The Impact of Protein Acetylation/Deacetylation on Systemic Lupus Erythematosus


Abstract
:1. Introduction
2. Systemic Lupus Erythematosus
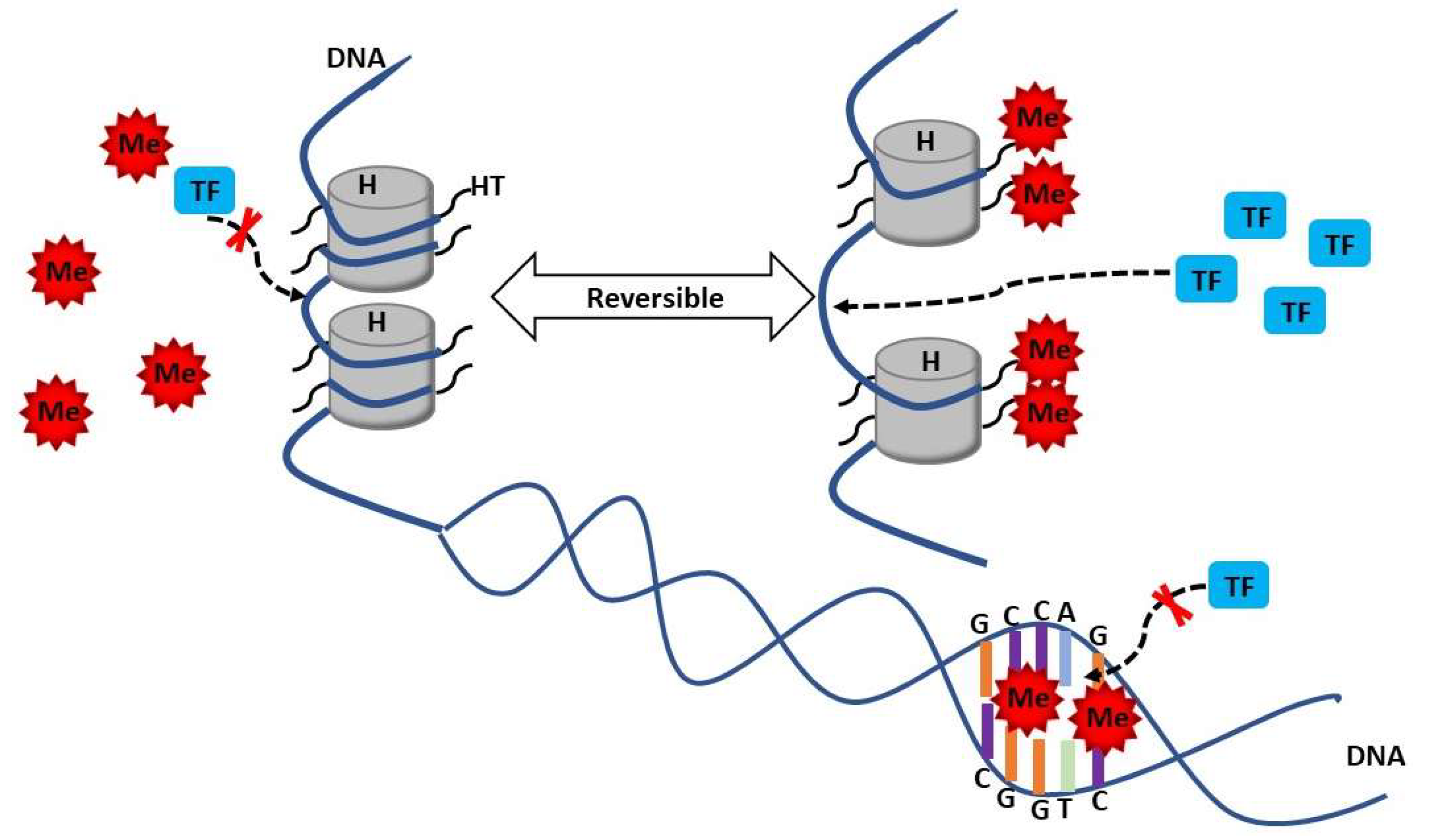
3. Methylation in SLE
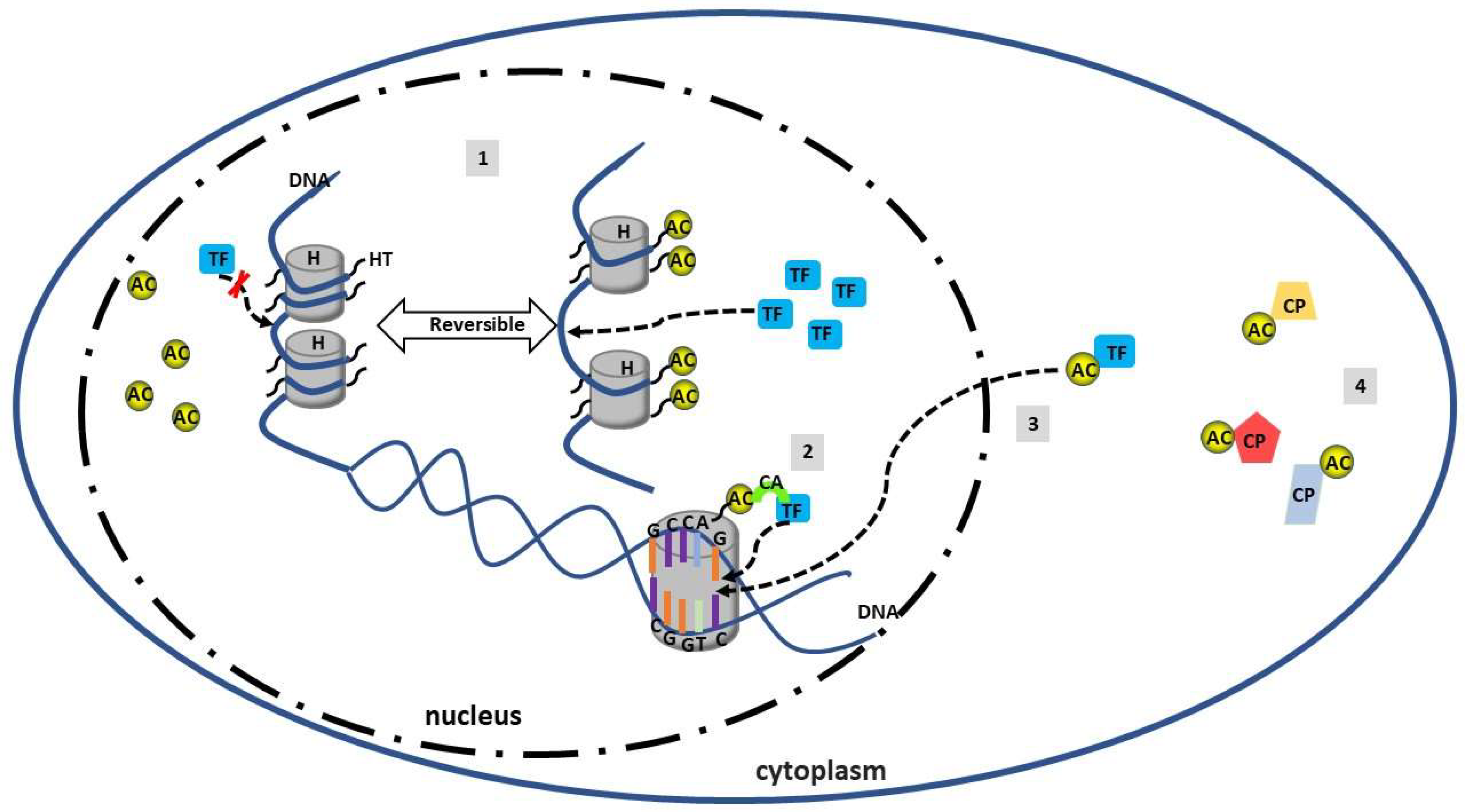
4. Acetylation in SLE
5. Metabolism and Epigenetic Crosstalk in Lupus
6. Summary
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| SLE | Systemic lupus erythematosus |
| MHC | Major histocompatibility complex |
| HDACs | Histone deacetylase |
| HATs | Histone acetyltransferase |
| MEF2 | Myocyte enhancer factor 2 |
| TFN I/II | Type I/II interferons |
| TNF | Tumor necrosis factors |
| BLys | B-lymphocyte stimulators |
| LN | Lupus nephritis |
| SNPs | Single-nucleotide polymorphisms |
| TLR | Toll like receptor |
| SLEDAI | SLE disease activity index |
| TSA | Trichostatin |
| SAHA | Suberonylanilide hydroxamix acid |
| PBMC | Peripheral blood mononuclear cell |
| NZB/W | New Zealand Black/White |
References
- Kaul, A.; Gordon, C.; Crow, M.K.; Touma, Z.; Urowitz, M.B.; van Vollenhoven, R.; Ruiz-Irastorza, G.; Hughes, G. Systemic lupus erythematosus. Nat. Rev. Dis. Prim. 2016, 2, 16039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almaani, S.; Meara, A.; Rovin, B.H. Update on Lupus Nephritis. Clin. J. Am. Soc. Nephrol. 2017, 12, 825–835. [Google Scholar] [CrossRef]
- Bernatsky, S.; Boivin, J.F.; Joseph, L.; Manzi, S.; Ginzler, E.; Gladman, D.D.; Urowitz, M.; Fortin, P.R.; Petri, M.; Barr, S.; et al. Mortality in systemic lupus erythematosus. Arthritis Rheum. 2006, 54, 2550–2557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedrich, C.M. Epigenetics in SLE. Curr. Rheumatol. Rep. 2017, 19, 58. [Google Scholar] [CrossRef] [PubMed]
- Franks, A.L.; Slansky, J.E. Multiple associations between a broad spectrum of autoimmune diseases, chronic inflammatory diseases and cancer. Anticancer Res. 2012, 32, 1119–1136. [Google Scholar]
- Saito, Y.; Saito, H.; Liang, G.; Friedman, J.M. Epigenetic alterations and microRNA misexpression in cancer and autoimmune diseases: A critical review. Clin. Rev. Allergy Immunol. 2014, 47, 128–135. [Google Scholar] [CrossRef]
- Jeffries, M.A. Epigenetic editing: How cutting-edge targeted epigenetic modification might provide novel avenues for autoimmune disease therapy. Clin. Immunol. 2018. [Google Scholar] [CrossRef]
- Jeffries, M.A.; Sawalha, A.H. Autoimmune disease in the epigenetic era: How has epigenetics changed our understanding of disease and how can we expect the field to evolve? Expert Rev Clin. Immunol. 2015, 11, 45–58. [Google Scholar] [CrossRef]
- Regna, N.L.; Vieson, M.D.; Gojmerac, A.M.; Luo, X.M.; Caudell, D.L.; Reilly, C.M. HDAC expression and activity is upregulated in diseased lupus-prone mice. Int. Immunopharmacol. 2015, 29, 494–503. [Google Scholar] [CrossRef]
- Reilly, C.M.; Regna, N.; Mishra, N. HDAC inhibition in lupus models. Mol. Med. 2011, 17, 417–425. [Google Scholar] [CrossRef]
- Zhang, Z.; Song, L.; Maurer, K.; Petri, M.A.; Sullivan, K.E. Global H4 acetylation analysis by ChIP-chip in systemic lupus erythematosus monocytes. Genes Immun. 2010, 11, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.A.; Nititham, J.; Elboudwarej, E.; Quach, H.L.; Taylor, K.E.; Barcellos, L.F.; Criswell, L.A. Genome-Wide Assessment of Differential DNA Methylation Associated with Autoantibody Production in Systemic Lupus Erythematosus. PLoS ONE 2015, 10, e0129813. [Google Scholar] [CrossRef] [PubMed]
- Coit, P.; Jeffries, M.; Altorok, N.; Dozmorov, M.G.; Koelsch, K.A.; Wren, J.D.; Merrill, J.T.; McCune, W.J.; Sawalha, A.H. Genome-wide DNA methylation study suggests epigenetic accessibility and transcriptional poising of interferon-regulated genes in naive CD4+ T cells from lupus patients. J. Autoimmun. 2013, 43, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Sawalha, A.H.; Jeffries, M.; Webb, R.; Lu, Q.; Gorelik, G.; Ray, D.; Osban, J.; Knowlton, N.; Johnson, K.; Richardson, B. Defective T-cell ERK signaling induces interferon-regulated gene expression and overexpression of methylation-sensitive genes similar to lupus patients. Genes Immun. 2008, 9, 368–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahren-Herlenius, M.; Dorner, T. Immunopathogenic mechanisms of systemic autoimmune disease. Lancet 2013, 382, 819–831. [Google Scholar] [CrossRef]
- Deng, Y.; Tsao, B.P. Genetic susceptibility to systemic lupus erythematosus in the genomic era. Nat. Rev. Rheumatol. 2010, 6, 683–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, W.H.; Cohen, P.L. Disturbances of apoptotic cell clearance in systemic lupus erythematosus. Arthritis Res. Ther. 2011, 13, 202. [Google Scholar] [CrossRef] [PubMed]
- Fadeel, B.; Xue, D.; Kagan, V. Programmed cell clearance: Molecular regulation of the elimination of apoptotic cell corpses and its role in the resolution of inflammation. Biochem. Biophys. Res. Commun. 2010, 396, 7–10. [Google Scholar] [CrossRef] [Green Version]
