Alternative mRNA Splicing in the Pathogenesis of Obesity

1
Department of Health Technology and Informatics, The Hong Kong Polytechnic University, Hong Kong, China
2
The State Key Laboratory of Pharmaceutical Biotechnology, Department of Medicine, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong, China
3
School of Medical and Health Sciences, Tung Wah College, Hong Kong, China
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(2), 632; https://doi.org/10.3390/ijms19020632
Submission received: 5 January 2018
/
Revised: 21 February 2018
/
Accepted: 21 February 2018
/
Published: 23 February 2018
(This article belongs to the Special Issue Pre-mRNA Splicing 2017)
Abstract
:Alternative mRNA splicing is an important mechanism in expansion of proteome diversity by production of multiple protein isoforms. However, emerging evidence indicates that only a limited number of annotated protein isoforms by alternative splicing are detected, and the coding sequence of alternative splice variants usually is only slightly different from that of the canonical sequence. Nevertheless, mis-splicing is associated with a large array of human diseases. Previous reviews mainly focused on hereditary and somatic mutations in cis-acting RNA sequence elements and trans-acting splicing factors. The importance of environmental perturbations contributed to mis-splicing is not assessed. As significant changes in exon skipping and splicing factors expression levels are observed with diet-induced obesity, this review focuses on several well-known alternatively spliced metabolic factors and discusses recent advances in the regulation of the expressions of splice variants under the pathophysiological conditions of obesity. The potential of targeting the alternative mRNA mis-splicing for obesity-associated diseases therapies will also be discussed.
1. Introduction
Alternative mRNA splicing plays a key role in enhancing protein diversity [1]. Based on a recent analysis of the statistics for annotated human nuclear genes in GeneBase 1.1, nearly 80% of human protein-coding genes produce more than one transcript [2]. On average, human protein-coding genes contain ~11 exons per transcript and produce 5.4 mRNAs per gene. The longest exon in human is exon 5 of the zinc finger and BTB domain containing 20 (ZBTB20) gene which has 24927 nucleotides (nt), and the shortest human exon is exon 2 of septin 7 (SEPT7) gene with only two nt [2]. Most mammalian pre-mRNAs contain short exonic sequences separated by longer intronic stretches.
Ninety-four percent of mammalian protein-coding exons exceed 51 nucleotides (nt) in length [3]. It was a greater challenge to recognize the micro-exons (≤51 nt) than for the longer exons by the process of splicing machinery [3]. The size of human introns also varies considerably from 30 nt [intron 9 of macrophage stimulating one like (MST1L) gene] to ~1,160,000 nt [intron 2 of roundabout guidance receptor 2 (ROBO2) gene] [2]. Minimal length of intron is required for maintaining efficient splicing [4]. However, unlike the exons, short introns (>100 nt) are common in human transcriptome [5,6]. It is noted that short introns promote the generation of protein isoforms and they are often alternatively spliced [6].
Existence of introns is still one of the biggest mysteries in biology. Huge energy is spent on the production of introns and maintenance of the highly accurate cleavage/ligation mechanism. It is a heavy burden on cells. It is generally accepted that alternative mRNA splicing of exons is mainly for the production of multiple protein isoforms from the same gene [1]. The development of high-throughput array and sequencing technologies entirely revolutionized the analysis of alternative splicing events by allowing an unbiased assessment [7]. Remarkably, only a limited number of annotated alternative isoforms are detected in proteomics studies [8]. In other words, despite multiple alternative mRNA transcripts per gene are detected, most genes encode only one protein isoform under most circumstances [9]. The phenomenon may be attributed to the rare expression of alternative mRNA transcripts that express only in particular tissues and developmental stages [10,11].
It is also hypothesized that alternative mRNA splicing increases the chance of removal of the important domains for modulating the protein-protein interactions. About 70% of the expressed alternative isoforms lost more than 60 amino acid residues [8]. Surprisingly, recent transcriptome analysis demonstrated highly alternative mRNA transcripts have similar coding sequences to that of the canonical isoforms, and the sites for protein-protein interaction are usually protected from alternative splicing-mediated removals [12]. Although further functional verification is required to confirm the bioinformatics study, the findings strongly suggested that, the main function of generating multiple protein isoforms by alternative mRNA splicing is not only for generating protein isoforms. Possibilities of putative functions such as mRNA transport, nonsense mediated decay, regulation of gene expression as well as mutational buffering are proposed [13].
Nevertheless, according to Human Gene Mutation Database, it was estimated that about 60% of disease-causing mutations are disturbing proper mRNA splicing [14]. Recent reviews have provided extensive information on splicing mechanisms [15,16,17], and the pathological consequences of mis-splicing that mutations in cis-acting RNA sequence elements and trans-acting splicing factors are described [18,19,20]. However, as splicing dysregulation needs not be directly linked with any genetic mutation, the number of splicing-related diseases shall be substantially underestimated. For example, spontaneous gene mutations in obese subjects with insulin resistance are found but the incidences are rare. Environmental factors play significant roles in their metabolic dysregulation [21,22], potentially by extensive changes in alternative RNA processing [23]. The number and extent of diseases related to splicing dysregulation, which have not been covered by the mutation databases, remain to be explored.
Obesity is a significant leading cause for metabolic diseases, especially for diabetes and cardiovascular problems. The major changes associated with obesity include positive energy balance and activation of immune system [24]. As alternative mRNA splicing is tightly regulated by signaling pathway to cope with the physiological changes [23], diet-induced obesity model can be applied in the investigations of the significance of alternative mRNA splicing in the pathogenesis of obesity. Indeed, significant changes in the expression level of splice variants and splicing factors in association with age and metabolic dysregulation in animal models, as well as in human populations, had already been reported [25,26,27,28,29,30]. Dysregulation of alternative mRNA splicing may stipulate an important driver of ageing process and metabolic diseases.
This review summarizes recent advances on a number of well-known alternative splicing regulated metabolic factors as examples to illustrate the contribution of alternative mRNA mis-splicing in metabolic dysregulation. The potential regulatory mechanism by various splicing factors altering the expression of those splice variants under pathophysiological conditions are included. The possibility by modulation of the expression level and activity of those splicing factors as potential therapeutic targets for obesity-associated metabolic complications will also be discussed.
2. Mis-Splicing of Metabolic Factors in Obesity
2.1. Insulin Receptor
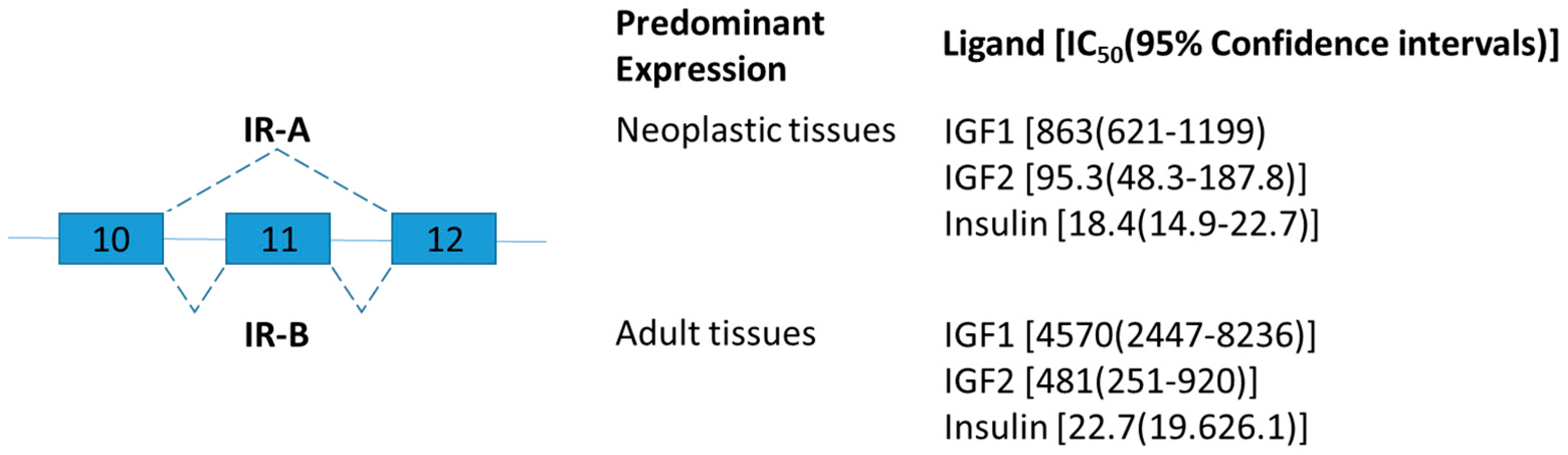
Insulin receptor belongs to a subfamily of receptor tyrosine kinases and plays an important role as regulators of cell growth, differentiation, and metabolism [31]. The human insulin receptor exists in two isoforms differing by the presence of exon 11 (Figure 1) [32]. Exon 11, encodes 12 amino acids in the C terminal of insulin receptor and is skipped in a developmental and tissue-specific manner [33]. In brief, insulin receptor type A (IR-A) lacking exon 11 is predominantly expressed during prenatal life for growth and fetal development, and IR-B is predominantly expressed in well-differentiated adult tissues such as the liver for metabolic insulin action [33]. IR-A and IR-B have similar binding affinity for insulin, but different affinity for insulin-like growth factor (IGF)-2 and proinsulin (Figure 1) [34].
Based on the information above, it was hypothesized that insulin sensitivity might be associated with the alteration of insulin receptor isoform expression. Many subsequent studies were performed to explore the correlation between the expression of insulin receptor variants and insulin resistance [36,37,38,39]. As sequence analysis revealed that the region of exons 9 through 12 of rhesus insulin receptor gene is very similar to that of humans, diabetic monkeys were used to explore the potential association between hyperinsulinemia and alternations in the insulin receptor mRNA splicing in 1994 [40]. This study provided the first direct evidence demonstrating that the hyperinsulinemic monkeys have higher levels of IR-A in muscle than those non-hyperinsulinemic controls [40]. The same team further studied the alternative splicing of insulin receptor in liver of normal, prediabetic, and diabetic monkeys. Increase of IR-A level was also observed in the liver of a diabetic monkey, which was significantly correlated with fasting plasma glucose and intravenous glucose disappearance rate [41].
Interestingly, in agreement with the findings in diabetic rhesus monkeys as mentioned above, two recent studies demonstrated that IR-B mRNA variant increased in response to weight loss by either low calorie diet [42] or bariatric surgery in human [43]. They demonstrated that the expression of adipose IR-B is negatively correlated with fasting insulin levels [42,43]. Although conflicting data was reported by other group [44] and the role of insulin receptor isoforms in noninsulin-dependent diabetes mellitus remained elusive, the studies raised the possibility of cellular metabolic status alters the ratio of splice variants. As splicing enhancer and silencer elements are responsible for the alternatively spliced insulin receptor intron 10 and exon 11 [45], several splicing factors (namely hnRNPA1, SF3A, and SFRS7) are proposed to have regulatory role in the exon inclusion of insulin receptor [42]. However, the findings were based on the correlation between the alternative splicing of insulin receptor and the expression of those splicing factors. The detailed molecular mechanism remains to be explored.
Further investigation revealed that Serine and Arginine Rich Splicing Factor 3 (SRSF3; also known as SRP20) play an important role in regulating the insulin receptor exon 11 skipping [45]. Overexpression of SFSR3 results in the inclusion of exon 11, and knockdown of SRSF3 leads to exon 11 skipping in hepatoma cells [45]. In addition, as identified by genomic analysis identified many genes are critical regulators for glucose and lipid homeostasis were mis-spliced in SRSF3HKO liver of SRSF3 liver specific knockout (SRSF3HKO) mice [46]. However, information for the expression level and activity of SRSF3 in obese and diabetes subjects is scarce. Further studies on the regulatory roles of SFSR3 in hepatic glucose and lipid homeostasis under the pathological condition of obese subjects are required.
2.2. Leptin Receptor
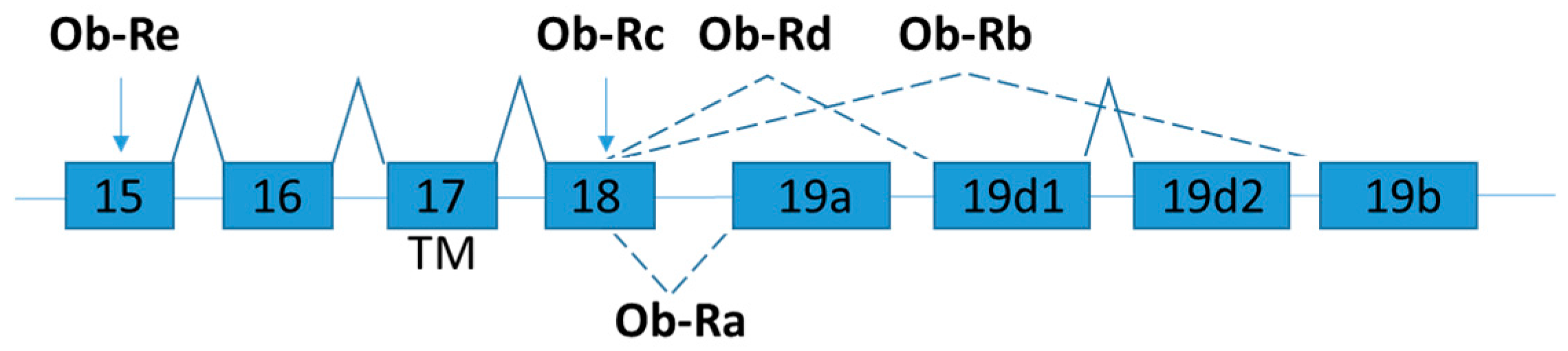
Leptin receptor (also known as obesity receptor, Ob-R) is expressed in several isoforms by alternative mRNA splicing (Figure 2) [47]. According to the structural differences, the isoforms are grouped into three classes: namely long, short, and secretory leptin receptors. All Ob-R isoforms have similar N-terminal extracellular ligand-binding domain. The long isoform (named as OB-Rb) is the full-length isoform, which is mainly expressed in the hypothalamus and immune cells and play important roles in energy homeostasis and immunity, respectively [48]. OB-Rb is the only isoform that can fully activate signal transduction. The development of the early obesity phenotype in db/db mice is due to the lacking of Ob-Rb [49].
