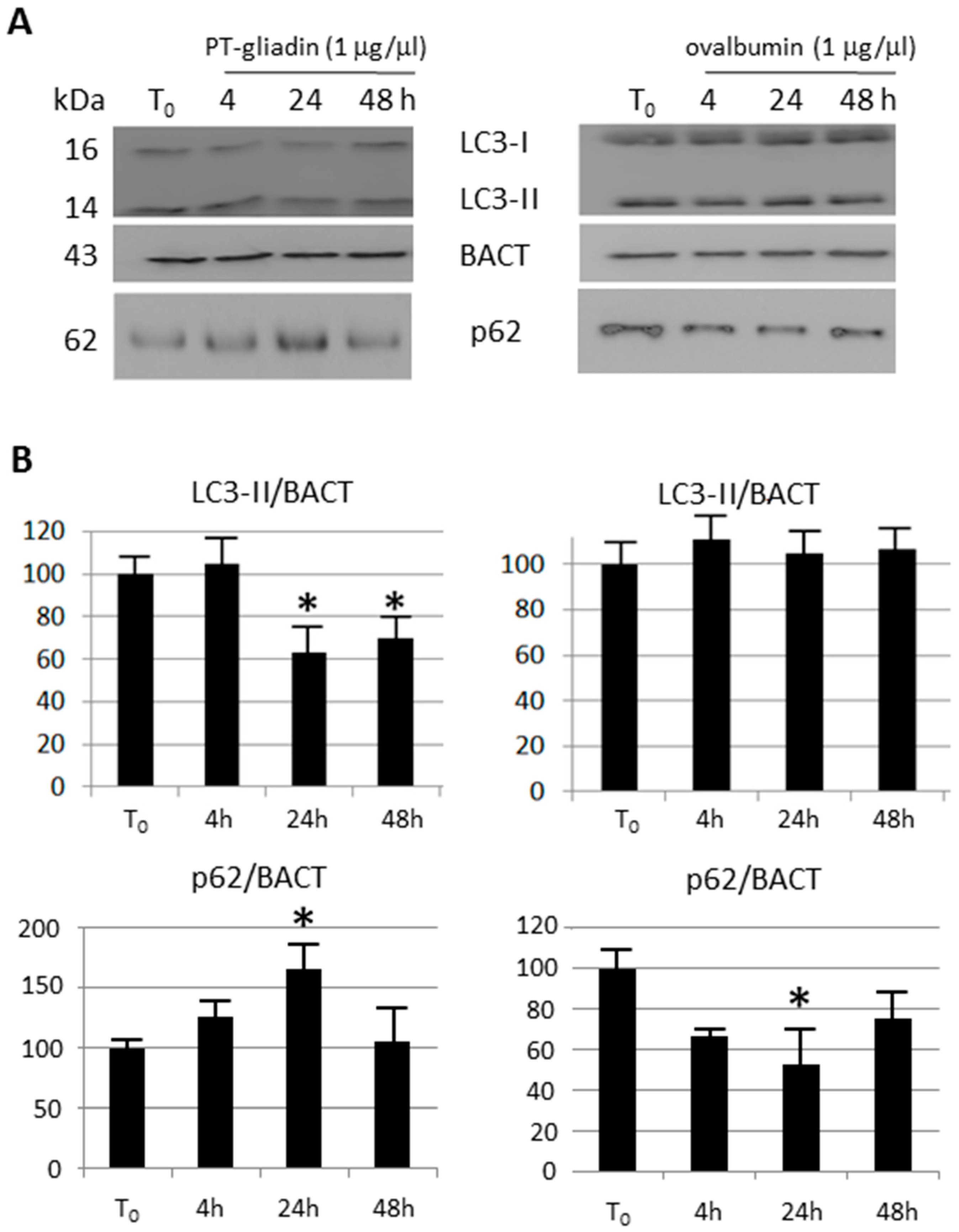
2.2. PT-Gliadin Affects Viability and Impairs Autophagy in Caco-2 Cells
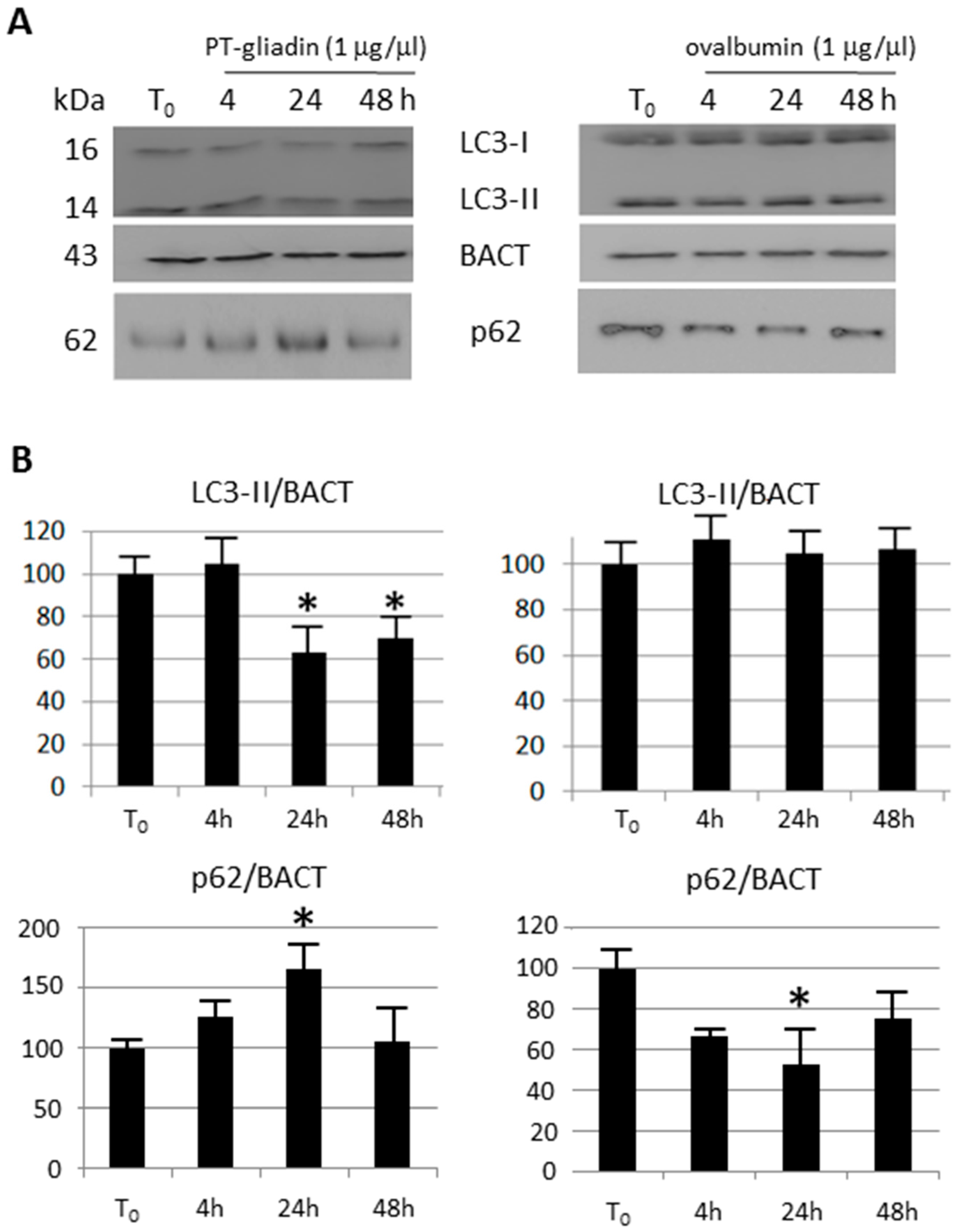
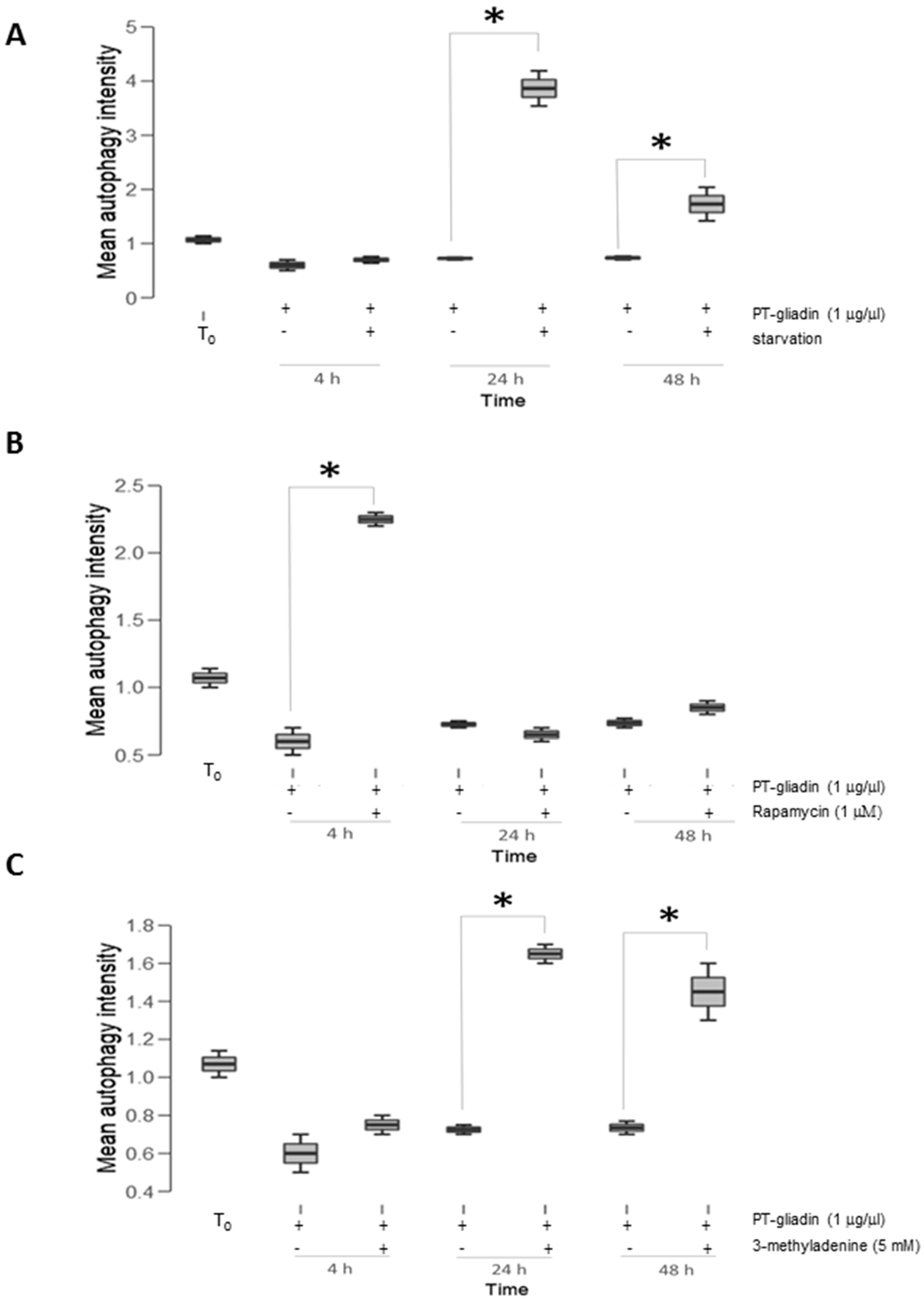
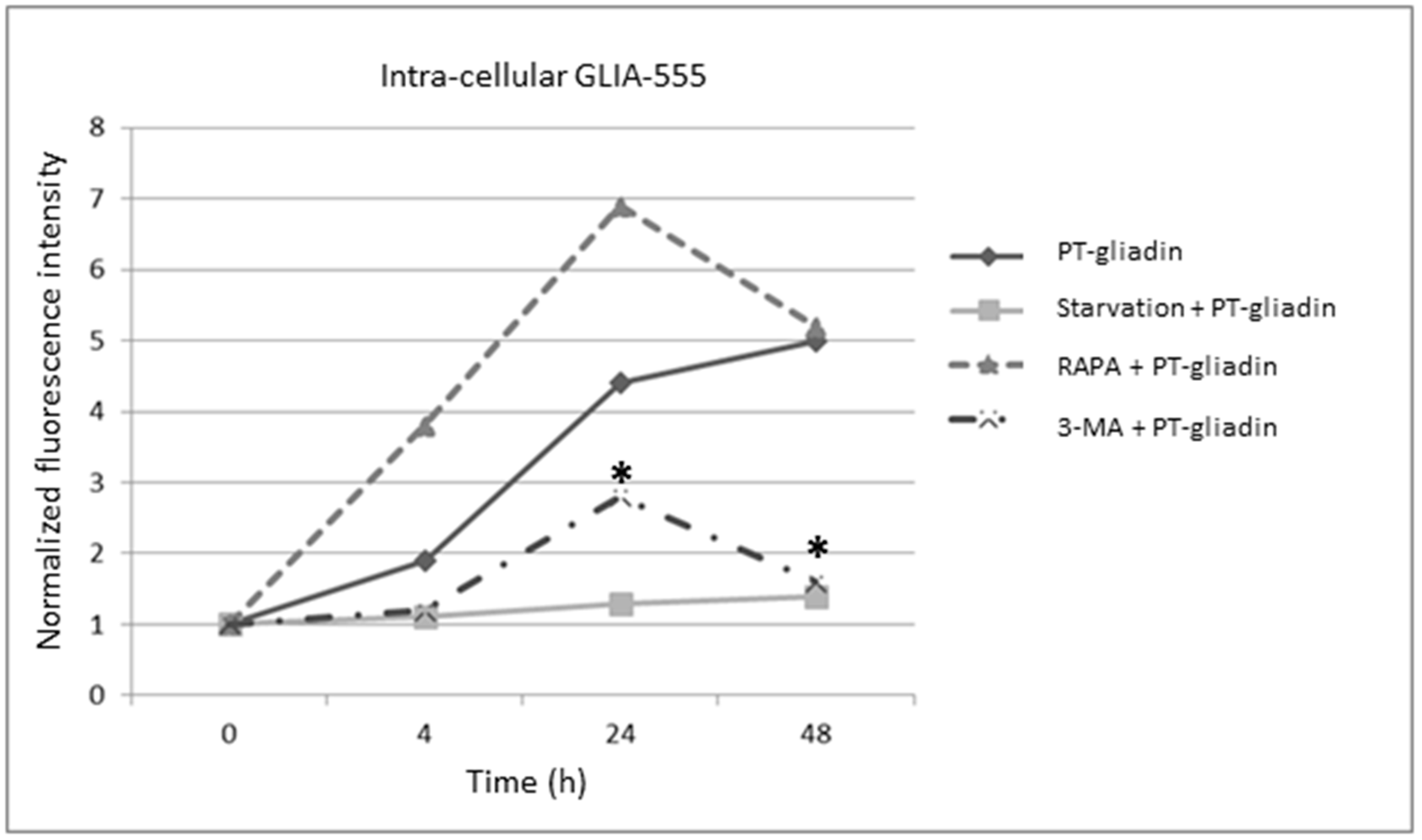
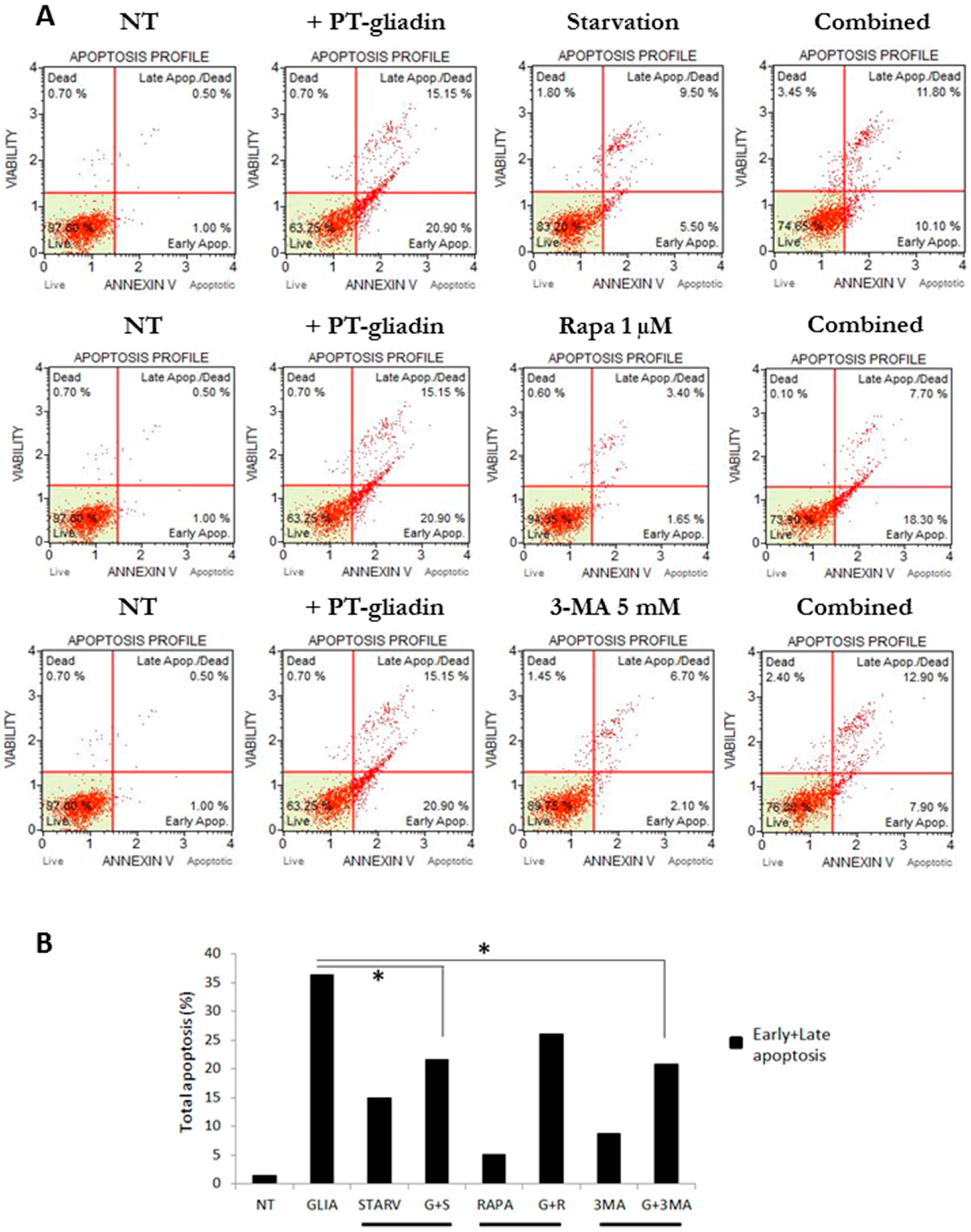
2.3. the Effects of Autophagy Modulation in Caco-2 Cells Treated with PT-Gliadin
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Autophagy Modulation Protocols
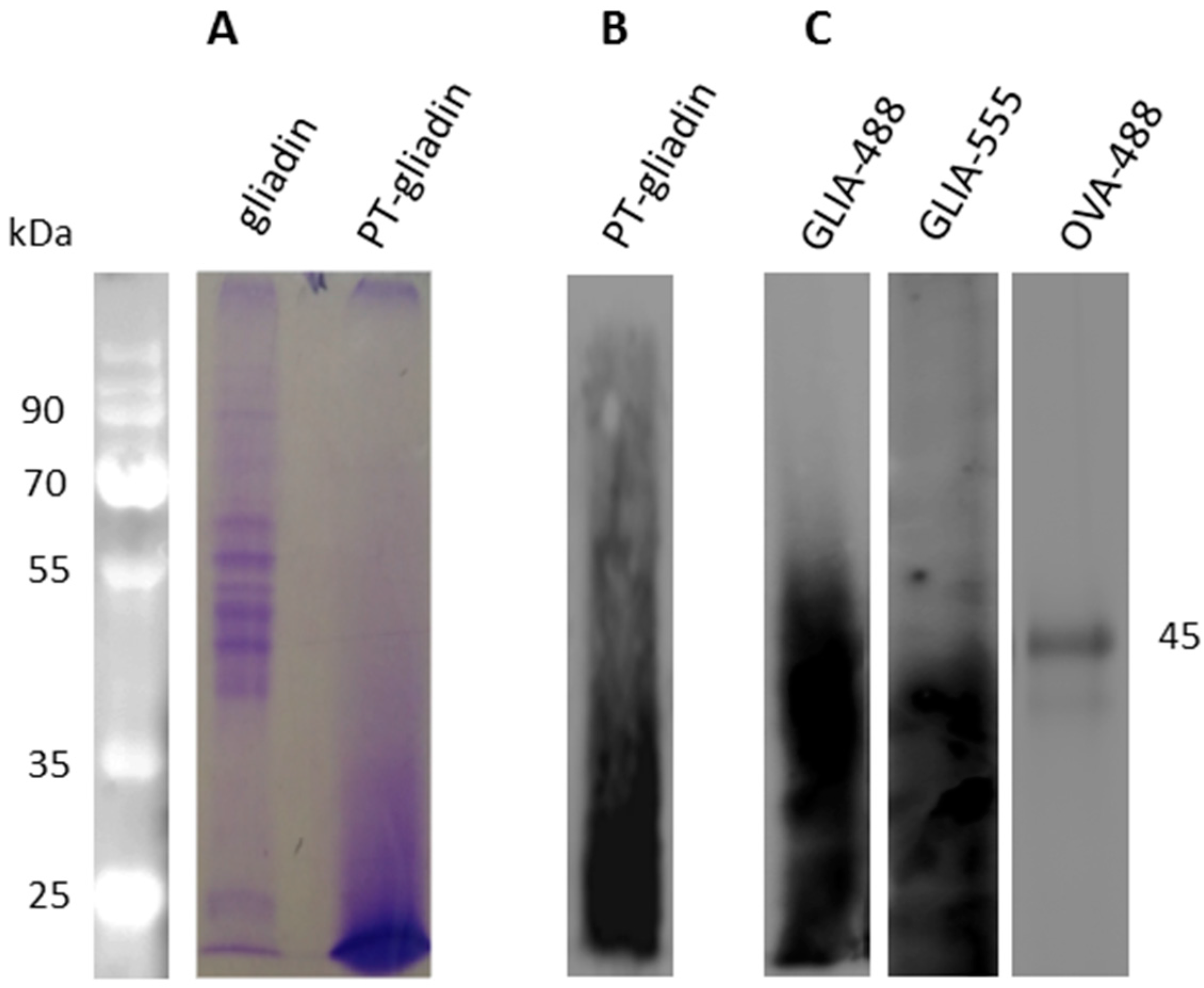
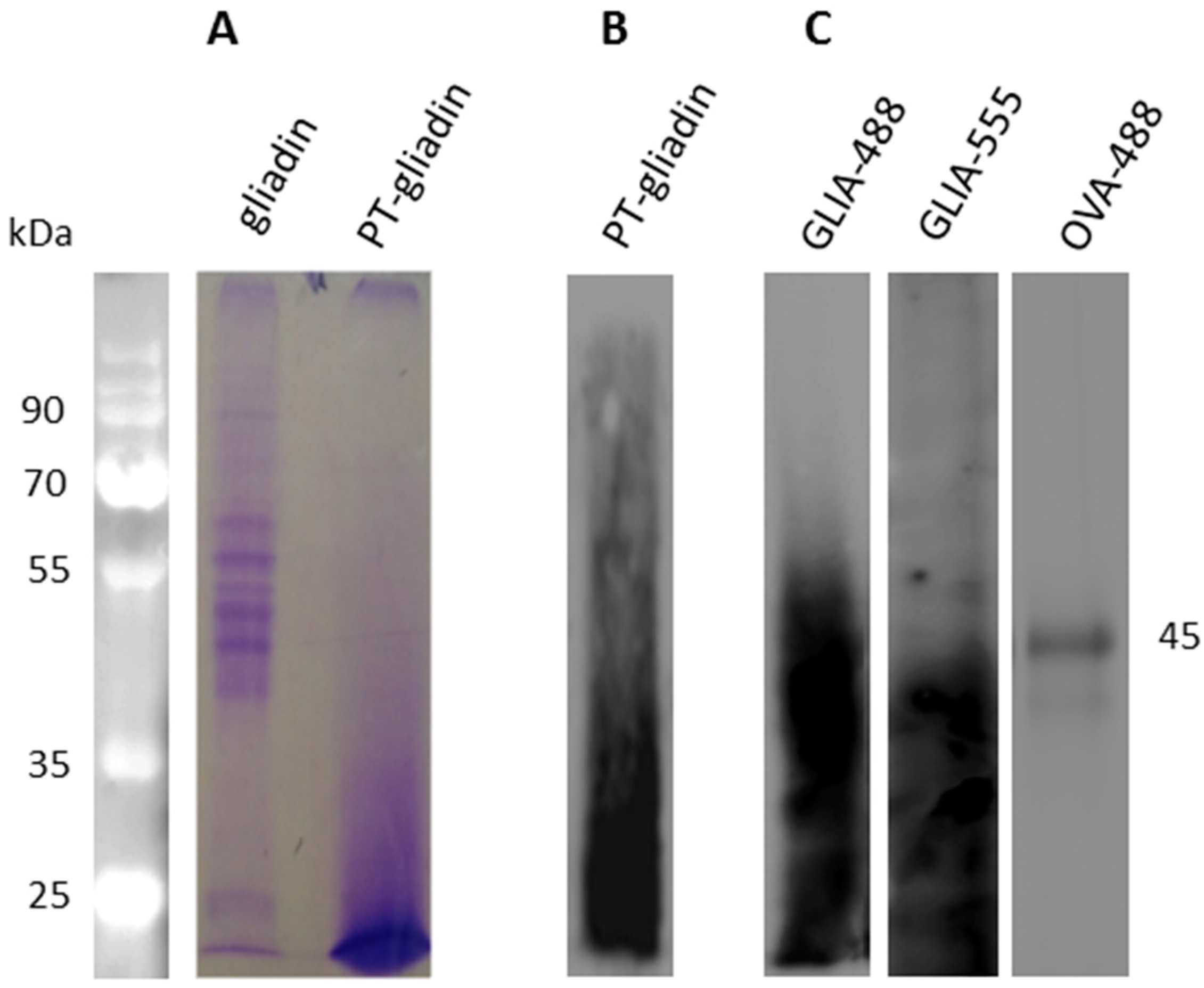
4.2. Peptic and Trypsin Digested Gliadin (PT-Gliadin) Preparation And Fluorescent Labeling
4.3. Transmission Electron Microscopy (TEM) Analysis of PT-Gliadin Aggregates in Caco-2 Cells
4.4. Immunofluorescence Analysis
4.5. Acridine Orange Staining
4.6. Lc3b-Gfp Autophagosome Analysis
4.7. Immunoblotting Analysis
4.8. Fluorimetric and Cytofluorimetric Analysis of Extra- And Intra-Cellular PT-Gliadin Content
4.9. Apoptosis and Autophagy Cytofluorimetric Analysis after PT-Gliadin Administration
4.10. Micronuclei Assay
4.11. BECN1 Sirna Assay and Transfection
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Deter, R.L.; De Duve, C. Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J. Cell Biol. 1967, 33, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, U.; Warmuth-Metz, M. Inhibition by insulin of cellular autophagy in proximal tubular cells of rat kidney. Am. J. Physiol. 1983, 244, E109–E114. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [PubMed]
