Role of the IL-23/IL-17 Axis in Psoriasis and Psoriatic Arthritis: The Clinical Importance of Its Divergence in Skin and Joints

 ,
,
Abstract
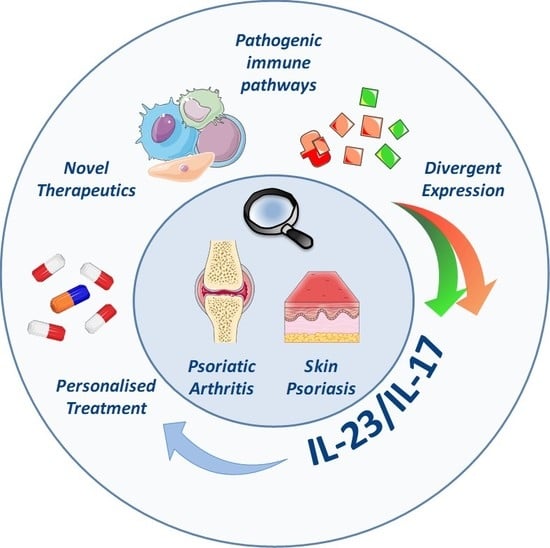
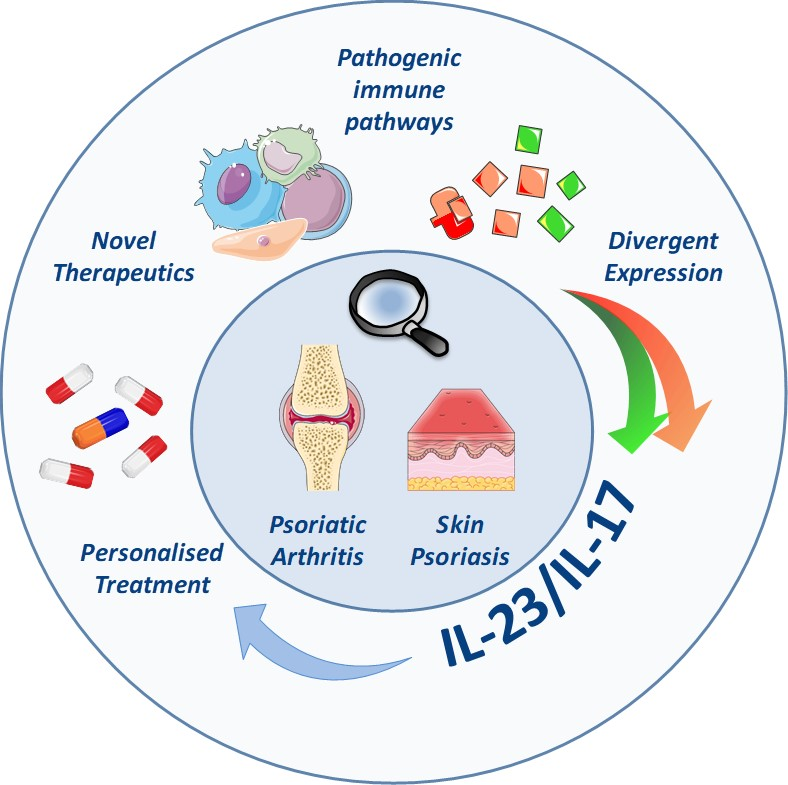
:
1. Introduction
2. Psoriasis and Psoriatic Arthritis, Same Disease?
2.1. Common and Disease-Specific Genetic Factors
2.2. The Role of Soluble Biomarkers in Psoriasis and Psoriatic Arthritis
2.3. Co-Morbidities and Complications of Psoriasis and Psoriatic Arthritis
2.4. Histological Features of the Affected Skin and Synovium
2.5. Pathogenic Immunologic Pathways Driving the Inflammation in Psoriasis and Psoriatic Arthritis
3. Pivotal Role of the IL-23/IL-17 Axis in Psoriasis and Psoriatic Arthritis
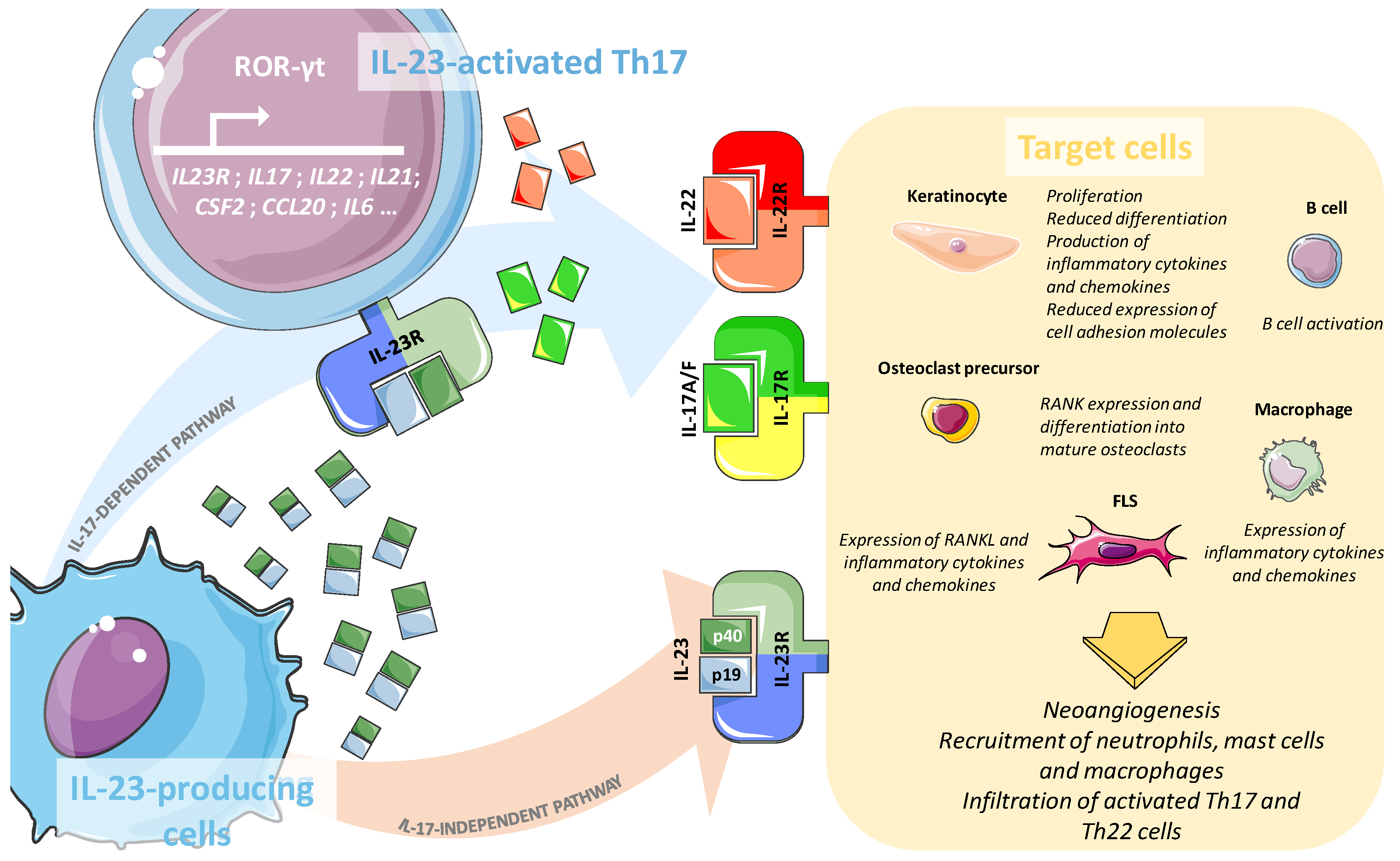
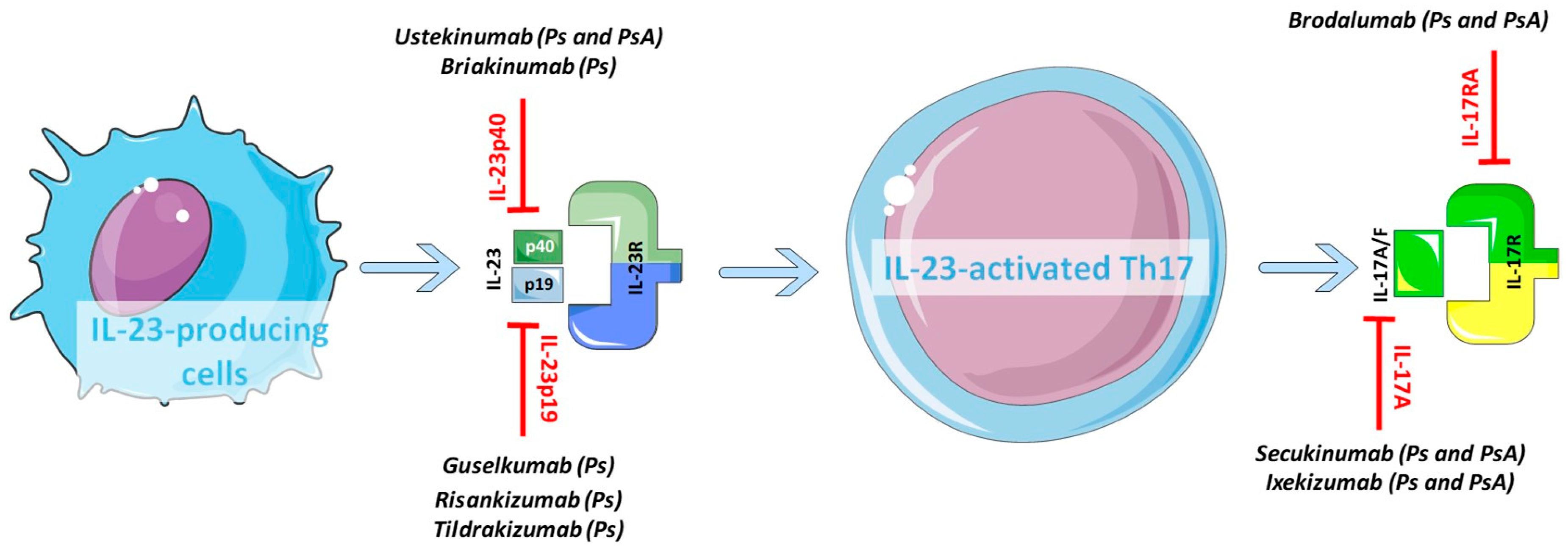
3.1. Key Cytokines and Receptors of the IL-23/IL-17 Axis
3.2. Role of the IL-23/IL-17 Axis in the Psoriatic Skin
3.3. Involvement of the IL-23/IL-17 Axis in Psoriatic Arthritis Synovium
4. Towards a Personalized Use of the Biologics in Psoriatic Arthritis
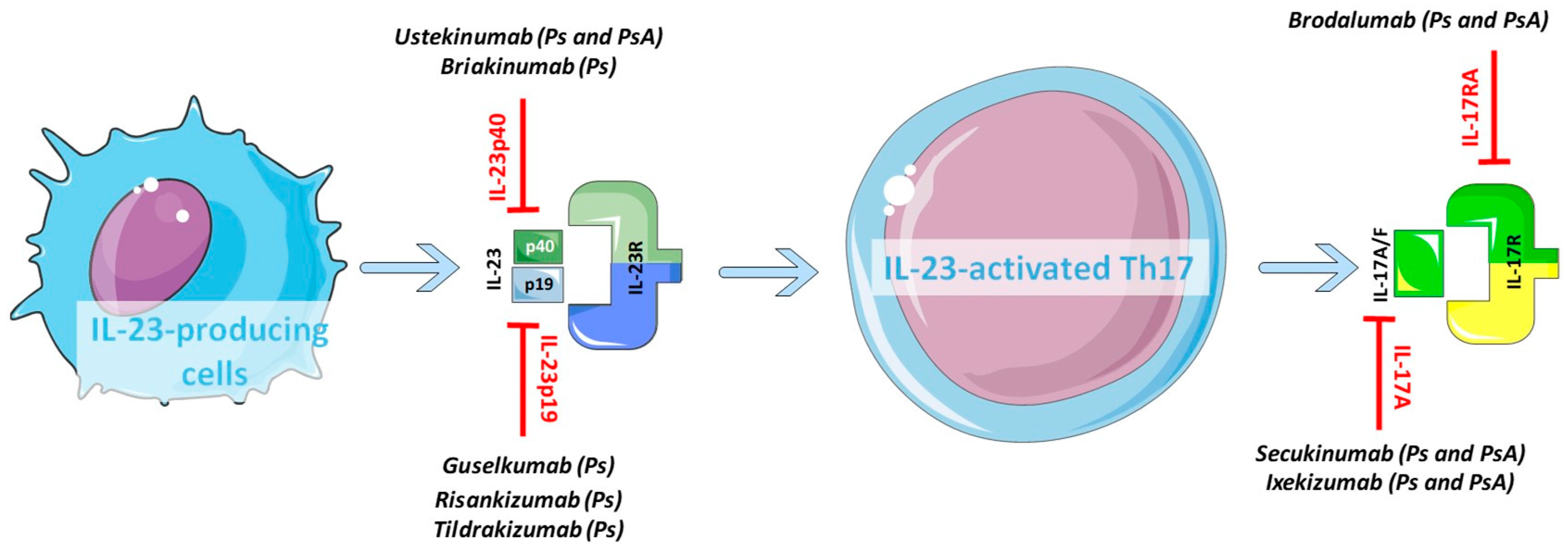
4.1. The Divergent Response to Biologics Targeting the IL-23/IL-17 Axis in Psoriatic Skin and Joints
4.2. Novel Perspectives and Future Drugs
4.2.1. Targeting ROR-γt
4.2.2. Targeting JAK/STAT
4.2.3. Targeting Multiple Cytokines and Novel Anti-Inflammatory Pathways
5. Conclusions
Author Contributions
Conflicts of Interest
Abbreviations
| Ps | Psoriasis |
| PsA | Psoriatic Arthritis |
| CASPAR | ClASsification criteria for Psoriatic Arthritis |
| RA | Rheumatoid Arthritis |
| Th | T helper |
| ROR-γt | Retinoic-acid-receptor-related Orphan Receptor-γt |
| HLA | Human Leukocyte Antigen |
| NF-κB | Nuclear Factor-Kappa B |
| TNF | Tumor Necrosis Factor |
| IFIH1 | Interferon-Induced with Helicase C Domain 1 |
| GWAS | Genome-Wide Association Studies |
| MTX | Methotrexate |
| PASI | Psoriasis Area and Severity Index |
| CRP | C-Reactive Protein |
| CXCL10 | C-X-C motif chemokine Ligand 10 |
| IBD | Inflammatory Bowel Disease |
| VEGF | Vascular Endothelial Growth Factor |
| PEAC | Pathobiology of Early Arthritis Cohort |
| APC | Antigen-Presenting Cells |
| CLA | Cutaneous Lymphocyte Antigen |
| ELS | Ectopic Lymphoid Structure |
| NK | Natural Killer |
| TGFβ | Transforming Growth Factor-β |
| IL-17R | IL-17 Receptors |
| JAK | Janus Kinase |
| STAT | Signal Transducer and Activator of Transcription |
| CCL20 | C-C motif chemokine Ligand 20 |
| CSF2 | Colony Stimulating Factor 2 |
| GM-CSF | Granulocyte Macrophage-Colony Stimulating Factor |
| NKT | Natural Killer T |
| ILC | Innate Lymphoid Cells |
| MAPK | Mitogen-Activated Protein Kinase |
| DC | Dendritic Cells |
| RANKL | Receptor Activator of Nuclear factor κB |
| K16 | Keratin 16 |
| IFNγ | Interferon γ |
| ACR | American College of Rheumatology |
| PGA | Physician Global Assessment |
| FDA | Food and Drugs Administration |
| DMARDs | Disease Modifying Anti-Rheumatic Drugs |
| NICE | National Institute for health and Care Excellence |
| Ust | Ustekinumab |
| Bri | Briakinumab |
| Gus | Guselkumab |
| Ada | Adalimumab |
| Ris | Risankizumab |
| Til | Tildrakizumab |
| Eta | Etanercept |
| Sec | Secukinumab |
| Ixe | Ixekizumab |
| Bro | Brodalumab |
| IGA | Investigator’s Global Assessment |
References
- Parisi, R.; Symmons, D.P.M.; Griffiths, C.E.M.; Ashcroft, D.M. Global Epidemiology of Psoriasis: A Systematic Review of Incidence and Prevalence. J. Investig. Dermatol. 2013, 133, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Ogdie, A.; Weiss, P. The Epidemiology of Psoriatic Arthritis. Rheum. Dis. Clin. N. Am. 2015, 41, 545–568. [Google Scholar] [CrossRef] [PubMed]
- Gladman, D.D.; Ang, M.; Su, L.; Tom, B.D.M.; Schentag, C.T.; Farewell, V.T. Cardiovascular morbidity in psoriatic arthritis. Ann. Rheum. Dis. 2009, 68, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Wilton, K.M.; Crowson, C.S.; Matteson, E.L. Malignancy incidence in patients with psoriatic arthritis: A comparison cohort-based incidence study. Clin. Rheumatol. 2016, 35, 2603–2607. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.K.; Maverakis, E.; Raychaudhuri, S.P. Diagnosis and classification of psoriasis. Autoimmun. Rev. 2014, 13, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.B.; Jerome, D.; Yeung, J. Diagnosis and management of psoriasis. Can. Fam. Phys. Med. Fam. Can. 2017, 63, 278–285. [Google Scholar]
- Ritchlin, C.T.; Colbert, R.A.; Gladman, D.D. Psoriatic Arthritis. N. Engl. J. Med. 2017, 376, 957–970. [Google Scholar] [CrossRef] [PubMed]
