Defining a Characteristic Gene Expression Set Responsible for Cancer Stem Cell-Like Features in a Sub-Population of Ewing Sarcoma Cells CADO-ES1


Abstract
:1. Introduction
2. Results
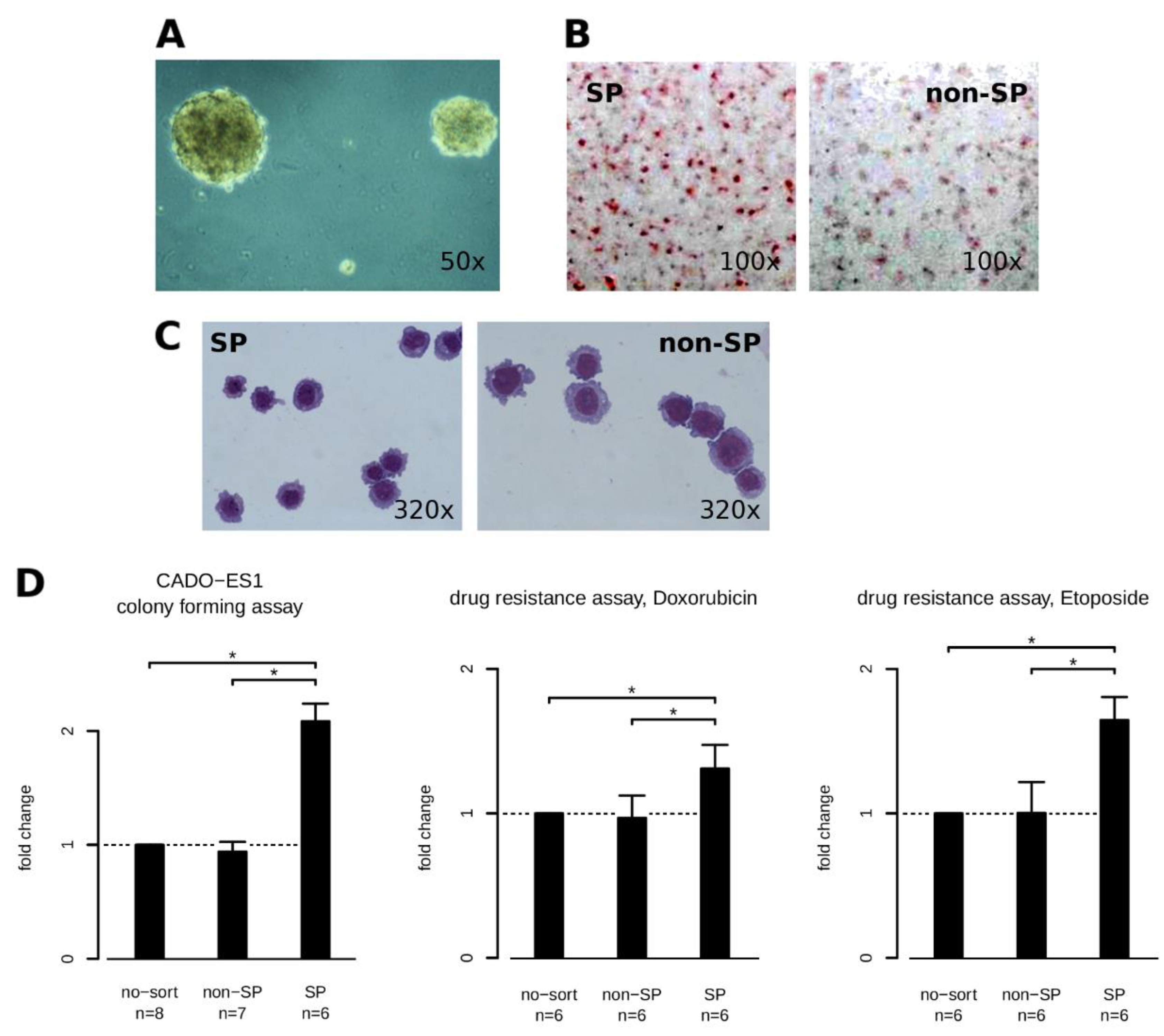
2.1. Defining a CADO-ES1 Side Population
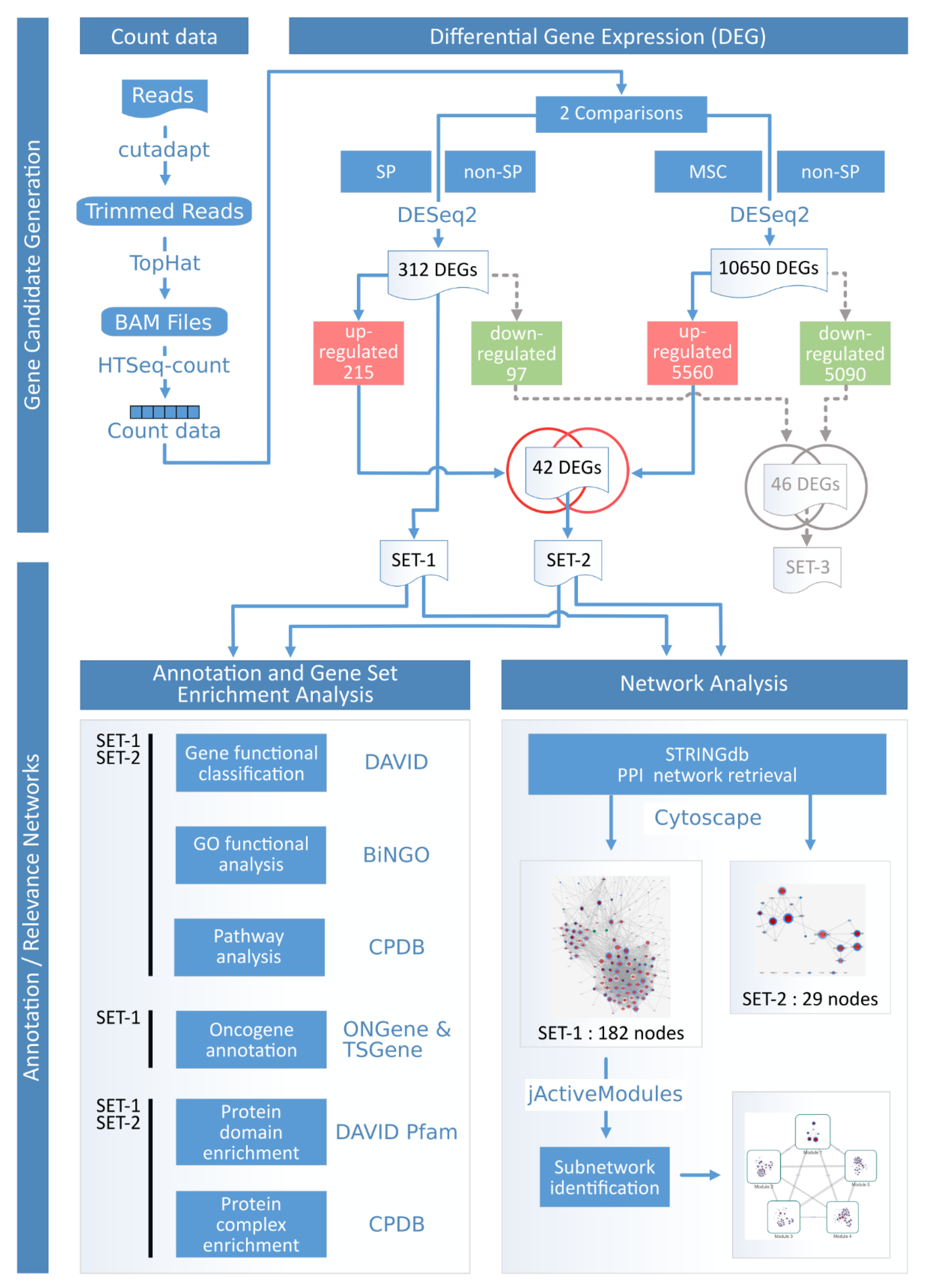
2.2. Double Differential Experimental