- Bah, A.; Vergne, I. Macrophage Autophagy and Bacterial Infections. Front. Immunol. 2017, 8, 1483. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, S.; Comincini, S. Autophagy and ionizing radiation in tumors: The “survive or not survive” dilemma. J. Cell. Physiol. 2013, 228, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Svenning, S.; Johansen, T. Regulation of selective autophagy: The p62/SQSTM1 paradigm. Essays Biochem. 2017, 61, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Colacurcio, D.J.; Pensalfini, A.; Jiang, Y.; Nixon, R.A. Dysfunction of autophagy and endosomal-lysosomal pathways: Roles in pathogenesis of Down syndrome and Alzheimer’s Disease. Free Radic. Biol. Med. 2018, 114, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Winslow, A.R.; Chen, C.W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. α-Synuclein impairs macroautophagy: Implications for Parkinson’ disease. J. Cell Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; de Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo recognition failure is responsible for inefficient au ophagy in Huntington’s disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Di Sabatino, A.; Corazza, G.R. Coeliac disease. Lancet 2009, 373, 1480–1493. [Google Scholar] [CrossRef]
- Cornell, H.J.; Townley, R.R. The toxicity of certain cereal proteins in coeliac disease. Gut 1974, 15, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Wieser, H. Relation between gliadin structure and coeliac toxicity. Acta Paediatr. Suppl. 