- Rouzaud, M.; Sevrain, M.; Villani, A.P.; Barnetche, T.; Paul, C.; Richard, M.-A.; Jullien, D.; Misery, L.; Le Maître, M.; Aractingi, S.; et al. Is there a psoriasis skin phenotype associated with psoriatic arthritis? Systematic literature review. J. Eur. Acad. Dermatol. Venereol. 2014, 28, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Acosta Felquer, M.L.; FitzGerald, O. Peripheral joint involvement in psoriatic arthritis patients. Clin. Exp. Rheumatol. 2015, 33, S26–S30. [Google Scholar] [PubMed]
- Moll, J.M.H.; Wright, V. Psoriatic arthritis. Semin. Arthritis Rheum. 1973, 3, 55–78. [Google Scholar] [CrossRef]
- Baraliakos, X.; Coates, L.C.; Braun, J. The involvement of the spine in psoriatic arthritis. Clin. Exp. Rheumatol. 2015, 33, S31–S35. [Google Scholar] [PubMed]
- Ritchlin, C.T.; Kavanaugh, A.; Gladman, D.D.; Mease, P.J.; Helliwell, P.; Boehncke, W.-H.; de Vlam, K.; Fiorentino, D.; Fitzgerald, O.; Gottlieb, A.B.; et al. Treatment recommendations for psoriatic arthritis. Ann. Rheum. Dis. 2009, 68, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-G.; Kim, S.; Lee, M.-G. The Origin of Skin Dendritic Cell Network and Its Role in Psoriasis. Int. J. Mol. Sci. 2017, 19, 42. [Google Scholar] [CrossRef] [PubMed]
- Piskin, G.; Sylva-Steenland, R.M.R.; Bos, J.D.; Teunissen, M.B.M. In Vitro and In Situ Expression of IL-23 by Keratinocytes in Healthy Skin and Psoriasis Lesions: Enhanced Expression in Psoriatic Skin. J. Immunol. 2006, 176, 1908–1915. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, B.S.; Kastelein, R.A.; Cua, D.J. Understanding the IL-23–IL-17 immune pathway. Trends Immunol. 2006, 27, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, I.; Padula, A.; D’Angelo, S.; Cutro, M.S. Psoriatic arthritis sine psoriasis. J. Rheumatol. Suppl. 2009, 83, 28–29. [Google Scholar] [CrossRef] [PubMed]
- Generali, E.; Ceribelli, A.; Stazi, M.A.; Selmi, C. Lessons learned from twins in autoimmune and chronic inflammatory diseases. J. Autoimmun. 2017, 83, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Eder, L.; Chandran, V.; Gladman, D.D. What have we learned about genetic susceptibility in psoriasis and psoriatic arthritis? Curr. Opin. Rheumatol. 2015, 27, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, M.; van der Lee, R.; Cheng, S.-C.; Johnson, M.D.; Kumar, V.; Ng, A.; Plantinga, T.S.; Smeekens, S.P.; Oosting, M.; Wang, X.; et al. The RIG-I-like helicase receptor MDA5 (IFIH1) is involved in the host defense against Candida infections. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liao, W.; Cargill, M.; Chang, M.; Matsunami, N.; Feng, B.-J.; Poon, A.; Callis-Duffin, K.P.; Catanese, J.J.; Bowcock, A.M.; et al. Carriers of Rare Missense Variants in IFIH1 Are Protected from Psoriasis. J. Investig. Dermatol. 2010, 130, 2768–2772. [Google Scholar] [CrossRef] [PubMed]
- Budu-Aggrey, A.; Bowes, J.; Stuart, P.E.; Zawistowski, M.; Tsoi, L.C.; Nair, R.; Jadon, D.R.; McHugh, N.; Korendowych, E.; Elder, J.T.; et al. A rare coding allele in IFIH1 is protective for psoriatic arthritis. Ann. Rheum. Dis. 2017, 76, 1321–1324. [Google Scholar] [CrossRef] [PubMed]
- Bowes, J.; Budu-Aggrey, A.; Huffmeier, U.; Uebe, S.; Steel, K.; Hebert, H.L.; Wallace, C.; Massey, J.; Bruce, I.N.; Bluett, J.; et al. Dense genotyping of immune-related susceptibility loci reveals new insights into the genetics of psoriatic arthritis. Nat. Commun. 2015, 6, 6046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berinstein, J.; Pollock, R.; Pellett, F.; Thavaneswaran, A.; Chandran, V.; Gladman, D.D. Association of variably expressed KIR3dl1 alleles with psoriatic disease. Clin. Rheumatol. 2017, 36, 2261–2266. [Google Scholar] [CrossRef] [PubMed]
- Bowes, J.; Loehr, S.; Budu-Aggrey, A.; Uebe, S.; Bruce, I.N.; Feletar, M.; Marzo-Ortega, H.; Helliwell, P.; Ryan, A.W.; Kane, D.; et al. PTPN22 is associated with susceptibility to psoriatic arthritis but not psoriasis: Evidence for a further PsA-specific risk locus. Ann. Rheum. Dis. 2015, 74, 1882–1885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, J.; Ogston, S.; Berg, J.; Palmer, C.; Fleming, C.; Kumar, V.; Foerster, J. HLA-Cw6-positive patients with psoriasis show improved response to methotrexate treatment. Clin. Exp. Dermatol. 2017, 42, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Talamonti, M.; Galluzzo, M.; Chimenti, S.; Costanzo, A. HLA-C*06 and response to ustekinumab in Caucasian patients with psoriasis: Outcome and long-term follow-up. J. Am. Acad. Dermatol. 2016, 74, 374–375. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.Y.P.C.; Barton, A.; Worthington, J.; Plant, D.; Griffiths, C.E.M.; Young, H.S.; Bradburn, P.; Thomson, W.; Silman, A.J.; Bruce, I.N. Investigating the role of the HLA-Cw*06 and HLA-DRB1 genes in susceptibility to psoriatic arthritis: Comparison with psoriasis and undifferentiated inflammatory arthritis. Ann. Rheum. Dis. 2008, 67, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Bowes, J.; Ashcroft, J.; Dand, N.; Jalali-najafabadi, F.; Bellou, E.; Ho, P.; Marzo-Ortega, H.; Helliwell, P.S.; Feletar, M.; Ryan, A.W.; et al. Cross-phenotype association mapping of the MHC identifies genetic variants that differentiate psoriatic arthritis from psoriasis. Ann. Rheum. Dis. 2017, 76, 1774–1779. [Google Scholar] [CrossRef] [PubMed]
- Gladman, D.D.; Anhorn, K.A.; Schachter, R.K.; Mervart, H. HLA antigens in psoriatic arthritis. J. Rheumatol. 1986, 13, 586–592. [Google Scholar] [PubMed]
- Yan, D.; Afifi, L.; Jeon, C.; Trivedi, M.; Chang, H.W.; Lee, K.; Liao, W. The metabolomics of psoriatic disease. Psoriasis Targets Ther. 2017, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kamleh, M.A.; Snowden, S.G.; Grapov, D.; Blackburn, G.J.; Watson, D.G.; Xu, N.; Ståhle, M.; Wheelock, C.E. LC–MS Metabolomics of Psoriasis Patients Reveals Disease Severity-Dependent Increases in Circulating Amino Acids That Are Ameliorated by Anti-TNFα Treatment. J. Proteome Res. 2015, 14, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Gladman, D.D. Recent advances in understanding and managing psoriatic arthritis. F1000Research 2016, 5, 2670. [Google Scholar] [CrossRef] [PubMed]
- Nedoszytko, B.; Sokołowska-Wojdyło, M.; Ruckemann-Dziurdzińska, K.; Roszkiewicz, J.; Nowicki, R.J. Chemokines and cytokines network in the pathogenesis of the inflammatory skin diseases: Atopic dermatitis, psoriasis and skin mastocytosis. Adv. Dermatol. Allergol. 2014, 2, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Onishi, R.M.; Gaffen, S.L. Interleukin-17 and its target genes: Mechanisms of interleukin-17 function in disease. Immunology 2010, 129, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, C.; Chaves, M.; Verardino, G.; Frade, A.P.; Coscarelli, P.G.; Bianchi, W.A.; Ramos-e-Silva, M.; Carneiro, S. Evaluation of fatigue and its correlation with quality of life index, anxiety symptoms, depression and activity of disease in patients with psoriatic arthritis. Clin. Cosmet. Investig. Dermatol. 2017, 10, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Haddad, A.; Zisman, D. Comorbidities in Patients with Psoriatic Arthritis. Rambam Maimonides Med. J. 2017, 8, e0004. [Google Scholar] [CrossRef] [PubMed]
- Augustin, M.; Vietri, J.; Tian, H.; Gilloteau, I. Incremental burden of cardiovascular comorbidity and psoriatic arthritis among adults with moderate-to-severe psoriasis in five European countries. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 1316–1323. [Google Scholar] [CrossRef] [PubMed]
- Chau, T.; Parsi, K.K.; Ogawa, T.; Kiuru, M.; Konia, T.; Li, C.-S.; Fung, M.A. Psoriasis or not? Review of 51 clinically confirmed cases reveals an expanded histopathologic spectrum of psoriasis. J. Cutan. Pathol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kruithof, E.; Baeten, D.; De Rycke, L.; Vandooren, B.; Foell, D.; Roth, J.; Cañete, J.D.; Boots, A.M.; Veys, E.M.; De Keyser, F. Synovial histopathology of psoriatic arthritis, both oligo- and polyarticular, resembles spondyloarthropathy more than it does rheumatoid arthritis. Arthritis Res. Ther. 2005, 7, R569–R580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chimenti, M.S.; Ballanti, E.; Perricone, C.; Cipriani, P.; Giacomelli, R.; Perricone, R. Immunomodulation in psoriatic arthritis: Focus on cellular and molecular pathways. Autoimmun. Rev. 2013, 12, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, O. Psoriatic arthritis synovial histopathology: Commentary on the article by Kruithof and colleagues. Arthritis Res. Ther. 2005, 7, 124–125. [Google Scholar] [CrossRef] [PubMed]
- Pitzalis, C.; Kelly, S.; Humby, F. New learnings on the pathophysiology of RA from synovial biopsies. Curr. Opin. Rheumatol. 2013, 25, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Canete, J.D.; Santiago, B.; Cantaert, T.; Sanmarti, R.; Palacin, A.; Celis, R.; Graell, E.; Gil-Torregrosa, B.; Baeten, D.; Pablos, J.L. Ectopic lymphoid neogenesis in psoriatic arthritis. Ann. Rheum. Dis. 2007, 66, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Pathobiology of Early Arthritis Cohort (PEAC). Available online: http://www.peac-mrc.mds.qmul.ac.uk/ (accessed on 1 September 2017).
