Benzothiophenone Derivatives Targeting Mutant Forms of Estrogen Receptor-α in Hormone-Resistant Breast Cancers

 ,
,
Abstract
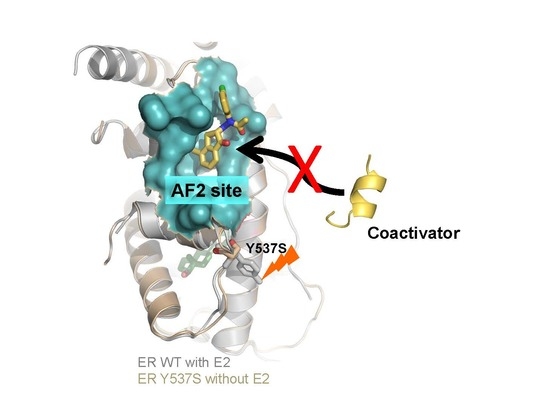
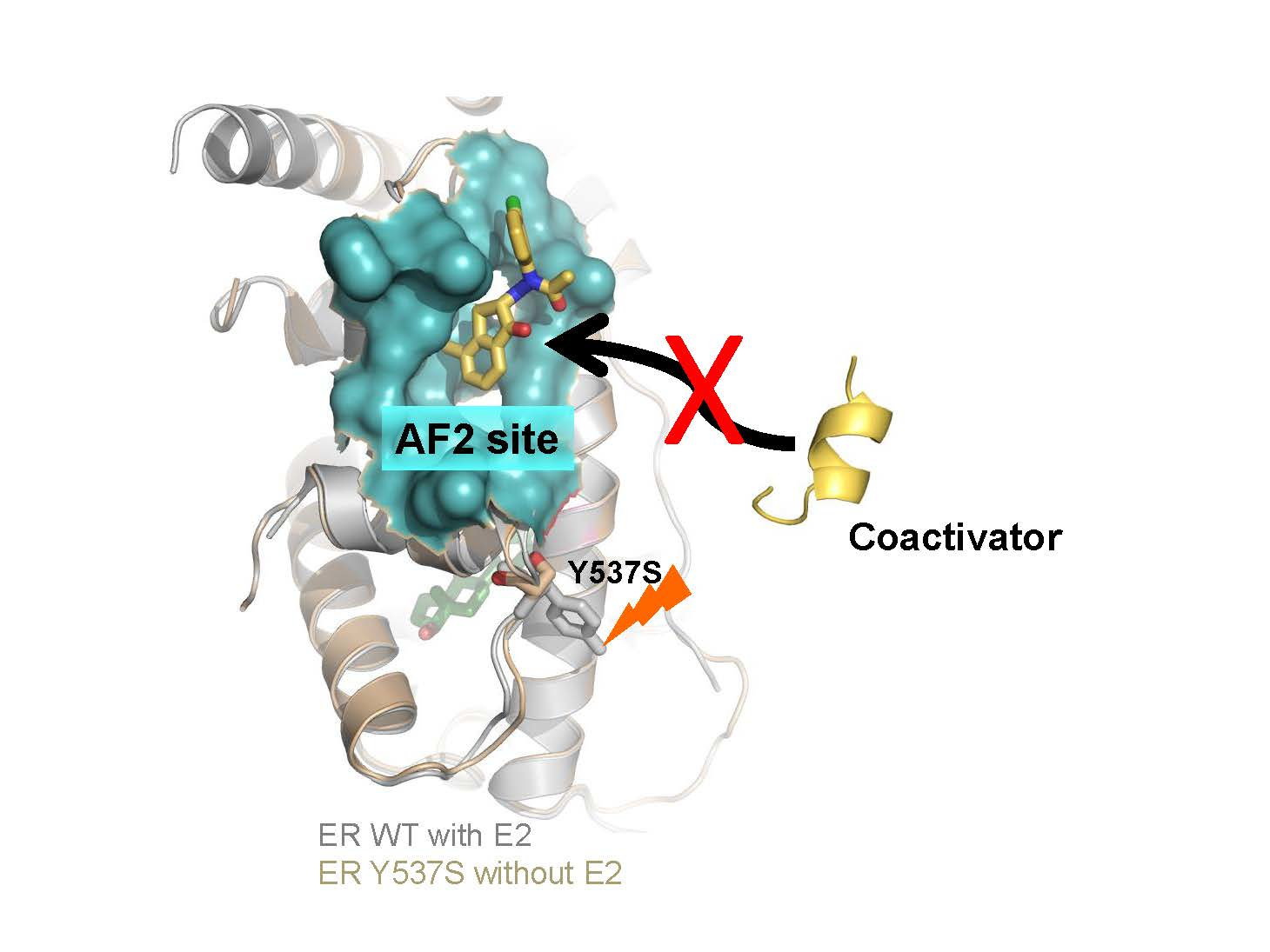
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
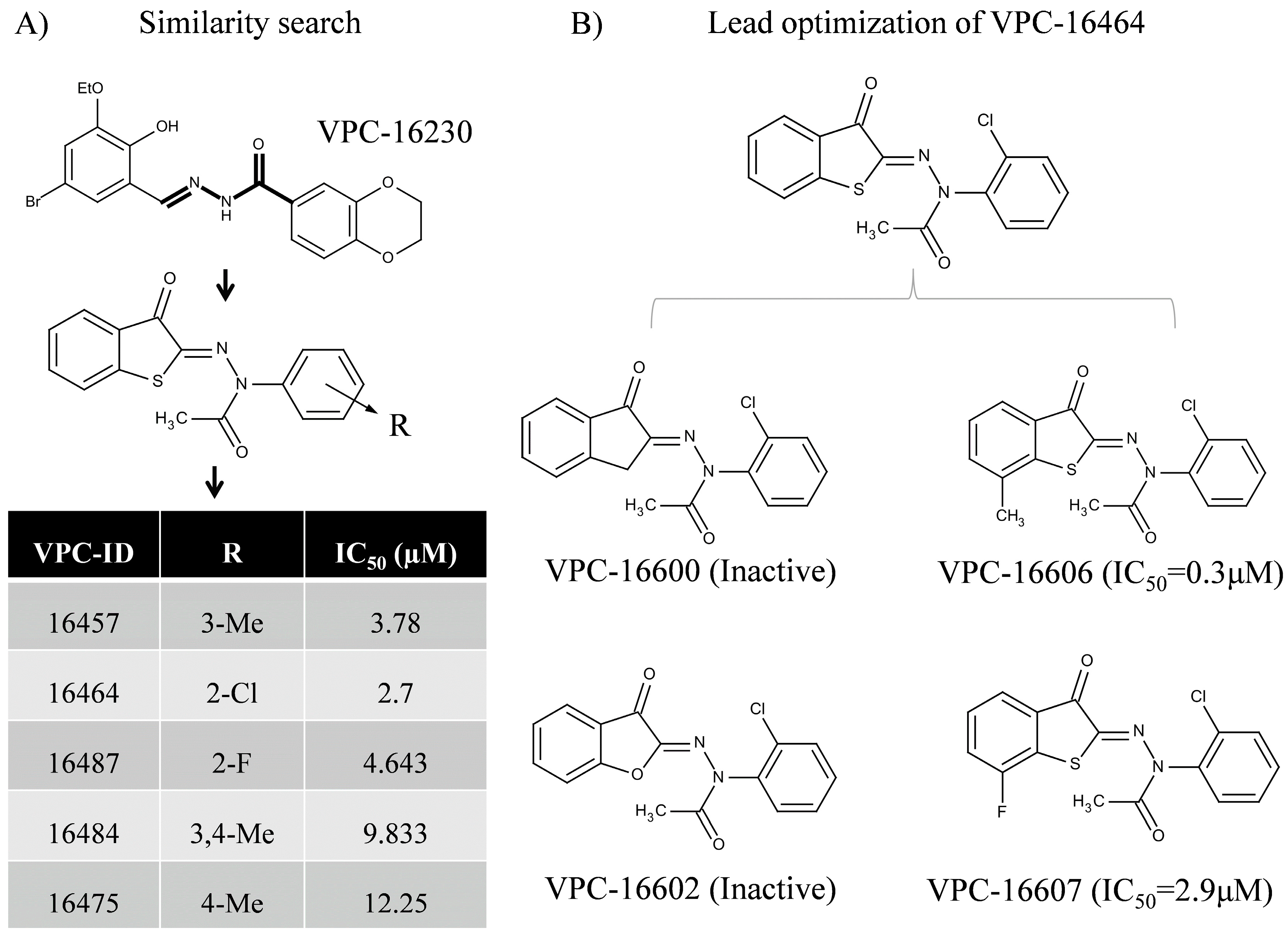
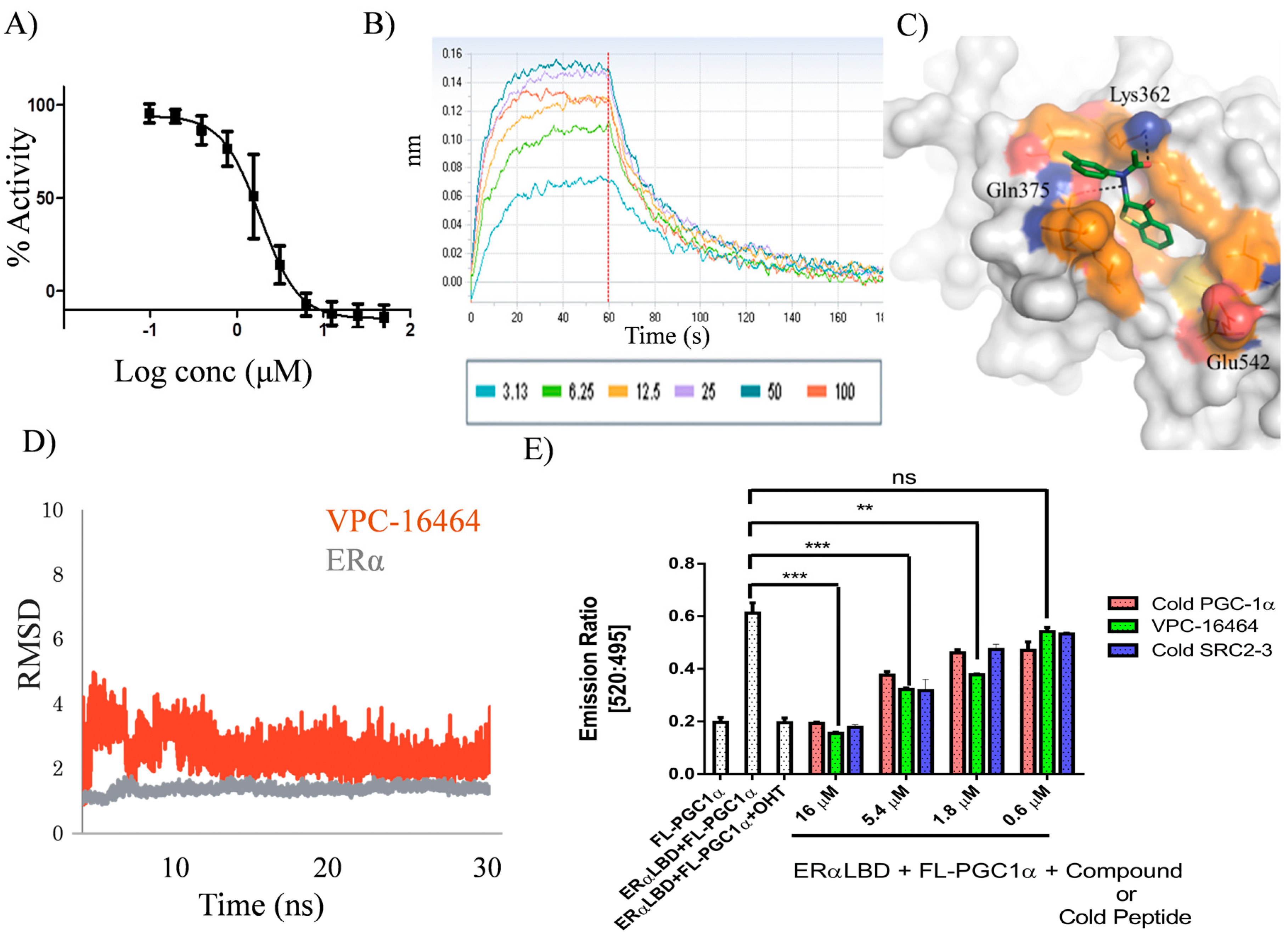
2.1. In Silico Identification and Experimental Evaluation of Benzothiophenone Analogues
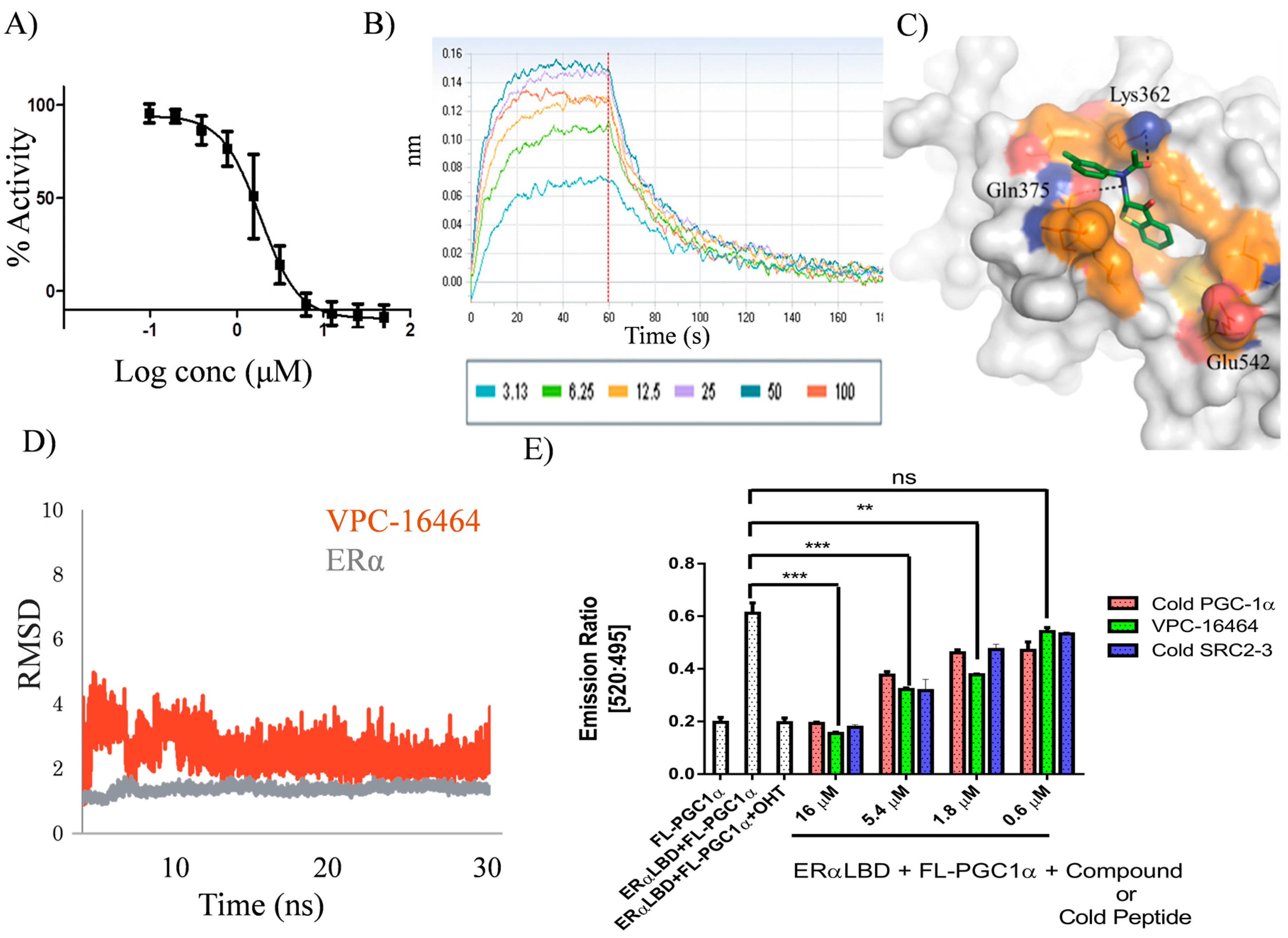
2.2. VPC-16464 Stably Binds to the AF2 Site during Molecular Dynamics Simulations
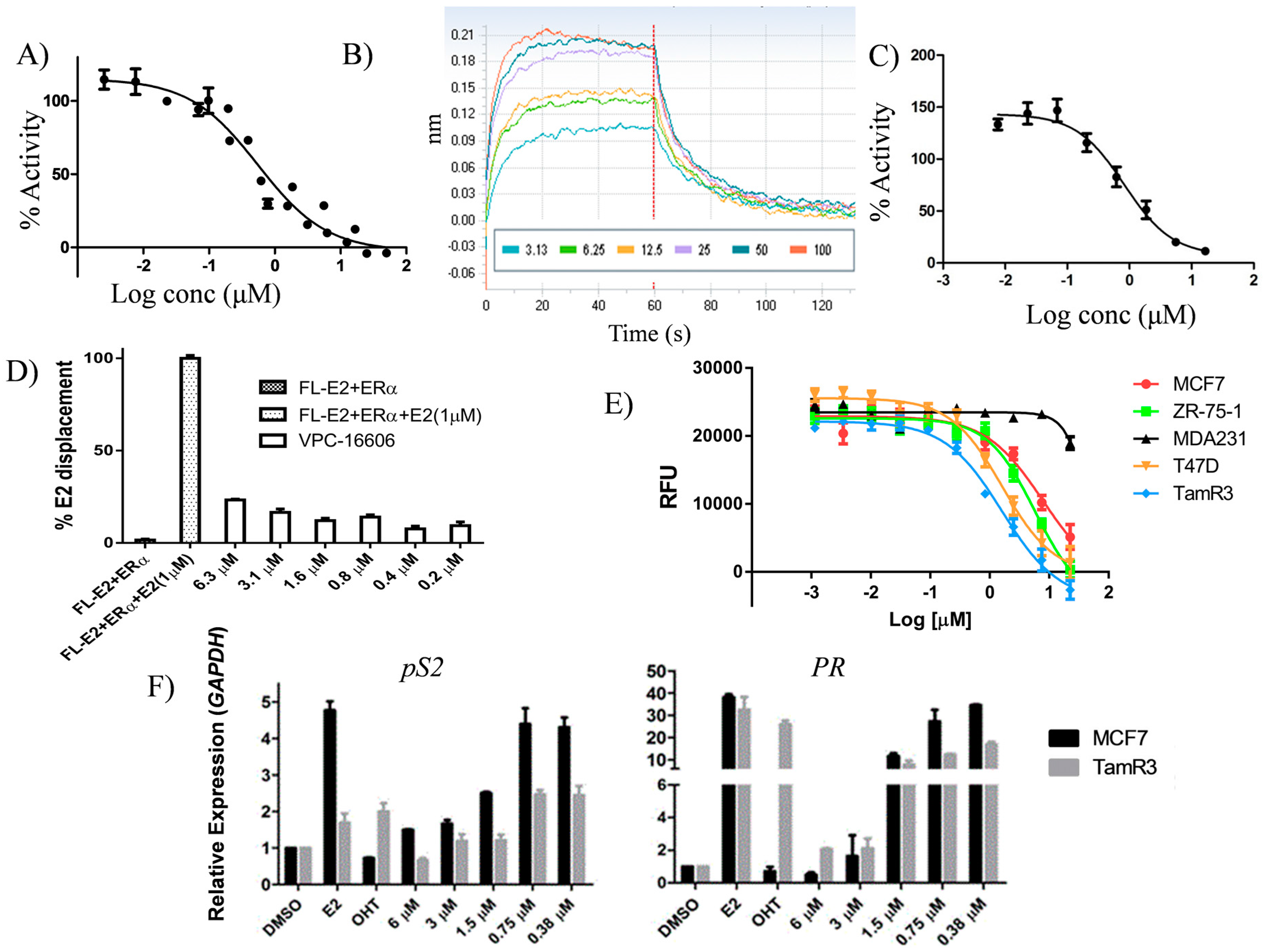
2.3. Lead Optimization of VPC-16464
2.4. VPC-16606 Blocks the Interactions between Co-Activators at the ERα AF2 Site
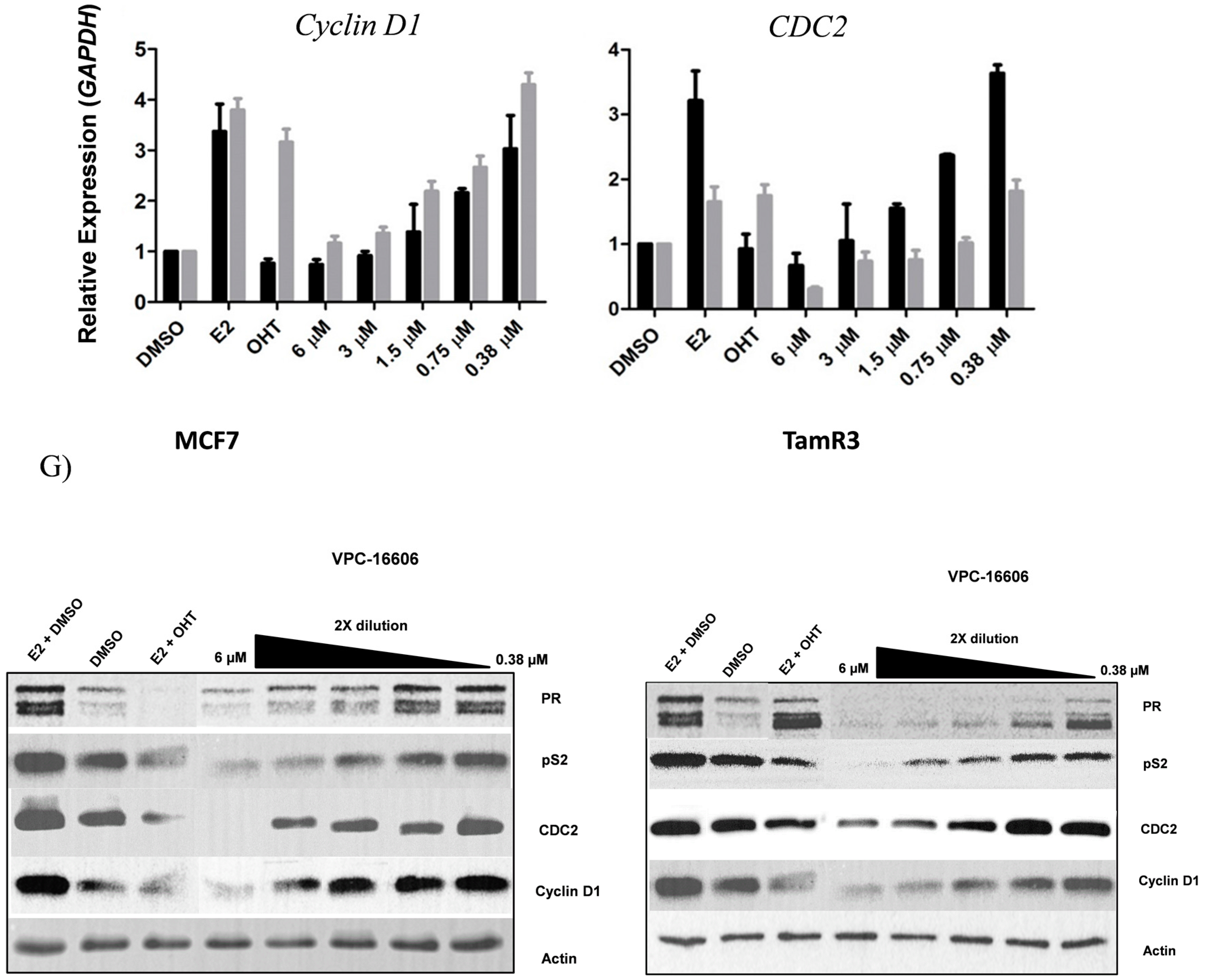
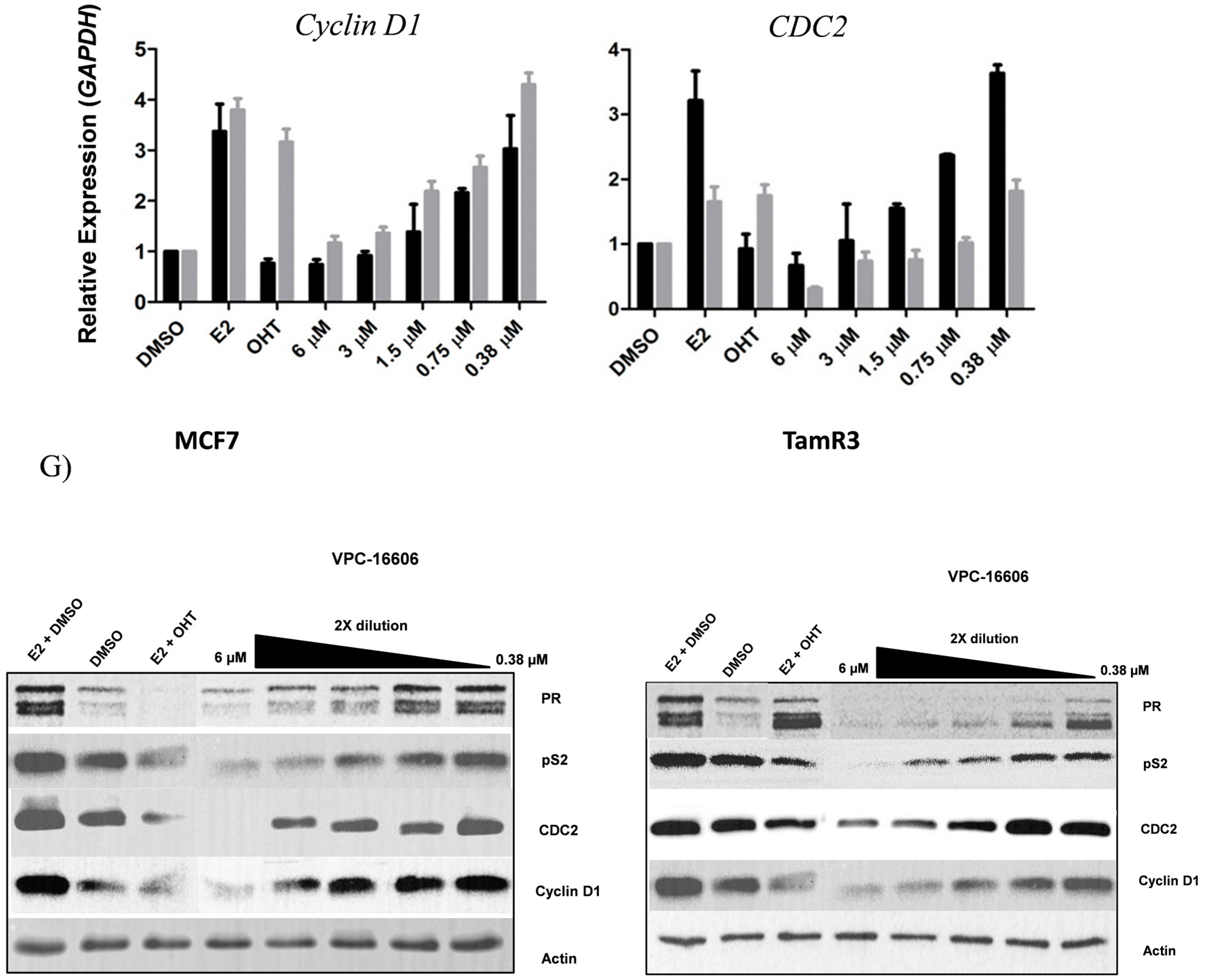
2.5. VPC-16606 Inhibits ERα-Dependent Cell Growth and Gene Expression
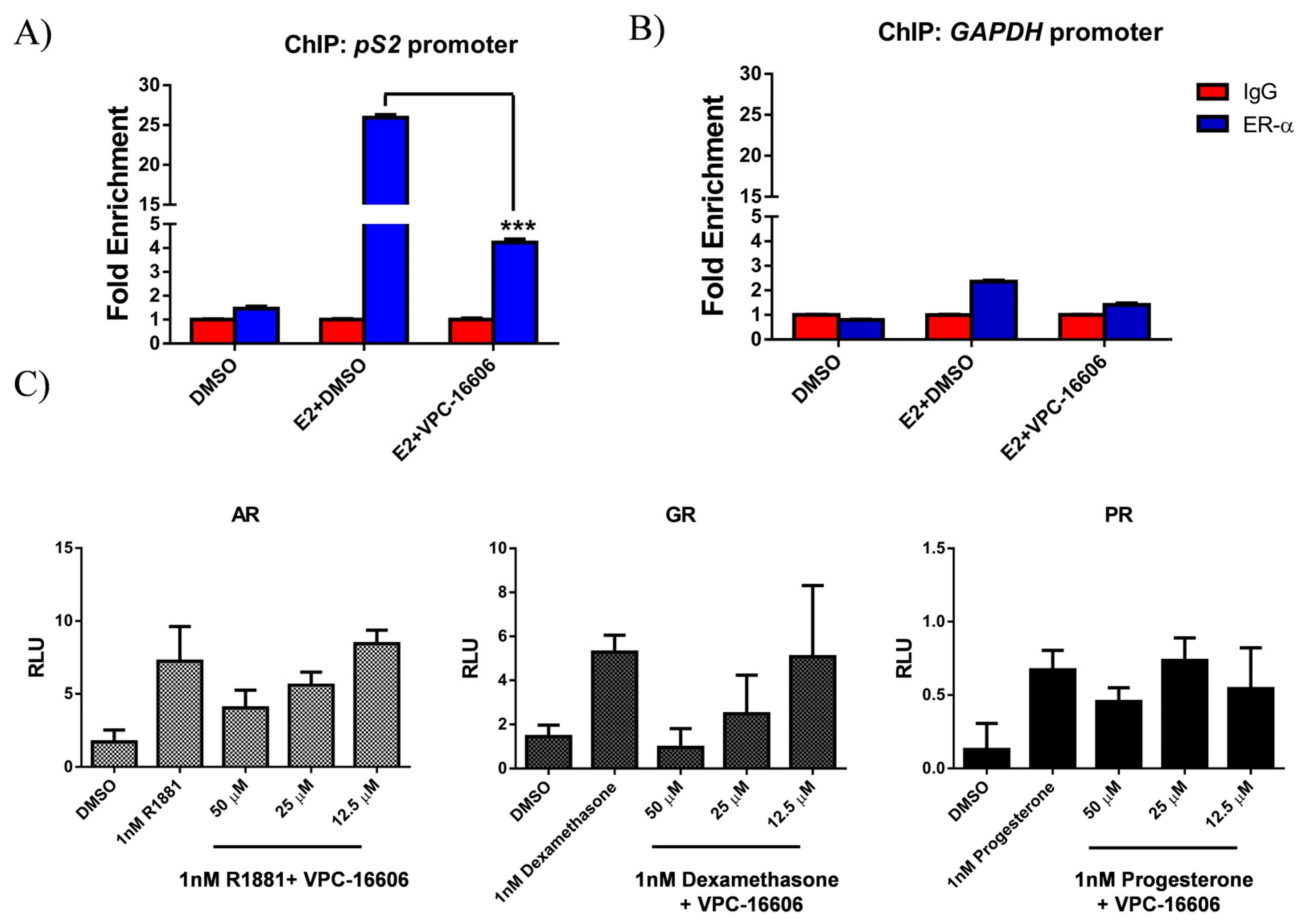
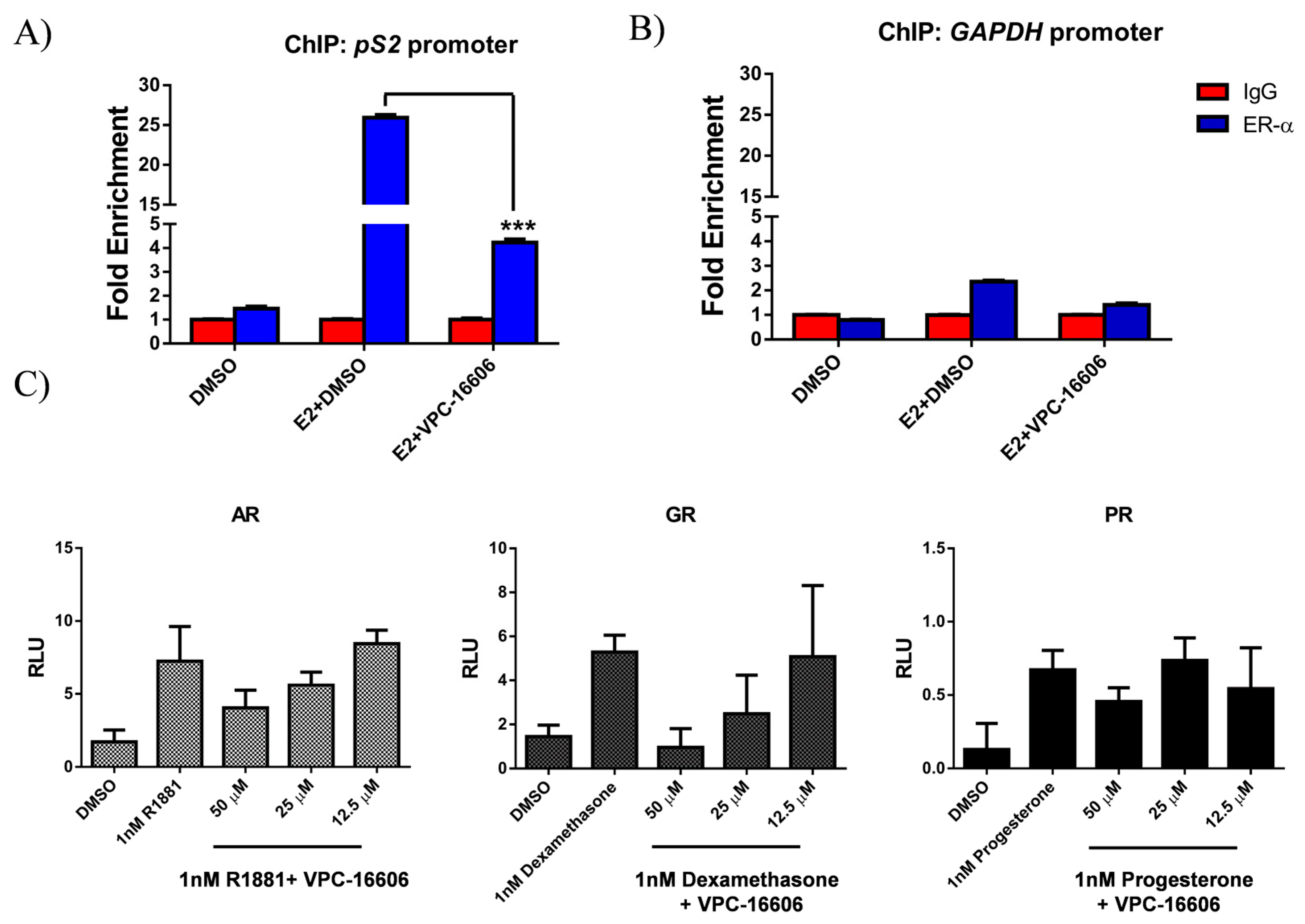
2.6. VPC-16606 Diminishes ERα Binding on Estrogen Response Elements (ERE)
2.7. VPC-16606 is Selective towards ERα
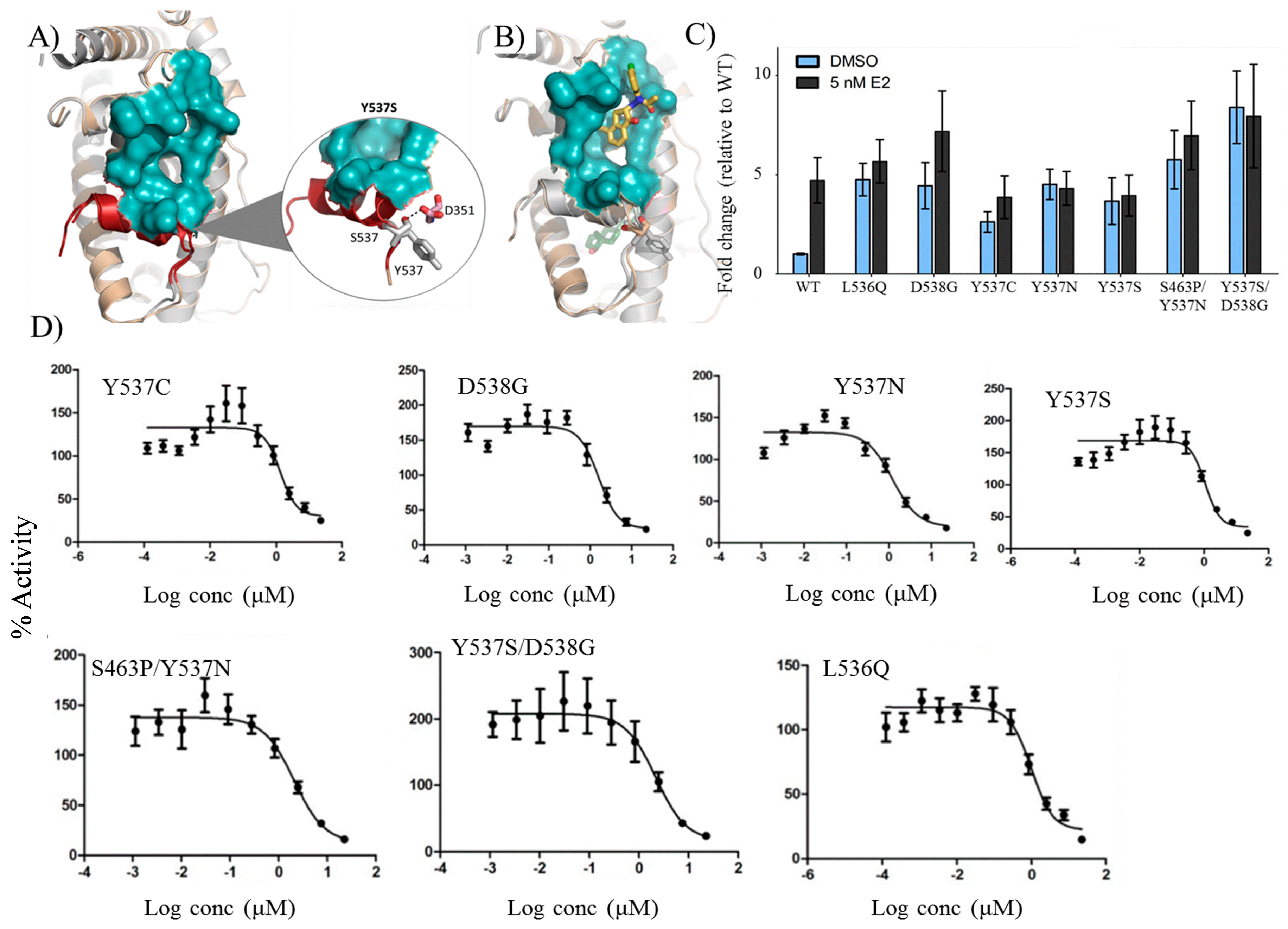
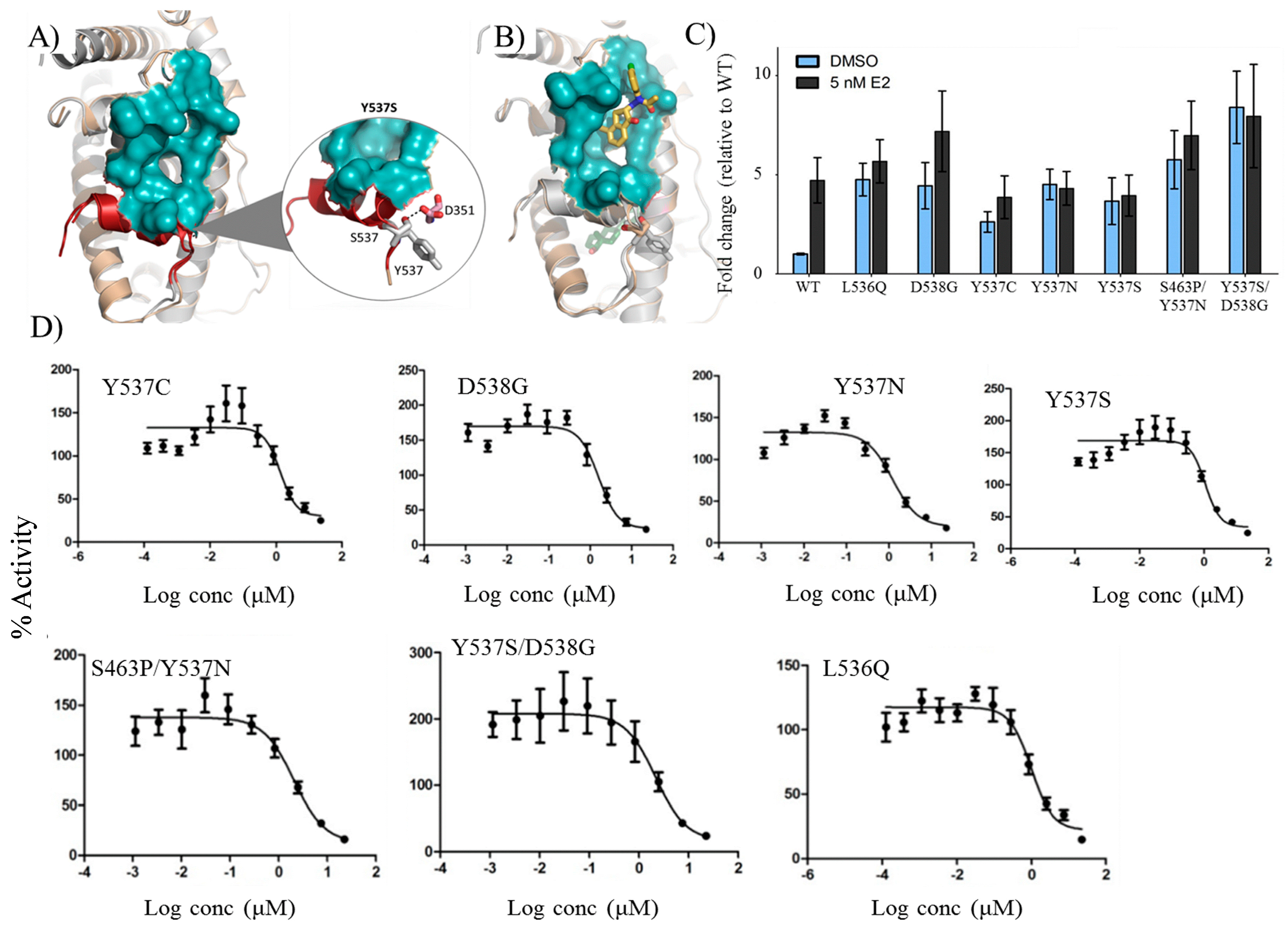
2.8. VPC-16606 Inhibits Clinically-Relevant Mutant Forms of ERα
3. Discussion
4. Materials and Methods
4.1. In Silico Modeling of AF2 Inhibitors
4.2. Cell Culture
4.3. Chemicals and Antibodies
4.4. Plasmids and Constructs:
4.5. Luciferase Transcriptional Assay
4.6. TR-FRET Assay
4.7. E2 Displacement Assay
4.8. BLI Assay
4.9. Cell Viability Assay
4.10. qRT-PCR