1996, 412, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.Y.; Quarsten, H.; Bergseng, E.; Khosla, C.; Sollid, L.M. Structural basis for HLA-DQ2-mediated presentation of gluten epitopes in celiac disease. Proc. Natl. Acad. Sci. USA 2004, 101, 4175–4179. [Google Scholar] [CrossRef] [PubMed]
- Rashtak, S.; Murray, J. Review article: Coeliac disease, new approaches to therapy. Aliment. Pharmacol. Ther. 2012, 35, 768–781. [Google Scholar] [CrossRef] [PubMed]
- Hall, N.; Rubin, G.; Charnock, A. Intentional and inadvertent non-adherence in adult coeliac disease. A cross-sectional survey. Appetite 2013, 68, 56–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubio-Tapia, A.; Murray, J. Classification and management of refractory coeliac disease. Gut 2010, 59, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Gujral, N.; Suh, J.W.; Sunwoo, H.H. Effect of anti-gliadin IgY antibody on epithelial intestinal integrity and inflammatory response induced by gliadin. BMC Immunol. 2015, 16, 41. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.G.; Veuthey, T.V.; Dodero, V.I. Self-organization of gliadin in aqueous media under physiological digestive pHs. Colloids Surf. B Biointerfaces 2016, 141, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Fassina, L.; Magenes, G.; Inzaghi, A.; Palumbo, S.; Allavena, G.; Miracco, C.; Pirtoli, L.; Biggiogera, M.; Comincini, S. AUTOCOUNTER, an ImageJ JavaScript to analyze LC3B-GFP expression dynamics in autophagy-induced astrocytoma cells. Eur. J. Histochem. 2012, 56, e44. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, C.; Rudloff, S.; Lochnit, G.; Arampatzi, S.; Maison, W.; Zimmer, KP. Epithelial transport of immunogenic and toxic gliadin peptides in vitro. PLoS ONE 2014, 9, e113932. [Google Scholar] [CrossRef] [PubMed]
- Ravanan, P.; Srikumar, I.F.; Talwar, P. Autophagy: The spotlight for cellular stress responses. Life Sci. 2017, 188, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Noda, T.; Ohsumi, Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 1998, 273, 3963–3966. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.T.; Tan, H.L.; Shui, G.; Bauvy, C.; Huang, Q.; Wenk, M.R.; Ong, C.N.; Codogno, P.; Shen, H.M. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 2010, 285, 10850–10861. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, I.J.; Raub, T.J.; Borchardt, R.T. Characterization of the human colon carcinoma cell line (Caco-2) as a model system for intestinal epithelial permeability. Gastroenterology 1989, 96, 736–749. [Google Scholar] [CrossRef]
- Sambuy, Y.; De Angelis, I.; Ranaldi, G.; Scarino, M.L.; Stammati, A.; Zucco, F. The Caco-2 cell line as a model of the intestinal barrier: Influence of cell and culture-related factors on Caco-2 cell functional characteristics. Cell Biol. Toxicol. 2005, 21, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Urade, R.; Sato, N.; Sugiyama, M. Gliadins from wheat grain: An overview, from primary structure to nanostructures of aggregates. Biophys. Rev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, N.; Shimoni, Y.; Altschuler, Y.; Levanony, H.; Volokita, M.; Galili, G. Wheat (Triticum aestivum L.) [gamma]-gliadin accumulates in dense protein bodies within the endoplasmic reticulum of yeast. Plant Physiol. 