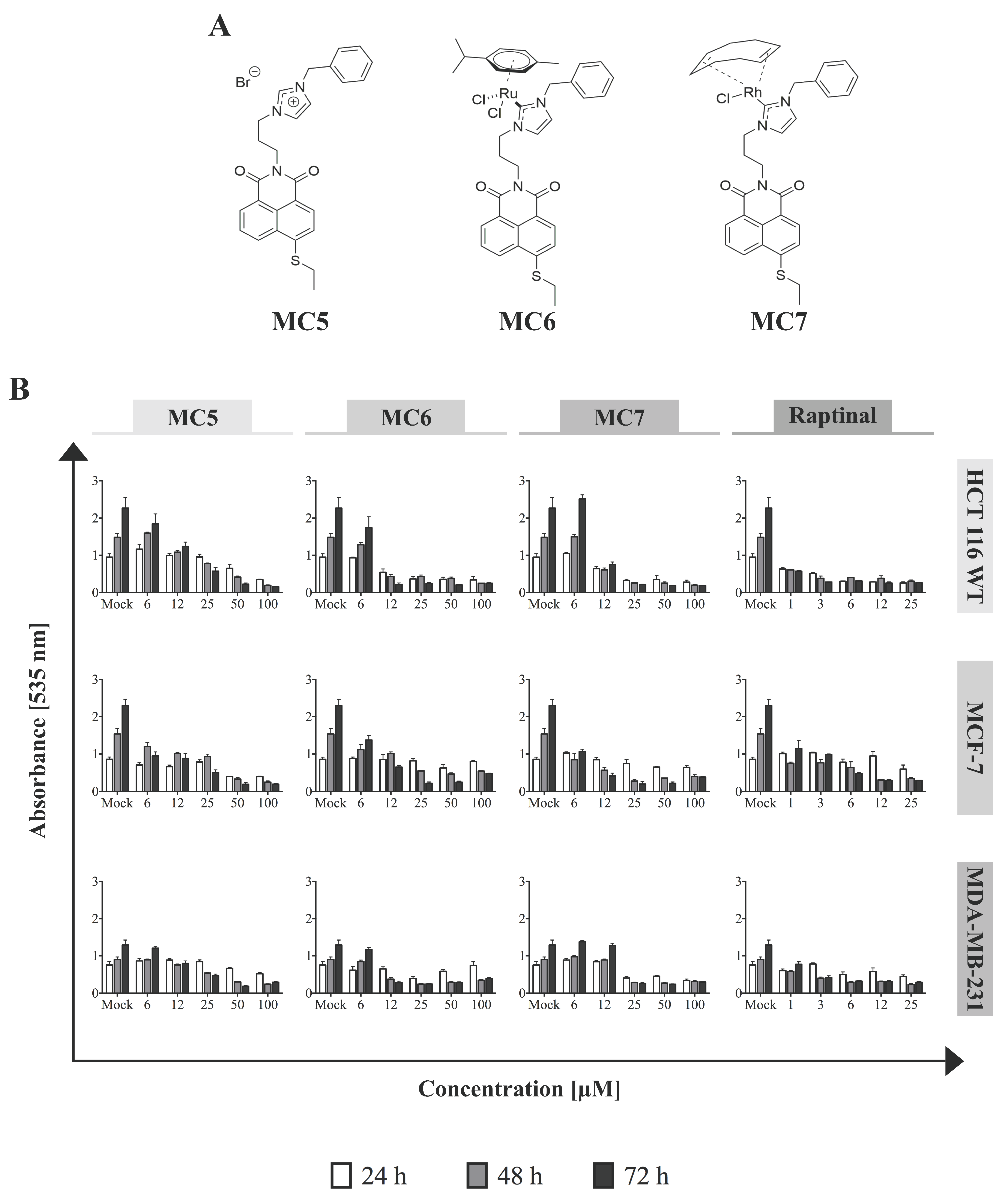
Figure 1.
Naphthalimide-
N-heterocyclic carbene (NHC) analogues exhibit cytotoxic effects against human breast- and colon cancer cells. (
A) Chemical structures of the compounds; (
B) Increasing concentrations of each of the complexes, as well as the rapid apoptosis inducer, raptinal [
16] (as positive control) were applied to the different cell lines and Sulforhodamine B (SRB) assay was performed after 24, 48, and 72 h of treatment. The Ru(II)- and Rh(I)-containing complexes show the highest and the least efficacy against HCT116 and MDA-MB-231, respectively, in most tested concentrations. 0.1% DMSO-treated cells served as mock. Data represent mean ± SD of three independent experiments, each was done in quadruplicates.
Figure 1.
Naphthalimide-
N-heterocyclic carbene (NHC) analogues exhibit cytotoxic effects against human breast- and colon cancer cells. (
A) Chemical structures of the compounds; (
B) Increasing concentrations of each of the complexes, as well as the rapid apoptosis inducer, raptinal [
16] (as positive control) were applied to the different cell lines and Sulforhodamine B (SRB) assay was performed after 24, 48, and 72 h of treatment. The Ru(II)- and Rh(I)-containing complexes show the highest and the least efficacy against HCT116 and MDA-MB-231, respectively, in most tested concentrations. 0.1% DMSO-treated cells served as mock. Data represent mean ± SD of three independent experiments, each was done in quadruplicates.
Figure 2.
Naphthalimide-NHC analogues induce cell cycle arrest and p21 expression in HCT116 CRC cells. (A) Representative histogram plots show the distribution of cell cycle phases in HCT116 cells treated with either 0.1% DMSO (as mock) or the three complexes, MC5, MC6, and MC7 at a concentration of 50, 12, and 25 μM, respectively for 24 h; (B) All of the analogues were found to induce a G1 phase arrest as compared to mock treatment. Comparison of the percentage cell population of G1, S, and G2/M phases between mock and each of the three complexes was performed by two-tailed student’s t-test. Error bars represent the SD of two biological replicates, one of which is depicted in (A); (C) p21 mRNA levels are up-regulated in response to 24 h of treatments, analyzed by qRT-PCR. Relative expression was calculated using the ∆∆ Ct method where the Ct values of p21 were normalized to those of the housekeeping gene (vinculin). Lower and upper ends of the bars indicate the minimum and maximum values, respectively, and the “+” in the middle represents the mean. Error bars ± SD; n = 4; (D) p21 protein levels upon 24 h of treatment with the three complexes at the indicated concentrations, determined by immunoblotting; (E) Densitometric quantification of p21 bands normalized to those of the loading control (vinculin). Error bars indicate the SEM of two biological replicates, one of which is presented in (D). Multiple comparisons were made using one-way ANOVA test and a post-hoc Tukey test. *, **, ***, and **** denote p-values less than or equal to 0.05, 0.01, 0.001, and 0.0001, respectively.
Figure 2.