- Jacob, C.; Lebrun-Julien, F.; Suter, U. How histone deacetylases control myelination. Mol. Neurobiol. 2011, 44, 303–312. [Google Scholar] [CrossRef]
- Mackay, M.; Oswald, M.; Sanchez-Guerrero, J.; Lichauco, J.; Aranow, C.; Kotkin, S.; Korsunsky, I.; Gregersen, P.K.; Diamond, B. Molecular signatures in systemic lupus erythematosus: Distinction between disease flare and infection. Lupus Sci. Med. 2016, 3, e000159. [Google Scholar] [CrossRef]
- Theofilopoulos, A.N.; Baccala, R.; Beutler, B.; Kono, D.H. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu. Rev. Immunol. 2005, 23, 307–336. [Google Scholar] [CrossRef] [PubMed]
- Hiraki, L.T.; Feldman, C.H.; Liu, J.; Alarcon, G.S.; Fischer, M.A.; Winkelmayer, W.C.; Costenbader, K.H. Prevalence, incidence, and demographics of systemic lupus erythematosus and lupus nephritis from 2000 to 2004 among children in the US Medicaid beneficiary population. Arthritis Rheum. 2012, 64, 2669–2676. [Google Scholar] [CrossRef] [PubMed]
- Mok, C.C.; Kwok, C.L.; Ho, L.Y.; Chan, P.T.; Yip, S.F. Life expectancy, standardized mortality ratios, and causes of death in six rheumatic diseases in Hong Kong, China. Arthritis Rheum. 2011, 63, 1182–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, L.; Leone, V.; Pilkington, C.; Tullus, K.; Rangaraj, S.; McDonagh, J.E.; Gardner-Medwin, J.; Wilkinson, N.; Riley, P.; Tizard, J.; et al. Disease activity, severity, and damage in the UK Juvenile-Onset Systemic Lupus Erythematosus Cohort. Arthritis Rheum. 2012, 64, 2356–2365. [Google Scholar] [CrossRef] [PubMed]
- Borchers, A.T.; Keen, C.L.; Shoenfeld, Y.; Gershwin, M.E. Surviving the butterfly and the wolf: Mortality trends in systemic lupus erythematosus. Autoimmun. Rev. 2004, 3, 423–453. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, M.A.; Habib, H.B.; Islam, M.; Ahmad, B.; Majid, S.; Saeed, W.; Shah, S.M.; Ahmad, A. Survival analysis and prognostic indicators of systemic lupus erythematosus in Pakistani patients. Lupus 2009, 18, 848–855. [Google Scholar] [CrossRef]
- Flower, C.; Hennis, A.J.; Hambleton, I.R.; Nicholson, G.D.; Liang, M.H.; Barbados National Lupus Registry Group. Systemic lupus erythematosus in an African Caribbean population: Incidence, clinical manifestations, and survival in the Barbados National Lupus Registry. Arthritis Care Res. 2012, 64, 1151–1158. [Google Scholar]
- Pons-Estel, G.J.; Serrano, R.; Plasin, M.A.; Espinosa, G.; Cervera, R. Epidemiology and management of refractory lupus nephritis. Autoimmun. Rev. 2011, 10, 655–663. [Google Scholar] [CrossRef]
- Baranowska-Daca, E.; Choi, Y.J.; Barrios, R.; Nassar, G.; Suki, W.N.; Truong, L.D. Nonlupus nephritides in patients with systemic lupus erythematosus: A comprehensive clinicopathologic study and review of the literature. Hum. Pathol. 2001, 32, 1125–1135. [Google Scholar] [CrossRef]
- Andrews, K.T.; Tran, T.N.; Wheatley, N.C.; Fairlie, D.P. Targeting histone deacetylase inhibitors for anti-malarial therapy. Curr. Top. Med. Chem. 2009, 9, 292–308. [Google Scholar] [CrossRef]
- Rullo, O.J.; Tsao, B.P. Recent insights into the genetic basis of systemic lupus erythematosus. Ann. Rheum. Dis. 2013, 72 (Suppl. 2), ii56–ii61. [Google Scholar] [CrossRef] [PubMed]
- Tsokos, G.C.; Kammer, G.M. Molecular aberrations in human systemic lupus erythematosus. Mol. Med. Today 2000, 6, 418–424. [Google Scholar] [CrossRef]
- Harley, J.B.; Moser, K.L.; Gaffney, P.M.; Behrens, T.W. The genetics of human systemic lupus erythematosus. Curr. Opin. Immunol. 1998, 10, 690–696. [Google Scholar] [CrossRef]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zampeli, E.; Klinman, D.M.; Gershwin, M.E.; Moutsopoulos, H.M. A comprehensive evaluation for the treatment of lupus nephritis. J. Autoimmun. 2017, 78, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tsokos, G.C.; Magrath, I.T.; Balow, J.E. Epstein-Barr virus induces normal B cell responses but defective suppressor T cell responses in patients with systemic lupus erythematosus. J. Immunol. 1983, 131, 1797–1801. [Google Scholar] [PubMed]
- Kang, I.; Quan, T.; Nolasco, H.; Park, S.H.; Hong, M.S.; Crouch, J.; Pamer, E.G.; Howe, J.G.; Craft, J. Defective control of latent Epstein-Barr virus infection in systemic lupus erythematosus. J. Immunol. 2004, 172, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Poole, B.D.; Scofield, R.H.; Harley, J.B.; James, J.A. Epstein-Barr virus and molecular mimicry in systemic lupus erythematosus. Autoimmunity 2006, 39, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Smith-Bouvier, D.L.; Divekar, A.A.; Sasidhar, M.; Du, S.; Tiwari-Woodruff, S.K.; King, J.K.; Arnold, A.P.; Singh, R.R.; Voskuhl, R.R. A role for sex chromosome complement in the female bias in autoimmune disease. J. Exp. Med. 2008, 205, 1099–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costenbader, K.H.; Feskanich, D.; Stampfer, M.J.; Karlson, E.W. Reproductive and menopausal factors and risk of systemic lupus erythematosus in women. Arthritis Rheum. 2007, 56, 1251–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, B. Primer: Epigenetics of autoimmunity. Nat. Clin. Pract. Rheumatol. 2007, 3, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L. The complex language of chromatin regulation during transcription. Nature 2007, 447, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Munshi, A.; Shafi, G.; Aliya, N.; Jyothy, A. Histone modifications dictate specific biological readouts. J. Genet. Genom. 2009, 36, 75–88. [Google Scholar] [CrossRef]
