New Insights into the Immune Molecular Regulation of the Pathogenesis of Acute Respiratory Distress Syndrome


 and
and 
Abstract
:1. Introduction
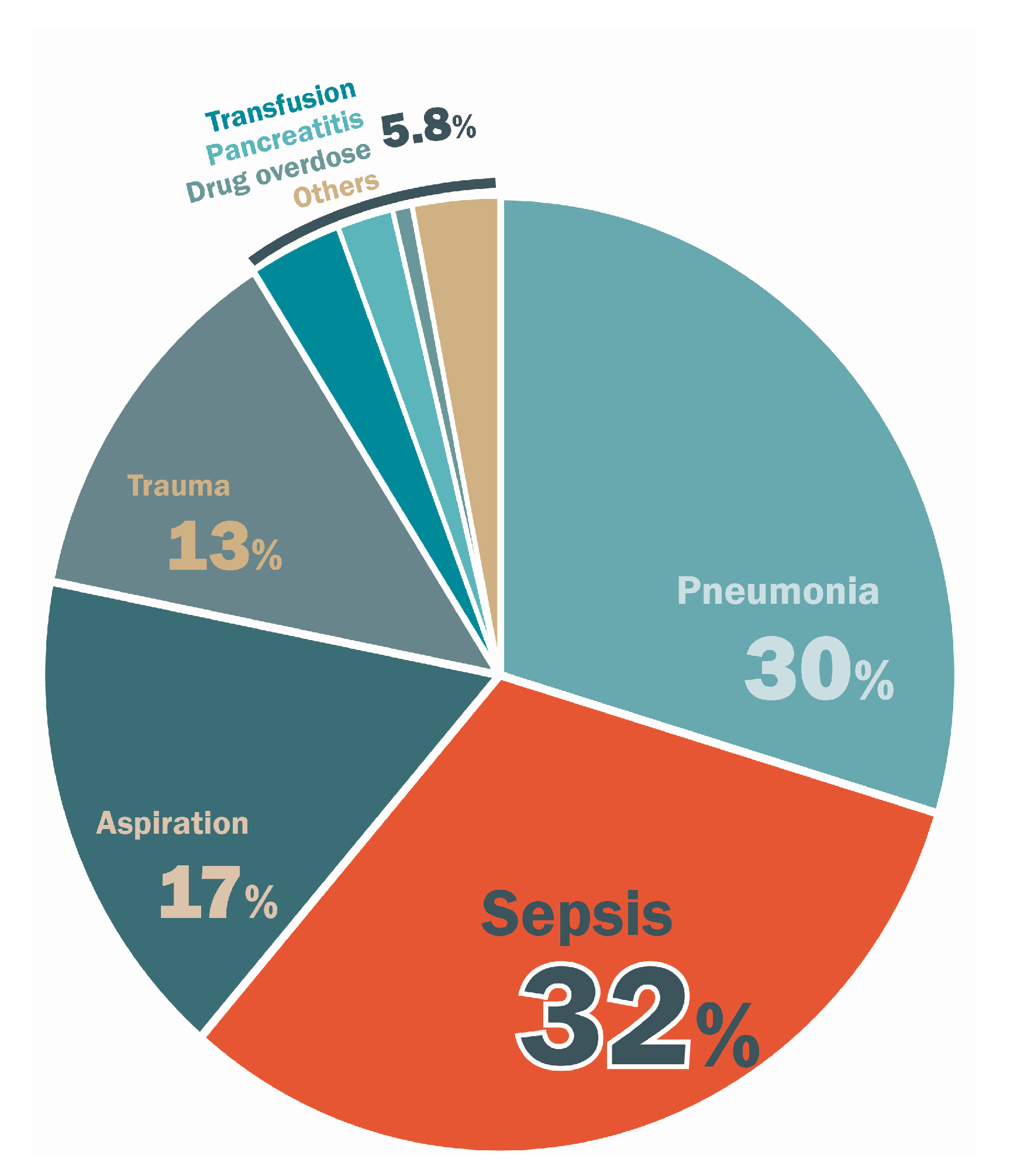
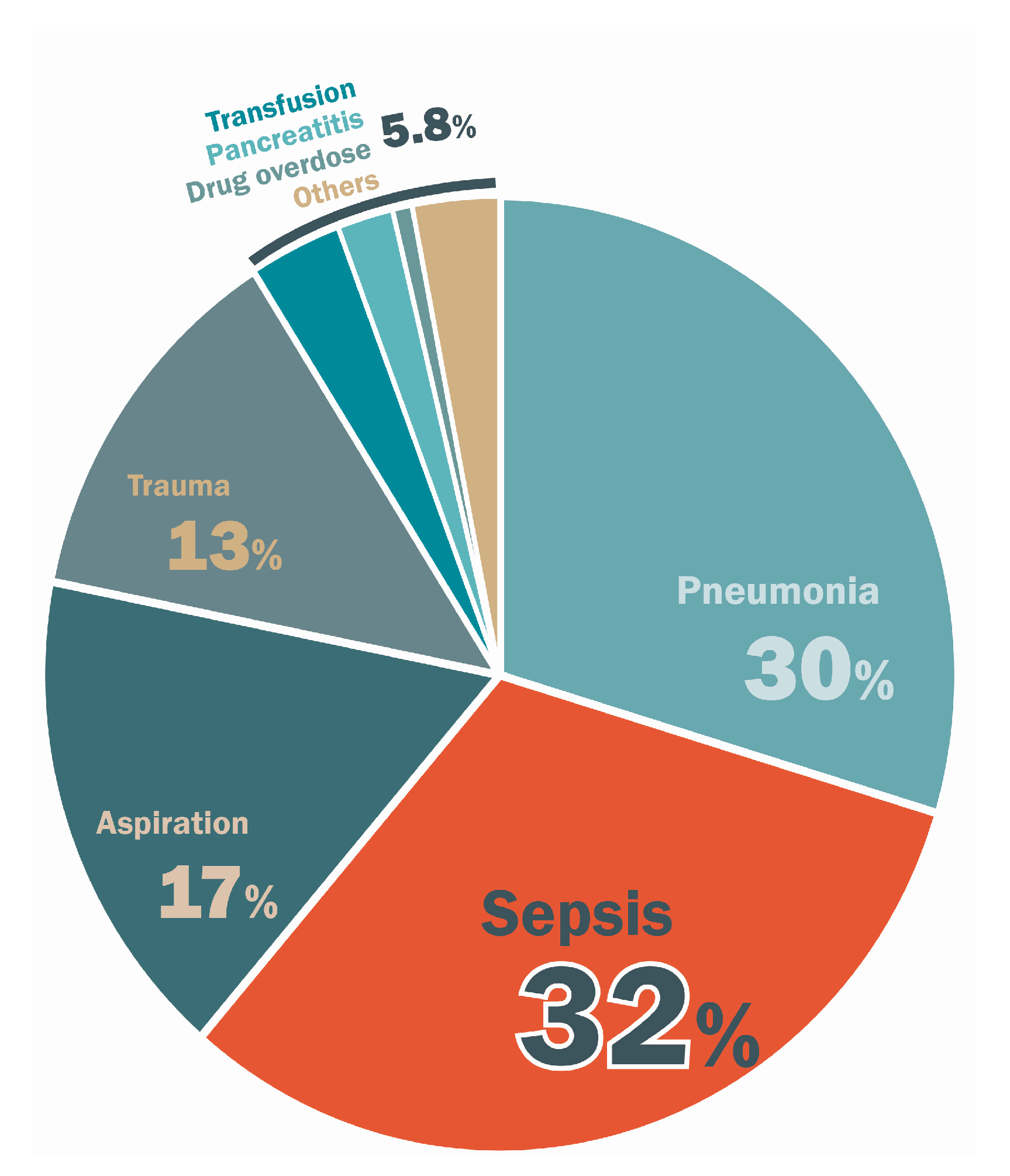
2. Epidemiologic and Clinical Features
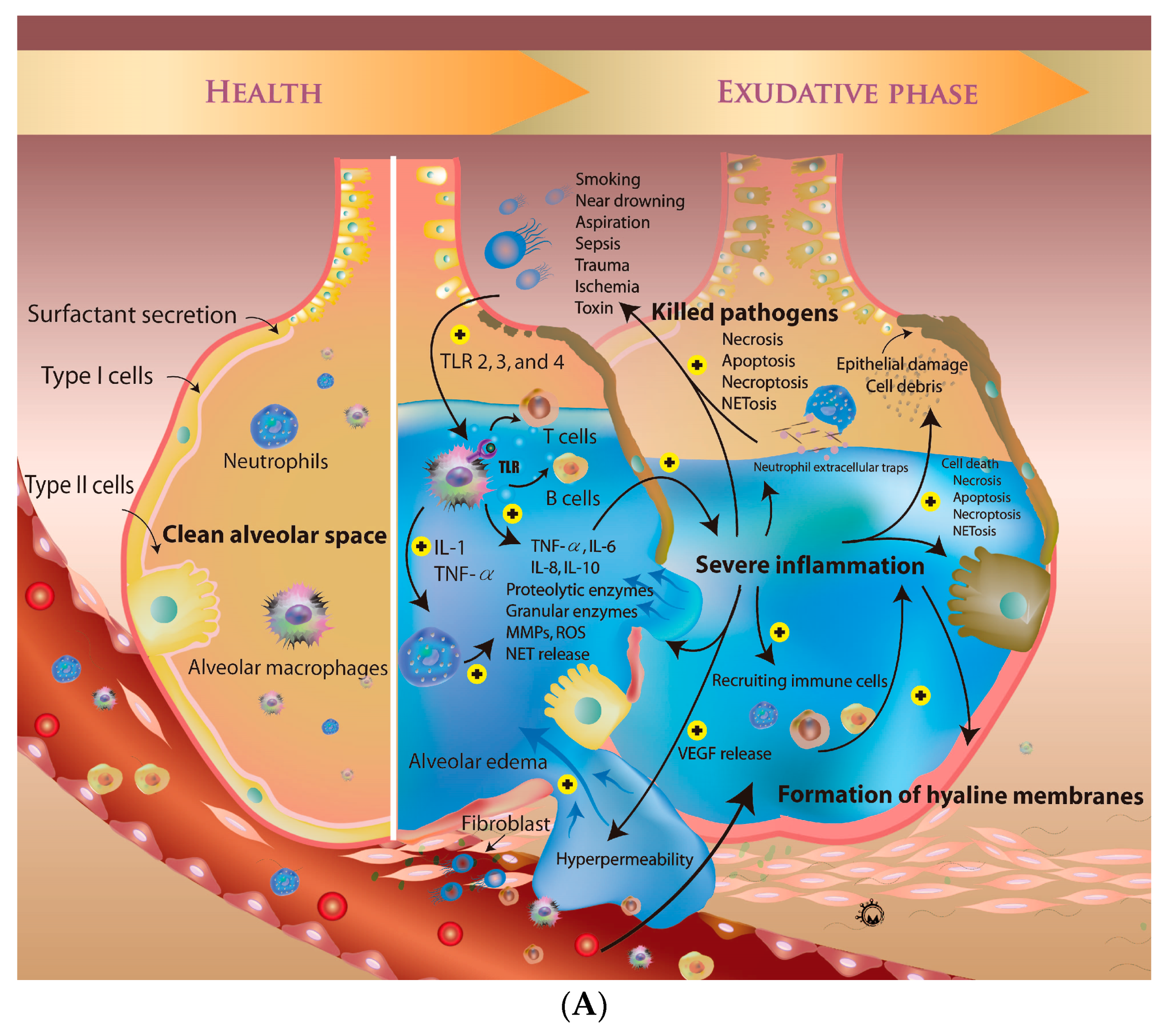
3. Overview of Pathogenesis of ARDS
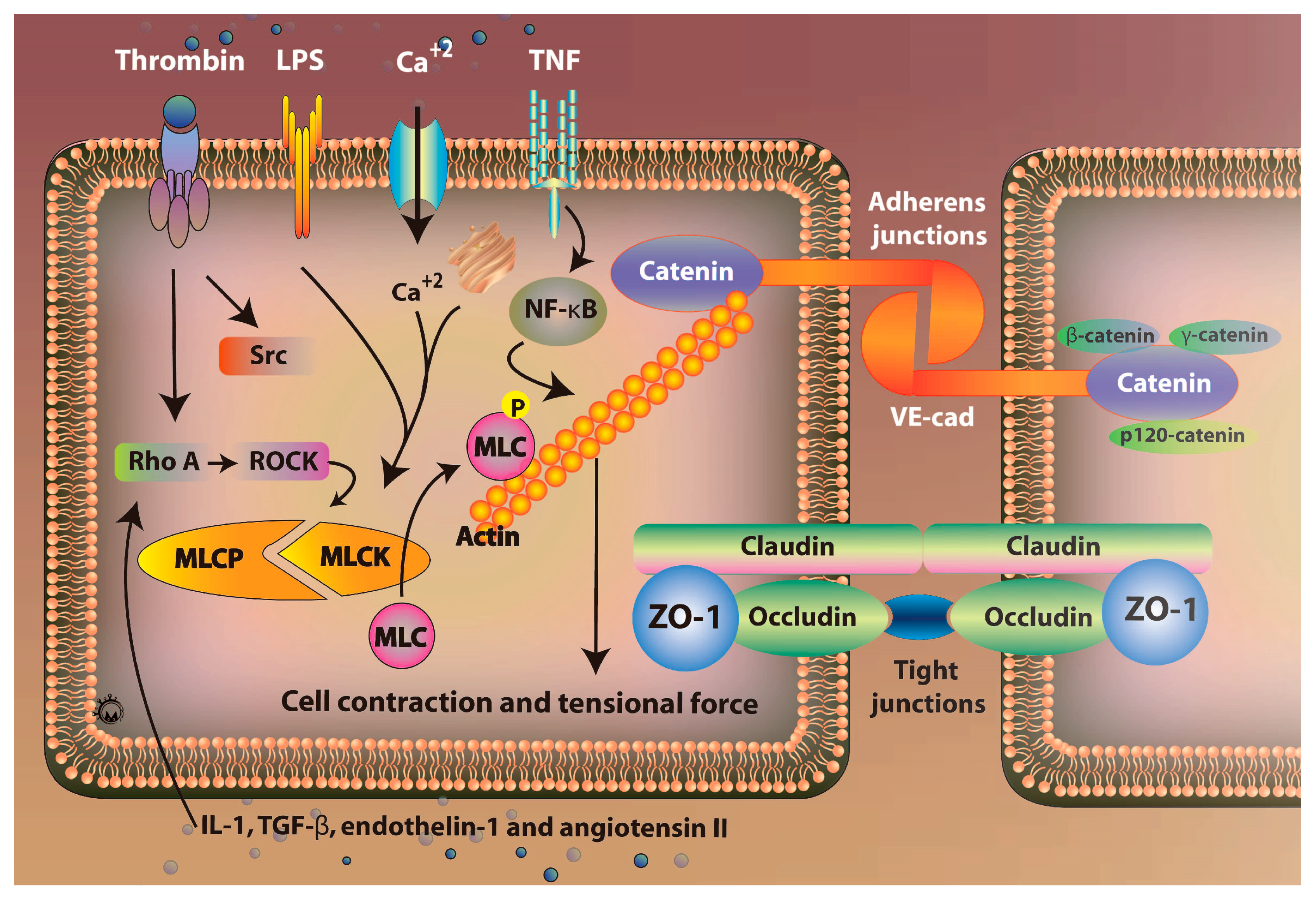
3.1. Mechanisms of Regulation of Vascular Permeability
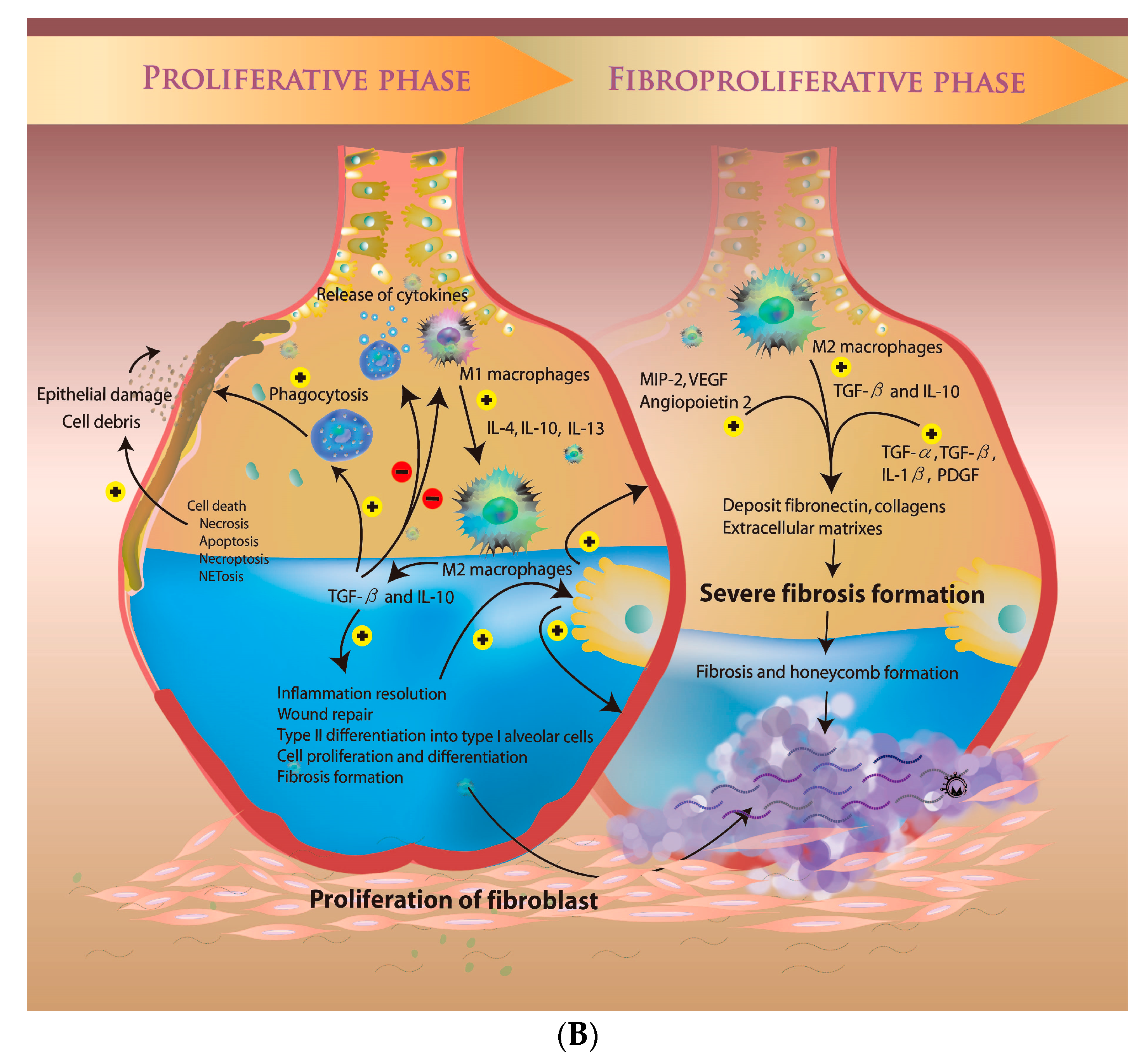
3.2. Microenvironment during the Proliferative Phase
3.3. The Fibroproliferative Phase of ARDS
4. The Micro-Inflammatory Response of ARDS
4.1. Role of Alveolar Macrophages in ARDS
4.2. Activated Neutrophils in ARDS
4.3. Role of T and B Cells in ARDS
5. Vascular Endothelial Barrier and Vascular Permeability
5.1. Role of Rho/ROCK Signaling Pathway
5.2. Role of Claudins Signaling Pathway
6. Therapeutic and Molecular Interventions
6.1. Corticosteroid
6.2. Angiotensin
6.3. mTOR
6.4. Neuromuscular Blockade
6.5. Vasodilators
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AECC | American-european consensus conference |
| AJ | Adherens junction |
| APC | Antigen-presenting cell |
| APCs | Antigen-presenting cells acute respiratory distress syndrome |
| AQP | Aquaporin |
| ARDS | Acute respiratory distress syndrome |
| CPAP | Continuous positive airway pressure |
| FiO2 | Inspiratory oxygen fraction |
| IL | Interleukin |
| iNO | Inhaled nitric oxide |
| MLC | Myosin light chain |
| MLKL | Mixed lineage kinase domain-like protein |
| MLCK | Myosin light chain kinase |
| MLCP | Myosin light chain phosphatase |
| NET | Eutrophil extracellular trap |
| NF-κB | Nuclear factor-κb |
| NKA | N+-K+-adenosine triphosphatase |
| PaO2 | Arterial oxygen tension |
| TNF | Tumor necrosis factor |
| PEEP | Positive end-expiratory pressure |
| RIP3 | Receptor-interacting protein kinase 3 |
| ROS | Reactive oxygen species |
| S1P | Sphingosine-1-phosphate |
| TGF | Transforming growth factor |
| TJs | Tight junctions |
| TNF | Tumor necrosis factor |
| VEGF | Vascular endothelial growth factor |
References
- Lee, K.Y. Pneumonia, Acute Respiratory Distress Syndrome, and Early Immune-Modulator Therapy. Int. J. Mol. Sci. 2017, 18, 388. [Google Scholar] [CrossRef] [PubMed]
- Ashbaugh, D.G.; Bigelow, D.B.; Petty, T.L.; Levine, B.E. Acute respiratory distress in adults. Lancet 1967, 2, 319–323. [Google Scholar] [CrossRef]
- Abrams, D.; Brodie, D. Extracorporeal membrane oxygenation for adult respiratory failure: 2017 update. Chest 2017, 152, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Confalonieri, M.; Salton, F.; Fabiano, F. Acute respiratory distress syndrome. Eur. Respir. Rev. 2017, 26. [Google Scholar] [CrossRef] [PubMed]
- Epelbaum, O.; Aronow, W.S. Mechanical ventilation in the acute respiratory distress syndrome. Hosp. Pract. 2017, 45, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; McAuley, D.F.; Ware, L.B. Clinical trials in acute respiratory distress syndrome: Challenges and opportunities. Lancet Respir. Med. 2017, 5, 524–534. [Google Scholar] [CrossRef]
- Meyer, N.J.; Calfee, C.S. Novel translational approaches to the search for precision therapies for acute respiratory distress syndrome. Lancet Respir. Med. 2017, 5, 512–523. [Google Scholar] [CrossRef]
- Brower, R.G.; Lanken, P.N.; MacIntyre, N.; Matthay, M.A.; Morris, A.; Ancukiewicz, M.; Schoenfeld, D.; Thompson, B.T. Higher versus lower positive end-expiratory pressures in patients with the acute respiratory distress syndrome. N. Engl. J. Med. 2004, 351, 327–336. [Google Scholar] [PubMed]
- Esteban, A.; Frutos-Vivar, F.; Muriel, A.; Ferguson, N.D.; Penuelas, O.; Abraira, V.; Raymondos, K.; Rios, F.; Nin, N.; Apezteguia, C.; et al. Evolution of mortality over time in patients receiving mechanical ventilation. Am. J. Respir. Crit. Care Med. 2013, 188, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Villar, J.; Blanco, J.; Anon, J.M.; Santos-Bouza, A.; Blanch, L.; Ambros, A.; Gandia, F.; Carriedo, D.; Mosteiro, F.; Basaldua, S.; et al. The Alien study: Incidence and outcome of acute respiratory distress syndrome in the era of lung protective ventilation. Intensive Care Med. 2011, 37, 1932–1941. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Calfee, C.S.; Paul, D.W.; Janz, D.R.; May, A.K.; Zhuo, H.; Bernard, G.R.; Matthay, M.A.; Ware, L.B.; Kangelaris, K.N. One-year mortality and predictors of death among hospital survivors of acute respiratory distress syndrome. Intensive Care Med. 2014, 40, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA 2016, 315, 788–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bersten, A.D.; Edibam, C.; Hunt, T.; Moran, J. Incidence and mortality of acute lung injury and the acute respiratory distress syndrome in three Australian states. Am. J. Respir. Crit. Care Med. 2002, 165, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Estenssoro, E.; Dubin, A.; Laffaire, E.; Canales, H.; Saenz, G.; Moseinco, M.; Pozo, M.; Gomez, A.; Baredes, N.; Jannello, G.; et al. Incidence, clinical course, and outcome in 217 patients with acute respiratory distress syndrome. Crit. Care Med. 2002, 30, 2450–2456. [Google Scholar] [CrossRef] [PubMed]
- MacCallum, N.S.; Evans, T.W. Epidemiology of acute lung injury. Curr. Opin. Crit. Care 2005, 11, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Rubenfeld, G.D.; Caldwell, E.; Peabody, E.; Weaver, J.; Martin, D.P.; Neff, M.; Stern, E.J.; Hudson, L.D. Incidence and outcomes of acute lung injury. N. Engl. J. Med. 2005, 353, 1685–1693. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.Y.; Hong, S.B. Sepsis and acute respiratory distress syndrome: Recent update. Tuberc. Respir. Dis. (Seoul) 2016, 79, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Hudson, L.D.; Milberg, J.A.; Anardi, D.; Maunder, R.J. Clinical risks for development of the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1995, 151, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Fanelli, V.; Vlachou, A.; Ghannadian, S.; Simonetti, U.; Slutsky, A.S.; Zhang, H. Acute respiratory distress syndrome: New definition, current and future therapeutic options. J. Thoracic Dis. 2013, 5, 326–334. [Google Scholar]
- Bernard, G.R.; Artigas, A.; Brigham, K.L.; Carlet, J.; Falke, K.; Hudson, L.; Lamy, M.; Legall, J.R.; Morris, A.; Spragg, R. The American-European consensus conference on Ards. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am. J. Respir. Crit. Care Med. 1994, 149, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, N.D.; Fan, E.; Camporota, L.; Antonelli, M.; Anzueto, A.; Beale, R.; Brochard, L.; Brower, R.; Esteban, A.; Gattinoni, L.; et al. The Berlin definition of Ards: An expanded rationale, justification, and supplementary material. Intensive Care Med. 2012, 38, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Walkey, A.J.; Summer, R.; Ho, V.; Alkana, P. Acute respiratory distress syndrome: Epidemiology and management approaches. Clin. Epidemiol. 2012, 4, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Greco, E.; Lupia, E.; Bosco, O.; Vizio, B.; Montrucchio, G. Platelets and multi-organ failure in sepsis. Int. J. Mol. Sci. 2017, 18, 2200. [Google Scholar] [CrossRef] [PubMed]
- Koh, Y. Update in acute respiratory distress syndrome. J. Intensive Care 2014, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Sharp, C.; Millar, A.B.; Medford, A.R. Advances in understanding of the pathogenesis of acute respiratory distress syndrome. Respiration 2015, 89, 420–434. [Google Scholar] [CrossRef] [PubMed]
- Ware, L.B.; Matthay, M.A. The acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1334–1349. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-Y.; Li, C.-J.; Hou, M.-F.; Chu, P.-Y. New insights into the role of inflammation in the pathogenesis of atherosclerosis. Int. J. Mol. Sci. 2017, 18, 2034. [Google Scholar] [CrossRef] [PubMed]
- Chavez, A.; Smith, M.; Mehta, D. Chapter six—New insights into the regulation of vascular permeability. In International Review of Cell and Molecular Biology; Jeon, K.W., Ed.; Academic Press: Cambridge, MA, USA, 2011; Volume 290, pp. 205–248. [Google Scholar]
- Fujishima, S. Pathophysiology and biomarkers of acute respiratory distress syndrome. J. Intensive Care 2014, 2, 32. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.G. Concepts in microvascular endothelial barrier regulation in health and disease. Microvasc. Res. 2009, 77, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Eklund, L.; Saharinen, P. Angiopoietin signaling in the vasculature. Exp. Cell Res. 2013, 319, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Dudek, S.M. Regulation of vascular permeability by sphingosine 1-phosphate. Microvasc. Res. 2009, 77, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Kendrick, S.F.W.; Jones, D.E.J. Mechanisms of innate immunity in sepsis. In Sepsis; Baudouin, S.V., Ed.; Springer: London, UK, 2008; pp. 5–10. [Google Scholar]