Naphthalimide-NHC analogues induce cell cycle arrest and p21 expression in HCT116 CRC cells. (A) Representative histogram plots show the distribution of cell cycle phases in HCT116 cells treated with either 0.1% DMSO (as mock) or the three complexes, MC5, MC6, and MC7 at a concentration of 50, 12, and 25 μM, respectively for 24 h; (B) All of the analogues were found to induce a G1 phase arrest as compared to mock treatment. Comparison of the percentage cell population of G1, S, and G2/M phases between mock and each of the three complexes was performed by two-tailed student’s t-test. Error bars represent the SD of two biological replicates, one of which is depicted in (A); (C) p21 mRNA levels are up-regulated in response to 24 h of treatments, analyzed by qRT-PCR. Relative expression was calculated using the ∆∆ Ct method where the Ct values of p21 were normalized to those of the housekeeping gene (vinculin). Lower and upper ends of the bars indicate the minimum and maximum values, respectively, and the “+” in the middle represents the mean. Error bars ± SD; n = 4; (D) p21 protein levels upon 24 h of treatment with the three complexes at the indicated concentrations, determined by immunoblotting; (E) Densitometric quantification of p21 bands normalized to those of the loading control (vinculin). Error bars indicate the SEM of two biological replicates, one of which is presented in (D). Multiple comparisons were made using one-way ANOVA test and a post-hoc Tukey test. *, **, ***, and **** denote p-values less than or equal to 0.05, 0.01, 0.001, and 0.0001, respectively.
![Ijms 19 03964 g002]()
Figure 3.
Total cellular reactive oxygen species (ROS) levels are moderately increased by naphthalimide-NHC analogues. (
A) HCT116 cells were treated with various concentrations of the respective compound for 24 h, after which flow cytometric analysis of ROS generation was performed using the superoxide indicator, dihydroethidium (DHE). A 24 h treatment with the gold(I) NHC complex, MC3 [
13] as well as the rapid apoptosis inducer, raptinal [
16] was included as positive control. Cellular ROS levels were found to be concentration-dependently induced in response to all the three complexes, with the metal-free ligand (MC5) showing the highest induction. Data were normalized to mock (0.1% DMSO) treatment. Error bars ± SD;
n = 4. Statistical significance between the respective treatment and mock was determined by two-tailed student’s
t-test. (
B) Representative density plots of one out of four biological replicates shown in (
A); (
C) ROS levels induced by MC6 (6 μM, 24 h) were found to be significantly decreased in HCT116 cells pre-treated for 1 h with the anti-oxidants,
N-acetyl-
l-cysteine (NAC) and glutathione (GSH), as well as the mitochondrial complex I inhibitor, rotenone at the concentrations indicated. A 2 h co-treatment with the mitochondrial uncoupling reagent (CCCP), and Ru(II) complex (MC6) caused a mild increase in the latter’s effects on ROS generation stained by DHE. Data were normalized to the values of mock (0.1% DMSO) as well as the corresponding single treatments. Statistical significance between MC6 in the absence/presence of each of the inhibitors was determined by two-tailed student’s
t-test. *, **, ***, and **** represent
p-values less than or equal to 0.05, 0.01, 0.001, and 0.0001, respectively.
Figure 3.
Total cellular reactive oxygen species (ROS) levels are moderately increased by naphthalimide-NHC analogues. (
A) HCT116 cells were treated with various concentrations of the respective compound for 24 h, after which flow cytometric analysis of ROS generation was performed using the superoxide indicator, dihydroethidium (DHE). A 24 h treatment with the gold(I) NHC complex, MC3 [
13] as well as the rapid apoptosis inducer, raptinal [
16] was included as positive control. Cellular ROS levels were found to be concentration-dependently induced in response to all the three complexes, with the metal-free ligand (MC5) showing the highest induction. Data were normalized to mock (0.1% DMSO) treatment. Error bars ± SD;
n = 4. Statistical significance between the respective treatment and mock was determined by two-tailed student’s
t-test. (
B) Representative density plots of one out of four biological replicates shown in (
A); (
C) ROS levels induced by MC6 (6 μM, 24 h) were found to be significantly decreased in HCT116 cells pre-treated for 1 h with the anti-oxidants,
N-acetyl-
l-cysteine (NAC) and glutathione (GSH), as well as the mitochondrial complex I inhibitor, rotenone at the concentrations indicated. A 2 h co-treatment with the mitochondrial uncoupling reagent (CCCP), and Ru(II) complex (MC6) caused a mild increase in the latter’s effects on ROS generation stained by DHE. Data were normalized to the values of mock (0.1% DMSO) as well as the corresponding single treatments. Statistical significance between MC6 in the absence/presence of each of the inhibitors was determined by two-tailed student’s
t-test. *, **, ***, and **** represent
p-values less than or equal to 0.05, 0.01, 0.001, and 0.0001, respectively.
![Ijms 19 03964 g003]()
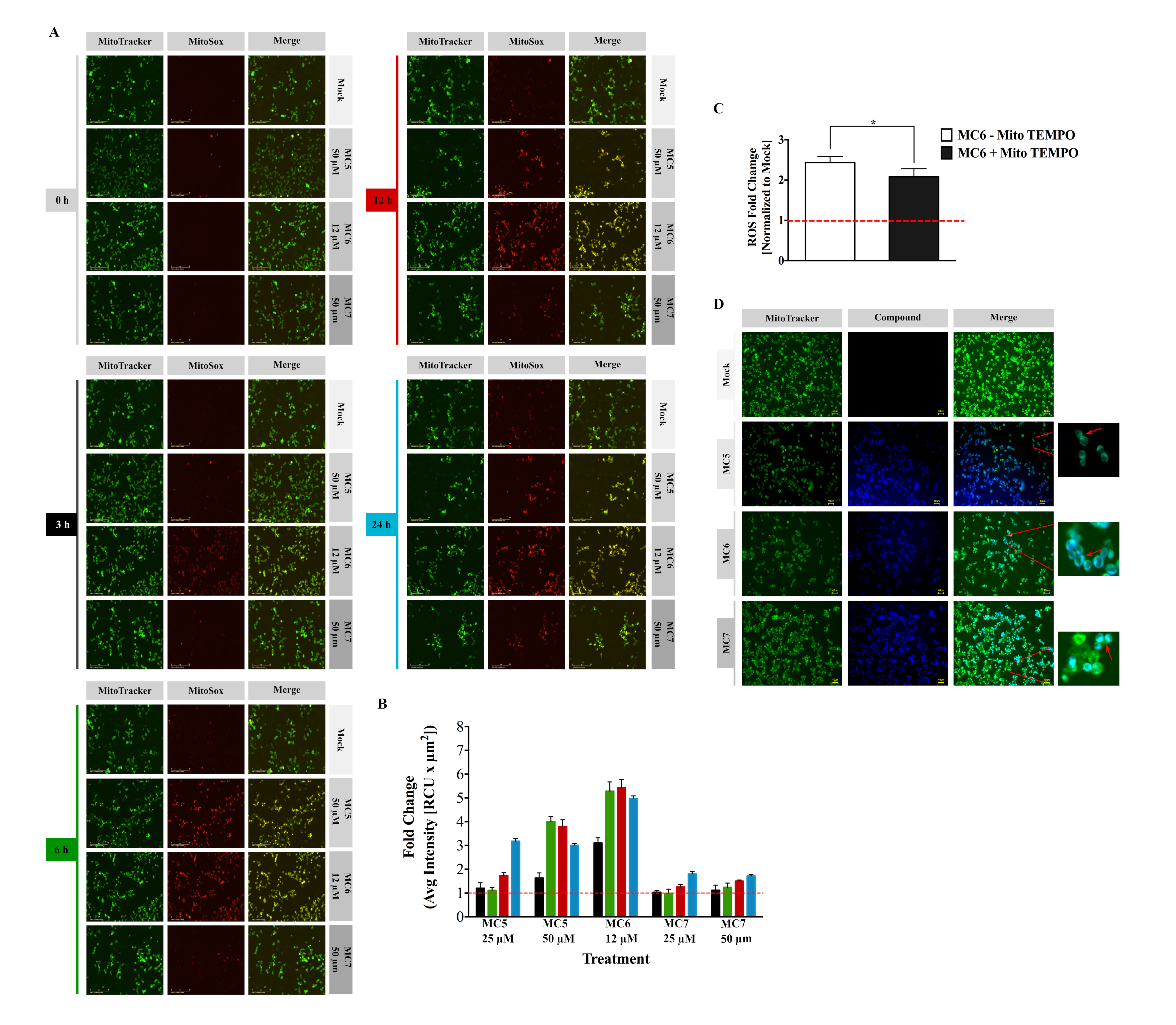
Figure 4.