1993, 102, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Paglin, S.; Hollister, T.; Delohery, T.; Hackett, N.; McMahill, M.; Sphicas, E.; Domingo, D.; Yahalom, J. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001, 61, 439–444. [Google Scholar] [PubMed]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed]
- Varshavsky, A. The Ubiquitin System, Autophagy, and Regulated Protein Degradation. Annu. Rev. Biochem. 2017, 86, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Cho, E.D.; Lee, K.W.; Kim, J.H.; Cho, S.G.; Lee, S.J. Autophagic failure promotes the exocytosis and intercellular transfer of α-synuclein. Exp. Mol. Med. 2013, 45, e22. [Google Scholar] [CrossRef] [PubMed]
- Claude-Taupin, A.; Jia, J.; Mudd, M.; Deretic, V. Autophagy’s secret life: Secretion instead of degradation. Essays Biochem. 2017, 61, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Martucci, S.; Fraser, J.S.; Biagi, F.; Corazza, G.R.; Ciclitira, P.J.; Ellis, H.J. Characterizing one of the DQ2 candidate epitopes in coeliac disease: A-gliadin 51–70 toxicity assessed using an organ culture system. Eur. J. Gastroenterol. Hepatol. 2003, 15, 1293–1298. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell. 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Shang, L.; Chen, S.; Du, F.; Li, S.; Zhao, L.; Wang, X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc. Natl. Acad. Sci. USA 2011, 108, 4788–4793. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Fang, Y.; Lou, H.; Wang, K.; Zheng, Y.; Lou, F.; Jin, W.; Xu, Y.; Chen, W.; Pan, H.; et al. p53 suppresses stress-induced cellular senescence via regulation of autophagy under the deprivation of serum. Mol. Med. Rep. 2015, 11, 1214–1220. [Google Scholar] [CrossRef] [PubMed]
- Ballou, L.M.; Lin, R.Z. Rapamycin and mTOR kinase inhibitors. J. Chem. Biol. 2008, 1, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Rajaguru, P.; Vaiphei, K.; Saikia, B.; Kochhar, R. Increased accumulation of dendritic cells in celiac disease associates with increased expression of autophagy protein LC3. Indian J. Pathol. Microbiol. 2013, 56, 342–348. [Google Scholar] [PubMed]
- Comincini, S.; Manai, F.; Meazza, C.; Pagani, S.; Martinelli, C.; Pasqua, N.; Pelizzo, G.; Biggiogera, M.; Bozzola, M. Identification of Autophagy-Related Genes and Their Regulatory miRNAs Associated with Celiac Disease in Children. Int. J. Mol. Sci. 2017, 18, 391. [Google Scholar] [CrossRef] [PubMed]
- Regeling, A.; Somasundaram, R.; de Haar, C.; van der Woude, C.J.; Braat, H.; Peppelenbosch, M.P. Role of defective autophagia and the intestinal flora in Crohn disease. Self Nonself. 