- Kalden, J.R. Defective phagocytosis of apoptotic cells: Possible explanation for the induction of autoantibodies in SLE. Lupus 1997, 6, 326–327. [Google Scholar] [CrossRef]
- Gaipl, U.S.; Kuhn, A.; Sheriff, A.; Munoz, L.E.; Franz, S.; Voll, R.E.; Kalden, J.R.; Herrmann, M. Clearance of apoptotic cells in human SLE. Curr. Dir. Autoimmun. 2006, 9, 173–187. [Google Scholar]
- Qiao, B.; Wu, J.; Chu, Y.W.; Wang, Y.; Wang, D.P.; Wu, H.S.; Xiong, S.D. Induction of systemic lupus erythematosus-like syndrome in syngeneic mice by immunization with activated lymphocyte-derived DNA. Rheumatology 2005, 44, 1108–1114. [Google Scholar] [CrossRef] [Green Version]
- Wen, Z.K.; Xu, W.; Xu, L.; Cao, Q.H.; Wang, Y.; Chu, Y.W.; Xiong, S.D. DNA hypomethylation is crucial for apoptotic DNA to induce systemic lupus erythematosus-like autoimmune disease in SLE-non-susceptible mice. Rheumatology 2007, 46, 1796–1803. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, K.; Richez, C.; Uccellini, M.B.; Richards, R.J.; Bonegio, R.G.; Akira, S.; Monestier, M.; Corley, R.B.; Viglianti, G.A.; Marshak-Rothstein, A.; et al. Requirement for DNA CpG content in TLR9-dependent dendritic cell activation induced by DNA-containing immune complexes. J. Immunol. 2009, 183, 3109–3117. [Google Scholar] [CrossRef]
- Richardson, B.; Scheinbart, L.; Strahler, J.; Gross, L.; Hanash, S.; Johnson, M. Evidence for impaired T cell DNA methylation in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum. 1990, 33, 1665–1673. [Google Scholar] [CrossRef]
- Liang, J.; Zhu, X.H.; Qin, H.H.; Lin, J.R.; Wang, D.Q.; Huang, L.; Luo, X.Q.; Xu, J.H. A correlation study on the effects of DNMT1 on methylation levels in CD4(+) T cells of SLE patients. Int. J. Clin. Exp. Med. 2015, 8, 19701–19708. [Google Scholar]
- Richardson, B.; Sawalha, A.H.; Ray, D.; Yung, R. Murine models of lupus induced by hypomethylated T cells (DNA hypomethylation and lupus.). Methods Mol. Biol. 2012, 900, 169–180. [Google Scholar] [PubMed]
- Lu, Q.; Kaplan, M.; Ray, D.; Ray, D.; Zacharek, S.; Gutsch, D.; Richardson, B. Demethylation of ITGAL (CD11a) regulatory sequences in systemic lupus erythematosus. Arthritis Rheum. 2002, 46, 1282–1291. [Google Scholar] [CrossRef] [PubMed]
- Vire, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, Y.B.; Pirrotta, V. Polycomb silencing mechanisms and the management of genomic programmes. Nat. Rev. Genet. 2007, 8, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Li, B.; Lin, B.; Lee, P.T.; Chung, T.H.; Tan, J.; Bi, C.; Lee, X.T.; Selvarajan, V.; Ng, S.B.; et al. EZH2 phosphorylation by JAK3 mediates a switch to noncanonical function in natural killer/T-cell lymphoma. Blood 2016, 128, 948–958. [Google Scholar] [CrossRef] [Green Version]
- Ho, L.; Crabtree, G.R. Chromatin remodelling during development. Nature 2010, 463, 474–484. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Long, H.; Liao, J.; Zhao, M.; Liang, G.; Wu, X.; Zhang, P.; Ding, S.; Luo, S.; Lu, Q. Inhibited expression of hematopoietic progenitor kinase 1 associated with loss of jumonji domain containing 3 promoter binding contributes to autoimmunity in systemic lupus erythematosus. J. Autoimmun. 2011, 37, 180–189. [Google Scholar] [CrossRef]
- Gray, S.G. Perspectives on epigenetic-based immune intervention for rheumatic diseases. Arthritis Res. Ther. 2013, 15, 207. [Google Scholar] [CrossRef]
- Tsou, P.S.; Coit, P.; Kilian, N.C.; Sawalha, A.H. EZH2 Modulates the DNA Methylome and Controls T Cell Adhesion Through Junctional Adhesion Molecule A in Lupus Patients. Arthritis Rheumatol. 2018, 70, 98–108. [Google Scholar] [CrossRef]
- Zhou, W.; Zhu, W.G. The changing face of HDAC inhibitor depsipeptide. Curr. Cancer Drug Targets 2009, 9, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Wu, A.; Tesmer, L.; Ray, D.; Yousif, N.; Richardson, B. Demethylation of CD40LG on the inactive X in T cells from women with lupus. J. Immunol. 2007, 179, 6352–6358. [Google Scholar] [CrossRef] [PubMed]