Mitochondrial ROS (mtROS) generation is strongly influenced by naphthalimide-NHC analogues. (A) Live cell imaging of mitochondrial superoxide generation stained with MitoSox Red and associated quantification (B) as described in the methods’ section. MitoTracker Green was used to indicate mitochondria; (C) The mitochondria-targeted anti-oxidant, Mito TEMPO, attenuated the ROS induced by 12 μM of MC6, determined by flow cytometric analysis of MitoSox Red staining. Mito TEMPO (10 μM) was pre-incubated with HCT116 cells 2 h before the exposure to MC6 for 24 h. Data are shown as mean ± SD of three biological replicates. Comparison of ROS fold change between the two groups was performed by two-tailed student’s t-test where a p-value less than or equal to 0.05 is denoted by *; (D) Fluorescence micrographs showing mitochondrial localization of the three complexes in HCT116 cells upon treatment with MC5 (50 μM), MC6 (12 μM), and MC7 (50 μM) for 4 h. Mitochondria were stained by MitoTracker Green. Scale bar: 40 μm. 0.1% DMSO was used as mock.
Figure 4.
Mitochondrial ROS (mtROS) generation is strongly influenced by naphthalimide-NHC analogues. (A) Live cell imaging of mitochondrial superoxide generation stained with MitoSox Red and associated quantification (B) as described in the methods’ section. MitoTracker Green was used to indicate mitochondria; (C) The mitochondria-targeted anti-oxidant, Mito TEMPO, attenuated the ROS induced by 12 μM of MC6, determined by flow cytometric analysis of MitoSox Red staining. Mito TEMPO (10 μM) was pre-incubated with HCT116 cells 2 h before the exposure to MC6 for 24 h. Data are shown as mean ± SD of three biological replicates. Comparison of ROS fold change between the two groups was performed by two-tailed student’s t-test where a p-value less than or equal to 0.05 is denoted by *; (D) Fluorescence micrographs showing mitochondrial localization of the three complexes in HCT116 cells upon treatment with MC5 (50 μM), MC6 (12 μM), and MC7 (50 μM) for 4 h. Mitochondria were stained by MitoTracker Green. Scale bar: 40 μm. 0.1% DMSO was used as mock.
![Ijms 19 03964 g004]()
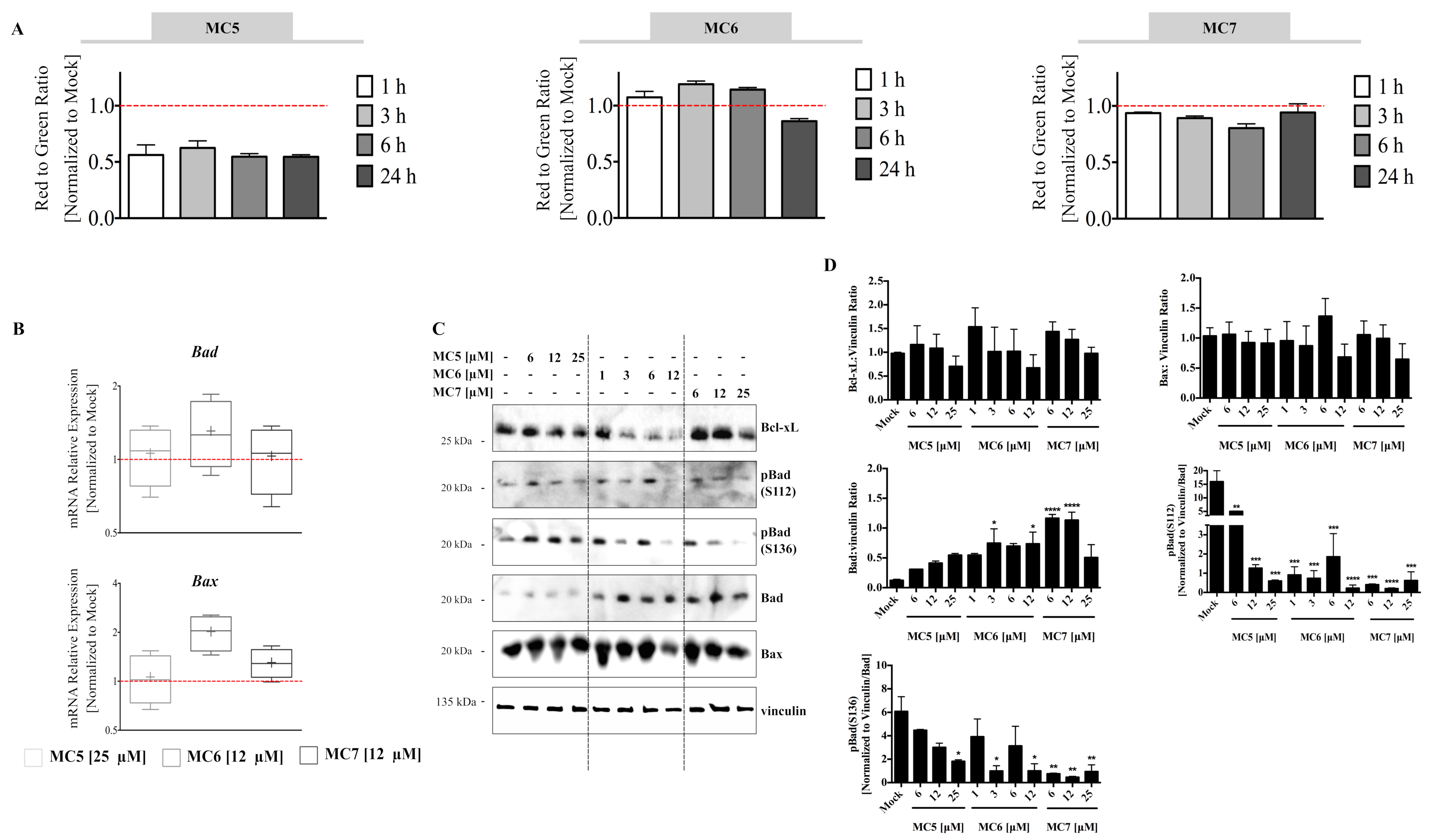
Figure 5.