- Remick, D.G. Pathophysiology of sepsis. Am. J. Pathol. 2007, 170, 1435–1444. [Google Scholar] [CrossRef] [PubMed]
- Barratt, S.; Medford, A.R.; Millar, A.B. Vascular endothelial growth factor in acute lung injury and acute respiratory distress syndrome. Respiration 2014, 87, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Azamfirei, L.; Gurzu, S.; Solomon, R.; Copotoiu, R.; Copotoiu, S.; Jung, I.; Tilinca, M.; Branzaniuc, K.; Corneci, D.; Szederjesi, J.; et al. Vascular endothelial growth factor: A possible mediator of endothelial activation in acute respiratory distress syndrome. Minerva Anestesiol. 2010, 76, 609–616. [Google Scholar] [PubMed]
- Ourradi, K.; Blythe, T.; Jarrett, C.; Barratt, S.L.; Welsh, G.I.; Millar, A.B. Vegf isoforms have differential effects on permeability of human pulmonary microvascular endothelial cells. Respir. Res. 2017, 18, 116. [Google Scholar] [CrossRef] [PubMed]
- Matute-Bello, G.; Liles, W.C.; Steinberg, K.P.; Kiener, P.A.; Mongovin, S.; Chi, E.Y.; Jonas, M.; Martin, T.R. Soluble fas ligand induces epithelial cell apoptosis in humans with acute lung injury (Ards). J. Immunol. 1999, 163, 2217–2225. [Google Scholar] [PubMed]
- Galani, V.; Tatsaki, E.; Bai, M.; Kitsoulis, P.; Lekka, M.; Nakos, G.; Kanavaros, P. The role of apoptosis in the pathophysiology of acute respiratory distress syndrome (Ards): An up-to-date cell-specific review. Pathol. Res. Pract. 2010, 206, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Fialkow, L.; Fochesatto Filho, L.; Bozzetti, M.C.; Milani, A.R.; Rodrigues Filho, E.M.; Ladniuk, R.M.; Pierozan, P.; de Moura, R.M.; Prolla, J.C.; Vachon, E.; et al. Neutrophil apoptosis: A marker of disease severity in sepsis and sepsis-induced acute respiratory distress syndrome. Crit. Care 2006, 10, R155. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Yao, D.C.; Yu, Y.Z.; Li, S.J.; Chen, B.J.; Hu, G.H.; Xi, C.; Wang, Z.H.; Wang, H.Y.; Li, J.H.; et al. Necrostatin-1 protects against Oleic acid-induced acute respiratory distress syndrome in rats. Biochem. Biophys. Res. Commun. 2016, 478, 1602–1608. [Google Scholar] [CrossRef] [PubMed]
- Qing, D.Y.; Conegliano, D.; Shashaty, M.G.; Seo, J.; Reilly, J.P.; Worthen, G.S.; Huh, D.; Meyer, N.J.; Mangalmurti, N.S. Red blood cells induce necroptosis of lung endothelial cells and increase susceptibility to lung inflammation. Am. J. Respir. Crit. Care Med. 2014, 190, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, T.; Li, H.; Liu, Q.; Zhang, Z.; Xie, W.; Feng, Y.; Socorburam, T.; Wu, G.; Xia, Z.; et al. Receptor interacting protein 3-mediated necroptosis promotes lipopolysaccharide-induced inflammation and acute respiratory distress syndrome in mice. PLoS ONE 2016, 11, e0155723. [Google Scholar] [CrossRef] [PubMed]
- Burnham, E.L.; Janssen, W.J.; Riches, D.W.H.; Moss, M.; Downey, G.P. The fibroproliferative response in acute respiratory distress syndrome: Mechanisms and clinical significance. Eur. Respir. J. 2014, 43, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Hamada, N.; Kuwano, K.; Yamada, M.; Hagimoto, N.; Hiasa, K.; Egashira, K.; Nakashima, N.; Maeyama, T.; Yoshimi, M.; Nakanishi, Y. Anti-vascular endothelial growth factor gene therapy attenuates lung injury and fibrosis in mice. J. Immunol. 2005, 175, 1224–1231. [Google Scholar] [CrossRef] [PubMed]
- Keane, M.P.; Belperio, J.A.; Moore, T.A.; Moore, B.B.; Arenberg, D.A.; Smith, R.E.; Burdick, M.D.; Kunkel, S.L.; Strieter, R.M. Neutralization of the Cxc chemokine, macrophage inflammatory protein-2, attenuates bleomycin-induced pulmonary fibrosis. J. Immunol. 1999, 162, 5511–5518. [Google Scholar] [PubMed]
- Ou, X.M.; Li, W.C.; Liu, D.S.; Li, Y.P.; Wen, F.Q.; Feng, Y.L.; Zhang, S.F.; Huang, X.Y.; Wang, T.; Wang, K.; et al. Vegfr-2 antagonist Su5416 attenuates bleomycin-induced pulmonary fibrosis in mice. Int. Immunopharmacol. 2009, 9, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.M.; Mammoto, T.; Schultz, A.; Yuan, H.T.; Christiani, D.; Karumanchi, S.A.; Sukhatme, V.P. Excess circulating angiopoietin-2 may contribute to pulmonary vascular leak in sepsis in humans. PLoS Med. 2006, 3, e46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Network, T.A.R.D.S. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1301–1308. [Google Scholar]