The short leptin isoforms include Ob-Ra, Ob-Rc, and Ob-Rd. Ob-Ra is the most common isoform which can be found in various tissues (e.g., lung and kidneys) [48]. Although the short leptin isoforms have also the transmembrane domain and a constant box 1 motif at the cytoplasmic domain which binds JAK kinases to activate signal transduction, the main functions of the short isoforms are for internalization and degradation of leptin [50]. Ob-Ra is significantly increased in db/db mice and accounts for their inability to respond to leptin signals [51].
The soluble isoform (Ob-Re or sOB-R) lacks the intracellular and cytoplasmic domains. Ob-Re is suggested to serve as a carrier protein that regulates serum leptin concentration by delaying the clearance of leptin and competitor with membrane receptors for the ligand binding [47]. dbPas/dbPas mice are grossly obese and exhibit hypercholesterolemia and hyperinsulinemia because soluble leptin receptor is absent [52,53]. The leptin and soluble leptin receptor levels in obese and weight-losing individuals are examined by soluble leptin receptor specific ELISA assay [54]. The expression level of soluble leptin receptor is inversely correlated with body mass index (BMI). After weight loss due to gastric restrictive surgery, the expression level of soluble leptin receptor slowly increased to normal level a year after surgery [54]. In contrast to mice, the human soluble OB-R is exclusively generated through proteolytic cleavage of the extracellular domain of membrane-anchored OB-R isoforms [55]. Mutations of leptin receptor gene are rare in humans [56], but polymorphisms in leptin receptor are reported [57]. It solicits further studies in the detailed mechanism on the generation of OB-R isoform and a fully evaluation of its physiological relevance in human [58].
2.3. Nuclear Receptor Corepressor
Nuclear Receptor Corepressor (NCoR) is one of the most extensively characterized transcriptional corepressors. NCoR mediates repression of nuclear receptors [thyroid hormone receptor, Liver X receptor (LXR), and peroxisome proliferator activated receptor (PPAR)] by recruitment of chromatin-modifying enzymes (e.g., histone deacetylase 3) [60]. A diverse series of corepressor protein variants of NCoR is generated by alternative mRNA splicing, and different splice variants can exert opposing transcriptional effects [61]. The difference between ω and δ splice variants is by the presence of exon 37, which encodes a third receptor interaction domain (RID) (Figure 3). NCoRω incorporates the exon 37 and has three RIDs [61]. The expression of NCoRω predominates in the preadipocyte, and overexpression of NCoRω inhibits adipose differentiation. In contrast, NCoRδ lacks exon 37 and has only two RIDs [61]. The expression of NCoRδ predominates in the mature adipocyte while its overexpression enhances adipose differentiation [61]. The number of RIDs regulates distinct panels of target genes.
As many NCoR interacting nuclear receptors are playing key roles in the regulation of both glucose and lipid metabolism [61], it was hypothesized that hormonal and nutritive events may regulate the alternative mRNA splicing of NCoR [62,63]. Dexamethasone, a synthetic derivative of a natural hormone that regulates glucose and lipid metabolism, was used to modulate the alternative mRNA splicing of NCoR in both cultured cells and mice [63]. Elevated dietary carbohydrates alter alternative NCoR mRNA splicing. In brief, fructose induced a shift from NCoRω to NCoRδ isoform at the mRNA level in Hepa1-6 hepatocytes, and a shift from NCoRδ to NCoRω was observed in liver tissue of high sucrose fed mice [63]. Although the data is relatively preliminary, the finding strongly supports the idea that hormonal and nutritive events can modulate alternative NCoR mRNA splicing. The precise mechanisms of the upstream nutrient-sensitive signaling pathways that regulate the alternative splicing of NCoR1 shall be further clarified.
To examine the role of switching between NCoRω and NCoRδ in mice, NCoRω splice-specific knockout (NCoRω−/−) mice was generated [62]. NCoRω−/− Mice exhibit greatly improved glucose sensitivity that is refractory to diet induced diabetes [62]. It is interesting to note that, as compared with other NCoR1 mouse model listed at Table 1, NCoRω−/− Mice display less severe and distinct phenotypes. Splice-specific knockout mice convincingly indicate that the NCoR variants regulate distinct target genes and hence different phenotypes. These results raise an important concern in determining the roles of genes that encode several variants by alternative splicing in vivo, the particular functions of the alternatively spliced variants could be overlooked by commonly-used whole-gene knockout strategy.
2.4. LMNA
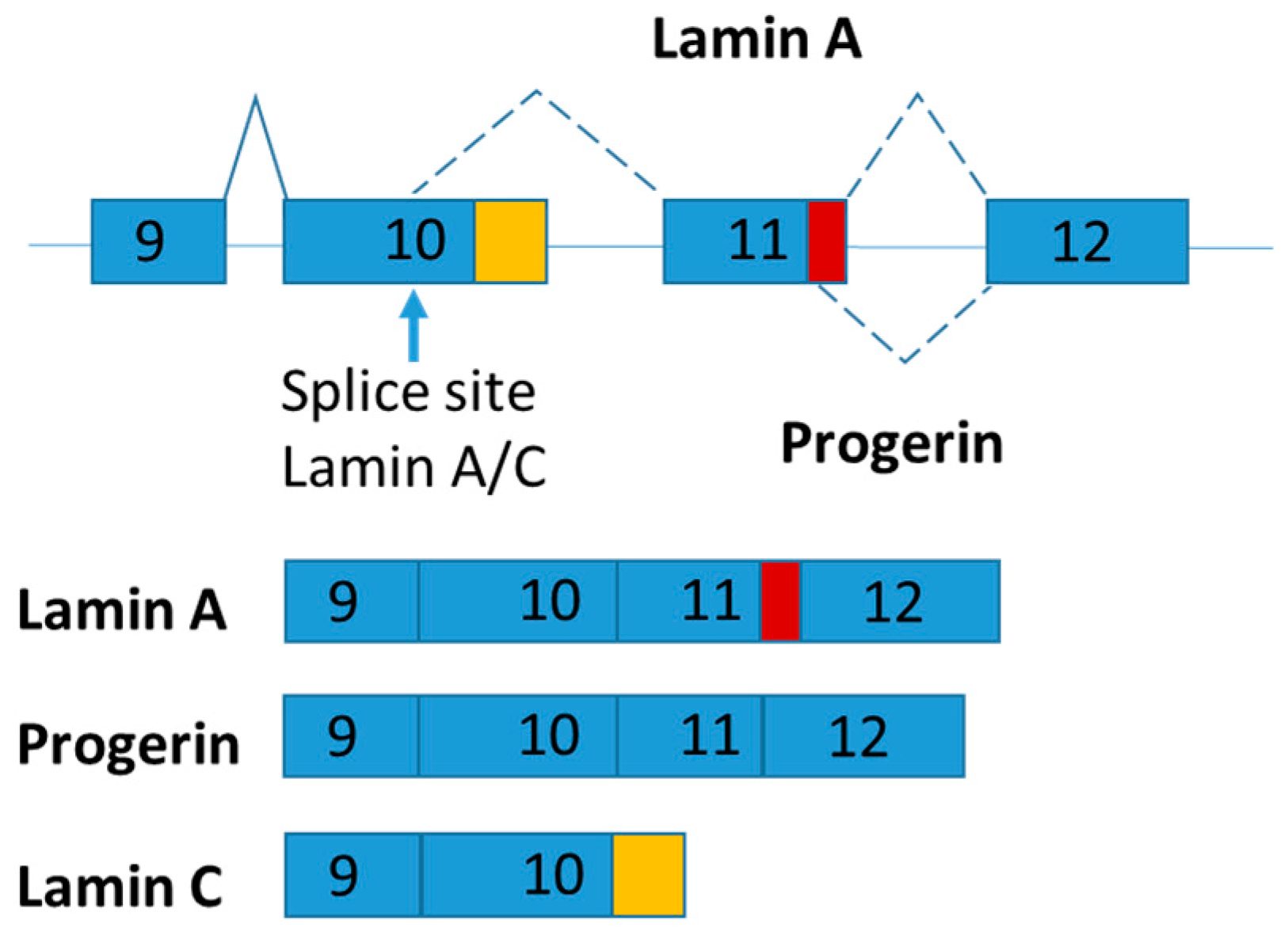
LMNA pre-mRNA produces three main isoforms (namely lamin A, progerin and lamin C) by alternative mRNA splicing (Figure 3). Lamins A and C are two major proteins produced from LMNA gene. The LMNA gene has 12 exons and generates lamins A and C by alternative splicing of exon 10 [68]. Both lamins A and C are nuclear intermediate-filament proteins. Progerin is an abnormal truncated version of lamin A protein with deletion of 50 amino acids near the C terminal by mutation (Figure 4). Progerin has also been found in cells and tissues from apparently healthy cells, although its expression is very low [69]. Continuous expression of progerin is suggested contributing to aging associated diseases [69]. Mutations in LMNA gene lead to several diseases called laminphathies (e.g., Emery-Dreifuss Muscular dystrophy and Hutchison-Gilford progeria syndrome) [70]. It was proposed that abnormalities in nuclear structure caused increased susceptibility to cellular damage by the “Mechanical-stress” hypothesis as well as the inappropriate interaction between nuclear envelop and chromatin components as explained the “Gene expression” hypothesis, respectively [71].
Interestingly, lamin C mRNA level was dramatically increased in subcutaneous adipose tissue of obese and type 2 diabetes patients [72]. Given that alternative mRNA splicing of LMNA is highly conserved throughout mammalian evolution, mouse model was used to explore the functions of LMNA isoforms [73]. Knock-in strategy was used to generate LMNA isoform specific expressing mice and found that lamin C and progerin are antagonistic in signaling adipose mitochondrial biogenesis and energy expenditure [74]. In brief, LmnaLCS/LCS mice exclusively expressing the lamin C isoform exhibit obese phenotypes with decreased energy metabolism and mitochondrial activity [74]. In contrast, progerin-expressing mice (LmnaG609G/+) present a higher energy metabolism and are lipodystrophic [74].
2.5. Lipin-1
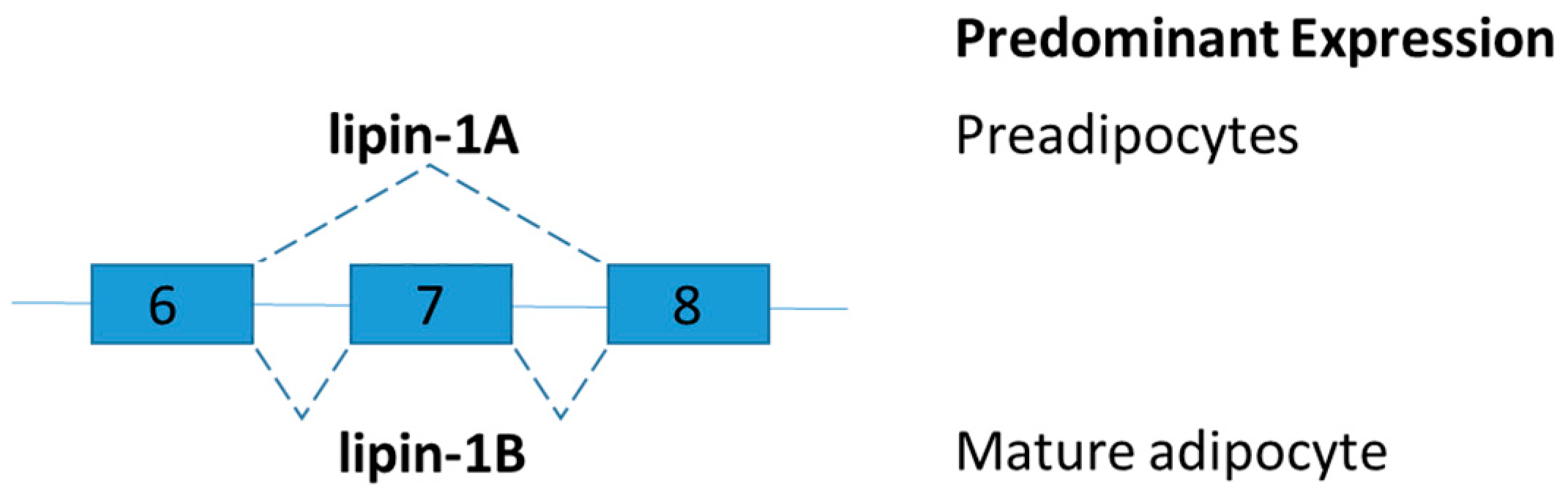
Lipin-1 is an inducible transcriptional coactivator which is required for adipocyte differentiation and lipid metabolism. Two lipin-1 protein isoforms (lipin-1A and lipin-1B) are generated by alternative mRNA splicing of the LPIN1 (Figure 5). Lipin-1B differs from lipin-1A by the presence of exon 7. The isoforms of LPIN1 serve distinct functions in adipocytes [75]. Lipin-1A is mainly expressed in early states of differentiation of preadipocytes, but lipin-1B expression increases with differentiation and is predominant in mature adipocytes. Lipin-1A induces the expression of adipogenic transcription factors peroxisome proliferator-activated receptor gamma (PPARγ) and CCAAT-enhancer-binding protein-alpha (C/EBPα), whereas lipin-1B more effectively induces lipogenic genes such as fatty acid synthase [75]. Studies on lipin-1-deficient mice and tissue-specific lipin-1 transgenic mice showed that lipin-1 is required for adipocyte differentiation. Lipin influences fat mass and energy balance in adipose tissues and skeletal muscle, respectively [76]. To further demonstrate the distinct role of lipin-1A and lipin-1B in vivo and disruption of the relative abundance of lipin-1A and lipin-1B, splice-specific knockout lipin-1A and lipin-1B mice shall be generated.