4.11. Mammalian Two Hybrid Assay
4.12. Western Blotting
4.13. Chromatin Immunoprecipitation (ChIP)
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ER-α | Estrogen receptor-α |
| AF2 | Activation function-2 |
| BCa | Breast cancer |
| LBD | Ligand binding domain |
| MD | Molecular dynamics |
| BLI | Biolayer interferometry |
References
- Available online: https://seer.cancer.gov (accessed on 4 January 2018).
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Dunnwald, L.K.; Rossing, M.A.; Li, C.I. Hormone receptor status, tumor characteristics, and prognosis: A prospective cohort of breast cancer patients. Breast Cancer Res. 2007, 9. [Google Scholar] [CrossRef] [PubMed]
- Niemeier, L.A.; Dabbs, D.J.; Beriwal, S.; Striebel, J.M.; Bhargava, R. Androgen receptor in breast cancer: Expression in estrogen receptor-positive tumors and in estrogen receptor-negative tumors with apocrine differentiation. Mod. Pathol. 2010, 23, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K. Drug therapy—Tamoxifen in the treatment of breast cancer. N. Engl. J. Med. 1998, 339, 1609–1618. [Google Scholar] [CrossRef] [PubMed]
- Burstein, H.J.; Temin, S.; Anderson, H.; Buchholz, T.A.; Davidson, N.E.; Gelmon, K.E.; Giordano, S.H.; Hudis, C.A.; Rowden, D.; Solky, A.J.; et al. Adjuvant Endocrine Therapy for Women with Hormone Receptor-Positive Breast Cancer: American Society of Clinical Oncology Clinical Practice Guideline Focused Update. J. Clin. Oncol. 2014, 32, 2255–2269. [Google Scholar] [CrossRef] [PubMed]