- Tremblay, L.N.; Slutsky, A.S. Ventilator-induced lung injury: From the bench to the bedside. Intensive Care Med. 2006, 32, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, P.; Rocco, P.R. Effects of Mechanical Ventilation on the Extracellular Matrix. Intensive Care Med 2008, 34, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, L.N.; Miatto, D.; Hamid, Q.; Govindarajan, A.; Slutsky, A.S. Injurious ventilation induces widespread pulmonary epithelial expression of tumor necrosis factor-alpha and interleukin-6 messenger RNA. Crit. Care Med. 2002, 30, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Capelozzi, V.L.; Allen, T.C.; Beasley, M.B.; Cagle, P.T.; Guinee, D.; Hariri, L.P.; Husain, A.N.; Jain, D.; Lantuejoul, S.; Larsen, B.T.; et al. Molecular and immune biomarkers in acute respiratory distress syndrome: A perspective from members of the pulmonary pathology society. Arch. Pathol. Lab. Med. 2017, 141, 1719–1727. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, N.R.; King, L.S.; D’Alessio, F.R. Diverse macrophage populations mediate acute lung inflammation and resolution. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L709–L725. [Google Scholar] [CrossRef] [PubMed]
- Han, S.H.; Mallampalli, R.K. The acute respiratory distress syndrome: From mechanism to translation. J. Immunol. 2015, 194, 855–860. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, F.R.; Craig, J.M.; Singer, B.D.; Files, D.C.; Mock, J.R.; Garibaldi, B.T.; Fallica, J.; Tripathi, A.; Mandke, P.; Gans, J.H.; et al. Enhanced resolution of experimental Ards through Il-4-mediated lung macrophage reprogramming. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L733–L746. [Google Scholar] [CrossRef] [PubMed]
- Norman, J.G.; Fink, G.W.; Franz, M.G. Acute pancreatitis induces intrapancreatic tumor necrosis factor gene expression. Arch. Surg. 1995, 130, 966–970. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J. The immunopathogenesis of sepsis. Nature 2002, 420, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Wrigge, H.; Stüber, F.; Putensen, C. Ventilator-associated systemic inflammation. In Yearbook of Intensive Care and Emergency Medicine 2001; Vincent, J.-L., Ed.; Springer: Berlin/Heidelberg, Germany, 2001; pp. 35–43. [Google Scholar]
- Park, W.Y.; Goodman, R.B.; Steinberg, K.P.; Ruzinski, J.T.; Radella, F., 2nd; Park, D.R.; Pugin, J.; Skerrett, S.J.; Hudson, L.D.; Martin, T.R. Cytokine balance in the lungs of patients with acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2001, 164, 1896–1903. [Google Scholar] [CrossRef] [PubMed]
- Siler, T.M.; Swierkosz, J.E.; Hyers, T.M.; Fowler, A.A.; Webster, R.O. Immunoreactive interleukin-1 in bronchoalveolar lavage fluid of high-risk patients and patients with the adult respiratory distress syndrome. Exp. Lung Res. 1989, 15, 881–894. [Google Scholar] [CrossRef] [PubMed]
- Giri, S.N.; Hyde, D.M.; Hollinger, M.A. Effect of antibody to transforming growth factor beta on bleomycin induced accumulation of lung collagen in mice. Thorax 1993, 48, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Ihn, H. Pathogenesis of fibrosis: Role of Tgf-Beta and Ctgf. Curr. Opin. Rheumatol. 2002, 14, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Shull, M.M.; Ormsby, I.; Kier, A.B.; Pawlowski, S.; Diebold, R.J.; Yin, M.; Allen, R.; Sidman, C.; Proetzel, G.; Calvin, D.; et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature 1992, 359, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Broekelmann, T.J.; Limper, A.H.; Colby, T.V.; McDonald, J.A. Transforming growth factor beta 1 is present at sites of extracellular matrix gene expression in human pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 1991, 88, 6642–6646. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.; O’Garra, A. Biological properties of interleukin 10. Immunol. Today 1992, 13, 198–200. [Google Scholar] [CrossRef]
- Smith, S.R.; Terminelli, C.; Kenworthy-Bott, L.; Calzetta, A.; Donkin, J. The cooperative effects of Tnf-alpha and Ifn-gamma are determining factors in the ability of Il-10 to protect mice from lethal endotoxemia. J. Leukoc. Biol. 1994, 55, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, D.F.; Zlotnik, A.; Mosmann, T.R.; Howard, M.; O’Garra, A. Il-10 inhibits cytokine production by activated macrophages. J. Immunol. 1991, 147, 3815–3822. [Google Scholar] [PubMed]