Naphthalimide-NHC analogues reduce, induce and have no clear effect on the Mitochondrial Membrane Potential (MMP) (Δψm) in HCT116 cells. (A) MMP was time-dependently decreased by MC5 (25 μM); it was increased in the earlier time points by MC6 (12 μM) followed by a decrease at 24 h; and was found to be mostly unaltered in response to MC7 (25 μM). Error bars are the SD of three biological replicates; (B) mRNA expression analysis of the pro-apoptotic Bcl-2 family members, Bad and Bax, after 24 h of treatment with the compounds at indicated concentrations. Relative expression was calculated by the ∆∆ Ct method where the Ct values of the target genes were normalized to those of vinculin. Lower and upper ends of the bars denote the minimum and maximum values, respectively and “+” in the middle represents the mean. Error bars ± SD; n = 4; (C) 24 h of treatment with the respective compound led to a concentration-dependent decrease and increase in Bcl-xL and Bad protein levels, respectively, while it had no clear effect on Bax protein expression. Additionally, different phosphorylated forms of Bad were found to decrease in response to treatment; (D) Densitometric quantification of Bcl-2 family members normalized to the respective loading control (vinculin). The values of phosphorylated Bad were normalized to those of vinculin as well as total protein levels. Data are expressed as mean ± SEM of two independent experiments, one of which is presented in (C). Statistical comparisons were made between mock (0.1% DMSO), and the respective treatment using two-tailed student’s t-test. *, **, ***, and **** represent p-values less than or equal to 0.05, 0.01, 0.001, and 0.0001, respectively.
Figure 5.
Naphthalimide-NHC analogues reduce, induce and have no clear effect on the Mitochondrial Membrane Potential (MMP) (Δψm) in HCT116 cells. (A) MMP was time-dependently decreased by MC5 (25 μM); it was increased in the earlier time points by MC6 (12 μM) followed by a decrease at 24 h; and was found to be mostly unaltered in response to MC7 (25 μM). Error bars are the SD of three biological replicates; (B) mRNA expression analysis of the pro-apoptotic Bcl-2 family members, Bad and Bax, after 24 h of treatment with the compounds at indicated concentrations. Relative expression was calculated by the ∆∆ Ct method where the Ct values of the target genes were normalized to those of vinculin. Lower and upper ends of the bars denote the minimum and maximum values, respectively and “+” in the middle represents the mean. Error bars ± SD; n = 4; (C) 24 h of treatment with the respective compound led to a concentration-dependent decrease and increase in Bcl-xL and Bad protein levels, respectively, while it had no clear effect on Bax protein expression. Additionally, different phosphorylated forms of Bad were found to decrease in response to treatment; (D) Densitometric quantification of Bcl-2 family members normalized to the respective loading control (vinculin). The values of phosphorylated Bad were normalized to those of vinculin as well as total protein levels. Data are expressed as mean ± SEM of two independent experiments, one of which is presented in (C). Statistical comparisons were made between mock (0.1% DMSO), and the respective treatment using two-tailed student’s t-test. *, **, ***, and **** represent p-values less than or equal to 0.05, 0.01, 0.001, and 0.0001, respectively.
![Ijms 19 03964 g005]()
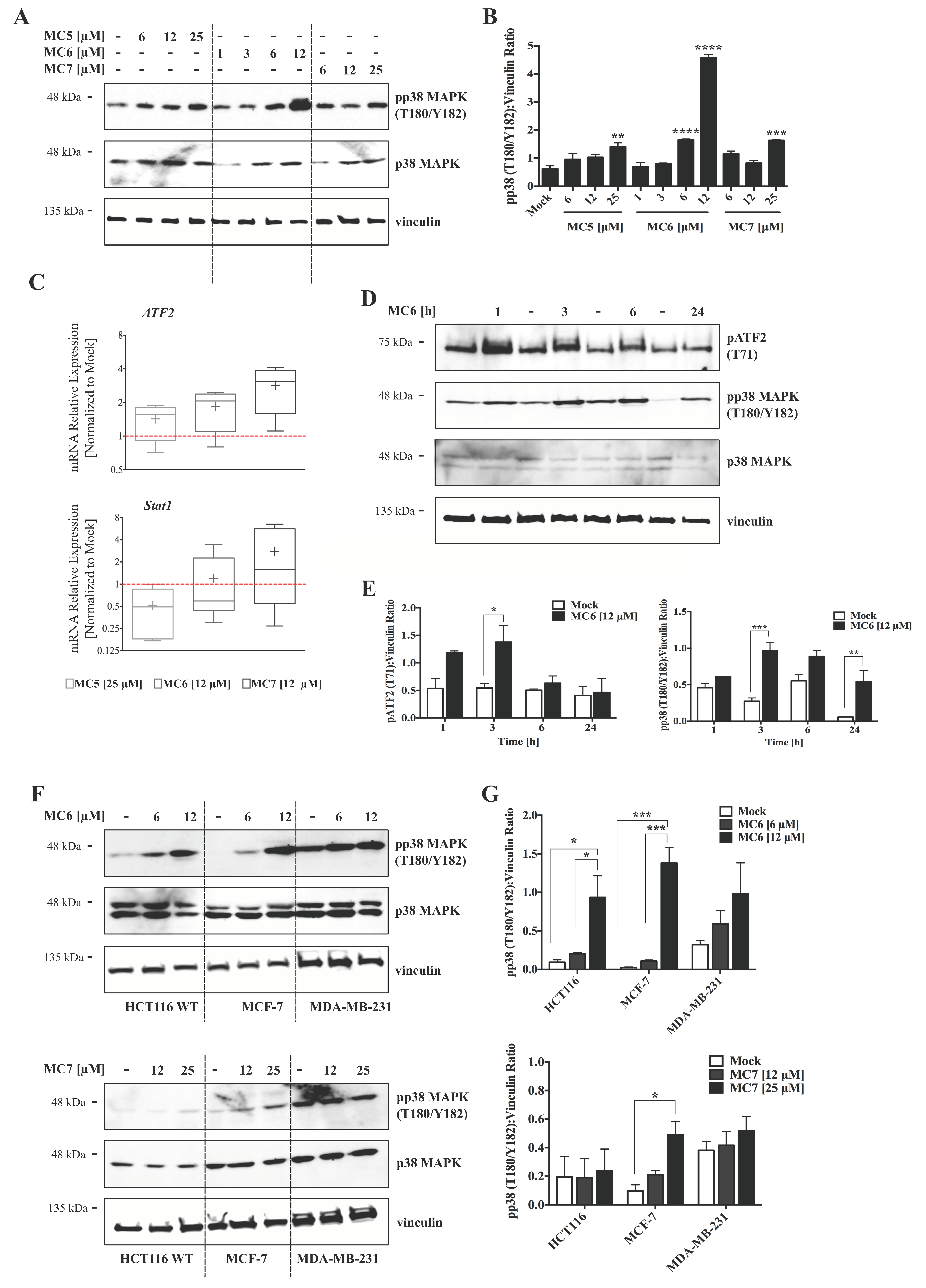
Figure 6.