- Lewis-Wambi, J.; Jordan, V.C. Treatment of Postmenopausal Breast Cancer with Selective Estrogen Receptor Modulators (SERMs). Breast Dis. 2005, 24, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Davies, C.; Godwin, J.; Gray, R.; Clarke, M.; Cutter, D.; Darby, S.; McGale, P.; Pan, H.C.; Taylor, C.; Wang, Y.C.; et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: Patient-level meta-analysis of randomised trials. Lancet 2011, 378, 771–784. [Google Scholar] [PubMed]
- Jeselsohn, R.; Yelensky, R.; Buchwalter, G.; Frampton, G.; Meric-Bernstam, F.; Gonzalez-Angulo, A.M.; Ferrer-Lozano, J.; Perez-Fidalgo, J.A.; Cristofanilli, M.; Gómez, H.; et al. Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin. Cancer Res. 2014, 20, 1757–1767. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Wu, Y.M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 2013, 45, 1446–1451. [Google Scholar] [CrossRef] [PubMed]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.; King, T.A.; et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 2013, 45, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.X.; Borg, A.; Wolf, D.M.; Oesterreich, S.; Fuqua, S.A.W. An estrogen receptor mutant with strong hormone-independent activity from a metastatic breast cancer. Cancer Res. 1997, 57, 1244–1249. [Google Scholar] [PubMed]