- Oelke, K.; Lu, Q.; Richardson, D.; Wu, A.; Deng, C.; Hanash, S.; Richardson, B. Overexpression of CD70 and overstimulation of IgG synthesis by lupus T cells and T cells treated with DNA methylation inhibitors. Arthritis Rheum. 2004, 50, 1850–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulff-Moller, C.J.; Asmar, F.; Liu, Y.; Svendsen, A.J.; Busato, F.; Gronbaek, K.; Tost, J.; Jacobsen, S. Twin DNA Methylation Profiling Reveals Flare-Dependent Interferon Signature and B Cell Promoter Hypermethylation in Systemic Lupus Erythematosus. Arthritis Rheumatol. 2018, 70, 878–890. [Google Scholar] [CrossRef] [PubMed]
- Ballestar, E.; Esteller, M.; Richardson, B.C. The epigenetic face of systemic lupus erythematosus. J. Immunol. 2006, 176, 7143–7147. [Google Scholar] [CrossRef] [PubMed]
- Cheung, W.L.; Briggs, S.D.; Allis, C.D. Acetylation and chromosomal functions. Curr. Opin. Cell Biol. 2000, 12, 326–333. [Google Scholar] [CrossRef]
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349–352. [Google Scholar] [CrossRef]
- Shahbazian, M.D.; Grunstein, M. Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 2007, 76, 75–100. [Google Scholar] [CrossRef]
- Ruthenburg, A.J.; Li, H.; Patel, D.J.; Allis, C.D. Multivalent engagement of chromatin modifications by linked binding modules. Nat. Rev. Mol. Cell Biol. 2007, 8, 983–994. [Google Scholar] [CrossRef] [Green Version]
- Kouzarides, T. Acetylation: A regulatory modification to rival phosphorylation? EMBO J. 2000, 19, 1176–1179. [Google Scholar] [CrossRef]
- Taunton, J.; Hassig, C.A.; Schreiber, S.L. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science 1996, 272, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Seto, E. The Rpd3/Hda1 family of lysine deacetylases: From bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 2008, 9, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Kurdistani, S.K.; Grunstein, M. Histone acetylation and deacetylation in yeast. Nat. Rev. Mol. Cell Biol. 2003, 4, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Seto, E. Collaborative spirit of histone deacetylases in regulating chromatin structure and gene expression. Curr. Opin. Genet. Dev. 2003, 13, 143–153. [Google Scholar] [CrossRef]
- Vega, R.B.; Matsuda, K.; Oh, J.; Barbosa, A.C.; Yang, X.; Meadows, E.; McAnally, J.; Pomajzl, C.; Shelton, J.M.; Richardson, J.A.; et al. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell 2004, 119, 555–566. [Google Scholar] [CrossRef]
- McKinsey, T.A.; Zhang, C.L.; Lu, J.; Olson, E.N. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 2000, 408, 106–111. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; McKinsey, T.A.; Nicol, R.L.; Olson, E.N. Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc. Natl. Acad. Sci. USA 2000, 97, 4070–4075. [Google Scholar] [CrossRef] [Green Version]
- Passier, R.; Zeng, H.; Frey, N.; Naya, F.J.; Nicol, R.L.; McKinsey, T.A.; Overbeek, P.; Richardson, J.A.; Grant, S.R.; Olson, E.N. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J. Clin. Investig. 2000, 105, 1395–1406. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.L.; McKinsey, T.A.; Chang, S.; Antos, C.L.; Hill, J.A.; Olson, E.N. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 2002, 110, 479–488. [Google Scholar] [CrossRef]
- Chang, S.; McKinsey, T.A.; Zhang, C.L.; Richardson, J.A.; Hill, J.A.; Olson, E.N. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol. Cell. Biol. 2004, 24, 8467–8476. [Google Scholar] [CrossRef]
- Chang, S.; Young, B.D.; Li, S.; Qi, X.; Richardson, J.A.; Olson, E.N. Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell 2006, 126, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fang, H.; Jiao, J.; Xu, W. The structure and function of histone deacetylases: The target for anti-cancer therapy. Curr. Med. Chem. 2008, 15, 2840–2849. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.D.; Cai, R.; Bhatia, U.; Asselbergs, F.A.; Song, C.; Terry, R.; Trogani, N.; Widmer, R.; Atadja, P.; Cohen, D. Isolation and characterization of a novel class II histone deacetylase, HDAC10. J. Biol. Chem. 2002, 277, 6656–6666. [Google Scholar] [CrossRef]
- Guardiola, A.R.; Yao, T.P. Molecular cloning and characterization of a novel histone deacetylase HDAC10. J. Biol. Chem. 2002, 277, 3350–3356. [Google Scholar] [CrossRef]
- Tang, X.; Gao, J.S.; Guan, Y.J.; McLane, K.E.; Yuan, Z.L.; Ramratnam, B.; Chin, Y.E. Acetylation-dependent signal transduction for type I interferon receptor. Cell 2007, 131, 93–105. [Google Scholar] [CrossRef]
- Zhang, X.; Yuan, Z.; Zhang, Y.; Yong, S.; Salas-Burgos, A.; Koomen, J.; Olashaw, N.; Parsons, J.T.; Yang, X.J.; Dent, S.R.; et al. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol. Cell 2007, 27, 197–213. [Google Scholar] [CrossRef]
- Kovacs, J.J.; Murphy, P.J.; Gaillard, S.; Zhao, X.; Wu, J.T.; Nicchitta, C.V.; Yoshida, M.; Toft, D.O.; Pratt, W.B.; Yao, T.P. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 2005, 18, 601–607. [Google Scholar] [CrossRef]