- Sala, M.; Elaissari, A.; Fessi, H. Advances in psoriasis physiopathology and treatments: Up to date of mechanistic insights and perspectives of novel therapies based on innovative skin drug delivery systems (ISDDS). J. Control. Release 2016, 239, 182–202. [Google Scholar] [CrossRef] [PubMed]
- Coates, L.C.; FitzGerald, O.; Helliwell, P.S.; Paul, C. Psoriasis, psoriatic arthritis, and rheumatoid arthritis: Is all inflammation the same? Semin. Arthritis Rheum. 2016, 46, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Fry, L.; Baker, B.S.; Powles, A.V.; Fahlen, A.; Engstrand, L. Is chronic plaque psoriasis triggered by microbiota in the skin? Br. J. Dermatol. 2013, 169, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Sticherling, M. Psoriasis and autoimmunity. Autoimmun. Rev. 2016, 15, 1167–1170. [Google Scholar] [CrossRef] [PubMed]
- Sumaria, N.; Roediger, B.; Ng, L.G.; Qin, J.; Pinto, R.; Cavanagh, L.L.; Shklovskaya, E.; Fazekas de St. Groth, B.; Triccas, J.A.; Weninger, W. Cutaneous immunosurveillance by self-renewing dermal γδ T cells. J. Exp. Med. 2011, 208, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare, A.; Di Meglio, P.; Nestle, F.O. The IL-23/Th17 axis in the immunopathogenesis of psoriasis. J. Investig. Dermatol. 2009, 129, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- FitzGerald, O.; Winchester, R. Psoriatic arthritis: From pathogenesis to therapy. Arthritis Res. Ther. 2009, 11, 214. [Google Scholar] [CrossRef] [PubMed]
- Tassiulas, I.; Duncan, S.R.; Centola, M.; Theofilopoulos, A.N.; Boumpas, D.T. Clonal characteristics of T cell infiltrates in skin and synovium of patients with psoriatic arthritis. Hum. Immunol. 1999, 60, 479–491. [Google Scholar] [CrossRef]
- Pitzalis, C.; Cauli, A.; Pipitone, N.; Smith, C.; Barker, J.; Marchesoni, A.; Yanni, G.; Panayi, G.S. Cutaneous lymphocyte antigen-positive T lymphocytes preferentially migrate to the skin but not to the joint in psoriatic arthritis. Arthritis Rheum. 1996, 39, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Dolcino, M.; Lunardi, C.; Ottria, A.; Tinazzi, E.; Patuzzo, G.; Puccetti, A. Crossreactive Autoantibodies Directed against Cutaneous and Joint Antigens Are Present in Psoriatic Arthritis. PLoS ONE 2014, 9, e115424. [Google Scholar] [CrossRef] [PubMed]
- Costello, P.J.; Winchester, R.J.; Curran, S.A.; Peterson, K.S.; Kane, D.J.; Bresnihan, B.; FitzGerald, O.M. Psoriatic arthritis joint fluids are characterized by CD8 and CD4 T cell clonal expansions appear antigen driven. J. Immunol. 2001, 166, 2878–2886. [Google Scholar] [CrossRef] [PubMed]
- Pitzalis, C.; Jones, G.W.; Bombardieri, M.; Jones, S.A. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat. Rev. Immunol. 2014, 14, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Humby, F.; Bombardieri, M.; Manzo, A.; Kelly, S.; Blades, M.C.; Kirkham, B.; Spencer, J.; Pitzalis, C. Ectopic lymphoid structures support ongoing production of class-switched autoantibodies in rheumatoid synovium. PLoS Med. 2009, 6, e1. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-G.; Jee, H.; Fuentes-Duculan, J.; Wu, W.H.; Byamba, D.; Kim, D.-S.; Kim, D.-Y.; Lew, D.-H.; Yang, W.-I.; Krueger, J.G.; et al. Dermal Clusters of Mature Dendritic Cells and T Cells Are Associated with the CCL20/CCR6 Chemokine System in Chronic Psoriasis. J. Investig. Dermatol. 2014, 134, 1462–1465. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M. Interleukin 17 is a chief orchestrator of immunity. Nat. Immunol. 2017, 18, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Justa, S.; Brucks, M.; Endres, J.; Fox, D.A.; Zhou, X.; Alnaimat, F.; Whitaker, B.; Wheeler, J.C.; Jones, B.H.; et al. Interleukin (IL)-17A, F and AF in inflammation: A study in collagen-induced arthritis and rheumatoid arthritis: IL-17 subtypes in inflammatory arthritis. Clin. Exp. Immunol. 2014, 177, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Toy, D.; Kugler, D.; Wolfson, M.; Bos, T.V.; Gurgel, J.; Derry, J.; Tocker, J.; Peschon, J. Cutting Edge: Interleukin 17 Signals through a Heteromeric Receptor Complex. J. Immunol. 2006, 177, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23–IL-17 immune axis: From mechanisms to therapeutic testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Rutz, S.; Eidenschenk, C.; Ouyang, W. IL-22, not simply a Th17 cytokine. Immunol. Rev. 2013, 252, 116–132. [Google Scholar] [CrossRef] [PubMed]
- Eyerich, S.; Eyerich, K.; Pennino, D.; Carbone, T.; Nasorri, F.; Pallotta, S.; Cianfarani, F.; Odorisio, T.; Traidl-Hoffmann, C.; Behrendt, H.; et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J. Clin. Investig. 2009. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Chen, S.; Qian, H.; Huang, W. Interleukin-23: As a drug target for autoimmune inflammatory diseases: IL-23 in autoimmune inflammatory diseases. Immunology 2012, 135, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Lubberts, E. The IL-23-IL-17 axis in inflammatory arthritis. Nat. Rev. Rheumatol. 2015, 11, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Korn, T.; Oukka, M.; Kuchroo, V.K. Induction and effector functions of TH17 cells. Nature 2008, 453, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.A.; Langrish, C.L.; Chen, Y.; Blumenschein, W.; McClanahan, T.; Kastelein, R.A.; Sedgwick, J.D.; Cua, D.J. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J. Exp. Med. 2003, 198, 1951–1957. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, R.L.; Born, W.K. Dermal γδ T cells—What have we learned? Cell. Immunol. 2015, 296, 62–69. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 2007, 7, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Trepicchio, W.L.; Oestreicher, J.L.; Pittman, D.; Wang, F.; Chamian, F.; Dhodapkar, M.; Krueger, J.G. Increased Expression of Interleukin 23 p19 and p40 in Lesional Skin of Patients with Psoriasis Vulgaris. J. Exp. Med. 2004, 199, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, X.; Liu, Z.; Yue, Q.; Liu, H. Expression of Th17 cytokines in skin lesions of patients with psoriasis. J. Huazhong Univ. Sci. Technol. 2007, 27, 330–332. [Google Scholar] [CrossRef] [PubMed]
- Brentano, F.; Ospelt, C.; Stanczyk, J.; Gay, R.E.; Gay, S.; Kyburz, D. Abundant expression of the interleukin (IL)23 subunit p19, but low levels of bioactive IL23 in the rheumatoid synovium: Differential expression and Toll-like receptor-(TLR) dependent regulation of the IL23 subunits, p19 and p40, in rheumatoid arthritis. Ann. Rheum. Dis. 2009, 68, 143–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeuchi, H.; Kuroiwa, T.; Hiramatsu, N.; Kaneko, Y.; Hiromura, K.; Ueki, K.; Nojima, Y. Expression of interleukin-22 in rheumatoid arthritis: Potential role as a proinflammatory cytokine. Arthritis Rheum. 2005, 52, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Van Baarsen, L.G.; Lebre, M.C.; van der Coelen, D.; Aarrass, S.; Tang, M.W.; Ramwadhdoebe, T.H.; Gerlag, D.M.; Tak, P.P. Heterogeneous expression pattern of interleukin 17A (IL-17A), IL-17F and their receptors in synovium of rheumatoid arthritis, psoriatic arthritis and osteoarthritis: Possible explanation for nonresponse to anti-IL-17 therapy? Arthritis Res. Ther. 2014, 16. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.R.; Blumenschein, W.; Murphy, E.; Diveu, C.; Wiekowski, M.; Abbondanzo, S.; Lucian, L.; Geissler, R.; Brodie, S.; Kimball, A.B.; et al. IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2–dependent mechanisms with implications for psoriasis pathogenesis. J. Exp. Med. 2006, 203, 2577–2587. [Google Scholar] [CrossRef] [PubMed]