- Hao, L.; Rizzo, P.; Osipo, C.; Pannuti, A.; Wyatt, D.; Cheung, L.W.K.; Sonenshein, G.; Osborne, B.A.; Miele, L. Notch-1 activates estrogen receptor-α-dependent transcription via IKKα in breast cancer cells. Oncogene 2010, 29, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Gururaj, A.E.; Vadlamudi, R.K.; Rayala, S.K. The clinical relevance of steroid hormone receptor corepressors. Clin. Cancer Res. 2005, 11, 2822–2831. [Google Scholar] [CrossRef] [PubMed]
- Habashy, H.O.; Powe, D.G.; Rakha, E.A.; Ball, G.; Macmillan, R.D.; Green, A.R.; Ellis, I.O. The prognostic significance of PELP1 expression in invasive breast cancer with emphasis on the ER-positive luminal-like subtype. Breast Cancer Res. Treat. 2010, 120, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Carlson, K.E.; Choi, I.; Gee, A.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A. Altered ligand binding properties and enhanced stability of a constitutively active estrogen receptor: Evidence that an open pocket conformation is required for ligand interaction. Biochemistry 1997, 36, 14897–14905. [Google Scholar] [CrossRef] [PubMed]
- Toy, W.; Weir, H.; Razavi, P.; Lawson, M.; Goeppert, A.U.; Mazzola, A.M.; Smith, A.; Wilson, J.; Morrow, C.; Wong, W.L.; et al. Activating ESR1 Mutations Differentially Affect the Efficacy of ER Antagonists. Cancer Discov. 2017, 7, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Fanning, S.W.; Mayne, C.G.; Dharmarjan, V.; Carlson, K.E.; Martin, T.A.; Novick, S.J.; Toy, W.; Green, B.; Panchamukhi, S.; Katzenellenbogen, B.S.; et al. Estrogen receptor α somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Bahreini, A.; Li, Z.Q.; Wang, P.L.; Levine, K.M.; Tasdemir, N.; Cao, L.; Weir, H.M.; Puhalla, S.L.; Davidson, N.E.; Stern, A.M.; et al. Mutation site and context dependent effects of ESR1 mutation in genome-edited breast cancer cell models. Breast Cancer Res. 2017, 19. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.L.; Tamrazi, A.; Collins, M.L.; Katzenellenbogen, J.A. Design, synthesis, and in vitro biological evaluation of small molecule inhibitors of estrogen receptor a coactivator binding. J. Med. Chem. 2004, 47, 600–611. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.B.; Collins, M.L.; Gunther, J.R.; Comninos, J.S.; Katzenellenbogen, J.A. Bicyclo 2.2.2 octanes: Close structural mimics of the nuclear receptor-binding motif of steroid receptor coactivators. Bioorg. Med. Chem. Lett. 2007, 17, 4118–4122. [Google Scholar] [CrossRef] [PubMed]
- Gunther, J.R.; Moore, T.W.; Cottins, M.L.; Katzenettenbogen, J.A. Amphipathic benzenes are designed inhibitors of the estrogen receptor α/steroid receptor coactivator interaction. ACS Chem. Biol. 2008, 3, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, G.; Gallo, D.; Cossy, J.; Laios, I.; Larsimont, D.; Laurent, G.; Jacquot, Y. Peptides Targeting Estrogen Receptor α-Potential Applications for Breast Cancer Treatment. Curr. Pharm. Des. 2011, 17, 2632–2653. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Munuganti, R.S.N.; Leblanc, E.; Lin, Y.L.; Leung, E.; Lallous, N.; Butler, M.; Cherkasov, A.; Rennie, P.S. In silico discovery and validation of potent small-molecule inhibitors targeting the activation function 2 site of human oestrogen receptor α. Breast Cancer Res. 2015, 17. [Google Scholar] [CrossRef] [PubMed]