Naphthalimide-NHC derivatives activate the p38 pathway in human breast- and colon cancer cell lines. (A) Immunoblots as well as densitometric quantification (B) showing the accumulation of pp38 mitogen activated protein kinase (MAPK) (T180/Y182) protein levels by the three complexes in HCT116 CRC cells upon 24 h treatment with increasing concentrations of the respective compound as indicated. The induction appeared to be more profound in case of the Ru(II) analogue, MC6. Data in (B) are presented as mean ± SEM of three independent experiments, one of those is shown in (A); (C) qRT-PCR analysis of p38-associated signaling molecules, ATF2 and Stat1 in HCT116 cells treated with the compounds at indicated concentrations for 24 h. Lower and upper ends of the bars denote the minimum and maximum values, respectively, with the “+” sign representing the mean of four biological replicates. Error bars ± SD; (D) Time-course analyses of pp38 MAPK (T180/Y182) as well as its down-stream effector, pATF2 (T71) in HCT116 cells treated with 12 μM of MC6, determined by immunoblotting; (E) Densitometric analyses of pATF2 (T71) and pp38 (T180/Y182) bands obtained from three independent experiments, one of which is depicted in (D). Error bars ± SEM; (F) Regulation of p38 MAPK signaling was compared among HCT116, MCF-7 and MDA-MB-231 cancer cell lines treated for 24 h with the metal-containing analogues (MC6 and MC7). In case of MDA-MB-231 where the basal levels of pp38 (T180/Y182) are high, treatments did not profoundly impact the molecule’s phosphorylation; (G) Densitometric quantifications illustrate no significant change in pp38 (T180/Y182) levels in MDA-MB-231 cells. Error bars ± SEM; n = 3. Statistical comparisons were made between mock (0.1% DMSO) and the respective treatment using two-tailed student’s t-test. p-values less than or equal to 0.05, 0.01, 0.001, and 0.0001 are indicated as *, **, ***, and ****, respectively.
Figure 6.
Naphthalimide-NHC derivatives activate the p38 pathway in human breast- and colon cancer cell lines. (A) Immunoblots as well as densitometric quantification (B) showing the accumulation of pp38 mitogen activated protein kinase (MAPK) (T180/Y182) protein levels by the three complexes in HCT116 CRC cells upon 24 h treatment with increasing concentrations of the respective compound as indicated. The induction appeared to be more profound in case of the Ru(II) analogue, MC6. Data in (B) are presented as mean ± SEM of three independent experiments, one of those is shown in (A); (C) qRT-PCR analysis of p38-associated signaling molecules, ATF2 and Stat1 in HCT116 cells treated with the compounds at indicated concentrations for 24 h. Lower and upper ends of the bars denote the minimum and maximum values, respectively, with the “+” sign representing the mean of four biological replicates. Error bars ± SD; (D) Time-course analyses of pp38 MAPK (T180/Y182) as well as its down-stream effector, pATF2 (T71) in HCT116 cells treated with 12 μM of MC6, determined by immunoblotting; (E) Densitometric analyses of pATF2 (T71) and pp38 (T180/Y182) bands obtained from three independent experiments, one of which is depicted in (D). Error bars ± SEM; (F) Regulation of p38 MAPK signaling was compared among HCT116, MCF-7 and MDA-MB-231 cancer cell lines treated for 24 h with the metal-containing analogues (MC6 and MC7). In case of MDA-MB-231 where the basal levels of pp38 (T180/Y182) are high, treatments did not profoundly impact the molecule’s phosphorylation; (G) Densitometric quantifications illustrate no significant change in pp38 (T180/Y182) levels in MDA-MB-231 cells. Error bars ± SEM; n = 3. Statistical comparisons were made between mock (0.1% DMSO) and the respective treatment using two-tailed student’s t-test. p-values less than or equal to 0.05, 0.01, 0.001, and 0.0001 are indicated as *, **, ***, and ****, respectively.