- Wolk, K.; Witte, E.; Wallace, E.; Döcke, W.-D.; Kunz, S.; Asadullah, K.; Volk, H.-D.; Sterry, W.; Sabat, R. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: A potential role in psoriasis. Eur. J. Immunol. 2006, 36, 1309–1323. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.-L.; Liang, S.; Li, J.; Napierata, L.; Brown, T.; Benoit, S.; Senices, M.; Gill, D.; Dunussi-Joannopoulos, K.; Collins, M.; et al. IL-22 is required for Th17 cell–mediated pathology in a mouse model of psoriasis-like skin inflammation. J. Clin. Investig. 2008. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Danilenko, D.M.; Valdez, P.; Kasman, I.; Eastham-Anderson, J.; Wu, J.; Ouyang, W. Interleukin-22, a TH17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature 2007, 445, 648–651. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kim, K.-W.; Cho, M.-L.; Ju, J.-H.; Kang, C.-M.; Oh, H.-J.; Min, J.-K.; Lee, S.-H.; Park, S.-H.; Kim, H.-Y. IL-23 induces receptor activator of NF-κB ligand expression in fibroblast-like synoviocytes via STAT3 and NF-κB signal pathways. Immunol. Lett. 2010, 127, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Cañete, J.D.; Celis, R.; Yeremenko, N.; Sanmartí, R.; van Duivenvoorde, L.; Ramírez, J.; Blijdorp, I.; García-Herrero, C.M.; Pablos, J.L.; Baeten, D.L. Ectopic lymphoid neogenesis is strongly associated with activation of the IL-23 pathway in rheumatoid synovitis. Arthritis Res. Ther. 2015, 17. [Google Scholar] [CrossRef] [PubMed]
- Alzabin, S.; Abraham, S.M.; Taher, T.E.; Palfreeman, A.; Hull, D.; McNamee, K.; Jawad, A.; Pathan, E.; Kinderlerer, A.; Taylor, P.C.; et al. Incomplete response of inflammatory arthritis to TNFα blockade is associated with the Th17 pathway. Ann. Rheum. Dis. 2012, 71, 1741–1748. [Google Scholar] [CrossRef] [PubMed]
- Belasco, J.; Louie, J.S.; Gulati, N.; Wei, N.; Nograles, K.; Fuentes-Duculan, J.; Mitsui, H.; Suárez-Fariñas, M.; Krueger, J.G. Comparative Genomic Profiling of Synovium Versus Skin Lesions in Psoriatic Arthritis: Genomic Profiling of Psoriatic Arthritis. Arthritis Rheumatol. 2015, 67, 934–944. [Google Scholar] [CrossRef] [PubMed]
- Celis, R.; Planell, N.; Fernández-Sueiro, J.L.; Sanmartí, R.; Ramírez, J.; González-Álvaro, I.; Pablos, J.L.; Cañete, J.D. Synovial cytokine expression in psoriatic arthritis and associations with lymphoid neogenesis and clinical features. Arthritis Res. Ther. 2012, 14, R93. [Google Scholar] [CrossRef] [PubMed]
- Fiocco, U.; Stramare, R.; Martini, V.; Coran, A.; Caso, F.; Costa, L.; Felicetti, M.; Rizzo, G.; Tonietto, M.; Scanu, A.; et al. Quantitative imaging by pixel-based contrast-enhanced ultrasound reveals a linear relationship between synovial vascular perfusion and the recruitment of pathogenic IL-17A-F+IL-23+ CD161+ CD4+ T helper cells in psoriatic arthritis joints. Clin. Rheumatol. 2017, 36, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Sherlock, J.P.; Joyce-Shaikh, B.; Turner, S.P.; Chao, C.-C.; Sathe, M.; Grein, J.; Gorman, D.M.; Bowman, E.P.; McClanahan, T.K.; Yearley, J.H.; et al. IL-23 induces spondyloarthropathy by acting on ROR-γt+ CD3+CD4−CD8− entheseal resident T cells. Nat. Med. 2012, 18, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Raychaudhuri, S.K.; Raychaudhuri, S.P. Functional role of IL-22 in psoriatic arthritis. Arthritis Res. Ther. 2012, 14, R65. [Google Scholar] [CrossRef] [PubMed]
- Benham, H.; Norris, P.; Goodall, J.; Wechalekar, M.D.; FitzGerald, O.; Szentpetery, A.; Smith, M.; Thomas, R.; Gaston, H. Th17 and Th22 cells in psoriatic arthritis and psoriasis. Arthritis Res. Ther. 2013, 15, R136. [Google Scholar] [CrossRef] [PubMed]
- Eberle, F.C.; Brück, J.; Holstein, J.; Hirahara, K.; Ghoreschi, K. Recent advances in understanding psoriasis. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Krueger, G.G.; Langley, R.G.; Leonardi, C.; Yeilding, N.; Guzzo, C.; Wang, Y.; Dooley, L.T.; Lebwohl, M. A Human Interleukin-12/23 Monoclonal Antibody for the Treatment of Psoriasis. N. Engl. J. Med. 2007, 356, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Kimball, A.B. Safety and Efficacy of ABT-874, a Fully Human Interleukin 12/23 Monoclonal Antibody, in the Treatment of Moderate to Severe Chronic Plaque Psoriasis: Results of a Randomized, Placebo-Controlled, Phase 2 Trial. Arch. Dermatol. 2008, 144, 200. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, C.L.; Kimball, A.B.; Papp, K.A.; Yeilding, N.; Guzzo, C.; Wang, Y.; Li, S.; Dooley, L.T.; Gordon, K.B. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-Week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet 2008, 371, 1665–1674. [Google Scholar] [CrossRef]
- Papp, K.A.; Langley, R.G.; Lebwohl, M.; Krueger, G.G.; Szapary, P.; Yeilding, N.; Guzzo, C.; Hsu, M.-C.; Wang, Y.; Li, S.; et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-Week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet 2008, 371, 1675–1684. [Google Scholar] [CrossRef]
- Langley, R.G.; Lebwohl, M.; Krueger, G.G.; Szapary, P.O.; Wasfi, Y.; Chan, D.; Hsu, M.C.; You, Y.; Poulin, Y.; Korman, N.; et al. Long-term efficacy and safety of ustekinumab, with and without dosing adjustment, in patients with moderate-to-severe psoriasis: Results from the PHOENIX 2 study through 5 years of follow-up. Br. J. Dermatol. 2015, 172, 1371–1383. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.A.; Griffiths, C.E.M.; Gordon, K.; Lebwohl, M.; Szapary, P.O.; Wasfi, Y.; Chan, D.; Hsu, M.-C.; Ho, V.; Ghislain, P.D.; et al. Long-term safety of ustekinumab in patients with moderate-to-severe psoriasis: Final results from 5 years of follow-up: Long-term ustekinumab safety in psoriasis. Br. J. Dermatol. 2013, 168, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Kimball, A.B.; Gordon, K.B.; Fakharzadeh, S.; Yeilding, N.; Szapary, P.O.; Schenkel, B.; Guzzo, C.; Li, S.; Papp, K.A. Long-term efficacy of ustekinumab in patients with moderate-to-severe psoriasis: Results from the PHOENIX 1 trial through up to 3 years: Long-term efficacy of ustekinumab in moderate-to-severe psoriasis. Br. J. Dermatol. 2012, 166, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Rich, P.; Bourcier, M.; Sofen, H.; Fakharzadeh, S.; Wasfi, Y.; Wang, Y.; Kerkmann, U.; Ghislain, P.-D.; Poulin, Y.; the PHOENIX 1 investigators. Ustekinumab improves nail disease in patients with moderate-to-severe psoriasis: Results from PHOENIX 1. Br. J. Dermatol. 2014, 170, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, A.; Menter, A.; Mendelsohn, A.; Shen, Y.-K.; Li, S.; Guzzo, C.; Fretzin, S.; Kunynetz, R.; Kavanaugh, A. Ustekinumab, a human interleukin 12/23 monoclonal antibody, for psoriatic arthritis: Randomised, double-blind, placebo-controlled, crossover trial. Lancet 2009, 373, 633–640. [Google Scholar] [CrossRef]