As mentioned in the introduction, a recent study demonstrated the downregulation of the expression of many RNA processing genes, including SFRS10 (also known as transformer 2 beta homolog TRA2B) in key metabolic organs, liver, and skeletal muscle of obese subjects [28]. Sfrs10 heterozygous mice were generated to explore the role of RNA splicing factor in obesity-related lipogenesis. Interestingly, LPIN1 is a splicing target of SFRS10. Reduced SFRS10 favors the expression of lipogenic lipin-1B [28]. Sirtuin 1 (SIRT1), the key coordinator of the metabolic response to caloric restriction, also plays a regulatory role in the expression of lipin-1B [77]. Ethanol inhibited the expression of SIRT1 which leads to reduced SFRS10 mRNA and protein expression levels in liver [77]. In agreement with previous findings, reduction of SFRS10 increases in expression of lipin-1B in parallel with a decrease in the lipin-1A isoform [28]. Hepatic SIRT1-SFRS10-LIPIN-1A/B axis was explored in the pathogenesis of alcoholic fatty liver disease [77]. Obesity-associated downregulation of SIRT1 in adipose tissues of obese subjects were reported [78,79]. It is interesting to further explore if the SIRT1-SFRS10-LIPIN-1A/B regulatory axis is conserved in adipose tissues.
3. The Expression Level of Splicing Factors Altered in Obese Subjects
A number of examples of alternative splicing being changed by hormonal or metabolic signals have been reported [80]. For example, insulin signaling pathway may regulate alternative mRNA splicing via phosphorylation of the splicing factors serine/arginine (SR)-rich proteins and also heterogeneous nuclear ribonucleoproteins (HNRNP). In addition to the post-translational modifications, recent studies demonstrated that expression of several RNA processing genes was altered in various organs of obese subjects [29,81,82]. Further investigation on the dysregulation of splicing machinery components of obesity may provide novel diagnostic and therapeutic tools for this pandemic non-communicable disease.
3.1. RNA Binding Protein, Fox-1 Homolog 2
RNA Binding Protein, Fox-1 Homolog 2 (RBFOX2), encodes an RNA binding protein which binds to a conserved element (U)GCAUG stretch in regulated exons or in flanking introns [83], and promotes recruitment of U1 snRNP to the 5′ Splice site leading to inclusion of the alternative exon in the mature transcript [84]. Recent studies demonstrated that RBFOX2 contributes to transcriptome changes under diabetic conditions [81,82]. A genome wide analysis on the alternative splicing profiles of the heart of Type 1 diabetic mouse was changed with a corresponding increase in RBFOX2 protein levels [81]. PKC was identified as regulators of alternative mRNA splicing via the phosphorylation of RBFOX2 [81]. In diabetic heart, the activation of PKC increases the phosphorylation of RBFOX2 which leads to reactivation of fetal alternative splicing programs [81]. Inhibition of PKC activity reduces the steady state levels of RBFOX2 protein [81].
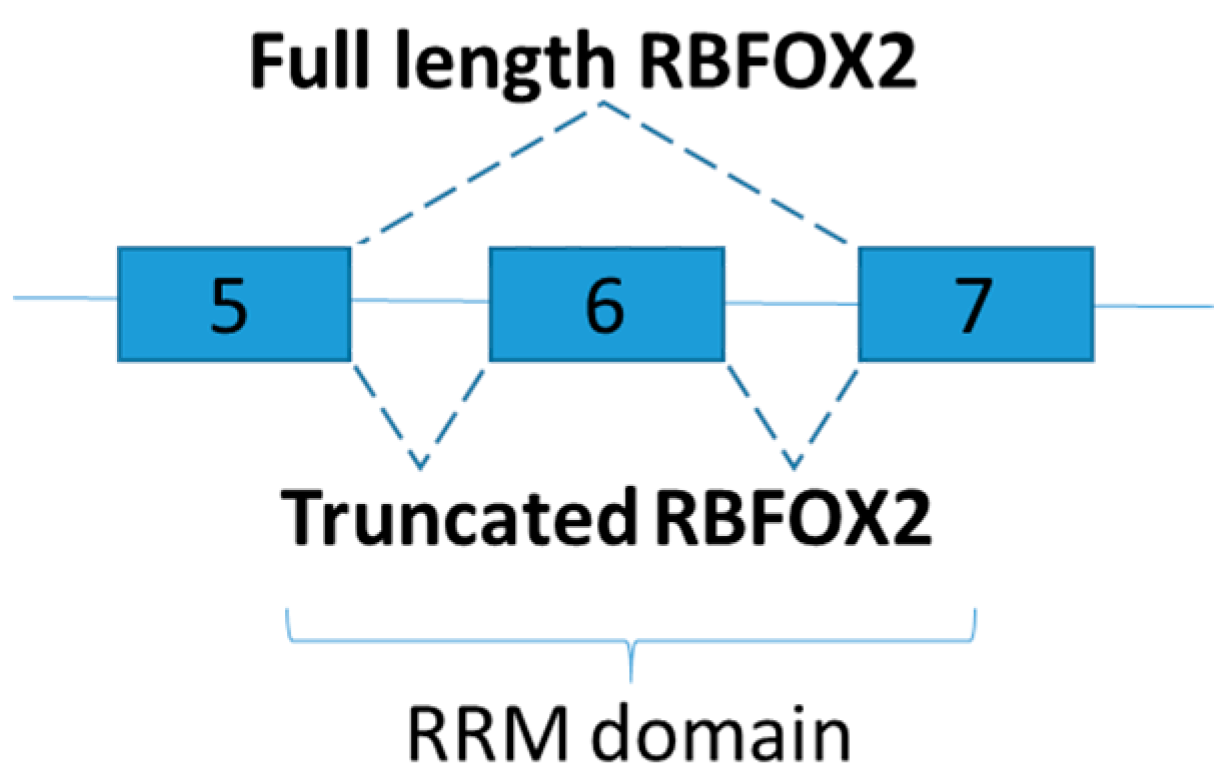
A subsequent investigation demonstrated that RBFOX2 targets more than 70% of mis-spliced pre-mRNAs of the diabetic hearts [82]. Consistent with the study mentioned above [81], RBFOX2 protein levels increased during myocardial differentiation, but alternative splicing activity of RBFOX2 in diabetic hearts is surprisingly low [82]. The phenomenon is explained by the increase of a dominant negative version, instead of the full-length, of RBFOX2 in cardiac tissues of diabetic samples [82]. Dominant negative version of RBFOX2 is generated via exclusion of exon 6, which encodes half to the RNA recognition motif (Figure 6). As a result, dominant negative RBFOX2 has lower RNA binding capability than its full-length RBFOX2, and calcium handling in diabetic hearts is adversely affected by inhibiting RBFOX2 dependent splicing [82]. Although RBFOX proteins facilitate a significant number of the splicing of micro-exons in muscle [3], only five genes relevant to skeletal muscle physiology were found mis-spliced in skeletal muscles of diabetic type 1 despite an elevated RBFOX1 protein levels [78]. A genome wide analysis on the alternative splicing profiles of the skeletal muscle shall be performed. In addition, it is interesting to further explore whether truncated RBFOX2 could be found in human as RRM domain of human RBFOX2 also located on exon 5 to 7 [85].
3.2. Neuro-Oncological Ventral Antigen (NOVA) Splicing Factors
Exon skipping is the most common form of HFD-induced mis-splicing [29]. The phenomenon seemed to be adipose tissue specific, as very few HFD-induced mis-splicing were detected in the liver [29]. As NOVA potential binding sites were identified on the mis-spliced pre-mRNAs, NOVA cross-linking immunoprecipitation followed by sequencing (CLIP-seq) was performed to confirm the direct involvement of NOVA proteins in the HFD-induced exon inclusion [29].
NOVA1 and NOVA2 are highly homologous neuron specific RNA-bind proteins and function as alternative splicing regulators. Both NOVA1 and NOVA2 proteins are found in adipocytes [29,86]. HFD treatment decreased NOVA expression in adipose tissues. Adipocyte-specific NOVA deficiency mice were generated to explore the contribution of NOVA to metabolic regulation [29]. Similar to HFD treatment, exon skipping was the most common form of mis-splicing due to NOVA deficiency [29].
Targeting thermogenesis in adipose tissues is a potential strategy treating obesity [87,88,89]. Adipocytes deficient in the NOVA splicing factors displayed increased thermogenesis [29]. The finding is in agreement with the study that demonstrated action of NOVA1 as a brown-adipogenic repressor and manifested the regulation of RNA-binding motif protein 4a (RBM4a) on the expression of NOVA1 [86]. A recent publication showed that body temperature cycles drive rhythmic SR protein phosphorylation to control an alternative splicing program [90]. Change of 1 °C in body temperature is sufficient to induce a concerted splicing switch in a large group of functionally related genes [90]. It is interesting to further explore if alternative mRNA splicing can function as negative feedback system in the regulation of thermogenesis.
4. Alternative Splicing as a Therapeutic Target for Obesity
The section above summarizes the emerging evidences of detection of alternative mRNA mis-splicing in obese subjects and discuss the potential of mis-splicing as the root cause of the metabolic dysregulations. Targeting mis-splicing to treat human diseases, gene therapy is one of the approaches to fix the errors in the splicing process raised by mutated cis- and trans-regulatory elements [91]. Therapeutic strategies by small molecule modulators for post-translational modification of splicing factors, antisense oligonucleotides, and trans-splicing have been proposed as potential therapies [92,93]. Several spliceosome inhibitors have been used to treat cancer [94] but exert pronounced cytotoxic effects resulting in abnormal alternative splicing [95,96].
Remarkably, many Food and Drug Administration (FDA) approved marketed drugs, including metformin, affect the alternative splicing machinery [97,98]. Metformin is the first-line anti-diabetic drug which reduces hepatic glucose production, increases intestinal glucose utilization, increases GLP-1 production and alters the composition of the gut microbiota [99]. The proposed underlying molecular mechanisms of metformin are very complicated [100]. A recent study tested the effect of metformin in the treatment of a “spliceopathy-associated” disease, myotonic dystrophy type I (also known as Steinert’s diseases) [98]. Metformin was proposed to act as a modifier of alternative mRNA splicing of a subset of genes mis-splicing in myotonic dystrophy type 1 by activation of AMPK and downregulation of the expression of the RNA-binding protein 3 (RBM3) [98]. It is interesting to explore whether metformin can also fix the pathological mis-splicing in obesity.
In the previous section, we discussed the importance of lamin A/C mRNA level in energy metabolism [72,74]. LMNA luciferase reporter assay was developed for the screening of small molecules modulating SR protein activity [101]. A small molecule named as ABX300 was identified which can abrogate diet-induced obesity by modulating LMNA isoforms via serine and arginine rich splicing factor 1 (SRSF1) in HFD-fed mice [102]. SRSF1 also known as alternative splicing factor 1 (ASF1) and tends to promote exon skipping [101]. A previous study demonstrated that an indole derivative IDC16 inhibits HIV pre-mRNA splicing via targeting SRSF1 [103]. Based on the structure of IDC16, ABX300 was developed. Treatment of ABX300 reversed the mis-splicing induced by HFD potentially by directly binding to and inhibition of SRSF1 [102]. ABX300 also altered the metabolic rate or energy expenditure of mice by promoting the expression of the genes that prevent fat gain or induce fat loss when mice are on HFD [102]. Most importantly, ABX300 did not have any adverse effect on lean mice of normal weight during the study [102]. As the amino acid sequences of human and mouse SRSF1 are 100% identical, it is interesting to explore the translational potential of ABX300 in anti-obesity treatment.