![Ijms 19 03964 g006]()
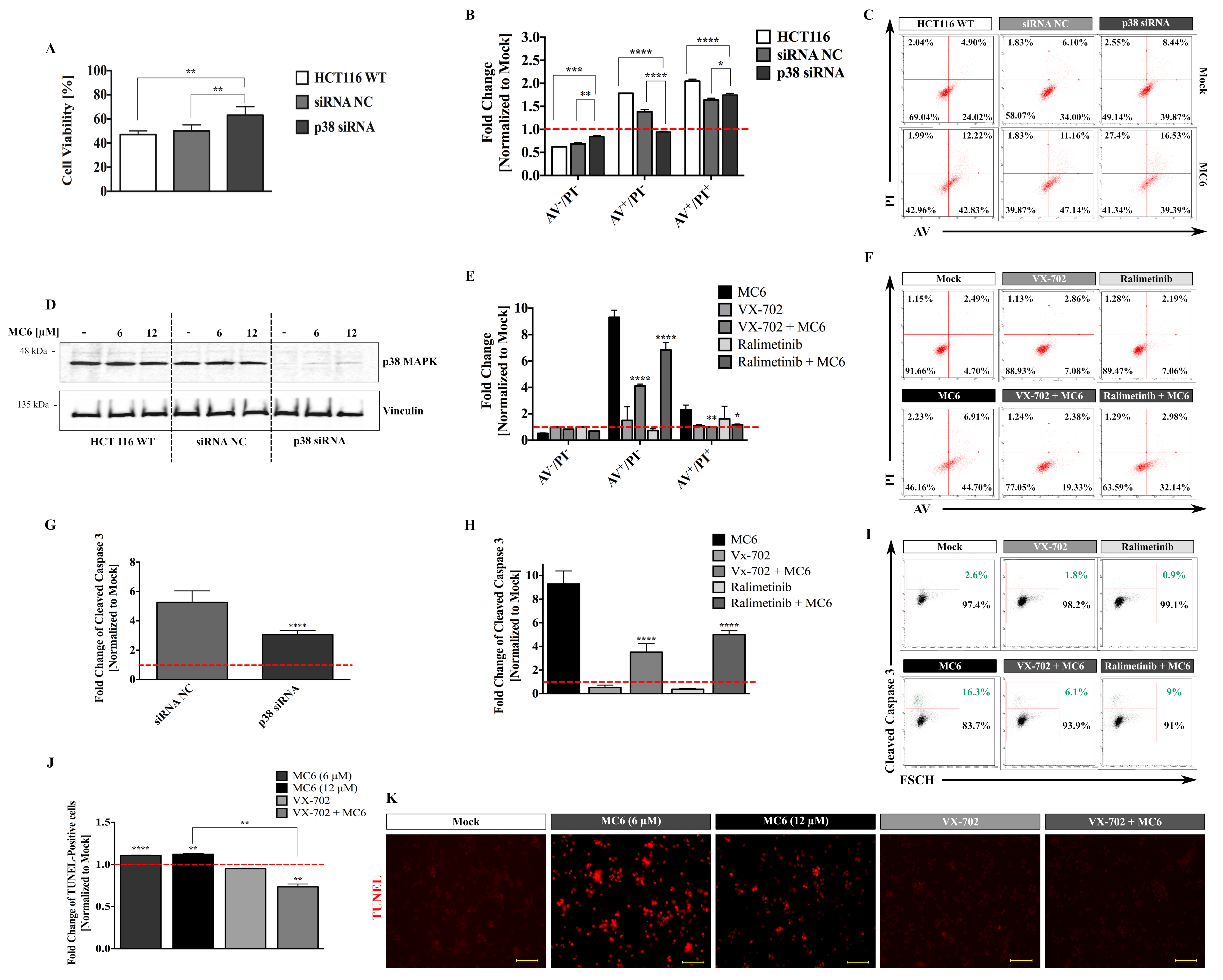
Figure 7.
p38 signaling appears to be crucial for the MC6-mediated cytotoxic- and pro-apoptotic effects. (A) siRNA-mediated repression of p38α was found to hamper the growth inhibitory effects of MC6 in HCT116 cells treated with 12 μM of the compound for 24 h, measured by SRB assay. Percentage cell viability was calculated by normalizing the values of MC6-treated cells to those of the corresponding mock (0.1% DMSO) treatments. Error bars ± SD; n = 3; (B) Knock-down of p38α attenuates the pro-apoptotic response to MC6 (12 μM, 24 h), assessed by flow cytometric analysis of AV/PI staining. Percentage cell population in each quadrant was normalized to the respective mock (0.1% DMSO) treatment. Error bars ± SD. Multiple comparisons were performed using two-way ANOVA followed by a post-hoc Tukey test; (C) Density plots representative of three biological replicates illustrate increased population of AV+/PI− and AV+/PI+ with treatment, however, to a lesser extent in case of cells transfected with anti-p38α siRNA; (D) Confirmation of knock-down efficiency, as determined by immunoblotting; (E) chemical inhibition of p38α abrogates the MC6-mediated apoptosis. HCT116 cells were treated with 12 μM of MC6 in the absence/presence of p38α inhibitors, VX-702 and Ralimetinib at a concentration of 0.5 μM for 24 h. Error bars ± SD, n = 3. Asterisks show significance in the amount of early- and late apoptotic population between cells treated with MC6 and each of the two inhibitors, and MC6 as a single agent, determined by two-tailed student’s t-test; (F) Representative density plots of one out of three biological replicates demonstrate reduced number of AV+/PI− and AV+/PI+ cells when p38α activity is inhibited; (G) Flow cytometric analysis of caspase 3 activation shows significantly less cleaved caspase 3 expression in cells transfected with anti-p38α siRNA as compared to that of the negative control. Error bars ± SD, n = 6. Statistical significance between the two groups was made using two-tailed student’s t-test; (H) Chemical inhibition of p38α was found to decrease the levels of active caspase 3 in a similar manner to that of p38α knock-down. Error bars ± SD, n = 6. Statistical comparison was performed between combination treatments and MC6 using two-tailed student’s t-test; (I) Representative density plots of one out of six biological replicates; (J) Detection of apoptotic cells using TUNEL assay. HCT116 cells were treated with the indicated concentrations of MC6 for 24 h in the absence/presence of VX-702 (0.5 μM). Statistical significance was calculated between mock and the respective treatment as well as MC6 as single agent and in combination with VX-702 using two-tailed student’s t-test; (K) Representative fluorescence images of TUNEL reaction. Scale bar: 100 μm. p-values less than or equal to 0.05, 0.01, 0.001, and 0.0001 are denoted as *, **, ***, and ****, respectively.
Figure 7.
p38 signaling appears to be crucial for the MC6-mediated cytotoxic- and pro-apoptotic effects. (A) siRNA-mediated repression of p38α was found to hamper the growth inhibitory effects of MC6 in HCT116 cells treated with 12 μM of the compound for 24 h, measured by SRB assay. Percentage cell viability was calculated by normalizing the values of MC6-treated cells to those of the corresponding mock (0.1% DMSO) treatments. Error bars ± SD; n = 3; (B) Knock-down of p38α attenuates the pro-apoptotic response to MC6 (12 μM, 24 h), assessed by flow cytometric analysis of AV/PI staining. Percentage cell population in each quadrant was normalized to the respective mock (0.1% DMSO) treatment. Error bars ± SD. Multiple comparisons were performed using two-way ANOVA followed by a post-hoc Tukey test; (C) Density plots representative of three biological replicates illustrate increased population of AV+/PI− and AV+/PI+ with treatment, however, to a lesser extent in case of cells transfected with anti-p38α siRNA; (D) Confirmation of knock-down efficiency, as determined by immunoblotting; (E) chemical inhibition of p38α abrogates the MC6-mediated apoptosis. HCT116 cells were treated with 12 μM of MC6 in the absence/presence of p38α inhibitors, VX-702 and Ralimetinib at a concentration of 0.5 μM for 24 h. Error bars ± SD, n = 3. Asterisks show significance in the amount of early- and late apoptotic population between cells treated with MC6 and each of the two inhibitors, and MC6 as a single agent, determined by two-tailed student’s t-test; (F) Representative density plots of one out of three biological replicates demonstrate reduced number of AV+/PI− and AV+/PI+ cells when p38α activity is inhibited; (G) Flow cytometric analysis of caspase 3 activation shows significantly less cleaved caspase 3 expression in cells transfected with anti-p38α siRNA as compared to that of the negative control. Error bars ± SD, n = 6. Statistical significance between the two groups was made using two-tailed student’s t-test; (H) Chemical inhibition of p38α was found to decrease the levels of active caspase 3 in a similar manner to that of p38α knock-down. Error bars ± SD, n = 6. Statistical comparison was performed between combination treatments and MC6 using two-tailed student’s t-test; (I) Representative density plots of one out of six biological replicates; (J) Detection of apoptotic cells using TUNEL assay. HCT116 cells were treated with the indicated concentrations of MC6 for 24 h in the absence/presence of VX-702 (0.5 μM). Statistical significance was calculated between mock and the respective treatment as well as MC6 as single agent and in combination with VX-702 using two-tailed student’s t-test; (K) Representative fluorescence images of TUNEL reaction. Scale bar: 100 μm. p-values less than or equal to 0.05, 0.01, 0.001, and 0.0001 are denoted as *, **, ***, and ****, respectively.