- Cherian, M.T.; Wilson, E.M.; Shapiro, D.J. A Competitive Inhibitor That Reduces Recruitment of Androgen Receptor to Androgen-responsive Genes. J. Biol. Chem. 2012, 287, 23368–23380. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15-Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Munuganti, R.; Hassona, M.; Leblanc, E.; Frewin, K.; Singh, K.; Ma, D.; Ban, F.; Hsing, M.; Adomat, H.; Lallous, N.; et al. Identification of a Potent Antiandrogen that Targets the BF3 Site of the Androgen Receptor and Inhibits Enzalutamide-Resistant Prostate Cancer. ACS Chem. Biol. 2014, 21, 1476–1485. [Google Scholar] [CrossRef] [PubMed]
- Munuganti, R.; Hassona, M.; Leblanc, E.; Frewin, K.; Singh, K.; Ma, D.; Ban, F.; Hsing, M.; Adomat, H.; Lallous, N.; et al. Targeting the Binding Function 3 (BF3) Site of the Androgen Receptor through Virtual Screening. 2. Development of 2-((2-phenoxyethyl) thio)-1H-benzimidazole Derivatives. J. Med. Chem. 2013, 56, 1136–1148. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.A.; Qin, L.; Tien, J.C.Y.; Young, L.S.; Xu, J.M. The function of steroid receptor coactivator-1 in normal tissues and cancer. Int. J. Biol. Sci. 2012, 8, 470–485. [Google Scholar] [CrossRef] [PubMed]