- Matsuyama, A.; Shimazu, T.; Sumida, Y.; Saito, A.; Yoshimatsu, Y.; Seigneurin-Berny, D.; Osada, H.; Komatsu, Y.; Nishino, N.; Khochbin, S.; et al. In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J. 2002, 21, 6820–6831. [Google Scholar] [CrossRef] [Green Version]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Hu, Q.; Kaufman, A.; D’Ercole, A.J.; Ye, P. Developmental expression of histone deacetylase 11 in the murine brain. J. Neurosci. Res. 2008, 86, 537–543. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, M.; Clarke, C.; Marks, P.A. Histone deacetylase inhibitors: Overview and perspectives. Mol. Cancer Res. 2007, 5, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Mohseni, J.; Zabidi-Hussin, Z.A.M.H.; Sasongko, T.H. Histone deacetylase inhibitors as potential treatment for spinal muscular atrophy. Genet. Mol. Biol. 2013, 36, 299–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conte, M.; Dell’Aversana, C.; Benedetti, R.; Petraglia, F.; Carissimo, A.; Petrizzi, V.B.; D’Arco, A.M.; Abbondanza, C.; Nebbioso, A.; Altucci, L. HDAC2 deregulation in tumorigenesis is causally connected to repression of immune modulation and defense escape. Oncotarget 2015, 6, 886–901. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.F.; Chen, Y.M.; Lin, C.Y.; Huang, H.L.; Yeh, H.; Chang, Y.T.; Huang, K.T.; Lin, M.C. Histone deacetylase 2 (HDAC2) attenuates lipopolysaccharide (LPS)-induced inflammation by regulating PAI-1 expression. J. Inflamm. 2018, 15, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, H.; Pallos, J.; Jacques, V.; Lau, A.; Tang, B.; Cooper, A.; Syed, A.; Purcell, J.; Chen, Y.; Sharma, S.; et al. Histone deacetylase (HDAC) inhibitors targeting HDAC3 and HDAC1 ameliorate polyglutamine-elicited phenotypes in model systems of Huntington’s disease. Neurobiol. Dis. 2012, 46, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, A.; Oehme, I.; Witt, O.; Oliveira, G.; Sippl, W.; Romier, C.; Pierce, R.J.; Jung, M. HDAC8: A multifaceted target for therapeutic interventions. Trends Pharmacol. Sci. 2015, 36, 481–492. [Google Scholar] [CrossRef]
- Wang, Z.; Qin, G.; Zhao, T.C. HDAC4: Mechanism of regulation and biological functions. Epigenomics 2014, 6, 139–150. [Google Scholar] [CrossRef]
- Mielcarek, M.; Zielonka, D.; Carnemolla, A.; Marcinkowski, J.T.; Guidez, F. HDAC4 as a potential therapeutic target in neurodegenerative diseases: A summary of recent achievements. Front. Cell. Neurosci. 2015, 9, 42. [Google Scholar] [CrossRef]
- Cao, C.; Wu, H.; Vasilatos, S.N.; Chandran, U.; Qin, Y.; Wan, Y.; Oesterreich, S.; Davidson, N.E.; Huang, Y. HDAC5-LSD1 axis regulates antineoplastic effect of natural HDAC inhibitor sulforaphane in human breast cancer cells. Int. J. Cancer 2018. [Google Scholar] [CrossRef]
- Shakespear, M.R.; Hohenhaus, D.M.; Kelly, G.M.; Kamal, N.A.; Gupta, P.; Labzin, L.I.; Schroder, K.; Garceau, V.; Barbero, S.; Iyer, A.; et al. Histone deacetylase 7 promotes Toll-like receptor 4-dependent proinflammatory gene expression in macrophages. J. Biol. Chem. 2013, 288, 25362–25374. [Google Scholar] [CrossRef] [PubMed]
- Seidel, C.; Schnekenburger, M.; Dicato, M.; Diederich, M. Histone deacetylase 6 in health and disease. Epigenomics 2015, 7, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Regna, N.L.; Vieson, M.D.; Luo, X.M.; Chafin, C.B.; Puthiyaveetil, A.G.; Hammond, S.E.; Caudell, D.L.; Jarpe, M.B.; Reilly, C.M. Specific HDAC6 inhibition by ACY-738 reduces SLE pathogenesis in NZB/W mice. Clin. Immunol. 2016, 162, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.; Brown, D.R.; Olorenshaw, I.M.; Kammer, G.M. Trichostatin A reverses skewed expression of CD154, interleukin-10, and interferon-gamma gene and protein expression in lupus T cells. Proc. Natl. Acad. Sci. USA 2001, 98, 2628–2633. [Google Scholar] [CrossRef] [PubMed]
- Nambiar, M.P.; Warke, V.G.; Fisher, C.U.; Tsokos, G.C. Effect of trichostatin A on human T cells resembles signaling abnormalities in T cells of patients with systemic lupus erythematosus: A new mechanism for TCR zeta chain deficiency and abnormal signaling. J. Cell. Biochem. 2002, 85, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.; Reilly, C.M.; Brown, D.R.; Ruiz, P.; Gilkeson, G.S. Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse. J. Clin. Investig. 2003, 111, 539–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reilly, C.M.; Mishra, N.; Miller, J.M.; Joshi, D.; Ruiz, P.; Richon, V.M.; Marks, P.A.; Gilkeson, G.S. Modulation of renal disease in MRL/lpr mice by suberoylanilide hydroxamic acid. J. Immunol. 2004, 173, 4171–4178. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Chen, E.; Li, Y.; Zhu, X.; Zhang, T.; Zhu, X. Effects of Arsenic Trioxide on INF-gamma Gene Expression in MRL/lpr Mice and Human Lupus. Biol. Trace Elem. Res. 2018, 184, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Garcia, B.A.; Busby, S.A.; Shabanowitz, J.; Hunt, D.F.; Mishra, N. Resetting the epigenetic histone code in the MRL-lpr/lpr mouse model of lupus by histone deacetylase inhibition. J. Proteome Res. 2005, 4, 2032–2042. [Google Scholar] [CrossRef]