Alternative splicing is regulated by differential splicing factors binding to cis-acting sequences in the pre-mRNA. Antisense oligonucleotides can act as “splice-switchers” by binding to splicing enhancer or splicing silencer elements on the pre-mRNA to modulate alternative splicing [104]. For instance, exon skipping can be induced by using antisense oligonucleotides that bind to the splicing enhancer sequence and create a steric hindrance that blocks the recruitment of stimulatory splicing factor. Vice versa, antisense oligonucleotides binding to splicing silencer elements can promote exon inclusion by preventing the recruitment of negative splicing factors [104].
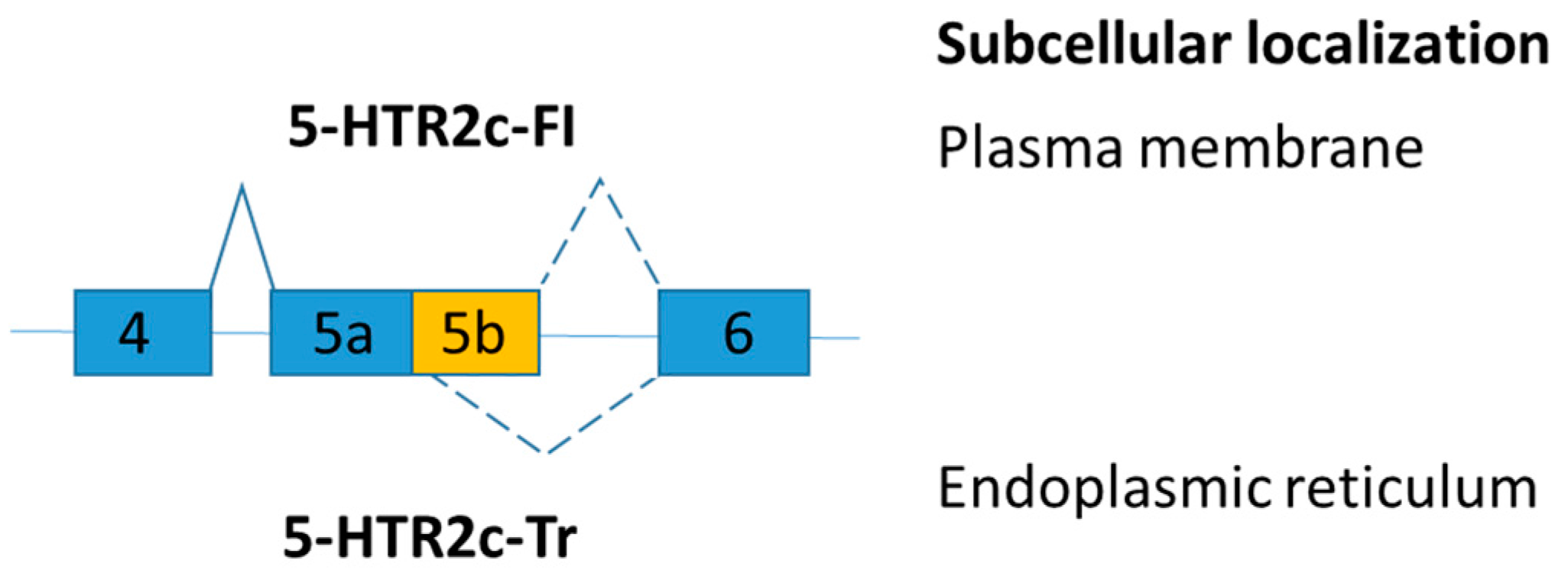
Serotonin 2C receptor (5-HTR2c) is involved in controlling appetite and food consumption. Alternative exon skipping generates a truncated 5-HTR2c protein isoform [105]. The transcript, including exon 5b, encodes the full-length serotonin 2C receptor (5-HTR2c-FI), but the transcript loss of exon 5b changes the amino acid reading frame and leads to the production of a truncated receptor (5-HTR2c-Tr) (Figure 7). The truncated serotonin 2C receptor (5-HTR2c-Tr) dimerizes with the full-length serotonin 2C receptor (5-HTR2c-FI) and prevents the full-length receptor to reach the plasma membrane [106]. Serotonin enhances satiety and hence reduces food intake [107]. As a result, endoplasmic reticulum retention of 5-HTR2c inhibits serotonin signaling [106].
A recent study demonstrated that a snoRNA, SNORD115, regulates the alternative mRNA splicing of serotonin 2C receptor [108] and is missing in patients with Prader-Willi syndrome [109]. The 18 nt oligonucleotide (complementarity against exon 5b and intron 5) promoted the inclusion of exon 5b into the pre-mRNA of 5-HTR2c, and reduced food uptake by increasing the ratio of 5-HTR2c-FI in a mouse model [110]. However, elevated anxiety and hypoactivity with overexpression of 5-HTR2c were reported when intracerebroventricular or carotid injection was used in the delivery of the oligonucleotide [110,111]. Improvement on oligonucleotide delivery is required and potential side effects of the oligonucleotide treatment shall be explored.
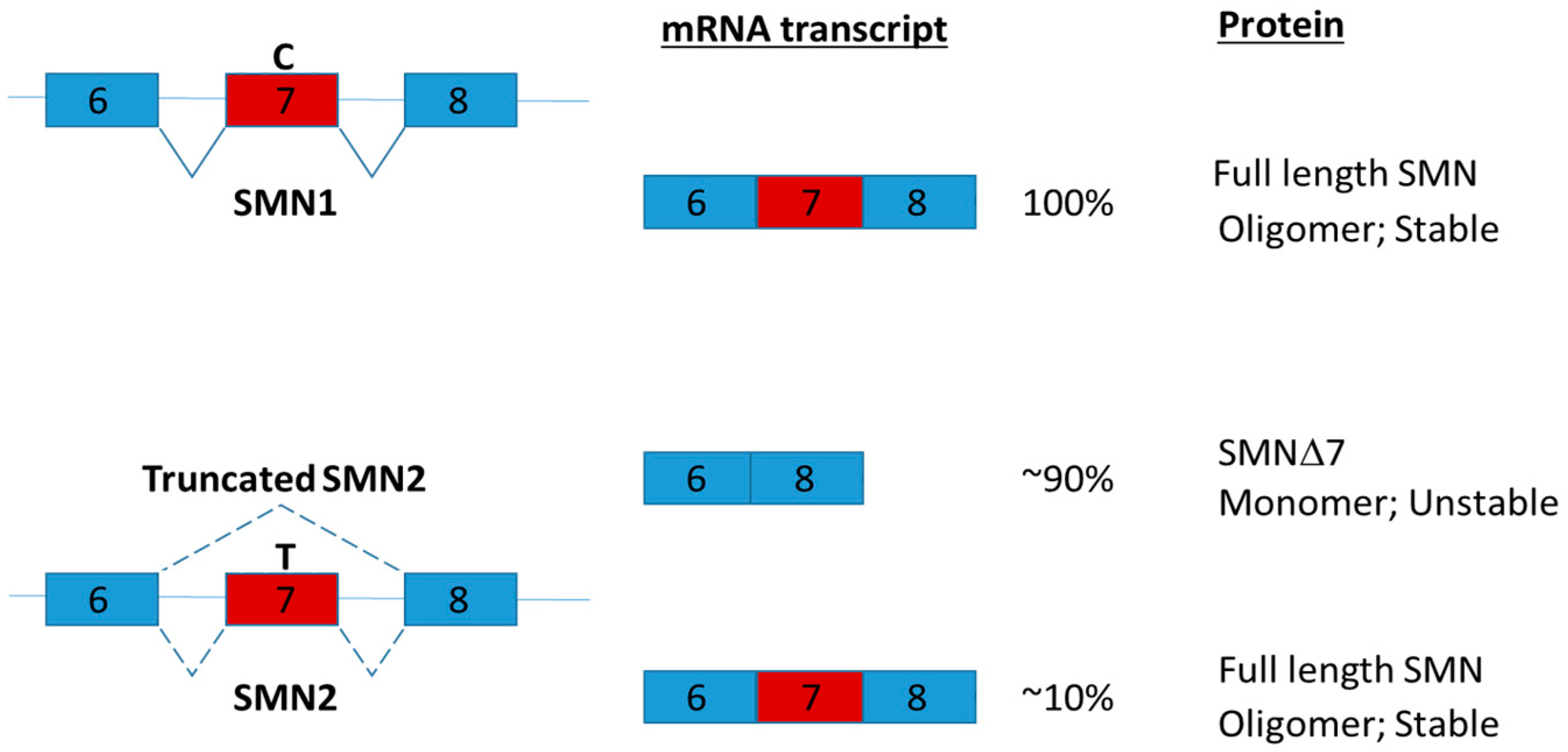
Intrathecal injection via lumber puncture is one of the promising ways to deliver antisense oligonucleotide to the central nervous system [112]. Nusinersen (marketed as Spinraza; a modified antisense oligonucleotide) was the first approved drug for Spinal Muscular Atrophy (SMA) in December 2016 [113]. SMA is a hereditary disease with global muscle atrophy caused by reduction of survival motor neuron (SMN) protein in spinal cord α-motor neurons [114]. SMN is involved in snRNP assembly, intron retention and DNA damage [115,116]. Reduction of SMN is proposed to disrupt the function of axons by affecting pre-mRNA splicing [117]. In human, two genes SMN1 and SMN2 encode SMN. SMN1 and SMN2 share almost identical amino acid sequences [118]. The critical difference between SMN1 and SMN2 is a single nucleotide difference in exon 7 which play a key role regulating the splicing of the genes [118]. Due to a single nucleotide change, the majority of SMN2 transcripts lack of exon 7 and produce truncated SMN proteins (Figure 8). The truncated SMN proteins are rapidly degraded and decrease the oligomerization efficiency [119]. Nusinersen promotes the inclusion of exon 7 by binding to intron splicing silencer-N1 and hence increasing the production of full length SMN2 protein [120]. Clinical trials in SMA patients have demonstrated that administration of nusinersen to the central nervous system using intrathecal injection significantly improved motor function [113].
As SMN is a ubiquitously expressed protein and metabolic dysregulations associated with SMA have been reported [123,124,125], heterozygous Smn-depleted [126] and severe SMA (Smn−/−; SMN2+/0) [127] mouse models had been employed to investigate the role of SMN in metabolism. Interestingly, metabolic function of Smn-depleted mice is indistinguishable from the wild type. However, after metabolically challenged with a high-fat diet, Smn(+/−)mice display abnormal localization of glucagon-producing alpha-cells within the pancreatic islets, increased number of insulin-producing beta cells, hyperinsulinemia and increased hepatic glucagon sensitivity [126]. In addition, in the severe SMA mouse model, subcutaneous administration of antisense oligonucleotide, which restored SMN expression, also restored the expression of hepatic IGF1 in SMA mice to normal levels [127]. The findings strongly suggested the importance of SMN in the metabolism of peripheral tissues. It is interesting to further explore the therapeutic potential of nusinersen in the treatment of metabolic diseases such as fatty acid metabolism and glucose homeostasis by systemic administration [128].
5. Conclusions
There is a growing interest in the role of alternative mRNA splicing in obese-related metabolic dysregulation. In Table 2, we summarized the splice variants described in this review known to play in metabolic diseases. The examples of alternative mRNA splicing contributed to obese-related diseases cited in this review are far from comprehensive. As recent studies demonstrated, dramatic change of splice variants is detected under different environmental and pathological conditions. Findings provide new information in the development of novel therapeutics by fixing splicing dysregulation. Further investigation on the mechanism of producing and functional consequence of the splice variants are required. The splice-specific KO mice and transgenic mice overexpressing particular splice variant are important tools for investigating unique function of each alternatively spliced variants.
Many approaches have been proposed to manipulate splicing. Use of antisense oligonucleotides is an attractive therapeutic approach for the treatment of diseases related to mis-splicing. The stability and delivery of antisense oligonucleotides are greatly improved by the recent advancement in chemically modifications of the oligonucleotides and delivery methods [104,129]. Numerous antisense oligonucleotides have progressed to human clinical trial for diseases such as muscular dystrophy [130]. While antisense oligonucleotides are still struggling their ways to the clinic, major challenges include off-target effects, efficacy, and immune system activation [131]. Interestingly, many marketed drugs also regulate the alternative splicing machinery, the contribution of those marketed drugs for the treatment of diseases in targeting alternative RNA splicing remains to be explored. Massive researches are still required in the future.
Acknowledgments
This work was supported by funding from National Natural Science Foundation of China (31271361) and financial support from the Hong Kong Polytechnic University (Internal grant 1.55.XX.99ZK and SFHMRF1718) (to C.M.W).