![Ijms 19 03964 g007]()
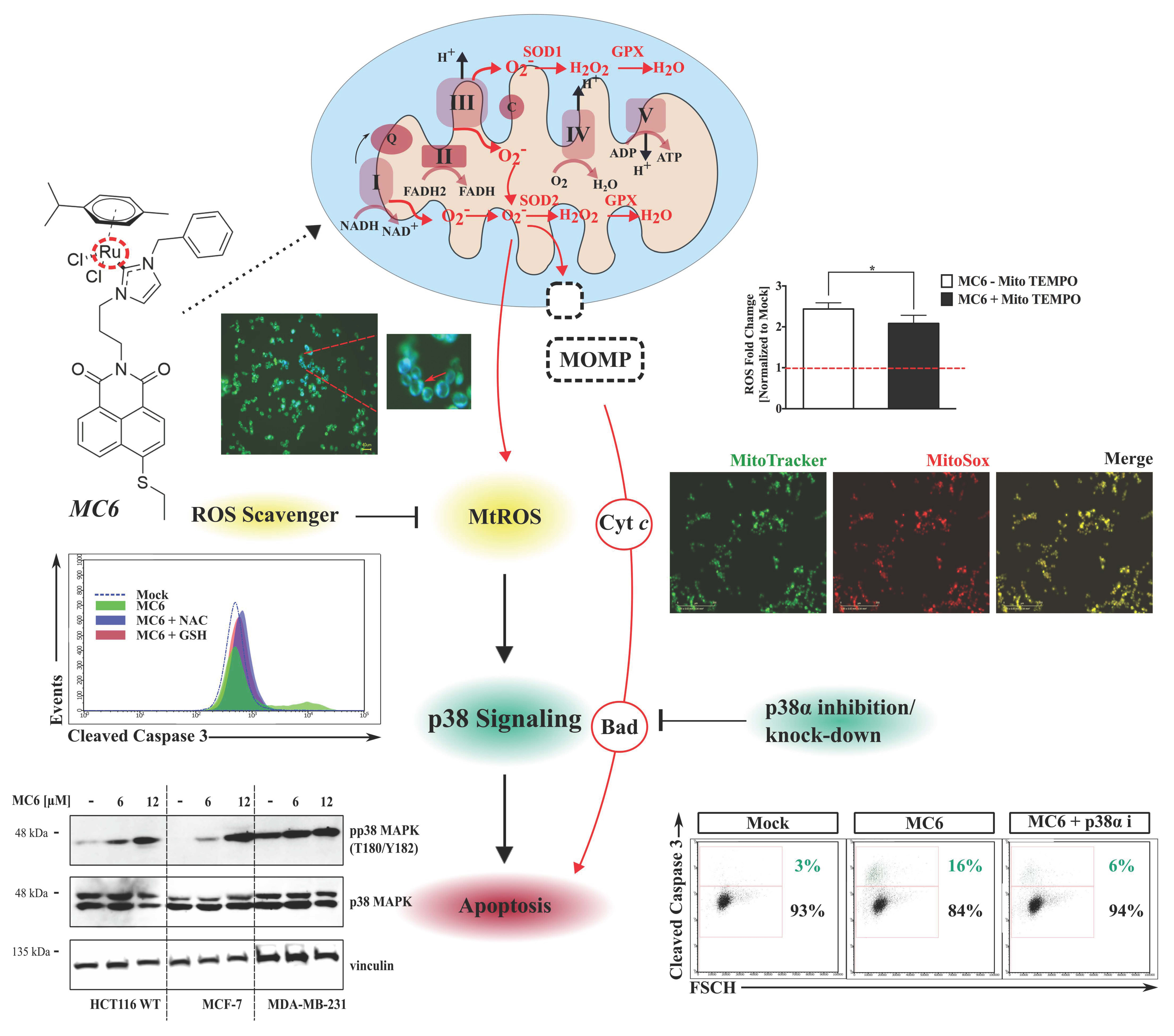
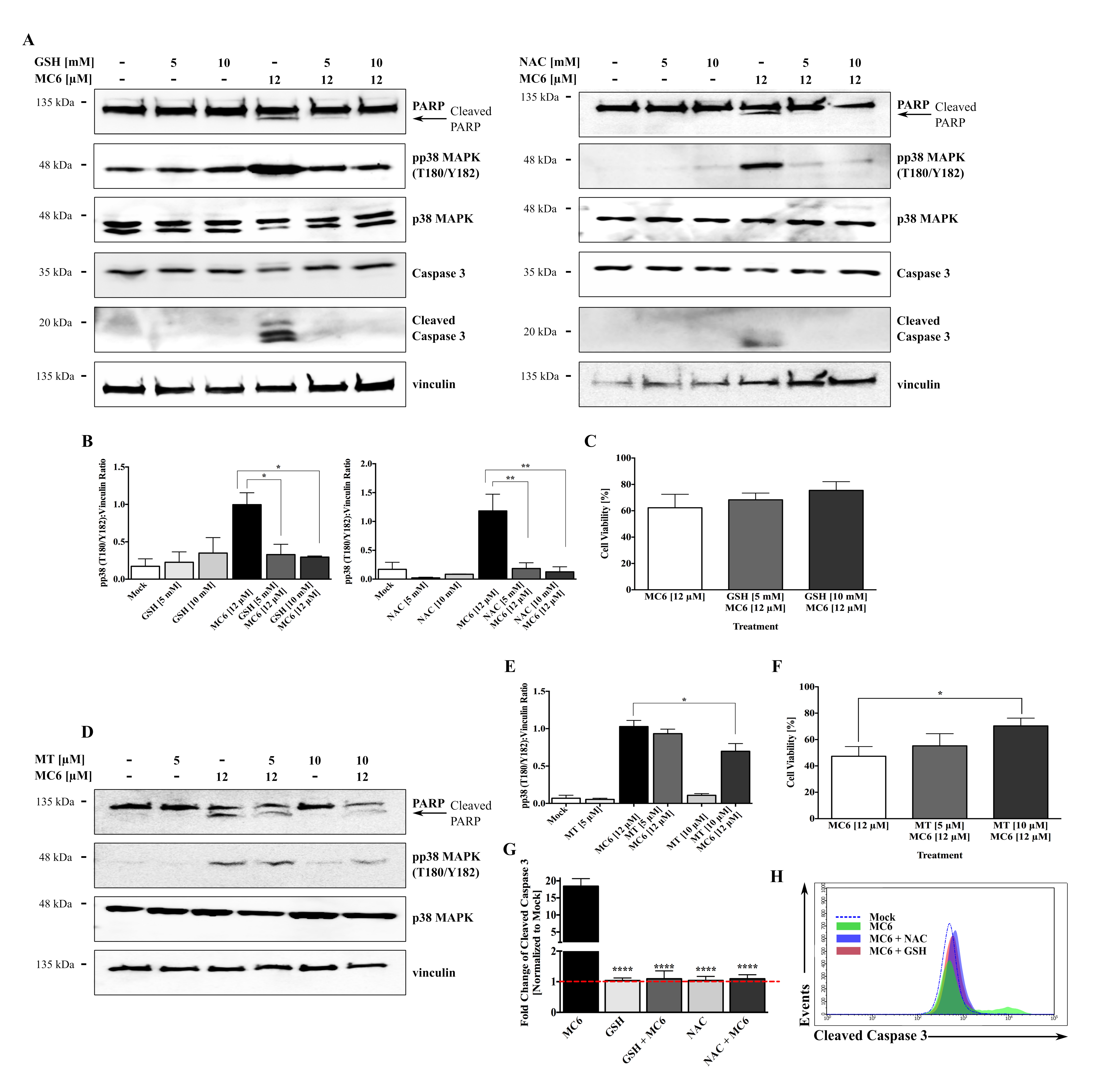
Figure 8.