- Lu, Z.P.; Ju, Z.L.; Shi, G.Y.; Zhang, J.W.; Sun, J. Histone deacetylase inhibitor Trichostatin A reduces anti-DNA autoantibody production and represses IgH gene transcription. Biochem. Biophys. Res. Commun. 2005, 330, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Reilly, C.M.; Thomas, M.; Gogal, R., Jr.; Olgun, S.; Santo, A.; Sodhi, R.; Samy, E.T.; Peng, S.L.; Gilkeson, G.S.; Mishra, N. The histone deacetylase inhibitor trichostatin A upregulates regulatory T cells and modulates autoimmunity in NZB/W F1 mice. J. Autoimmun. 2008, 31, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Waibel, M.; Christiansen, A.J.; Hibbs, M.L.; Shortt, J.; Jones, S.A.; Simpson, I.; Light, A.; O’Donnell, K.; Morand, E.F.; Tarlinton, D.M.; et al. Manipulation of B-cell responses with histone deacetylase inhibitors. Nat. Commun. 2015, 6, 6838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, N.; Long, H.; Zhao, M.; Yin, H.; Lu, Q. Aberrant expression pattern of histone acetylation modifiers and mitigation of lupus by SIRT1-siRNA in MRL/lpr mice. Scand. J. Rheumatol. 2009, 38, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Leung, Y.T.; Shi, L.; Maurer, K.; Song, L.; Zhang, Z.; Petri, M.; Sullivan, K.E. Interferon regulatory factor 1 and histone H4 acetylation in systemic lupus erythematosus. Epigenetics 2015, 10, 191–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Sun, Y.; Gao, F.; Wu, X.; Tang, J.; Yin, H.; Luo, Y.; Richardson, B.; Lu, Q. Epigenetics and SLE: RFX1 downregulation causes CD11a and CD70 overexpression by altering epigenetic modifications in lupus CD4+ T cells. J. Autoimmun. 2010, 35, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Cao, Q.; Reilly, C.M.; Young, N.L.; Garcia, B.A.; Mishra, N. Histone deacetylase 9 deficiency protects against effector T cell-mediated systemic autoimmunity. J. Biol. Chem. 2011, 286, 28833–28843. [Google Scholar] [CrossRef] [PubMed]
- Regna, N.L.; Chafin, C.B.; Hammond, S.E.; Puthiyaveetil, A.G.; Caudell, D.L.; Reilly, C.M. Class I and II histone deacetylase inhibition by ITF2357 reduces SLE pathogenesis in vivo. Clin. Immunol. 2014, 151, 29–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, A.O.; Wood, M.A. Does stress remove the HDAC brakes for the formation and persistence of long-term memory? Neurobiol. Learn. Mem. 2014, 112, 61–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.; Liao, X.; Vieson, M.D.; Chen, M.; Scott, R.; Kazmierczak, J.; Luo, X.M.; Reilly, C.M. Selective HDAC6 inhibition decreases early stage of lupus nephritis by down-regulating both innate and adaptive immune responses. Clin. Exp. Immunol. 2018, 191, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Pieterse, E.; Hofstra, J.; Berden, J.; Herrmann, M.; Dieker, J.; van der Vlag, J. Acetylated histones contribute to the immunostimulatory potential of neutrophil extracellular traps in systemic lupus erythematosus. Clin. Exp. Immunol. 2015, 179, 68–74. [Google Scholar] [CrossRef]
- Valenzuela-Fernandez, A.; Cabrero, J.R.; Serrador, J.M.; Sanchez-Madrid, F. HDAC6: A key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 2008, 18, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Vieson, M.D.; Gojmerac, A.M.; Khan, D.; Dai, R.; van Duzer, J.H.; Mazitschek, R.; Caudell, D.L.; Liao, X.; Luo, X.M.; Reilly, C.M. Treatment with a selective histone deacetylase 6 inhibitor decreases lupus nephritis in NZB/W mice. Histol. Histopathol. 2017, 32, 1317–1332. [Google Scholar] [PubMed]
- Lennard Richard, M.L.; Nowling, T.K.; Brandon, D.; Watson, D.K.; Zhang, X.K. Fli-1 controls transcription from the MCP-1 gene promoter, which may provide a novel mechanism for chemokine and cytokine activation. Mol. Immunol. 2015, 63, 566–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, E.; Karam, E.; Williams, S.; Watson, D.K.; Gilkeson, G.; Zhang, X.K. Fli-1 transcription factor affects glomerulonephritis development by regulating expression of monocyte chemoattractant protein-1 in endothelial cells in the kidney. Clin. Immunol. 2012, 145, 201–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, S.; Zhang, X.K. The Friend leukaemia virus integration 1 (Fli-1) transcription factor affects lupus nephritis development by regulating inflammatory cell infiltration into the kidney. Clin. Exp. Immunol. 2014, 177, 102–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lennard Richard, M.L.; Brandon, D.; Lou, N.; Sato, S.; Caldwell, T.; Nowling, T.K.; Gilkeson, G.; Zhang, X.K. Acetylation impacts Fli-1-driven regulation of granulocyte colony stimulating factor. Eur. J. Immunol. 2016, 46, 2322–2332. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Tan, Y.; Peng, Q.; Huang, C.; Guo, Y.; Liang, G.; Zhu, B.; Huang, Y.; Liu, A.; Wang, Z.; et al. IL-6/STAT3 pathway induced deficiency of RFX1 contributes to Th17-dependent autoimmune diseases via epigenetic regulation. Nat. Commun. 2018, 9, 583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Long, H.; Chang, C.; Zhao, M.; Lu, Q. Crosstalk between metabolism and epigenetic modifications in autoimmune diseases: A comprehensive overview. Cell. Mol. Life Sci. 2018, 75, 3353–3369. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, D.; Perl, A. Metabolic control of T cell activation and death in SLE. Autoimmun. Rev. 2009, 8, 184–189. [Google Scholar] [CrossRef] [Green Version]