Author Contributions
Mabel Yau, Lu Xu, and Chi-Ming Wong wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 5-HTR2c | Serotonin 2C receptor |
| HFD | High fat diet |
| IR | Insulin receptor |
| KO | Knockout |
| NCoR | Nuclear Receptor Corepressor |
| NOVA | Neuro-Oncological Ventral Antigen |
| nt | Nucleotides |
| Ob-R | Obesity receptor |
| RBFOX2 | RNA Binding Protein, Fox-1 Homolog 2 |
| SIRT1 | Sirtuin 1 |
| SMA | Spinal muscular atrophy |
| SMN | Survival motor neuron |
| SR | serine/arginine |
| SRSF1 | Serine and Arginine Rich Splicing Factor 1 |
| SRSF3 | Serine and Arginine Rich Splicing Factor 3 |
| LDL | Low-density lipoprotein |
References
- Black, D.L. Protein diversity from alternative splicing: A challenge for bioinformatics and post-genome biology. Cell 2000, 103, 367–370. [Google Scholar] [CrossRef]
- Piovesan, A.; Caracausi, M.; Antonaros, F.; Pelleri, M.C.; Vitale, L. GeneBase 1.1: A tool to summarize data from NCBI gene datasets and its application to an update of human gene statistics. Database 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.I.; Sanchez-Pulido, L.; Haerty, W.; Ponting, C.P. RBFOX and PTBP1 proteins regulate the alternative splicing of micro-exons in human brain transcripts. Genome Res. 2015, 25, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wieringa, B.; Hofer, E.; Weissmann, C. A minimal intron length but no specific internal sequence is required for splicing the large rabbit beta-globin intron. Cell 1984, 37, 915–925. [Google Scholar] [CrossRef]
- Shimada, M.K.; Sasaki-Haraguchi, N.; Mayeda, A. Identification and Validation of Evolutionarily Conserved Unusually Short Pre-mRNA Introns in the Human Genome. Int. J. Mol. Sci. 2015, 16, 10376–10388. [Google Scholar] [CrossRef] [PubMed]
- Abebrese, E.L.; Ali, S.H.; Arnold, Z.R.; Andrews, V.M.; Armstrong, K.; Burns, L.; Crowder, H.R.; Day, R.T., Jr.; Hsu, D.G.; Jarrell, K.; et al. Identification of human short introns. PLoS ONE 2017, 12, e0175393. [Google Scholar] [CrossRef] [PubMed]
- Casamassimi, A.; Federico, A.; Rienzo, M.; Esposito, S.; Ciccodicola, A. Transcriptome Profiling in Human Diseases: New Advances and Perspectives. Int. J. Mol. Sci. 2017, 18, 1652. [Google Scholar] [CrossRef] [PubMed]
- Tress, M.L.; Abascal, F.; Valencia, A. Alternative Splicing May Not Be the Key to Proteome Complexity. Trends Biochem. Sci. 2017, 42, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Ezkurdia, I.; Rodriguez, J.M.; Carrillo-de Santa Pau, E.; Vazquez, J.; Valencia, A.; Tress, M.L. Most highly expressed protein-coding genes have a single dominant isoform. J. Proteom. Res. 2015, 14, 1880–1887. [Google Scholar] [CrossRef] [PubMed]
- Yabas, M.; Elliott, H.; Hoyne, G.F. The Role of Alternative Splicing in the Control of Immune Homeostasis and Cellular Differentiation. Int. J. Mol. Sci. 2015, 17, 3. [Google Scholar] [CrossRef] [PubMed]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Colantoni, A.; Bianchi, V.; Gherardini, P.F.; Tomba, G.S.; Ausiello, G.; Helmer-Citterich, M.; Ferre, F. Alternative splicing tends to avoid partial removals of protein-protein interaction sites. BMC Genom. 2013, 14, 379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, B.S.; Choi, S.S. Introns: The Functional Benefits of Introns in Genomes. Genom. Inform. 2015, 13, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Tazi, J.; Bakkour, N.; Stamm, S. Alternative splicing and disease. Biochim. Biophys. Acta 2009, 1792, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Rio, D.C. Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef] [PubMed]
- Pan, K.; Lee, J.T.; Huang, Z.; Wong, C.M. Coupling and coordination in gene expression processes with pre-mRNA splicing. Int. J. Mol. Sci. 2015, 16, 5682–5696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Liu, J.; Huang, B.O.; Xu, Y.M.; Li, J.; Huang, L.F.; Lin, J.; Zhang, J.; Min, Q.H.; Yang, W.M.; et al. Mechanism of alternative splicing and its regulation. Biomed. Rep. 2015, 3, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Sterne-Weiler, T.; Sanford, J.R. Exon identity crisis: Disease-causing mutations that disrupt the splicing code. Genome Biol. 2014, 15, 201. [Google Scholar] [CrossRef] [PubMed]
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Chabot, B.; Shkreta, L. Defective control of pre-messenger RNA splicing in human disease. J. Cell Biol. 2016, 212, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Ussar, S.; Fujisaka, S.; Kahn, C.R. Interactions between host genetics and gut microbiome in diabetes and metabolic syndrome. Mol. Metab. 2016, 5, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kim, S.H.; Lee, M.S.; Kim, M.S. Epigenetic modification by dietary factors: Implications in metabolic syndrome. Mol. Asp. Med. 2017, 54, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.L.; Watza, D.; Findley, A.; Alazizi, A.; Wen, X.; Pai, A.A.; Pique-Regi, R.; Luca, F. Environmental perturbations lead to extensive directional shifts in RNA processing. PLoS Genet. 2017, 13, e1006995. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, V.E.; Kwo, J. Obesity: Physiologic changes and implications for preoperative management. BMC Anesthesiol. 2015, 15, 97. [Google Scholar] [CrossRef] [PubMed]
- Holly, A.C.; Melzer, D.; Pilling, L.C.; Fellows, A.C.; Tanaka, T.; Ferrucci, L.; Harries, L.W. Changes in splicing factor expression are associated with advancing age in man. Mech. Ageing Dev. 2013, 134, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.P.; Pilling, L.C.; Emond, F.; Flurkey, K.; Harrison, D.E.; Yuan, R.; Peters, L.L.; Kuchel, G.A.; Ferrucci, L.; Melzer, D.; et al. Changes in the expression of splicing factor transcripts and variations in alternative splicing are associated with lifespan in mice and humans. Aging Cell 2016, 15, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Latorre, E.; Harries, L.W. Splicing regulatory factors, ageing and age-related disease. Ageing Res. Rev. 2017, 36, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Pihlajamaki, J.; Lerin, C.; Itkonen, P.; Boes, T.; Floss, T.; Schroeder, J.; Dearie, F.; Crunkhorn, S.; Burak, F.; Jimenez-Chillaron, J.C.; et al. Expression of the splicing factor gene SFRS10 is reduced in human obesity and contributes to enhanced lipogenesis. Cell Metab. 2011, 14, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Vernia, S.; Edwards, Y.J.; Han, M.S.; Cavanagh-Kyros, J.; Barrett, T.; Kim, J.K.; Davis, R.J. An alternative splicing program promotes adipose tissue thermogenesis. Elife 2016, 5, e17672. [Google Scholar] [CrossRef] [PubMed]
- Kaminska, D.; Kakela, P.; Nikkola, E.; Venesmaa, S.; Ilves, I.; Herzig, K.H.; Kolehmainen, M.; Karhunen, L.; Kuusisto, J.; Gylling, H.; et al. Regulation of alternative splicing in human obesity loci. Obesity 2016, 24, 2033–2037. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Ebina, Y.; Ellis, L.; Jarnagin, K.; Edery, M.; Graf, L.; Clauser, E.; Ou, J.H.; Masiarz, F.; Kan, Y.W.; Goldfine, I.D.; et al. The human insulin receptor cDNA: The structural basis for hormone-activated transmembrane signalling. Cell 1985, 40, 747–758. [Google Scholar] [CrossRef]
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr. Rev. 2009, 30, 586–623. [Google Scholar] [CrossRef] [PubMed]
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr. Rev. 2017, 38, 379–431. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.; Norgaard-Pedersen, D.; Brandt, J.; Pettersson, I.; Slaaby, R. IGF1 and IGF2 specificities to the two insulin receptor isoforms are determined by insulin receptor amino acid 718. PLoS ONE 2017, 12, e0178885. [Google Scholar] [CrossRef] [PubMed]
- Sesti, G.; D’Alfonso, R.; Vargas Punti, M.D.; Frittitta, L.; Trischitta, V.; Liu, Y.Y.; Borboni, P.; Longhi, R.; Montemurro, A.; Lauro, R. Peptide-based radioimmunoassay for the two isoforms of the human insulin receptor. Diabetologia 1995, 38, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Sell, S.M.; Reese, D.; Ossowski, V.M. Insulin-inducible changes in insulin receptor mRNA splice variants. J. Biol. Chem. 1994, 269, 30769–30772. [Google Scholar] [PubMed]
- Mosthaf, L.; Vogt, B.; Haring, H.U.; Ullrich, A. Altered expression of insulin receptor types A and B in the skeletal muscle of non-insulin-dependent diabetes mellitus patients. Proc. Natl. Acad. Sci. USA 1991, 88, 4728–4730. [Google Scholar] [CrossRef] [PubMed]
- Sbraccia, P.; D’Adamo, M.; Leonetti, F.; Caiola, S.; Iozzo, P.; Giaccari, A.; Buongiorno, A.; Tamburrano, G. Chronic primary hyperinsulinaemia is associated with altered insulin receptor mRNA splicing in muscle of patients with insulinoma. Diabetologia 1996, 39, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Bodkin, N.L.; Ortmeyer, H.K.; Hansen, B.C.; Shuldiner, A.R. Hyperinsulinemia is associated with altered insulin receptor mRNA splicing in muscle of the spontaneously obese diabetic rhesus monkey. J. Clin. Investig. 1994, 94, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Bodkin, N.L.; Ortmeyer, H.K.; Zenilman, M.E.; Webster, N.J.; Hansen, B.C.; Shuldiner, A.R. Altered insulin receptor messenger ribonucleic acid splicing in liver is associated with deterioration of glucose tolerance in the spontaneously obese and diabetic rhesus monkey: Analysis of controversy between monkey and human studies. J. Clin. Endocrinol. Metab. 1996, 81, 1552–1556. [Google Scholar] [PubMed]
- Kaminska, D.; Hamalainen, M.; Cederberg, H.; Kakela, P.; Venesmaa, S.; Miettinen, P.; Ilves, I.; Herzig, K.H.; Kolehmainen, M.; Karhunen, L.; et al. Adipose tissue INSR splicing in humans associates with fasting insulin level and is regulated by weight loss. Diabetologia 2014, 57, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Besic, V.; Shi, H.; Stubbs, R.S.; Hayes, M.T. Aberrant liver insulin receptor isoform a expression normalises with remission of type 2 diabetes after gastric bypass surgery. PLoS ONE 2015, 10, e0119270. [Google Scholar] [CrossRef] [PubMed]
- Escribano, O.; Beneit, N.; Rubio-Longas, C.; Lopez-Pastor, A.R.; Gomez-Hernandez, A. The Role of Insulin Receptor Isoforms in Diabetes and Its Metabolic and Vascular Complications. J. Diabetes Res. 2017, 2017, 1403206. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Talukdar, I.; Webster, N.J. SRp20 and CUG-BP1 modulate insulin receptor exon 11 alternative splicing. Mol. Cell. Biol. 2009, 29, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Jumaa, H.; Webster, N.J. Splicing factor SRSF3 is crucial for hepatocyte differentiation and metabolic function. Nat. Commun. 2013, 4, 1336. [Google Scholar] [CrossRef] [PubMed]