- Kasama, T.; Strieter, R.M.; Lukacs, N.W.; Burdick, M.D.; Kunkel, S.L. Regulation of neutrophil-derived chemokine expression by Il-10. J. Immunol. 1994, 152, 3559–3569. [Google Scholar] [PubMed]
- Lo, C.-J.; Fu, M.; Cryer, H.G. Interleukin 10 inhibits alveolar macrophage production of inflammatory mediators involved in adult respiratory distress syndrome. J. Surg. Res. 1998, 79, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, L.; Millar, A.B. Relative production of tumour necrosis factor alpha and interleukin 10 in adult respiratory distress syndrome. Thorax 1997, 52, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.J.; Cohen, A.B.; Nagao, S.; Griffith, D.; Maunder, R.J.; Martin, T.R.; Weiner-Kronish, J.P.; Sticherling, M.; Christophers, E.; Matthay, M.A. Elevated levels of nap-1/interleukin-8 are present in the airspaces of patients with the adult respiratory distress syndrome and are associated with increased mortality. Am. Rev. Respir. Dis. 1992, 146, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Olson, T.S.; Ley, K. Chemokines and chemokine receptors in leukocyte trafficking. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 283, R7–R28. [Google Scholar] [CrossRef] [PubMed]
- Puneet, P.; Moochhala, S.; Bhatia, M. Chemokines in acute respiratory distress syndrome. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 288, L3–L15. [Google Scholar] [CrossRef] [PubMed]
- Cummings, C.J.; Martin, T.R.; Frevert, C.W.; Quan, J.M.; Wong, V.A.; Mongovin, S.M.; Hagen, T.R.; Steinberg, K.P.; Goodman, R.B. Expression and function of the chemokine receptors Cxcr1 and Cxcr2 in sepsis. J. Immunol. 1999, 162, 2341–2346. [Google Scholar] [PubMed]
- Chishti, A.D.; Dark, J.H.; Kesteven, P.; Powell, H.; Snowden, C.; Shenton, B.K.; Kirby, J.A.; Baudouin, S.V. Expression of chemokine receptors Cxcr1 and Cxcr2 during cardiopulmonary bypass. J. Thoracic Cardiovasc. Surg. 2001, 122, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Stillie, R.; Farooq, S.M.; Gordon, J.R.; Stadnyk, A.W. The functional significance behind expressing two Il-8 receptor types on Pmn. J. Leukoc. Biol. 2009, 86, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Summers, C.; Singh, N.R.; White, J.F.; Mackenzie, I.M.; Johnston, A.; Solanki, C.; Balan, K.K.; Peters, A.M.; Chilvers, E.R. Pulmonary retention of primed neutrophils: A novel protective host response, which is impaired in the acute respiratory distress syndrome. Thorax 2014, 69, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Grommes, J.; Soehnlein, O. Contribution of neutrophils to acute lung injury. Mol. Med. 2011, 17, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Buczek-Thomas, J.A.; Lucey, E.C.; Stone, P.J.; Chu, C.L.; Rich, C.B.; Carreras, I.; Goldstein, R.H.; Foster, J.A.; Nugent, M.A. Elastase mediates the release of growth factors from lung in vivo. Am. J. Respir. Cell Mol. Biol. 2004, 31, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yamashita, C.; Zemans, R.L.; Briones, N.; Van Linden, A.; Downey, G.P. Leukocyte elastase induces lung epithelial apoptosis via a Par-1–, Nf-κb–, and P53-dependent pathway. Am. J. Respir. Cell Mol. Biol. 2009, 41, 742–755. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.C.; Lin, H.C.; Liu, C.Y.; Wang, C.H.; Hwang, T.; Huang, T.T.; Lin, C.H.; Kuo, H.P. Neutrophil elastase induces Il-8 synthesis by lung epithelial cells via the mitogen-activated protein kinase pathway. J. Biomed. Sci. 2004, 11, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Witherden, I.R.; Vanden Bon, E.J.; Goldstraw, P.; Ratcliffe, C.; Pastorino, U.; Tetley, T.D. Primary human alveolar type II epithelial cell chemokine release: Effects of cigarette smoke and neutrophil elastase. Am. J. Respir. Cell Mol. Biol. 2004, 30, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, M.J. Neutrophil extracellular traps (nets): Double-edged swords of innate immunity. J. Immunol. 2012, 189, 2689–2695. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Dejana, E. Endothelial cell-cell junctions: Happy together. Nat. Rev. Mol. Cell Biol. 2004, 5, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Wennerberg, K.; Rossman, K.L.; Der, C.J. The ras superfamily at a glance. J. Cell Sci. 2005, 118, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Bustelo, X.R.; Sauzeau, V.; Berenjeno, I.M. Gtp-binding proteins of the Rho/Rac family: Regulation, effectors and functions in vivo. Bioessays 2007, 29, 356–370. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 2001, 1, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Amano, M.; Nakayama, M.; Kaibuchi, K. Rho-kinase/rock: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton 2010, 67, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Sawafuji, M.; Ishizaka, A.; Kohno, M.; Koh, H.; Tasaka, S.; Ishii, Y.; Kobayashi, K. Role of rho-kinase in reexpansion pulmonary edema in rabbits. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 289, L946–L953. [Google Scholar] [CrossRef] [PubMed]