- Perl, A. mTOR activation is a biomarker and a central pathway to autoimmune disorders, cancer, obesity, and aging. Ann. N. Y. Acad. Sci. 2015, 1346, 33–44. [Google Scholar] [CrossRef] [Green Version]
- Shah, D.; Sah, S.; Nath, S.K. Interaction between glutathione and apoptosis in systemic lupus erythematosus. Autoimmun. Rev. 2013, 12, 741–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Sivakumar, R.; Titov, A.A.; Choi, S.C.; Morel, L. Metabolic Factors that Contribute to Lupus Pathogenesis. Crit. Rev. Immunol. 2016, 36, 75–98. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Li, D.; Li, X.; Ren, J.; Xu, J.; Ding, L.; Dou, H.; Hou, Y. mTOR inhibitor INK128 attenuates systemic lupus erythematosus by regulating inflammation-induced CD11b(+)Gr1(+) cells. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1865, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, D.; Perl, A. mTOR signaling: A central pathway to pathogenesis in systemic lupus erythematosus? Discov. Med. 2010, 9, 173–178. [Google Scholar] [PubMed]
- Li, X.; Zhang, X.; Pan, Y.; Shi, G.; Ren, J.; Fan, H.; Dou, H.; Hou, Y. mTOR regulates NLRP3 inflammasome activation via reactive oxygen species in murine lupus. Acta Biochim. Biophys. Sin. 2018, 50, 888–896. [Google Scholar] [CrossRef]
- Oaks, Z.; Winans, T.; Huang, N.; Banki, K.; Perl, A. Activation of the Mechanistic Target of Rapamycin in SLE: Explosion of Evidence in the Last Five Years. Curr. Rheumatol. Rep. 2016, 18, 73. [Google Scholar] [CrossRef]
- Huang, N.; Perl, A. Metabolism as a Target for Modulation in Autoimmune Diseases. Trends Immunol. 2018, 39, 562–576. [Google Scholar] [CrossRef]
- Lai, Z.W.; Hanczko, R.; Bonilla, E.; Caza, T.N.; Clair, B.; Bartos, A.; Miklossy, G.; Jimah, J.; Doherty, E.; Tily, H.; et al. N-acetylcysteine reduces disease activity by blocking mammalian target of rapamycin in T cells from systemic lupus erythematosus patients: A randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2012, 64, 2937–2946. [Google Scholar] [CrossRef] [Green Version]
- Badawy, A.A. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. 2017, 10. [Google Scholar] [CrossRef]
- Boros, F.A.; Bohar, Z.; Vecsei, L. Genetic alterations affecting the genes encoding the enzymes of the kynurenine pathway and their association with human diseases. Mutat. Res. 2018, 776, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Oxenkrug, G.F. Tryptophan kynurenine metabolism as a common mediator of genetic and environmental impacts in major depressive disorder: The serotonin hypothesis revisited 40 years later. Isr. J. Psychiatry Relat. Sci. 2010, 47, 56–63. [Google Scholar] [PubMed]
- Perl, A. Oxidative stress in the pathology and treatment of systemic lupus erythematosus. Nat. Rev. Rheumatol. 2013, 9, 674–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perl, A. Activation of mTOR (mechanistic target of rapamycin) in rheumatic diseases. Nat. Rev. Rheumatol. 2016, 12, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Perl, A. Mechanistic target of rapamycin complex 1 expands Th17 and IL-4+ CD4-CD8-double-negative T cells and contracts regulatory T cells in systemic lupus erythematosus. J. Immunol. 2014, 192, 4134–4144. [Google Scholar] [CrossRef] [PubMed]
- Takasaka, N.; Araya, J.; Hara, H.; Ito, S.; Kobayashi, K.; Kurita, Y.; Wakui, H.; Yoshii, Y.; Yumino, Y.; Fujii, S.; et al. Autophagy induction by SIRT6 through attenuation of insulin-like growth factor signaling is involved in the regulation of human bronchial epithelial cell senescence. J. Immunol. 2014, 192, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Etchegaray, J.P.; Zhong, L.; Mostoslavsky, R. The histone deacetylase SIRT6: At the crossroads between epigenetics, metabolism and disease. Curr. Top. Med. Chem. 2013, 13, 2991–3000. [Google Scholar] [CrossRef]
- Chen, H.; Fan, M.; Pfeffer, L.M.; Laribee, R.N. The histone H3 lysine 56 acetylation pathway is regulated by target of rapamycin (TOR) signaling and functions directly in ribosomal RNA biogenesis. Nucleic Acids Res. 2012, 40, 6534–6546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birner, A.; Platzer, M.; Bengesser, S.A.; Dalkner, N.; Fellendorf, F.T.; Queissner, R.; Pilz, R.; Rauch, P.; Maget, A.; Hamm, C.; et al. Increased breakdown of kynurenine towards its neurotoxic branch in bipolar disorder. PLoS ONE 2017, 12, e0172699. [Google Scholar] [CrossRef]