- Gorska, E.; Popko, K.; Stelmaszczyk-Emmel, A.; Ciepiela, O.; Kucharska, A.; Wasik, M. Leptin receptors. Eur. J. Med. Res. 2010, 15, 50–54. [Google Scholar] [PubMed]
- Fei, H.; Okano, H.J.; Li, C.; Lee, G.H.; Zhao, C.; Darnell, R.; Friedman, J.M. Anatomic localization of alternatively spliced leptin receptors (Ob-R) in mouse brain and other tissues. Proc. Natl. Acad. Sci. USA 1997, 94, 7001–7005. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.H.; Proenca, R.; Montez, J.M.; Carroll, K.M.; Darvishzadeh, J.G.; Lee, J.I.; Friedman, J.M. Abnormal splicing of the leptin receptor in diabetic mice. Nature 1996, 379, 632–635. [Google Scholar] [CrossRef] [PubMed]
- Uotani, S.; Bjorbaek, C.; Tornoe, J.; Flier, J.S. Functional properties of leptin receptor isoforms: Internalization and degradation of leptin and ligand-induced receptor downregulation. Diabetes 1999, 48, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Sahai, A.; Malladi, P.; Pan, X.; Paul, R.; Melin-Aldana, H.; Green, R.M.; Whitington, P.F. Obese and diabetic db/db mice develop marked liver fibrosis in a model of nonalcoholic steatohepatitis: Role of short-form leptin receptors and osteopontin. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G1035–G1043. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Ioffe, E.; Fidahusein, N.; Connolly, E.; Friedman, J.M. Absence of soluble leptin receptor in plasma from dbPas/dbPas and other db/db mice. J. Biol. Chem. 1998, 273, 10078–10082. [Google Scholar] [CrossRef] [PubMed]
- Aubert, R.; Herzog, J.; Camus, M.C.; Guenet, J.L.; Lemonnier, D. Description of a new model of genetic obesity: The dbPas mouse. J. Nutr. 1985, 115, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Van Dielen, F.M.; van’t Veer, C.; Buurman, W.A.; Greve, J.W. Leptin and soluble leptin receptor levels in obese and weight-losing individuals. J. Clin. Endocrinol. Metab. 2002, 87, 1708–1716. [Google Scholar] [CrossRef] [PubMed]
- Maamra, M.; Bidlingmaier, M.; Postel-Vinay, M.C.; Wu, Z.; Strasburger, C.J.; Ross, R.J. Generation of human soluble leptin receptor by proteolytic cleavage of membrane-anchored receptors. Endocrinology 2001, 142, 4389–4393. [Google Scholar] [CrossRef] [PubMed]
- Clement, K.; Vaisse, C.; Lahlou, N.; Cabrol, S.; Pelloux, V.; Cassuto, D.; Gourmelen, M.; Dina, C.; Chambaz, J.; Lacorte, J.M.; et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature 1998, 392, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Cornelis, M.C.; Kraft, P.; Qi, L.; van Dam, R.M.; Girman, C.J.; Laurie, C.C.; Mirel, D.B.; Gong, H.; Sheu, C.C.; et al. Genome-wide association study identifies polymorphisms in LEPR as determinants of plasma soluble leptin receptor levels. Hum. Mol. Genet. 2010, 19, 1846–1855. [Google Scholar] [CrossRef] [PubMed]
- Schaab, M.; Kratzsch, J. The soluble leptin receptor. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ceccarini, G.; Eisenstein, M.; Tan, K.; Friedman, J.M. Phenotypic effects of an induced mutation of the ObRa isoform of the leptin receptor. Mol. Metab. 2013, 2, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Guenther, M.G.; Barak, O.; Lazar, M.A. The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol. Cell. Biol. 2001, 21, 6091–6101. [Google Scholar] [CrossRef] [PubMed]
- Goodson, M.L.; Mengeling, B.J.; Jonas, B.A.; Privalsky, M.L. Alternative mRNA splicing of corepressors generates variants that play opposing roles in adipocyte differentiation. J. Biol. Chem. 2011, 286, 44988–44999. [Google Scholar] [CrossRef] [PubMed]
- Goodson, M.L.; Young, B.M.; Snyder, C.A.; Schroeder, A.C.; Privalsky, M.L. Alteration of NCoR corepressor splicing in mice causes increased body weight and hepatosteatosis without glucose intolerance. Mol. Cell. Biol. 2014, 34, 4104–4114. [Google Scholar] [CrossRef] [PubMed]
- Snyder, C.A.; Goodson, M.L.; Schroeder, A.C.; Privalsky, M.L. Regulation of corepressor alternative mRNA splicing by hormonal and metabolic signaling. Mol. Cell. Endocrinol. 2015, 413, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Alenghat, T.; Meyers, K.; Mullican, S.E.; Leitner, K.; Adeniji-Adele, A.; Avila, J.; Bucan, M.; Ahima, R.S.; Kaestner, K.H.; Lazar, M.A. Nuclear receptor corepressor and histone deacetylase 3 govern circadian metabolic physiology. Nature 2008, 456, 997–1000. [Google Scholar] [CrossRef] [PubMed]
- Astapova, I.; Vella, K.R.; Ramadoss, P.; Holtz, K.A.; Rodwin, B.A.; Liao, X.H.; Weiss, R.E.; Rosenberg, M.A.; Rosenzweig, A.; Hollenberg, A.N. The nuclear receptor corepressor (NCoR) controls thyroid hormone sensitivity and the set point of the hypothalamic-pituitary-thyroid axis. Mol. Endocrinol. 2011, 25, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Williams, E.G.; Mouchiroud, L.; Canto, C.; Fan, W.; Downes, M.; Heligon, C.; Barish, G.D.; Desvergne, B.; Evans, R.M.; et al. NCoR1 is a conserved physiological modulator of muscle mass and oxidative function. Cell 2011, 147, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Fan, W.; Xu, J.; Lu, M.; Yamamoto, H.; Auwerx, J.; Sears, D.D.; Talukdar, S.; Oh, D.; Chen, A.; et al. Adipocyte NCoR knockout decreases PPARgamma phosphorylation and enhances PPARgamma activity and insulin sensitivity. Cell 2011, 147, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Worman, H.J. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J. Biol. Chem. 1993, 268, 16321–16326. [Google Scholar] [PubMed]
- Revechon, G.; Viceconte, N.; McKenna, T.; Sola Carvajal, A.; Vrtacnik, P.; Stenvinkel, P.; Lundgren, T.; Hultenby, K.; Franco, I.; Eriksson, M. Rare progerin-expressing preadipocytes and adipocytes contribute to tissue depletion over time. Sci. Rep. 2017, 7, 4405. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.; Comai, L. Recent advances in understanding the role of lamins in health and disease. F1000Research 2016, 5, 2536. [Google Scholar] [CrossRef] [PubMed]
- Worman, H.J.; Courvalin, J.C. How do mutations in lamins A and C cause disease? J. Clin. Investig. 2004, 113, 349–351. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.; Chacon, M.R.; Gutierrez, C.; Vilarrasa, N.; Gomez, J.M.; Caubet, E.; Megia, A.; Vendrell, J. LMNA mRNA expression is altered in human obesity and type 2 diabetes. Obesity 2008, 16, 1742–1748. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kieckhaefer, J.E.; Cao, K. Mouse models of laminopathies. Aging Cell 2013, 12, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Mejia, I.C.; de Toledo, M.; Chavey, C.; Lapasset, L.; Cavelier, P.; Lopez-Herrera, C.; Chebli, K.; Fort, P.; Beranger, G.; Fajas, L.; et al. Antagonistic functions of LMNA isoforms in energy expenditure and lifespan. EMBO Rep. 2014, 15, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Peterfy, M.; Phan, J.; Reue, K. Alternatively spliced lipin isoforms exhibit distinct expression pattern, subcellular localization, and role in adipogenesis. J. Biol. Chem. 2005, 280, 32883–32889. [Google Scholar] [CrossRef] [PubMed]
- Phan, J.; Reue, K. Lipin, a lipodystrophy and obesity gene. Cell Metab. 2005, 1, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Hu, M.; Liang, X.; Ajmo, J.M.; Li, X.; Bataller, R.; Odena, G.; Stevens, S.M., Jr.; You, M. Deletion of SIRT1 from hepatocytes in mice disrupts lipin-1 signaling and aggravates alcoholic fatty liver. Gastroenterology 2014, 146, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Gillum, M.P.; Kotas, M.E.; Erion, D.M.; Kursawe, R.; Chatterjee, P.; Nead, K.T.; Muise, E.S.; Hsiao, J.J.; Frederick, D.W.; Yonemitsu, S.; et al. SirT1 regulates adipose tissue inflammation. Diabetes 2011, 60, 3235–3245. [Google Scholar] [CrossRef] [PubMed]
- Kurylowicz, A.; Owczarz, M.; Polosak, J.; Jonas, M.I.; Lisik, W.; Jonas, M.; Chmura, A.; Puzianowska-Kuznicka, M. SIRT1 and SIRT7 expression in adipose tissues of obese and normal-weight individuals is regulated by microRNAs but not by methylation status. Int. J. Obes. 2016, 40, 1635–1642. [Google Scholar] [CrossRef] [PubMed]
- Chew, S.L. Alternative splicing of mRNA as a mode of endocrine regulation. Trends Endocrinol. Metab. TEM 1997, 8, 405–413. [Google Scholar] [CrossRef]
- Verma, S.K.; Deshmukh, V.; Liu, P.; Nutter, C.A.; Espejo, R.; Hung, M.L.; Wang, G.S.; Yeo, G.W.; Kuyumcu-Martinez, M.N. Reactivation of fetal splicing programs in diabetic hearts is mediated by protein kinase C signaling. J. Biol. Chem. 2013, 288, 35372–35386. [Google Scholar] [CrossRef] [PubMed]
- Nutter, C.A.; Jaworski, E.A.; Verma, S.K.; Deshmukh, V.; Wang, Q.; Botvinnik, O.B.; Lozano, M.J.; Abass, I.J.; Ijaz, T.; Brasier, A.R.; et al. Dysregulation of RBFOX2 Is an Early Event in Cardiac Pathogenesis of Diabetes. Cell Rep. 2016, 15, 2200–2213. [Google Scholar] [CrossRef] [PubMed]
- Kuroyanagi, H. Fox-1 family of RNA-binding proteins. Cell. Mol. Life Sci. CMLS 2009, 66, 3895–3907. [Google Scholar] [CrossRef] [PubMed]
- Misra, A.; Ou, J.; Zhu, L.J.; Green, M.R. Global Promotion of Alternative Internal Exon Usage by mRNA 3' End Formation Factors. Mol. Cell 2015, 58, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Arya, A.D.; Wilson, D.I.; Baralle, D.; Raponi, M. RBFOX2 protein domains and cellular activities. Biochem. Soc. Trans. 2014, 42, 1180–1183. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.C.; Lu, Y.H.; Liu, Y.R.; Lin, Y.J. RBM4a-regulated splicing cascade modulates the differentiation and metabolic activities of brown adipocytes. Sci. Rep. 2016, 6, 20665. [Google Scholar] [CrossRef] [PubMed]
- Schlein, C.; Heeren, J. Implications of thermogenic adipose tissues for metabolic health. Best Pract. Res. Clin. Endocrinol. Metab. 2016, 30, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Plutzky, J. Brown Fat and Browning for the Treatment of Obesity and Related Metabolic Disorders. Diabetes Metab. J. 2016, 40, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhong, L.; Lee, J.T.H.; Zhang, J.; Wu, D.; Geng, L.; Wang, Y.; Wong, C.M.; Xu, A. The FGF21-CCL11 Axis Mediates Beiging of White Adipose Tissues by Coupling Sympathetic Nervous System to Type 2 Immunity. Cell Metab. 2017, 26, 493–508. [Google Scholar] [CrossRef] [PubMed]
- Preussner, M.; Goldammer, G.; Neumann, A.; Haltenhof, T.; Rautenstrauch, P.; Muller-McNicoll, M.; Heyd, F. Body Temperature Cycles Control Rhythmic Alternative Splicing in Mammals. Mol. Cell 2017, 67, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Sune-Pou, M.; Prieto-Sanchez, S.; Boyero-Corral, S.; Moreno-Castro, C.; El Yousfi, Y.; Sune-Negre, J.M.; Hernandez-Munain, C.; Sune, C. Targeting Splicing in the Treatment of Human Disease. Genes 2017, 8, 87. [Google Scholar] [CrossRef] [PubMed]