2010, 1, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Cadwell, K.; Liu, J.Y.; Brown, S.L.; Miyoshi, H.; Loh, J.; Lennerz, J.K.; Kishi, C.; Kc, W.; Carrero, J.A.; Hunt, S.; et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008, 456, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Alirezaei, M.; Kemball, C.C.; Flynn, C.T.; Wood, M.R.; Whitton, J.L.; Kiosses, W.B. Short-term fasting induces profound neuronal autophagy. Autophagy 2010, 6, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Mattson, M.P. Fasting: Molecular mechanisms and clinical applications. Cell Metab. 2014, 19, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Di Biase, S.; Longo, V.D. Fasting-induced differential stress sensitization in cancer treatment. Mol. Cell. Oncol. 2015, 3, e1117701. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, G.J. Therapeutic strategies of drug repositioning targeting autophagy to induce cancer cell death: From pathophysiology to treatment. J. Hematol. Oncol. 2017, 9, 67. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, N.; Osera, C.; Fassina, L.; Amadio, M.; Angeletti, F.; Morini, M.; Magenes, G.; Venturini, L.; Biggiogera, M.; Ricevuti, G.; et al. Autophagy is modulated in human neuroblastoma cells through direct exposition to low frequency electromagnetic fields. J. Cell. Physiol. 2014, 229, 1776–1786. [Google Scholar] [CrossRef] [PubMed]
- Sbalchiero, E.; Azzalin, A.; Palumbo, S.; Barbieri, G.; Arias, A.; Simonelli, L.; Ferretti, L.; Comincini, S. Altered cellular distribution and sub-cellular sorting of doppel (Dpl) protein in human astrocytoma cell lines. Cell. Oncol. 2008, 30, 337–347. [Google Scholar] [PubMed]