- Paszti-Gere, E.; Csibrik-Nemeth, E.; Szeker, K.; Csizinszky, R.; Jakab, C.; Galfi, P. Acute oxidative stress affects Il-8 and Tnf-alpha expression in Ipec-J2 porcine epithelial cells. Inflammation 2012, 35, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Soini, Y. Claudins in lung diseases. Respir. Res. 2011, 12, 70. [Google Scholar] [CrossRef] [PubMed]
- Overgaard, C.E.; Daugherty, B.L.; Mitchell, L.A.; Koval, M. Claudins: Control of barrier function and regulation in response to oxidant stress. Antioxid. Redox. Signal. 2011, 15, 1179–1193. [Google Scholar] [CrossRef] [PubMed]
- Figueira, E.R.; Bacchella, T.; Coelho, A.M.; Sampietre, S.N.; Molan, N.A.; Leitao, R.M.; Machado, M.C. Timing-dependent protection of hypertonic saline solution administration in experimental liver ischemia/reperfusion injury. Surgery 2010, 147, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.; Verin, A.D.; Black, S.M.; Catravas, J.D. Regulators of endothelial and epithelial barrier integrity and function in acute lung injury. Biochem. Pharmacol. 2009, 77, 1763–1772. [Google Scholar] [CrossRef] [PubMed]
- Rokkam, D.; Lafemina, M.J.; Lee, J.W.; Matthay, M.A.; Frank, J.A. Claudin-4 levels are associated with intact alveolar fluid clearance in human lungs. Am. J. Pathol. 2011, 179, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, A.B.; Stager, M.A.; Carrico, C.J.; Hudson, L.D. Causes of mortality in patients with the adult respiratory distress syndrome. Am. Rev. Respir. Dis. 1985, 132, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.C.; Coalson, J.J.; Smith, J.D.; Johanson, W.G., Jr. Multiple organ system failure and infection in adult respiratory distress syndrome. Ann. Internal Med. 1983, 99, 293–298. [Google Scholar] [CrossRef]
- Anzueto, A.; Baughman, R.P.; Guntupalli, K.K.; Weg, J.G.; Wiedemann, H.P.; Raventos, A.A.; Lemaire, F.; Long, W.; Zaccardelli, D.S.; Pattishall, E.N. Aerosolized surfactant in adults with sepsis-induced acute respiratory distress syndrome. Exosurf acute respiratory distress syndrome sepsis study group. N. Engl. J. Med. 1996, 334, 1417–1421. [Google Scholar] [CrossRef] [PubMed]
- Khilnani, G.; Hadda, V. Corticosteroids and Ards: A review of treatment and prevention evidence. Lung India 2011, 28, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.W.; Zimmerman, J.L.; Dellinger, R.P.; Straube, R.C.; Criner, G.J.; Davis, K., Jr.; Kelly, K.M.; Smith, T.C.; Small, R.J. Low-dose inhaled nitric oxide in patients with acute lung injury: A randomized controlled trial. JAMA 2004, 291, 1603–1609. [Google Scholar] [CrossRef] [PubMed]
- Milberg, J.A.; Davis, D.R.; Steinberg, K.P.; Hudson, L.D. Improved survival of patients with acute respiratory distress syndrome (Ards): 1983–1993. JAMA 1995, 273, 306–309. [Google Scholar] [CrossRef] [PubMed]
- Abel, S.J.; Finney, S.J.; Brett, S.J.; Keogh, B.F.; Morgan, C.J.; Evans, T.W. Reduced mortality in association with the acute respiratory distress syndrome (Ards). Thorax 1998, 53, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Needham, D.M.; Colantuoni, E.; Mendez-Tellez, P.A.; Dinglas, V.D.; Sevransky, J.E.; Dennison Himmelfarb, C.R.; Desai, S.V.; Shanholtz, C.; Brower, R.G.; Pronovost, P.J. Lung protective mechanical ventilation and two year survival in patients with acute lung injury: Prospective cohort study. BMJ 2012, 344, e2124. [Google Scholar] [CrossRef] [PubMed]
- Manzano, F.; Fernandez-Mondejar, E.; Colmenero, M.; Poyatos, M.E.; Rivera, R.; Machado, J.; Catalan, I.; Artigas, A. Positive-end expiratory pressure reduces incidence of ventilator-associated pneumonia in nonhypoxemic patients. Crit. Care Med. 2008, 36, 2225–2231. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Pelosi, P.; Crotti, S.; Valenza, F. Effects of positive end-expiratory pressure on regional distribution of tidal volume and recruitment in adult respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1995, 151, 1807–1814. [Google Scholar] [CrossRef] [PubMed]