- Douglas, A.G.; Wood, M.J. RNA splicing: Disease and therapy. Brief. Funct. Genom. 2011, 10, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Le, K.Q.; Prabhakar, B.S.; Hong, W.J.; Li, L.C. Alternative splicing as a biomarker and potential target for drug discovery. Acta Pharmacol. Sin. 2015, 36, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Effenberger, K.A.; Urabe, V.K.; Jurica, M.S. Modulating splicing with small molecular inhibitors of the spliceosome. Wiley Interdiscip. Rev. RNA 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Salton, M.; Misteli, T. Small Molecule Modulators of Pre-mRNA Splicing in Cancer Therapy. Trends Mol. Med. 2016, 22, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.O.; Morris, J.C.; Oltean, S.; Donaldson, L.F. Pharmacology of Modulators of Alternative Splicing. Pharmacol. Rev. 2017, 69, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Younis, I.; Berg, M.; Kaida, D.; Dittmar, K.; Wang, C.; Dreyfuss, G. Rapid-response splicing reporter screens identify differential regulators of constitutive and alternative splicing. Mol. Cell. Biol. 2010, 30, 1718–1728. [Google Scholar] [CrossRef] [PubMed]
- Laustriat, D.; Gide, J.; Barrault, L.; Chautard, E.; Benoit, C.; Auboeuf, D.; Boland, A.; Battail, C.; Artiguenave, F.; Deleuze, J.F.; et al. In Vitro and In Vivo Modulation of Alternative Splicing by the Biguanide Metformin. Mol. Ther. Nucleic Acids 2015, 4, e262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Foretz, M. Revisiting the mechanisms of metformin action in the liver. Ann. d’Endocrinol. 2013, 74, 123–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Krainer, A.R. Emerging functions of SRSF1, splicing factor and oncoprotein, in RNA metabolism and cancer. Mol. Cancer Res. MCR 2014, 12, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Santo, J.; Lopez-Herrera, C.; Apolit, C.; Bareche, Y.; Lapasset, L.; Chavey, C.; Capozi, S.; Mahuteau-Betzer, F.; Najman, R.; Fornarelli, P.; et al. Pharmacological modulation of LMNA SRSF1-dependent splicing abrogates diet-induced obesity in mice. Int. J. Obes. 2017, 41, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Bakkour, N.; Lin, Y.L.; Maire, S.; Ayadi, L.; Mahuteau-Betzer, F.; Nguyen, C.H.; Mettling, C.; Portales, P.; Grierson, D.; Chabot, B.; et al. Small-molecule inhibition of HIV pre-mRNA splicing as a novel antiretroviral therapy to overcome drug resistance. PLoS Pathog. 2007, 3, 1530–1539. [Google Scholar] [CrossRef] [PubMed]
- Havens, M.A.; Hastings, M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef] [PubMed]
- Isles, A.R. Htr2c Splice Variants and 5HT2CR-Mediated Appetite. Trends Endocrinol. Metab. TEM 2017, 28, 542–544. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.B.; Ramond, F.; Farrington, D.T.; Aguiar, A.S., Jr.; Chevarin, C.; Berthiau, A.S.; Caussanel, S.; Lanfumey, L.; Herrick-Davis, K.; Hamon, M.; et al. RNA splicing and editing modulation of 5-HT(2C) receptor function: Relevance to anxiety and aggression in VGV mice. Mol. Psychiatry 2013, 18, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Halford, J.C.; Harrold, J.A. 5-HT(2C) receptor agonists and the control of appetite. Handb. Exp. Pharmacol. 2012, 209, 349–356. [Google Scholar]
- Kishore, S.; Stamm, S. The snoRNA HBII-52 regulates alternative splicing of the serotonin receptor 2C. Science 2006, 311, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Galiveti, C.R.; Raabe, C.A.; Konthur, Z.; Rozhdestvensky, T.S. Differential regulation of non-protein coding RNAs from Prader-Willi Syndrome locus. Sci. Rep. 2014, 4, 6445. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Shen, M.; Gresch, P.J.; Ghamari-Langroudi, M.; Rabchevsky, A.G.; Emeson, R.B.; Stamm, S. Oligonucleotide-induced alternative splicing of serotonin 2C receptor reduces food intake. EMBO Mol. Med. 2016, 8, 878–894. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Stevenson, P.L.; Carter, R.N.; Maccoll, G.; French, K.L.; Simons, J.P.; Al-Shawi, R.; Kelly, V.; Chapman, K.E.; Holmes, M.C. Overexpression of 5-HT2C receptors in forebrain leads to elevated anxiety and hypoactivity. Eur. J. Neurosci. 2009, 30, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Hache, M.; Swoboda, K.J.; Sethna, N.; Farrow-Gillespie, A.; Khandji, A.; Xia, S.; Bishop, K.M. Intrathecal Injections in Children With Spinal Muscular Atrophy: Nusinersen Clinical Trial Experience. J. Child Neurol. 2016, 31, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Paton, D.M. Nusinersen: Antisense oligonucleotide to increase SMN protein production in spinal muscular atrophy. Drugs Today 2017, 53, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Liu, Y.H.; Sahashi, K.; Rigo, F.; Bennett, C.F.; Krainer, A.R. Motor neuron cell-nonautonomous rescue of spinal muscular atrophy phenotypes in mild and severe transgenic mouse models. Genes Dev. 2015, 29, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Sivaramakrishnan, M.; McCarthy, K.D.; Campagne, S.; Huber, S.; Meier, S.; Augustin, A.; Heckel, T.; Meistermann, H.; Hug, M.N.; Birrer, P.; et al. Binding to SMN2 pre-mRNA-protein complex elicits specificity for small molecule splicing modifiers. Nature commun. 2017, 8, 1476. [Google Scholar] [CrossRef] [PubMed]
- Jangi, M.; Fleet, C.; Cullen, P.; Gupta, S.V.; Mekhoubad, S.; Chiao, E.; Allaire, N.; Bennett, C.F.; Rigo, F.; Krainer, A.R.; et al. SMN deficiency in severe models of spinal muscular atrophy causes widespread intron retention and DNA damage. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E2347–E2356. [Google Scholar] [CrossRef] [PubMed]
- Burghes, A.H.; Beattie, C.E. Spinal muscular atrophy: Why do low levels of survival motor neuron protein make motor neurons sick? Nat. Rev. Neurosci. 2009, 10, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Monani, U.R.; Lorson, C.L.; Parsons, D.W.; Prior, T.W.; Androphy, E.J.; Burghes, A.H.; McPherson, J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999, 8, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Burnett, B.G.; Munoz, E.; Tandon, A.; Kwon, D.Y.; Sumner, C.J.; Fischbeck, K.H. Regulation of SMN protein stability. Mol. Cell. Biol. 2009, 29, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Maharshi, V.; Hasan, S. Nusinersen: The First Option beyond Supportive Care for Spinal Muscular Atrophy. Clin. Drug Investig. 2017, 37, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Swoboda, K.J.; Prior, T.W.; Scott, C.B.; McNaught, T.P.; Wride, M.C.; Reyna, S.P.; Bromberg, M.B. Natural history of denervation in SMA: Relation to age, SMN2 copy number, and function. Ann. Neurol. 2005, 57, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Scoto, M.; Finkel, R.S.; Mercuri, E.; Muntoni, F. Therapeutic approaches for spinal muscular atrophy (SMA). Gene Ther. 2017, 24, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Tein, I.; Sloane, A.E.; Donner, E.J.; Lehotay, D.C.; Millington, D.S.; Kelley, R.I. Fatty acid oxidation abnormalities in childhood-onset spinal muscular atrophy: Primary or secondary defect(s)? Pediatr. Neurol. 1995, 12, 21–30. [Google Scholar] [CrossRef]
- Crawford, T.O.; Sladky, J.T.; Hurko, O.; Besner-Johnston, A.; Kelley, R.I. Abnormal fatty acid metabolism in childhood spinal muscular atrophy. Ann. Neurol. 1999, 45, 337–343. [Google Scholar] [CrossRef]
- Bowerman, M.; Swoboda, K.J.; Michalski, J.P.; Wang, G.S.; Reeks, C.; Beauvais, A.; Murphy, K.; Woulfe, J.; Screaton, R.A.; Scott, F.W.; et al. Glucose metabolism and pancreatic defects in spinal muscular atrophy. Ann. Neurol. 2012, 72, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Bowerman, M.; Michalski, J.P.; Beauvais, A.; Murray, L.M.; DeRepentigny, Y.; Kothary, R. Defects in pancreatic development and glucose metabolism in SMN-depleted mice independent of canonical spinal muscular atrophy neuromuscular pathology. Hum. Mol. Genet. 2014, 23, 3432–3444. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Sahashi, K.; Rigo, F.; Hung, G.; Horev, G.; Bennett, C.F.; Krainer, A.R. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 2011, 478, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Simone, C.; Ramirez, A.; Bucchia, M.; Rinchetti, P.; Rideout, H.; Papadimitriou, D.; Re, D.B.; Corti, S. Is spinal muscular atrophy a disease of the motor neurons only: Pathogenesis and therapeutic implications? Cell. Mol. Life Sci. 2016, 73, 1003–1020. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. [Google Scholar] [CrossRef] [PubMed]
- Voit, T.; Topaloglu, H.; Straub, V.; Muntoni, F.; Deconinck, N.; Campion, G.; De Kimpe, S.J.; Eagle, M.; Guglieri, M.; Hood, S.; et al. Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): An exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol. 2014, 13, 987–996. [Google Scholar] [CrossRef]
- Reautschnig, P.; Vogel, P.; Stafforst, T. The notorious R.N.A. in the spotlight-drug or target for the treatment of disease. RNA Biol. 2017, 14, 651–668. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic figure representing insulin receptor (IR) splice variants. Exons 10–12 are represented by boxes. Alternative splicing sites are depicted by connections with dashed lines. The insulin receptor isoform A (IR-A) lacks exon 11 which codes for a 12-amino acid segment present at the C terminal of the alpha chain of the isoform B (IR-B). The ligand binding affinity of human IR-A and IR-B is expressed as IC50 values in picomol [35].
Figure 1.
Schematic figure representing insulin receptor (IR) splice variants. Exons 10–12 are represented by boxes. Alternative splicing sites are depicted by connections with dashed lines. The insulin receptor isoform A (IR-A) lacks exon 11 which codes for a 12-amino acid segment present at the C terminal of the alpha chain of the isoform B (IR-B). The ligand binding affinity of human IR-A and IR-B is expressed as IC50 values in picomol [35].
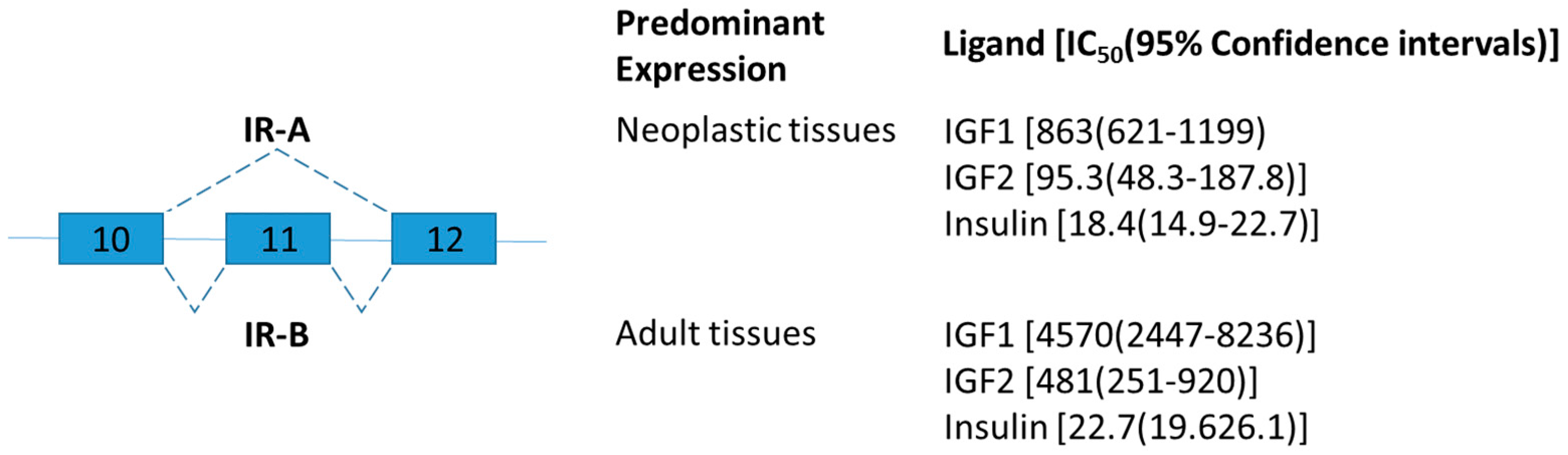
Figure 2.
Schematic figure representing leptin receptor (Ob-R) splice variants. Exons are represented by boxes. Alternative splicing sites are depicted by connections with dashed lines. Exon 17 encodes the trans-membrane domain (TM) [59].
Figure 2.
Schematic figure representing leptin receptor (Ob-R) splice variants. Exons are represented by boxes. Alternative splicing sites are depicted by connections with dashed lines. Exon 17 encodes the trans-membrane domain (TM) [59].
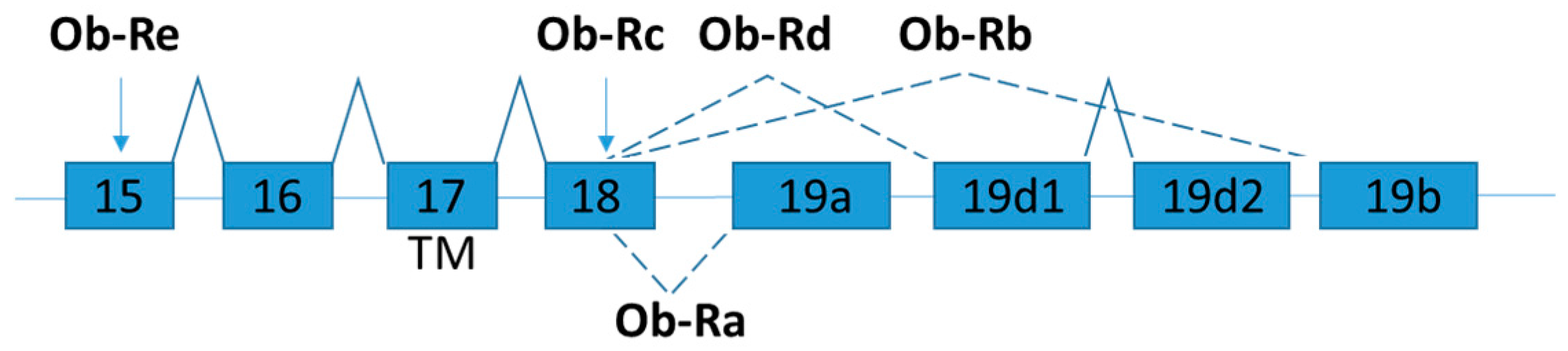
Figure 3.
Schematic figure representing nuclear receptor corepressor splice variants NCoRω and NCoRδ. Generation of NCoRω and NCoRδ splice variants are by alternatively splicing at two different 5′ splice donor sites on exon 37. The relative positions of receptor interaction domains (RIDs) coded by exon 37 and 38 are highlighted.
Figure 3.