- Barbieri, G.; Palumbo, S.; Gabrusiewicz, K.; Azzalin, A.; Marchesi, N.; Spedito, A.; Biggiogera, M.; Sbalchiero, E.; Mazzini, G.; Miracco, C.; et al. Silencing of cellular prion protein (PrPC) expression by DNA-antisense oligonucleotides induces autophagy-dependent cell death in glioma cells. Autophagy 2011, 7, 840–853. [Google Scholar] [CrossRef] [PubMed]
- ImageJ Software. Available online: http://rsbweb.nih.gov/ij (accessed on 15 September 2017).
- Fenech, M. The in vitro micronucleus technique. Mutat. Res. 2000, 455, 81–95. [Google Scholar] [CrossRef]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manai, F.; Azzalin, A.; Gabriele, F.; Martinelli, C.; Morandi, M.; Biggiogera, M.; Bozzola, M.; Comincini, S. The In Vitro Effects of Enzymatic Digested Gliadin on the Functionality of the Autophagy Process. Int. J. Mol. Sci. 2018, 19, 635. https://doi.org/10.3390/ijms19020635
Manai F, Azzalin A, Gabriele F, Martinelli C, Morandi M, Biggiogera M, Bozzola M, Comincini S. The In Vitro Effects of Enzymatic Digested Gliadin on the Functionality of the Autophagy Process. International Journal of Molecular Sciences. 2018; 19(2):635. https://doi.org/10.3390/ijms19020635
Chicago/Turabian StyleManai, Federico, Alberto Azzalin, Fabio Gabriele, Carolina Martinelli, Martina Morandi, Marco Biggiogera, Mauro Bozzola, and Sergio Comincini. 2018. "The In Vitro Effects of Enzymatic Digested Gliadin on the Functionality of the Autophagy Process" International Journal of Molecular Sciences 19, no. 2: 635. https://doi.org/10.3390/ijms19020635