- Caironi, P.; Cressoni, M.; Chiumello, D.; Ranieri, M.; Quintel, M.; Russo, S.G.; Cornejo, R.; Bugedo, G.; Carlesso, E.; Russo, R.; et al. Lung opening and closing during ventilation of acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2010, 181, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Meade, M.O.; Cook, D.J.; Guyatt, G.H.; Slutsky, A.S.; Arabi, Y.M.; Cooper, D.J.; Davies, A.R.; Hand, L.E.; Zhou, Q.; Thabane, L.; et al. Ventilation strategy using low tidal volumes, recruitment maneuvers, and high positive end-expiratory pressure for acute lung injury and acute respiratory distress syndrome: A randomized controlled trial. JAMA 2008, 299, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Lamontagne, F.; Brower, R.; Meade, M. Corticosteroid therapy in acute respiratory distress syndrome. CMAJ 2013, 185, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Belvitch, P.; Dudek, S.M. Corticosteroids and Ards: The debate continues. Crit. Care Med. 2013, 41, 1813–1814. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.T. Corticosteroids in Ards. N. Engl. J. Med. 2006, 355, 316–319. [Google Scholar]
- Greaves, M.W. Anti-inflammatory action of corticosteroids. Postgrad. Med. J. 1976, 52, 631–633. [Google Scholar] [CrossRef] [PubMed]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory action of glucocorticoids—New mechanisms for old drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Meduri, G.U.; Marik, P.E.; Chrousos, G.P.; Pastores, S.M.; Arlt, W.; Beishuizen, A.; Bokhari, F.; Zaloga, G.; Annane, D. Steroid treatment in Ards: A critical appraisal of the Ards network trial and the recent literature. Intensive Care Med. 2008, 34, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.M.; Craig, J.C.; Eslick, G.D.; Seppelt, I.; McLean, A.S. Use of corticosteroids in acute lung injury and acute respiratory distress syndrome: A systematic review and meta-analysis. Crit. Care Med. 2009, 37, 1594–1603. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, K.P.; Hudson, L.D.; Goodman, R.B.; Hough, C.L.; Lanken, P.N.; Hyzy, R.; Thompson, B.T.; Ancukiewicz, M. Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. N. Engl. J. Med. 2006, 354, 1671–1684. [Google Scholar] [PubMed]
- Bernard, G.R.; Luce, J.M.; Sprung, C.L.; Rinaldo, J.E.; Tate, R.M.; Sibbald, W.J.; Kariman, K.; Higgins, S.; Bradley, R.; Metz, C.A.; et al. High-dose corticosteroids in patients with the adult respiratory distress syndrome. N. Engl. J. Med. 1987, 317, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Fyhrquist, F.; Metsarinne, K.; Tikkanen, I. Role of angiotensin II in blood pressure regulation and in the pathophysiology of cardiovascular disorders. J. Hum. Hypertens. 1995, 9 (Suppl. 5), S19–S24. [Google Scholar] [PubMed]
- Cooper, C.L.; Shaffer, J.E.; Malik, K.U. Mechanism of action of angiotensin ii and bradykinin on prostaglandin synthesis and vascular tone in the isolated rat kidney. Effect of ca++ antagonists and calmodulin inhibitors. Circ. Res. 1985, 56, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Puscas, I.; Coltau, M.; Gilau, L.; Baican, M.; Pasca, R.; Domuta, G.; Hecht, A. The mechanism of action of angiotensin II is dependent on direct activation of vascular smooth muscle carbonic anhydrase I. Int. J. Clin. Lab. Res. 2000, 30, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Chawla, L.S.; Busse, L.W.; Brasha-Mitchell, E.; Alotaibi, Z. The use of angiotensin II in distributive shock. Crit. Care 2016, 20, 137. [Google Scholar] [CrossRef] [PubMed]
- Orfanos, S.E.; Armaganidis, A.; Glynos, C.; Psevdi, E.; Kaltsas, P.; Sarafidou, P.; Catravas, J.D.; Dafni, U.G.; Langleben, D.; Roussos, C. Pulmonary capillary endothelium-bound angiotensin-converting enzyme activity in acute lung injury. Circulation 2000, 102, 2011–2018. [Google Scholar] [CrossRef] [PubMed]
- Elder, J. The renin-angiotensin system: A potential therapeutic target in Ards? Thorax 2005, 60, 821. [Google Scholar]
- Zhao, Y.; Zhang, X.; Song, Z.; Qi, D.; Deng, X.; Xia, J.; He, J.; Deng, W.; Zhong, X.; Zhang, C.; et al. Rapamycin Attenuates Acute Lung Injury Induced by Lps through Inhibition of Th17 Cell Proliferation in Mice. Sci. Rep. 2016, 6, 20156. [Google Scholar]