Schematic figure representing nuclear receptor corepressor splice variants NCoRω and NCoRδ. Generation of NCoRω and NCoRδ splice variants are by alternatively splicing at two different 5′ splice donor sites on exon 37. The relative positions of receptor interaction domains (RIDs) coded by exon 37 and 38 are highlighted.

Figure 4.
Schematic figure representing LMNA splice variants. The part of exon 11 specific for lamin A is marked in red and the part of 10 specific for lamin C is marked in orange.
Figure 4.
Schematic figure representing LMNA splice variants. The part of exon 11 specific for lamin A is marked in red and the part of 10 specific for lamin C is marked in orange.
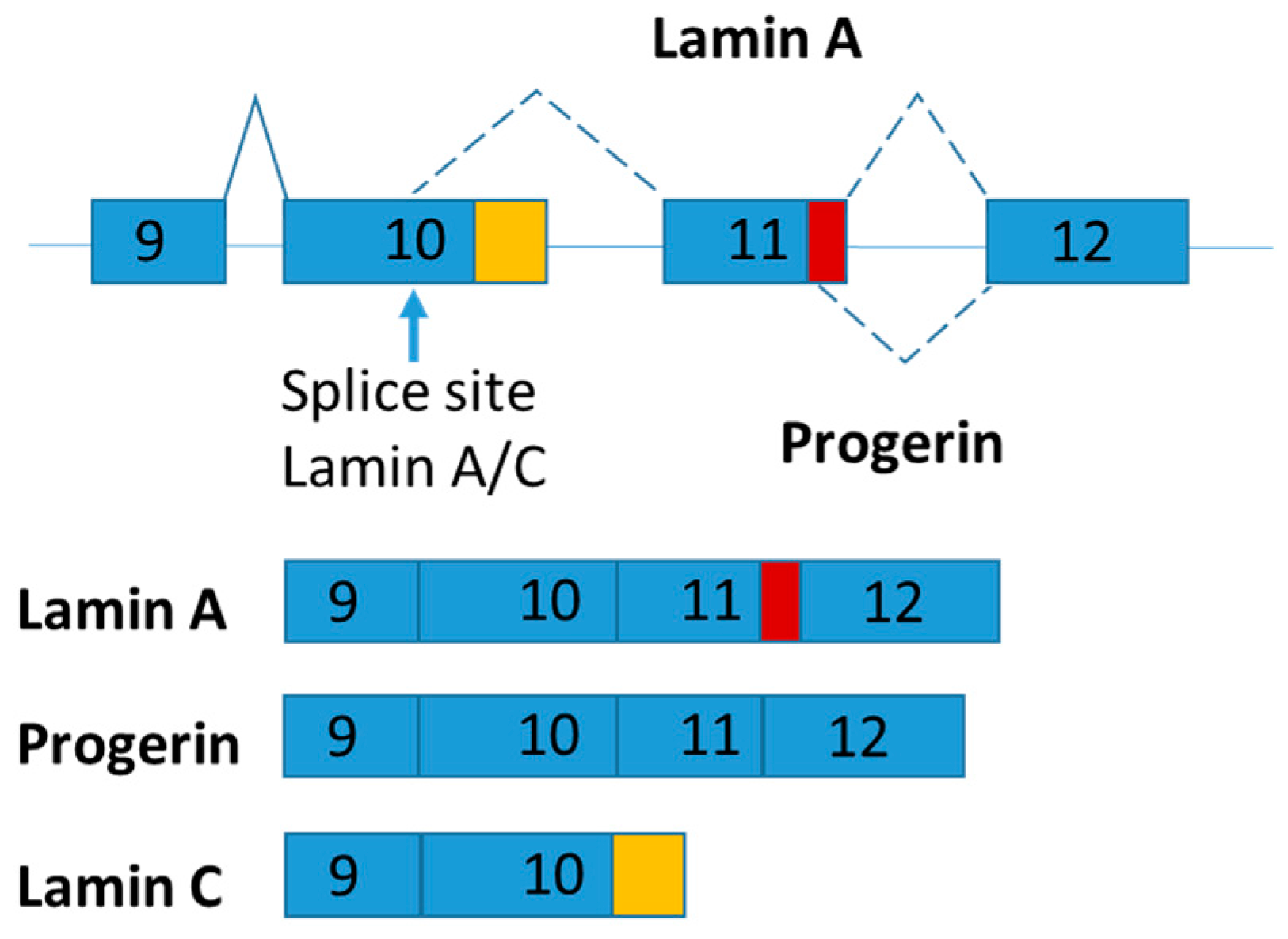
Figure 5.
Schematic figure representing lipin splice variants. Lipin-1B differs from lipin-1A by the presence of exon 7. The isoforms of LPIN1 serve distinct functions in adipocytes.
Figure 5.
Schematic figure representing lipin splice variants. Lipin-1B differs from lipin-1A by the presence of exon 7. The isoforms of LPIN1 serve distinct functions in adipocytes.
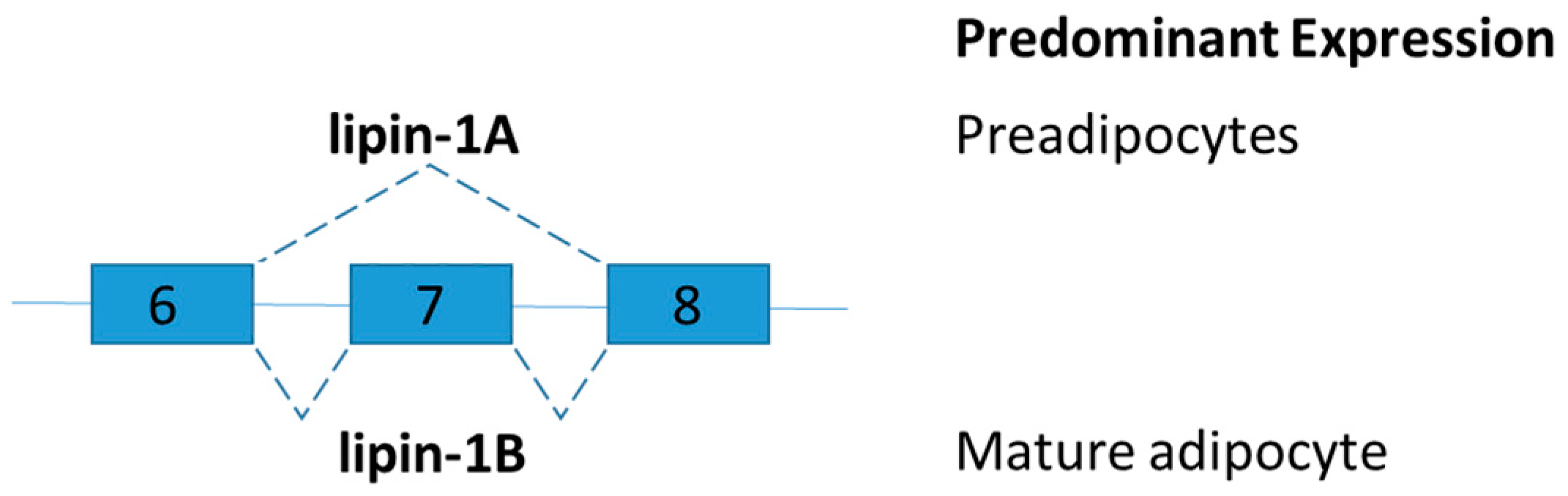
Figure 6.
Schematic figure representing RBFOX2 splice variants in the diabetic hearts of mice. As part of RNA recognition motif (RRM) domain is removed, exclusion of exon 6 generates dominant negative version of RBFOX2.
Figure 6.
Schematic figure representing RBFOX2 splice variants in the diabetic hearts of mice. As part of RNA recognition motif (RRM) domain is removed, exclusion of exon 6 generates dominant negative version of RBFOX2.

Figure 7.
Schematic figure representing serotonin 2C receptor splice variants. The splice variants have very different subcellular distribution.
Figure 7.
Schematic figure representing serotonin 2C receptor splice variants. The splice variants have very different subcellular distribution.

Figure 8.
Human SMN1 and SMN2 genes are located respectively in the telomeric and centromeric region of chromosome 5. The single nucleotide difference in exon 7 (C or T as indicated) of SMN1 and SMN2 gene affects their splicing. The single nucleotide change from C to T drastically reduces the efficiency of exon 7 inclusion and increase the production of the truncated mRNAs and proteins [121]. SMN has been shown to self-associate and functions as an oligomer [117]. The stability of SMN protein is highly influenced by oligomerization [119]. The splice variant lacking exon 7 (SMNΔ7) impairs oligomerization leading to rapid degradation of SMN [119]. SMN2 is present in all SMA patients. Splice intervention therapies promote SMN2 exon 7 retention offer a promising approach for SMA therapy by increasing the amount of full-length SMN2 transcript [122].
Figure 8.
Human SMN1 and SMN2 genes are located respectively in the telomeric and centromeric region of chromosome 5. The single nucleotide difference in exon 7 (C or T as indicated) of SMN1 and SMN2 gene affects their splicing. The single nucleotide change from C to T drastically reduces the efficiency of exon 7 inclusion and increase the production of the truncated mRNAs and proteins [121]. SMN has been shown to self-associate and functions as an oligomer [117]. The stability of SMN protein is highly influenced by oligomerization [119]. The splice variant lacking exon 7 (SMNΔ7) impairs oligomerization leading to rapid degradation of SMN [119]. SMN2 is present in all SMA patients. Splice intervention therapies promote SMN2 exon 7 retention offer a promising approach for SMA therapy by increasing the amount of full-length SMN2 transcript [122].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary of the different Nuclear Receptor Corepressor 1 (NCoR1) mouse model.
| Mouse Model | Key Metabolic Phenotypes | Proposed Mechanism | Reference |
|---|---|---|---|
| DADm: single amino acid substitution (Y478A) in the Ncor1 DAD domain that is unable to associate with or activate Hdac3 | Reduced weight and whole-body fat, Increased oxygen consumption and heat production Increased insulin sensitivity | Increased lipid consumption and obesity-resistant metabolic phenotype | [64] |
| NCoR ID : contains only 1 RID–N1 and thus would be unable to interact with the thyroid hormone receptor | Reduced body weight with a tendency for lower body fat content Increased oxygen consumption | Increased peripheral sensitivity to thyroid hormone | [65] |
| Muscle-specific KO | Increased of both muscle mass and of mitochondrial number and activity Reduced LDL cholesterol Improved insulin sensitivity Decreased in the respiratory exchange ratio | Increased muscle quantity and oxidative profile | [66] |
| Adipocyte-specific KO | Increased adipocyte hyperplasia Reduced inflammation in adipose tissue Increased insulin sensitivity in major metabolic organs (liver, fat and muscle) | Decreased inflammation contributing to the enhancement of insulin sensitivity | [67] |
| NCoRω−/−: NCoRω splice-specific knockout | Increased glucose tolerance Enhanced insulin resistance Increased the size of adipocytes Enhanced liver steatosis Elevated total serum cholesterol level and LDL complexes Reduced in the levels of circulating triglycerides and free fatty acids | Retention of the NCoRω splice variant counteracts prodiabetic physiology in the animals on the HFD | [62] |
Table 2.
Summary of the splice variants described in this review known to play a role in metabolic diseases.
Table 2.
Summary of the splice variants described in this review known to play a role in metabolic diseases.
| Gene | Changes of Variant Level in Pathological Conditions of Obese Subject | Key Changes in Metabolic Phenotypes | References |
|---|---|---|---|
| Insulin receptor (IR) | Increase IR-A (skipping of exon 11) in the liver | Correlate with fasting plasma glucose and insulin level | [41,42,43] |
| Leptin receptor (OB-R) | Decrease soluble OB-R * | Correlate with body mass index | [52,53,54] |
| LMNA | Increase Lamin C in subcutaneous adipose tissue | Correlate with type 2 diabetes | [72] |
| Lipin-1 | Increase Lipin 1-A (skipping of 7) in liver | Cause alcoholic fatty liver disease | [76] |
| RNA Binding Protein, Fox-1 Homolog 2 (RBFOX2) | Increase truncated RBFOX2 (skipping of exon 6) in heart | Lower calcium handling in diabetic heart | [81] |
| Serotonin 2C receptor (5-HTR2c) | Increase truncated 5-HTR2c (skipping of exon 5b) in brain | Reduce satiety and enhance food intake | [109] |
Remark: * The mechanisms of the soluble OB-R production in human and mouse are different [55].
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wong, C.-M.; Xu, L.; Yau, M.Y.-C. Alternative mRNA Splicing in the Pathogenesis of Obesity. Int. J. Mol. Sci. 2018, 19, 632. https://doi.org/10.3390/ijms19020632
AMA Style
Wong C-M, Xu L, Yau MY-C. Alternative mRNA Splicing in the Pathogenesis of Obesity. International Journal of Molecular Sciences. 2018; 19(2):632. https://doi.org/10.3390/ijms19020632
Chicago/Turabian StyleWong, Chi-Ming, Lu Xu, and Mabel Yin-Chun Yau. 2018. "Alternative mRNA Splicing in the Pathogenesis of Obesity" International Journal of Molecular Sciences 19, no. 2: 632. https://doi.org/10.3390/ijms19020632
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.