MC6-induced cytotoxicity and pro-apoptotic effects are mediated via the ROS-p38 signaling axis. (A) Treatment with GSH and NAC at the indicated concentrations 1 h prior to the addition of MC6 (12 μM) for 24 h blocked the activation of p38 as well as cleavages of caspase 3 and PARP, detected by immunoblotting; (B) Densitometric analyses show a significant reduction in the MC6-induced pp38 (T180/Y182) accumulation in the presence of anti-oxidants. Error bars indicate the SEM of three independent experiments, one of those is presented in (A); (C) Increased cellular survival of HCT116 cells pre-treated with either 5 or 10 mM of GSH 1 h before the addition of MC6 (12 μM) for 24 h, as determined by SRB assay. Data represent mean ± SD of three biological replicates, normalized to mock (0.1% DMSO) and the respective GSH treatment; (D–F) HCT116 cells pre-incubated with either 5 or 10 μM of Mito TEMPO (MT) for 2 h were treated with MC6 (12 μM) for 24 h. The mitochondria-targeted ROS scavenger was found to attenuate the MC6-mediated p38 activation as well as PARP cleavage at the highest used concentration (10 μM), as detected by immunoblotting (D) and the associated densitometric quantification (E), obtained from three independent experiments. Error bars ± SEM. Additionally, it rescued the MC6-mediated cytotoxic effects, as determined by SRB assay (F). Percentage cell viability of MC6-treated cells was normalized to mock (0.1% DMSO) and the respective MT treatment. Error bars ± SD; n = 3; (G) Flow cytometric analysis of caspase 3 activation illustrates significantly lower levels of the cleaved form of the protein in cells pre-treated for 1 h with either GSH (10 mM) or NAC (10 mM) as compared to that of MC6-treated cells (12 μM, 24 h). Error bars ± SD, n = 3; (H) Representative histogram of one out of three biological replicates presented in (G) demonstrates a left-ward shift in caspase 3 activity in the presence of ROS scavengers. Statistical significance between the MC6-treated cells in the absence/presence of anti-oxidants was calculated using two-tailed student’s t-test. *, **, ***, and **** on the figures represent p-values that are less than or equal to 0.05, 0.01, 0.001, and 0.0001, respectively.
Figure 8.
MC6-induced cytotoxicity and pro-apoptotic effects are mediated via the ROS-p38 signaling axis. (A) Treatment with GSH and NAC at the indicated concentrations 1 h prior to the addition of MC6 (12 μM) for 24 h blocked the activation of p38 as well as cleavages of caspase 3 and PARP, detected by immunoblotting; (B) Densitometric analyses show a significant reduction in the MC6-induced pp38 (T180/Y182) accumulation in the presence of anti-oxidants. Error bars indicate the SEM of three independent experiments, one of those is presented in (A); (C) Increased cellular survival of HCT116 cells pre-treated with either 5 or 10 mM of GSH 1 h before the addition of MC6 (12 μM) for 24 h, as determined by SRB assay. Data represent mean ± SD of three biological replicates, normalized to mock (0.1% DMSO) and the respective GSH treatment; (D–F) HCT116 cells pre-incubated with either 5 or 10 μM of Mito TEMPO (MT) for 2 h were treated with MC6 (12 μM) for 24 h. The mitochondria-targeted ROS scavenger was found to attenuate the MC6-mediated p38 activation as well as PARP cleavage at the highest used concentration (10 μM), as detected by immunoblotting (D) and the associated densitometric quantification (E), obtained from three independent experiments. Error bars ± SEM. Additionally, it rescued the MC6-mediated cytotoxic effects, as determined by SRB assay (F). Percentage cell viability of MC6-treated cells was normalized to mock (0.1% DMSO) and the respective MT treatment. Error bars ± SD; n = 3; (G) Flow cytometric analysis of caspase 3 activation illustrates significantly lower levels of the cleaved form of the protein in cells pre-treated for 1 h with either GSH (10 mM) or NAC (10 mM) as compared to that of MC6-treated cells (12 μM, 24 h). Error bars ± SD, n = 3; (H) Representative histogram of one out of three biological replicates presented in (G) demonstrates a left-ward shift in caspase 3 activity in the presence of ROS scavengers. Statistical significance between the MC6-treated cells in the absence/presence of anti-oxidants was calculated using two-tailed student’s t-test. *, **, ***, and **** on the figures represent p-values that are less than or equal to 0.05, 0.01, 0.001, and 0.0001, respectively.
![Ijms 19 03964 g008]()


 and
and 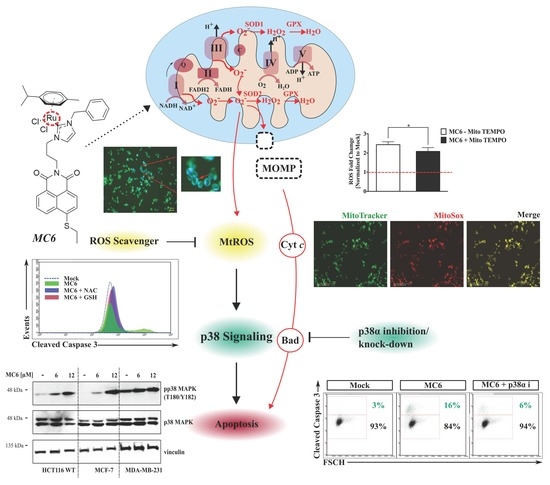
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