- Duplessis, T.T.; Williams, C.C.; Hill, S.M.; Rowan, B.G. Phosphorylation of Estrogen Receptor α at Serine 118 Directs Recruitment of Promoter Complexes and Gene-Specific Transcription. Endocrinology 2011, 152, 2517–2526. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chirgadze, N.Y.; Briggs, S.L.; Khan, S.; Jensen, E.V.; Burris, T.P. A second binding site for hydroxytamoxifen within the coactivator-binding groove of estrogen receptor β. Proc. Natl. Acad. Sci. USA 2006, 103, 9908–9911. [Google Scholar] [CrossRef] [PubMed]
- Barkhem, T.; Carlsson, B.; Nilsson, Y.; Enmark, E.; Gustafsson, J.A.; Nilsson, S. Differential response of estrogen receptor α and estrogen receptor β to partial estrogen agonists/antagonists. Mol. Pharmacol. 1998, 54, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Haldosen, L.A.; Zhao, C.Y.; Dahlman-Wright, K. Estrogen receptor β in breast cancer. Mol. Cell. Endocrinol. 2014, 382, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Leygue, E.; Murphy, L.C. A bi-faceted role of estrogen receptor β in breast cancer. Endocr. Relat. Cancer 2013, 20, R127–R139. [Google Scholar] [CrossRef] [PubMed]
- Lallous, N.; Leblanc, E.; Munuganti, R.S.N.; Hassona, M.D.H.; Al Nakouzi, N.; Awrey, S.; Morin, H.; Roshan-Moniri, M.; Singh, K.; Lawn, S.; et al. Targeting Binding Function-3 of the Androgen Receptor Blocks Its Co-Chaperone Interactions, Nuclear Translocation, and Activation. Mol. Cancer Ther. 2016, 15, 2936–2945. [Google Scholar] [CrossRef] [PubMed]