- Yang, Q.; Guan, K.L. Expanding Mtor Signaling. Cell Res 2007, 17, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.D.; Pollizzi, K.N.; Heikamp, E.B.; Horton, M.R. Regulation of Immune Responses by Mtor. Annu. Rev. Immunol. 2012, 30, 39–68. [Google Scholar] [CrossRef] [PubMed]
- Papazian, L.; Forel, J.-M.; Gacouin, A.; Penot-Ragon, C.; Perrin, G.; Loundou, A.; Jaber, S.; Arnal, J.-M.; Perez, D.; Seghboyan, J.-M.; et al. Neuromuscular blockers in early acute respiratory distress syndrome. N. Engl. J. Med. 2010, 363, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, R.M.; McAuley, D.F. Acute respiratory distress syndrome. Lancet 2016, 388, 2416–2430. [Google Scholar] [CrossRef] [Green Version]
- Umbrello, M.; Formenti, P.; Bolgiaghi, L.; Chiumello, D. Current concepts of Ards: A narrative review. Int. J. Mol. Sci. 2017, 18, 64. [Google Scholar] [CrossRef] [PubMed]
- Dellinger, R.P.; Zimmerman, J.L.; Taylor, R.W.; Straube, R.C.; Hauser, D.L.; Criner, G.J.; Davis, K., Jr.; Hyers, T.M.; Papadakos, P. Effects of inhaled nitric oxide in patients with acute respiratory distress syndrome: Results of a randomized phase II trial. Inhaled nitric oxide in Ards study group. Crit. Care Med. 1998, 26, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Troncy, E.; Collet, J.P.; Shapiro, S.; Guimond, J.G.; Blair, L.; Ducruet, T.; Francoeur, M.; Charbonneau, M.; Blaise, G. Inhaled nitric oxide in acute respiratory distress syndrome: A pilot randomized controlled study. Am. J. Respir. Crit. Care Med. 1998, 157, 1483–1488. [Google Scholar] [CrossRef] [PubMed]
- Michael, J.R.; Barton, R.G.; Saffle, J.R.; Mone, M.; Markewitz, B.A.; Hillier, K.; Elstad, M.R.; Campbell, E.J.; Troyer, B.E.; Whatley, R.E.; et al. Inhaled nitric oxide versus conventional therapy: Effect on oxygenation in Ards. Am. J. Respir. Crit. Care Med. 1998, 157, 1372–1380. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, N.K.J.; Burns, K.E.A.; Friedrich, J.O.; Granton, J.T.; Cook, D.J.; Meade, M.O. Effect of nitric oxide on oxygenation and mortality in acute lung injury: Systematic review and meta-analysis. BMJ 2007, 334, 779. [Google Scholar] [CrossRef] [PubMed]
- Heard, S.O.; Longtine, K.; Toth, I.; Puyana, J.C.; Potenza, B.; Smyrnios, N. The influence of liposome-encapsulated prostaglandin E1 on hydrogen peroxide concentrations in the exhaled breath of patients with the acute respiratory distress syndrome. Anesth. Analg. 1999, 89, 353–357. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AECC Definition from 1994 [20] | Berlin Definition from 2012 [21] | |
|---|---|---|
| Timing | Acute onset | Within 1 week of a known clinical insult or new/worsening respiratory symptoms |
| Chest imaging | Bilateral infiltrates seen on frontal chest radiograph | Chest X-ray or CT scan: Bilateral opacities not fully explained by effusions, lobar/lung collapse, or nodules |
| Origin of edema | Pulmonary artery wedge pressure ≤18 mmHg when measured, or no clinical evidence of left atrial hypertension | Respiratory failure not fully explained by cardiac failure or fluid overload; objective assessment (e.g., echocardiography) required to exclude hydrostatic edema if no risk factor presents |
| Oxygenation | Acute lung injury criteria: PaO2/FiO2 ≤ 300 mmHg (regardless of PEEP level) | Mild ARDS: 200 < PaO2/FiO2 ≤ 300 with PEEP or CPAP ≥ 5 cmH2O |
| ARDS criteria: PaO2/FiO2 ≤ 200 mmHg (regardless of PEEP level) | Moderate ARDS: 100 < PaO2/FiO2 ≤ 200 with PEEP ≥ 5 cmH2O | |
| Severe ARDS: PaO2/FiO2: ≤ 100 with PEEP ≥ 5 cmH2O |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.-Y.; Chen, C.-S.; Yiang, G.-T.; Cheng, Y.-L.; Yong, S.-B.; Wu, M.-Y.; Li, C.-J. New Insights into the Immune Molecular Regulation of the Pathogenesis of Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2018, 19, 588. https://doi.org/10.3390/ijms19020588
Yang C-Y, Chen C-S, Yiang G-T, Cheng Y-L, Yong S-B, Wu M-Y, Li C-J. New Insights into the Immune Molecular Regulation of the Pathogenesis of Acute Respiratory Distress Syndrome. International Journal of Molecular Sciences. 2018; 19(2):588. https://doi.org/10.3390/ijms19020588
Chicago/Turabian StyleYang, Chin-Yao, Chien-Sheng Chen, Giou-Teng Yiang, Yeung-Leung Cheng, Su-Boon Yong, Meng-Yu Wu, and Chia-Jung Li. 2018. "New Insights into the Immune Molecular Regulation of the Pathogenesis of Acute Respiratory Distress Syndrome" International Journal of Molecular Sciences 19, no. 2: 588. https://doi.org/10.3390/ijms19020588