- McInnes, I.B.; Kavanaugh, A.; Gottlieb, A.B.; Puig, L.; Rahman, P.; Ritchlin, C.; Brodmerkel, C.; Li, S.; Wang, Y.; Mendelsohn, A.M.; et al. Efficacy and safety of ustekinumab in patients with active psoriatic arthritis: 1 year results of the phase 3, multicentre, double-blind, placebo-controlled PSUMMIT 1 trial. Lancet 2013, 382, 780–789. [Google Scholar] [CrossRef]
- Ritchlin, C.; Rahman, P.; Kavanaugh, A.; McInnes, I.B.; Puig, L.; Li, S.; Wang, Y.; Shen, Y.-K.; Doyle, M.K.; Mendelsohn, A.M.; et al. Efficacy and safety of the anti-IL-12/23 p40 monoclonal antibody, ustekinumab, in patients with active psoriatic arthritis despite conventional non-biological and biological anti-tumour necrosis factor therapy: 6-month and 1-year results of the phase 3, multicentre, double-blind, placebo-controlled, randomised PSUMMIT 2 trial. Ann. Rheum. Dis. 2014, 73, 990–999. [Google Scholar] [CrossRef] [PubMed]
- Kavanaugh, A.; Puig, L.; Gottlieb, A.B.; Ritchlin, C.; You, Y.; Li, S.; Song, M.; Randazzo, B.; Rahman, P.; McInnes, I.B. Efficacy and safety of ustekinumab in psoriatic arthritis patients with peripheral arthritis and physician-reported spondylitis: Post-hoc analyses from two phase III, multicentre, double-blind, placebo-controlled studies (PSUMMIT-1/PSUMMIT-2). Ann. Rheum. Dis. 2016, 75, 1984–1988. [Google Scholar] [CrossRef] [PubMed]
- Kavanaugh, A.; Ritchlin, C.; Rahman, P.; Puig, L.; Gottlieb, A.B.; Li, S.; Wang, Y.; Noonan, L.; Brodmerkel, C.; Song, M.; et al. Ustekinumab, an anti-IL-12/23 p40 monoclonal antibody, inhibits radiographic progression in patients with active psoriatic arthritis: Results of an integrated analysis of radiographic data from the phase 3, multicentre, randomised, double-blind, placebo-controlled PSUMMIT-1 and PSUMMIT-2 trials. Ann. Rheum. Dis. 2014, 73, 1000–1006. [Google Scholar] [CrossRef] [PubMed]
- Coates, L.C.; Kavanaugh, A.; Mease, P.J.; Soriano, E.R.; Laura Acosta-Felquer, M.; Armstrong, A.W.; Bautista-Molano, W.; Boehncke, W.-H.; Campbell, W.; Cauli, A.; et al. Group for Research and Assessment of Psoriasis and Psoriatic Arthritis 2015 Treatment Recommendations for Psoriatic Arthritis. Arthritis Rheumatol. 2016, 68, 1060–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlieb, A.B.; Leonardi, C.; Kerdel, F.; Mehlis, S.; Olds, M.; Williams, D.A. Efficacy and safety of briakinumab vs. etanercept and placebo in patients with moderate to severe chronic plaque psoriasis. Br. J. Dermatol. 2011, 165, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Reich, K.; Langley, R.G.; Papp, K.A.; Ortonne, J.-P.; Unnebrink, K.; Kaul, M.; Valdes, J.M. A 52-week trial comparing briakinumab with methotrexate in patients with psoriasis. N. Engl. J. Med. 2011, 365, 1586–1596. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.B.; Langley, R.G.; Gottlieb, A.B.; Papp, K.A.; Krueger, G.G.; Strober, B.E.; Williams, D.A.; Gu, Y.; Valdes, J.M. A phase III, randomized, controlled trial of the fully human IL-12/23 mAb briakinumab in moderate-to-severe psoriasis. J. Investig. Dermatol. 2012, 132, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Langley, R.G.; Papp, K.; Gottlieb, A.B.; Krueger, G.G.; Gordon, K.B.; Williams, D.; Valdes, J.; Setze, C.; Strober, B. Safety results from a pooled analysis of randomized, controlled phase II and III clinical trials and interim data from an open-label extension trial of the interleukin-12/23 monoclonal antibody, briakinumab, in moderate to severe psoriasis: Pooled safety analysis of briakinumab in psoriasis. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 1252–1261. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Guselkumab: First Global Approval. Drugs 2017, 77, 1487–1492. [Google Scholar] [CrossRef] [PubMed]
- Sofen, H.; Smith, S.; Matheson, R.T.; Leonardi, C.L.; Calderon, C.; Brodmerkel, C.; Li, K.; Campbell, K.; Marciniak, S.J.; Wasfi, Y.; et al. Guselkumab (an IL-23–specific mAb) demonstrates clinical and molecular response in patients with moderate-to-severe psoriasis. J. Allergy Clin. Immunol. 2014, 133, 1032–1040. [Google Scholar] [CrossRef] [PubMed]
- Blauvelt, A.; Papp, K.A.; Griffiths, C.E.M.; Randazzo, B.; Wasfi, Y.; Shen, Y.-K.; Li, S.; Kimball, A.B. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: Results from the phase III, double-blinded, placebo- and active comparator-controlled VOYAGE 1 trial. J. Am. Acad. Dermatol. 2017, 76, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Reich, K.; Armstrong, A.W.; Foley, P.; Song, M.; Wasfi, Y.; Randazzo, B.; Li, S.; Shen, Y.-K.; Gordon, K.B. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: Results from the phase III, double-blind, placebo- and active comparator-controlled VOYAGE 2 trial. J. Am. Acad. Dermatol. 2017, 76, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Langley, R.G.; Tsai, T.-F.; Flavin, S.; Song, M.; Randazzo, B.; Wasfi, Y.; Jiang, J.; Li, S.; Puig, L. Efficacy and safety of guselkumab in patients with psoriasis who have an inadequate response to ustekinumab: Results of the randomized, double-blind, phase III NAVIGATE trial. Br. J. Dermatol. 2017. [Google Scholar] [CrossRef]
- Singh, S.; Kroe-Barrett, R.R.; Canada, K.A.; Zhu, X.; Sepulveda, E.; Wu, H.; He, Y.; Raymond, E.L.; Ahlberg, J.; Frego, L.E.; et al. Selective targeting of the IL23 pathway: Generation and characterization of a novel high-affinity humanized anti-IL23A antibody. mAbs 2015, 7, 778–791. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.A.; Blauvelt, A.; Bukhalo, M.; Gooderham, M.; Krueger, J.G.; Lacour, J.-P.; Menter, A.; Philipp, S.; Sofen, H.; Tyring, S.; et al. Risankizumab versus Ustekinumab for Moderate-to-Severe Plaque Psoriasis. N. Engl. J. Med. 2017, 376, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.; Thaçi, D.; Reich, K.; Riedl, E.; Langley, R.G.; Krueger, J.G.; Gottlieb, A.B.; Nakagawa, H.; Bowman, E.P.; Mehta, A.; et al. Tildrakizumab (MK-3222), an anti-interleukin-23p19 monoclonal antibody, improves psoriasis in a phase IIb randomized placebo-controlled trial. Br. J. Dermatol. 2015, 173, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Reich, K.; Papp, K.A.; Blauvelt, A.; Tyring, S.K.; Sinclair, R.; Thaçi, D.; Nograles, K.; Mehta, A.; Cichanowitz, N.; Li, Q.; et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): Results from two randomised controlled, phase 3 trials. Lancet 2017, 390, 276–288. [Google Scholar] [CrossRef]