- Wilson, V.S.; Bobseine, K.; Gray, L.E. Development and characterization of a cell line that stably expresses an estrogen-responsive luciferase reporter for the detection of estrogen receptor agonist and antagonists. Toxicol. Sci. 2004, 81, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Dalal, K.; Roshan-Moniri, M.; Sharma, A.; Li, H.; Ban, F.; Hessein, M.; Hsing, M.; Singh, K.; LeBlanc, E.; Dehm, S.; et al. Selectively Targeting the DNA-binding Domain of the Androgen Receptor as a Prospective Therapy for Prostate Cancer. J. Biol. Chem. 2014, 289, 26417–26429. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, K.; Munuganti, R.S.N.; Lallous, N.; Dalal, K.; Yoon, J.S.; Sharma, A.; Yamazaki, T.; Cherkasov, A.; Rennie, P.S. Benzothiophenone Derivatives Targeting Mutant Forms of Estrogen Receptor-α in Hormone-Resistant Breast Cancers. Int. J. Mol. Sci. 2018, 19, 579. https://doi.org/10.3390/ijms19020579
Singh K, Munuganti RSN, Lallous N, Dalal K, Yoon JS, Sharma A, Yamazaki T, Cherkasov A, Rennie PS. Benzothiophenone Derivatives Targeting Mutant Forms of Estrogen Receptor-α in Hormone-Resistant Breast Cancers. International Journal of Molecular Sciences. 2018; 19(2):579. https://doi.org/10.3390/ijms19020579
Chicago/Turabian StyleSingh, Kriti, Ravi S. N. Munuganti, Nada Lallous, Kush Dalal, Ji Soo Yoon, Aishwariya Sharma, Takeshi Yamazaki, Artem Cherkasov, and Paul S. Rennie. 2018. "Benzothiophenone Derivatives Targeting Mutant Forms of Estrogen Receptor-α in Hormone-Resistant Breast Cancers" International Journal of Molecular Sciences 19, no. 2: 579. https://doi.org/10.3390/ijms19020579