- Chen, Y.; Guillemin, G.J. Kynurenine pathway metabolites in humans: Disease and healthy States. Int. J. Tryptophan Res. 2009, 2. [Google Scholar] [CrossRef]
- Braidy, N.; Guillemin, G.J.; Grant, R. Effects of Kynurenine Pathway Inhibition on NAD Metabolism and Cell Viability in Human Primary Astrocytes and Neurons. Int. J. Tryptophan Res. 2011, 4, 29–37. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Histone Deacetylases (HDAC) Classification | Enzymatic Activity | Mechanism of Action | Location | Substrates | HDAC Inhibitor | Autoimmunity and Systemic Lupus Erythematosus (SLE) Involvement |
|---|---|---|---|---|---|---|
| Class I. | ||||||
| HDAC1 | Enhanced when incorporated into complexes | 1 class I catalytic domain | Nucleus | p53, RB, MyoD, NF-kB, DNMTI, DNMT3a, MBD2, Sp1, BRCA1, MeCP2, ATM, Smad7 [61,92] | Valproic acid, phenylbutyrate, MS-275, Romidepsin, Suberoylanilide Hydroxamic Acid [93] | Overexpression of HDAC1 increases the activity of the 3’-IgH enhancers. HDAC1 is recruited to the IgH enhancer region, and TSA treatment of B cells reduced the production of anti-DNA autoantibodies. |
| HDAC2 | Enhanced when incorporated into complexes | 1 class I catalytic domain | Nucleus | RB, NF-kB, BRCA1, DNMTI [61] | Valproic Acid, phenylbutyrate, Suberoylanilide Hydroxamic Acid, MS-275, Romidepsin [93,94,95] | Critical for transcriptional regulation, cell cycle progression and developmental processes. |
| HDAC3 | Enhanced when incorporated into complexes | 1 class I catalytic domain | Nucleus/Cytoplasm | RB, NF-kB, Smad7, Stat3, SRY [61] | Valproic Acid, Suberoylanilide Hydroxamic Acid, MS-275 [93,96] | HDAC3 gene expression is decreased in SLE monocytes, involved in macrophage polarization. |
| HDAC8 | Fully active in isolation | 1 class I catalytic domain | Nucleus | Not Reported | Suberoylanilide Hydroxamic Acid, Resveratrol, APHA, Curcumin [93,97] | Downregulate the expression of pro-inflammatory cytokines (TNF-alpha, TGF-beta, IL-1beta, and IL-6). |
| Class IIa. | ||||||
| HDAC4 | Weak enzymatic activity in isolation | 1 class II catalytic domain | Nucleus/Cytoplasm | GCMa, GATA-1, HP-1 [92,98,99] | Not reported | Role in pro-inflammatory gene expression. |
| HDAC5 | Weak enzymatic activity in isolation | 1 class II catalytic domain | Nucleus/Cytoplasm | GCMa, Smad7, HP-1 [92,100] | TSA [93] | HDAC5 mRNA expression is enhanced in inflammatory states. |
| HDAC7 | Weak enzymatic activity in isolation | 1 class II catalytic domain | Nucleus/Cytoplasm | PLAG1, PLAG2 [92,101] | Not reported | Promotes inflammatory responses in macrophages, regulates TLR responses in macrophages, regulates LPS signaling. |
| HDAC9 | Weak enzymatic activity in isolation | 1 class II catalytic domain | Nucleus/Cytoplasm | Not Reported | Suberoylanilide Hydroxamic Acid, MS-275 [93] | Regulates Foxp3-dependent suppression. Increase in Treg cells—decrease in suppressive activity. HDAC9 inhibition may benefit SLE patients as shown in MRL/lpr mice. |
| Class IIb. | ||||||
| HDAC6 | Acts on structural proteins | 2 class II catalytic domains with 1215 amino acids. SE14 repeats. BUZ is ZnF domain | Mainly cytoplasmic | Smad7, α-Tubulin, Hsp90 [61,102] | M344 [92,93,103] | HDAC6 is overexpressed in SLE—causes an increased B cell development and response. Inhibition causes reduced germinal center B cells, T follicular cells and IFN-gamma secreting cells. |
| HDAC10 | Not measurable | 2 class II catalytic domains | Nucleus/Cytoplasm | Not reported | Not reported | Overexpressed in B cells from the spleen. |
| Class IV. | ||||||
| HDAC11 | Regulates immune activation and immune tolerance | 1 class IV catalytic domain | Nucleus | Not reported | Not reported | Gene expression is decreased in SLE monocytes, negative transcriptional regulator |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, J.; Panther, E.; Liao, X.; Grammer, A.C.; Lipsky, P.E.; Reilly, C.M. The Impact of Protein Acetylation/Deacetylation on Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2018, 19, 4007. https://doi.org/10.3390/ijms19124007
Ren J, Panther E, Liao X, Grammer AC, Lipsky PE, Reilly CM. The Impact of Protein Acetylation/Deacetylation on Systemic Lupus Erythematosus. International Journal of Molecular Sciences. 2018; 19(12):4007. https://doi.org/10.3390/ijms19124007
Chicago/Turabian StyleRen, Jingjing, Eric Panther, Xiaofeng Liao, Amrie C. Grammer, Peter E. Lipsky, and Chris M. Reilly. 2018. "The Impact of Protein Acetylation/Deacetylation on Systemic Lupus Erythematosus" International Journal of Molecular Sciences 19, no. 12: 4007. https://doi.org/10.3390/ijms19124007