- Langley, R.G.; Elewski, B.E.; Lebwohl, M.; Reich, K.; Griffiths, C.E.M.; Papp, K.; Puig, L.; Nakagawa, H.; Spelman, L.; Sigurgeirsson, B.; et al. Secukinumab in Plaque Psoriasis—Results of Two Phase 3 Trials. N. Engl. J. Med. 2014, 371, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Strober, B.; Sigurgeirsson, B.; Popp, G.; Sinclair, R.; Krell, J.; Stonkus, S.; Septe, M.; Elewski, B.E.; Gottlieb, A.B.; Zhao, Y.; et al. Secukinumab improves patient-reported psoriasis symptoms of itching, pain, and scaling: Results of two phase 3, randomized, placebo-controlled clinical trials. Int. J. Dermatol. 2016, 55, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.J.; McInnes, I.B.; Kirkham, B.; Kavanaugh, A.; Rahman, P.; van der Heijde, D.; Landewé, R.; Nash, P.; Pricop, L.; Yuan, J.; et al. Secukinumab Inhibition of Interleukin-17A in Patients with Psoriatic Arthritis. N. Engl. J. Med. 2015, 373, 1329–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strand, V.; Mease, P.; Gossec, L.; Elkayam, O.; van den Bosch, F.; Zuazo, J.; Pricop, L.; Mpofu, S. Secukinumab improves patient-reported outcomes in subjects with active psoriatic arthritis: Results from a randomised phase III trial (FUTURE 1). Ann. Rheum. Dis. 2017, 76, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Kavanaugh, A.; Mease, P.J.; Reimold, A.M.; Tahir, H.; Rech, J.; Hall, S.; Geusens, P.; Wang, Z.; Pricop, L.; Mpofu, S.; et al. Secukinumab for Long-Term Treatment of Psoriatic Arthritis: A Two-Year Followup From a Phase III, Randomized, Double-Blind Placebo-Controlled Study: PsA and Long-Term Treatment With Secukinumab. Arthritis Care Res. 2017, 69, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Van der Heijde, D.; Landewé, R.B.; Mease, P.J.; McInnes, I.B.; Conaghan, P.G.; Pricop, L.; Ligozio, G.; Richards, H.B.; Mpofu, S. Brief Report: Secukinumab Provides Significant and Sustained Inhibition of Joint Structural Damage in a Phase III Study of Active Psoriatic Arthritis. Arthritis Rheumatol. 2016, 68, 1914–1921. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Mease, P.J.; Kirkham, B.; Kavanaugh, A.; Ritchlin, C.T.; Rahman, P.; van der Heijde, D.; Landewé, R.; Conaghan, P.G.; Gottlieb, A.B.; et al. Secukinumab, a human anti-interleukin-17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015, 386, 1137–1146. [Google Scholar] [CrossRef]
- Sanford, M.; McKeage, K. Secukinumab: First Global Approval. Drugs 2015, 75, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Kimball, A.B.; Luger, T.; Gottlieb, A.; Puig, L.; Kaufmann, R.; Nikaï, E.; Zhu, B.; Edson-Heredia, E.; Carlier, H.; Lin, C.-Y.; et al. Impact of ixekizumab on psoriasis itch severity and other psoriasis symptoms: Results from 3 phase III psoriasis clinical trials. J. Am. Acad. Dermatol. 2016, 75, 1156–1161. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.B.; Blauvelt, A.; Papp, K.A.; Langley, R.G.; Luger, T.; Ohtsuki, M.; Reich, K.; Amato, D.; Ball, S.G.; Braun, D.K.; et al. Phase 3 Trials of Ixekizumab in Moderate-to-Severe Plaque Psoriasis. N. Engl. J. Med. 2016, 375, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.J.; van der Heijde, D.; Ritchlin, C.T.; Okada, M.; Cuchacovich, R.S.; Shuler, C.L.; Lin, C.-Y.; Braun, D.K.; Lee, C.H.; Gladman, D.D.; et al. Ixekizumab, an interleukin-17A specific monoclonal antibody, for the treatment of biologic-naive patients with active psoriatic arthritis: Results from the 24-week randomised, double-blind, placebo-controlled and active (adalimumab)-controlled period of the phase III trial SPIRIT-P1. Ann. Rheum. Dis. 2017, 76, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.A.; Reich, K.; Paul, C.; Blauvelt, A.; Baran, W.; Bolduc, C.; Toth, D.; Langley, R.G.; Cather, J.; Gottlieb, A.B.; et al. A prospective phase III, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasis. Br. J. Dermatol. 2016, 175, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Lebwohl, M.; Strober, B.; Menter, A.; Gordon, K.; Weglowska, J.; Puig, L.; Papp, K.; Spelman, L.; Toth, D.; Kerdel, F.; et al. Phase 3 Studies Comparing Brodalumab with Ustekinumab in Psoriasis. N. Engl. J. Med. 2015, 373, 1318–1328. [Google Scholar] [CrossRef] [PubMed]
- Astorri, E.; Nerviani, A.; Bombardieri, M.; Pitzalis, C. Towards a stratified targeted approach with biologic treatments in rheumatoid arthritis: Role of synovial pathobiology. Curr. Pharm. Des. 2015, 21, 2216–2224. [Google Scholar] [CrossRef] [PubMed]
- Strober, B.E.; Crowley, J.J.; Yamauchi, P.S.; Olds, M.; Williams, D.A. Efficacy and safety results from a phase III, randomized controlled trial comparing the safety and efficacy of briakinumab with etanercept and placebo in patients with moderate to severe chronic plaque psoriasis. Br. J. Dermatol. 2011, 165, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.J.; Genovese, M.C.; Greenwald, M.W.; Ritchlin, C.T.; Beaulieu, A.D.; Deodhar, A.; Newmark, R.; Feng, J.; Erondu, N.; Nirula, A. Brodalumab, an anti-IL17RA monoclonal antibody, in psoriatic arthritis. N. Engl. J. Med. 2014, 370, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Skepner, J.; Ramesh, R.; Trocha, M.; Schmidt, D.; Baloglu, E.; Lobera, M.; Carlson, T.; Hill, J.; Orband-Miller, L.A.; Barnes, A.; et al. Pharmacologic Inhibition of ROR t Regulates Th17 Signature Gene Expression and Suppresses Cutaneous Inflammation In Vivo. J. Immunol. 2014, 192, 2564–2575. [Google Scholar] [CrossRef] [PubMed]
- Welsch, K.; Holstein, J.; Laurence, A.; Ghoreschi, K. Targeting JAK/STAT signalling in inflammatory skin diseases with small molecule inhibitors. Eur. J. Immunol. 2017, 47, 1096–1107. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; McGarry, T.; Orr, C.; McCormick, J.; Veale, D.J.; Fearon, U. Tofacitinib regulates synovial inflammation in psoriatic arthritis, inhibiting STAT activation and induction of negative feedback inhibitors. Ann. Rheum. Dis. 2016, 75, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.; Hall, S.; FitzGerald, O.; van der Heijde, D.; Merola, J.F.; Avila-Zapata, F.; Cieślak, D.; Graham, D.; Wang, C.; Menon, S.; et al. Tofacitinib or Adalimumab versus Placebo for Psoriatic Arthritis. N. Engl. J. Med. 2017, 377, 1537–1550. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Ren, G.; Xu, L.; Wang, Q.; Qi, J.; Wang, W.; Zhou, B.; Han, X.; Sun, C.; Wu, Q.; et al. Therapeutic efficacy of three bispecific antibodies on collagen-induced arthritis mouse model. Int. Immunopharmacol. 2014, 21, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Teng, X.; Hu, Z.; Wei, X.; Wang, Z.; Guan, T.; Liu, N.; Liu, X.; Ye, N.; Deng, G.; Luo, C.; et al. IL-37 Ameliorates the Inflammatory Process in Psoriasis by Suppressing Proinflammatory Cytokine Production. J. Immunol. 2014, 192, 1815–1823. [Google Scholar] [CrossRef] [PubMed]
- Boutet, M.-A.; Najm, A.; Bart, G.; Brion, R.; Touchais, S.; Trichet, V.; Layrolle, P.; Gabay, C.; Palmer, G.; Blanchard, F.; et al. IL-38 overexpression induces anti-inflammatory effects in mice arthritis models and in human macrophages in vitro. Ann. Rheum. Dis. 2017. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, G.; Koenders, M.; Kalabokis, V.; Kim, J.; Tan, A.C.; Garlanda, C.; Mantovani, A.; Dagna, L.; Joosten, L.A.B.; Dinarello, C.A. Treating experimental arthritis with the innate immune inhibitor interleukin-37 reduces joint and systemic inflammation. Rheumatology 2016, 55, 2220–2229. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Jiang, B.; Deng, J.; Du, J.; Xiong, W.; Guan, Y.; Wen, Z.; Huang, K.; Huang, Z. IL-37 Alleviates Rheumatoid Arthritis by Suppressing IL-17 and IL-17-Triggering Cytokine Production and Limiting Th17 Cell Proliferation. J. Immunol. 2015, 194, 5110–5119. [Google Scholar] [CrossRef] [PubMed]
- Nold-Petry, C.A.; Lo, C.Y.; Rudloff, I.; Elgass, K.D.; Li, S.; Gantier, M.P.; Lotz-Havla, A.S.; Gersting, S.W.; Cho, S.X.; Lao, J.C.; et al. IL-37 requires the receptors IL-18Rα and IL-1R8 (SIGIRR) to carry out its multifaceted anti-inflammatory program upon innate signal transduction. Nat. Immunol. 2015, 16, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Van de Veerdonk, F.L.; Stoeckman, A.K.; Wu, G.; Boeckermann, A.N.; Azam, T.; Netea, M.G.; Joosten, L.A.B.; van der Meer, J.W.M.; Hao, R.; Kalabokis, V.; et al. IL-38 binds to the IL-36 receptor and has biological effects on immune cells similar to IL-36 receptor antagonist. Proc. Natl. Acad. Sci. USA 2012, 109, 3001–3005. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Agents | Name of the Study and Reference | Condition | Trial Phase | Treatment Arm | Control Arm | Primary Endpoint | |
|---|---|---|---|---|---|---|---|
| Skin PASI75 at Week 12 Unless Otherwise Specified | Joint-Related ACR20 Unless Otherwise Specified | ||||||
| Anti-IL12/23p40 | |||||||
| Ustekinumab (CNTO 1275, Stelara) | PHOENIX 1 NCT00267969 [92] | Psoriasis | III | Ust 45 mg (n = 255) Ust 90 mg (n = 256) | Placebo (n = 255) | Ust 45 mg 67.1% Ust 90 mg 66.4% Placebo 3.1% | - |
| PHOENIX 2 NCT00307437 [93,95] | Psoriasis | III | Ust 45 mg (n = 409) Ust 90 mg (n = 411) | Placebo (n = 410) | Ust 45 mg 66.7% Ust 90 mg 75.7% Placebo 3.7% | - | |
| PSUMMIT1 NCT01009086 [99] | PsA | III | Ust 45 mg (n = 205) Ust 90 mg (n = 204) | Placebo (n = 206) | Ust 45 mg 57.2% Ust 90 mg 62.4% Placebo 11.0% | At week 24: Ust 45 mg 42.4% Ust 90 mg 49.5% Placebo 22.8% | |
| PSUMMIT 2 NCT01077362201 [100] | PsA | III | Ust 45 mg (n = 103) Ust 90 mg (n = 105) | Placebo (n = 104) | At week 24: Ust 45 mg 51.3% Ust 90 mg 55.6% Placebo 5% | At week 24: Ust 45 mg 43.7% Ust 90 mg 43.8% Placebo 20.2% | |
| Briakinumab (ABT-874, Ozespa) | NCT00691964 [104] | Psoriasis | III | Bri 200 mg × 2 then 100 mg (n = 138) | Etanercept 50 mg twice-weekly (n = 141) Placebo (n = 68) | Bri 81.9% Eta 56.0% Placebo 7.4% | - |
| NCT00679731 [105] | Psoriasis | III | Bri 200 mg × 2 then 100 mg (n = 154) | MTX 5 to 25 mg weekly (n = 163) | (At week 24) Bri 81.8% MTX 39.9% | - | |
| NCT00710580 [131] | Psoriasis | III | Bri 200 mg × 2 then 100 mg (n = 138) | Etanercept 50 mg twice-weekly (n = 141) Placebo (n = 68) | Bri 80.6% Eta 39.6% Placebo 6.9% | - | |
| NCT00570986 [106] | Psoriasis | III | Bri 200 mg × 2 then 100 mg (n = 981) | Placebo (484) | Bri 80.7% Placebo 4.5% | - | |
| Anti-IL23p19 | |||||||
| Guselkumab (CNTO 1959; Tremfya) | NAVIGATE NCT02203032 [112] | Psoriasis | III | Ust non-responder patients at Week 12 (total n = 268): Gus 100 mg (n = 135) Ust (n = 133) | Ust responder patients at Week 12 (n = 585) | Visits at which patients achieved IGA 0/1 and >2-grade improvement: Gus 1.5 ± 1.6 Ust 0.7 ± 1.3 | - |
| VOYAGE-1 NCT02207231 [110] | Psoriasis | III | Gus 100 mg (n = 329) | Placebo (weeks 0, 4, 12) then Gus 100 mg (weeks 16, 20) (n = 174) Ada 80 mg (n = 334) | PASI75 at week 16: Gus 91.2% Placebo 5.7% Ada 73.1% | - | |
| VOYAGE-2 NCT02207244 [111] | Psoriasis | III | Gus 100 mg (n = 496) | Placebo (weeks 0, 4, 12) then Gus 100 mg (weeks 16, 20) (n = 248) Ada 80 mg (n = 248) | PASI75 at week 16: Gus 86.3% Placebo 8.1% Ada 68.5% | - | |
| Risankizumab (BI 655066) | NCT02054481 [114] | Psoriasis | II | Ris 18 mg (n = 43) Ris 90 mg (n = 41) Ris 180 mg (n = 42) | Ust (n = 40) | PASI90 at week 12: Ris 90 and 180 mg 77% Ust 40% | - |
| UltIMMa-1 NCT02684370 (Not yet completed) | Psoriasis | III | Ris | Ust Placebo | Not yet completed | - | |
| Tildrakizumab (MK-3222) | reSURFACE-1 NCT01722331 [116] | Psoriasis | III | Til 100 mg (n = 309) Til 200 mg (n = 308) | Placebo (n = 155) | Til 100 mg 62% Til 200 mg 64% Placebo 6% | - |
| reSURFACE-2 NCT01729754 [116] | Psoriasis | III | Til 100 mg (n = 307) Til 200 mg (n = 314) | Placebo (n = 156) Eta (n = 313) | Til 100 mg 61% Til 200 mg 66% Placebo 6% Eta 48% | - | |
| Anti-IL17A | |||||||
| Secukinumab (AIN457, Cosentyx) | ERASURE NCT01365455 [118] | Psoriasis | III | Sec 150 mg (n = 245) Sec 300 mg (n = 245) | Placebo (n = 248) | Sec 150 mg 71.6% Sec 300 mg 81.6% Placebo 4.5% | - |
| FIXTURE NCT01358578 [118] | Psoriasis | III | Sec 150 mg (n = 327) Sec 300 mg (n = 327) | Placebo (n = 326) | Sec 150 mg 67.0% Sec 300 mg 77.1% Placebo 4.9% | - | |
| FUTURE1 NCT01392326 [119] | PsA | III | Sec 75 mg (n = 202) Sec 150 mg (n = 202) | Placebo (n = 202) | PASI75 at week 24: Sec 75 mg 64.8% Sec 150 mg 61.1% Placebo 8.3% | ACR20 at week 24: Sec 75 mg 50% Sec 150 mg 50.5% Placebo 17.3% | |
| FUTURE2 NCT01752634 [123] | PsA | III | Sec 75 mg (n = 99) Sec 150 mg (n = 100) Sec 300 mg (n = 100) | Placebo (n = 98) | PASI75 at week 24: Sec 150 mg 48% Sec 300 mg 63% Placebo 16% | ACR20 at week 24: Sec 75 mg 29% Sec 150 mg 51% Sec 300 mg 54% Placebo 15% | |
| Ixekizumab (LY2439821, Taltz) | UNCOVER-1 NCT01474512 [126] | Psoriasis | III | Ixe 80 mg q2wk (n = 433) Ixe 80 mg q4wk (n = 432) | Placebo (n = 431) | Ixe 80 mg q2wk 89.1% Ixe 80 mg q4wk 82.6% Placebo 3.9% | - |
| UNCOVER-2 NCT01597245 [126] | Psoriasis | III | Ixe 80 mg q2wk (n = 351) Ixe 80 mg q4wk (n = 347) | Eta 50 mg twice-weekly (n = 358) Placebo (n = 168) | Ixe q2wk 89.7% Ixe q4wk 77.5% Eta 41.6% Placebo 2.4% | - | |
| UNCOVER-3 NCT01646177 [126] | Psoriasis | III | Ixe 80 mg q2wk (n = 385) Ixe 80 mg q4wk (n = 386) | Eta 50 mg twice-weekly (n = 382) Placebo (n = 193) | Ixe q2wk 87.3% Ixe q4wk 84.2% Eta 53.4% Placebo 7.3% | - | |
| SPIRIT-P1 NCT01695239 [127] | PsA | III | Ixe 80 mg q2wk (n = 103) Ixe 80 mg q4wk (n = 107) | Ada 40 mg q2wk (n = 101) Placebo (n = 106) | PASI75 at week 24: Ixe q2wk 79.7% Ixe q4wk 71.2% Ada 54.4% Placebo 10.4% | ACR20 at week 24: Ixe q2wk 62.1% Ixe q4wk 57.9% Ada 57.4% Placebo 30.2% | |
| Anti-IL17RA | |||||||
| Brodalumab (AMG 827, Siliq) | AMAGINE-2 NCT01708603 [129] | Psoriasis | III | Bro 140 mg (n = 610) Bro 210 mg (n = 612) | Ust 45 mg (n = 300) Placebo (n = 309) | Bro 140 mg 67% Bro 210 mg 86% Ust 70% Placebo 8% | - |
| AMAGINE-3 NCT01708629 [132] | Psoriasis | III | Bro 140 mg (n = 629) Bro 210 mg (n = 624) | Ustekinumab 45 mg (n = 313) Placebo (n = 315) | Bro 140 mg 69% Bro 210 mg 85% Ust 69% Placebo 6% | - | |
| NCT01516957 | PsA | II | Bro 280 mg (n = 56) Bro 140 mg (n = 57) | Placebo (n = 55) | - | Bro 280 mg 39% Bro 140 mg 37% Placebo 18% | |
| AMVISION-2 NCT02024646 (Not yet completed) | PsA | III | Bro 140 mg Bro 210 mg | Placebo | Not yet completed | Not yet completed | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boutet, M.-A.; Nerviani, A.; Gallo Afflitto, G.; Pitzalis, C. Role of the IL-23/IL-17 Axis in Psoriasis and Psoriatic Arthritis: The Clinical Importance of Its Divergence in Skin and Joints. Int. J. Mol. Sci. 2018, 19, 530. https://doi.org/10.3390/ijms19020530
Boutet M-A, Nerviani A, Gallo Afflitto G, Pitzalis C. Role of the IL-23/IL-17 Axis in Psoriasis and Psoriatic Arthritis: The Clinical Importance of Its Divergence in Skin and Joints. International Journal of Molecular Sciences. 2018; 19(2):530. https://doi.org/10.3390/ijms19020530
Chicago/Turabian StyleBoutet, Marie-Astrid, Alessandra Nerviani, Gabriele Gallo Afflitto, and Costantino Pitzalis. 2018. "Role of the IL-23/IL-17 Axis in Psoriasis and Psoriatic Arthritis: The Clinical Importance of Its Divergence in Skin and Joints" International Journal of Molecular Sciences 19, no. 2: 530. https://doi.org/10.3390/ijms19020530