Design and Short Read Sequencing
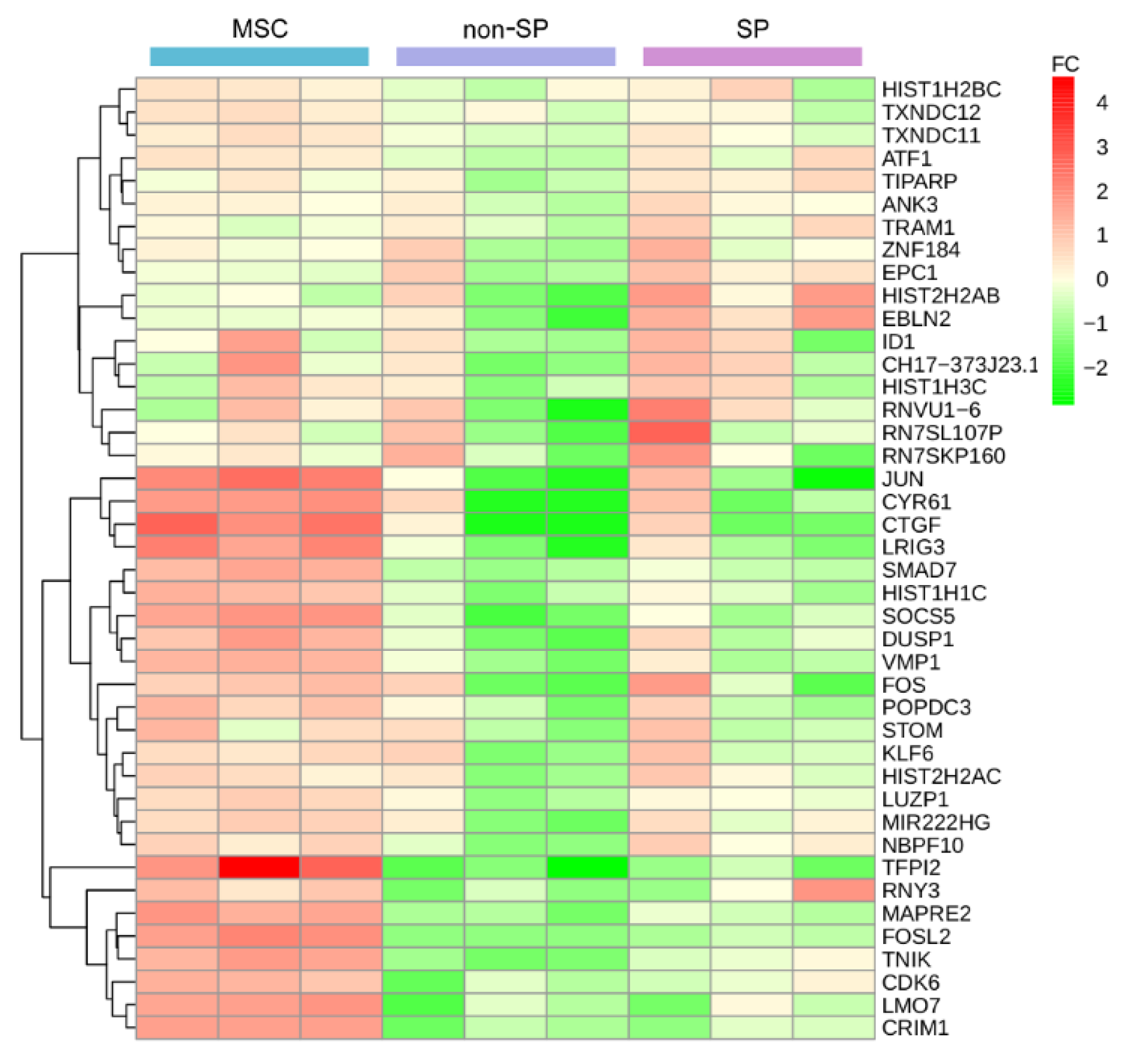
2.3. The Identification of Tumor Driver Gene-Sets
2.4. Functional and Pathway Enrichment Analysis
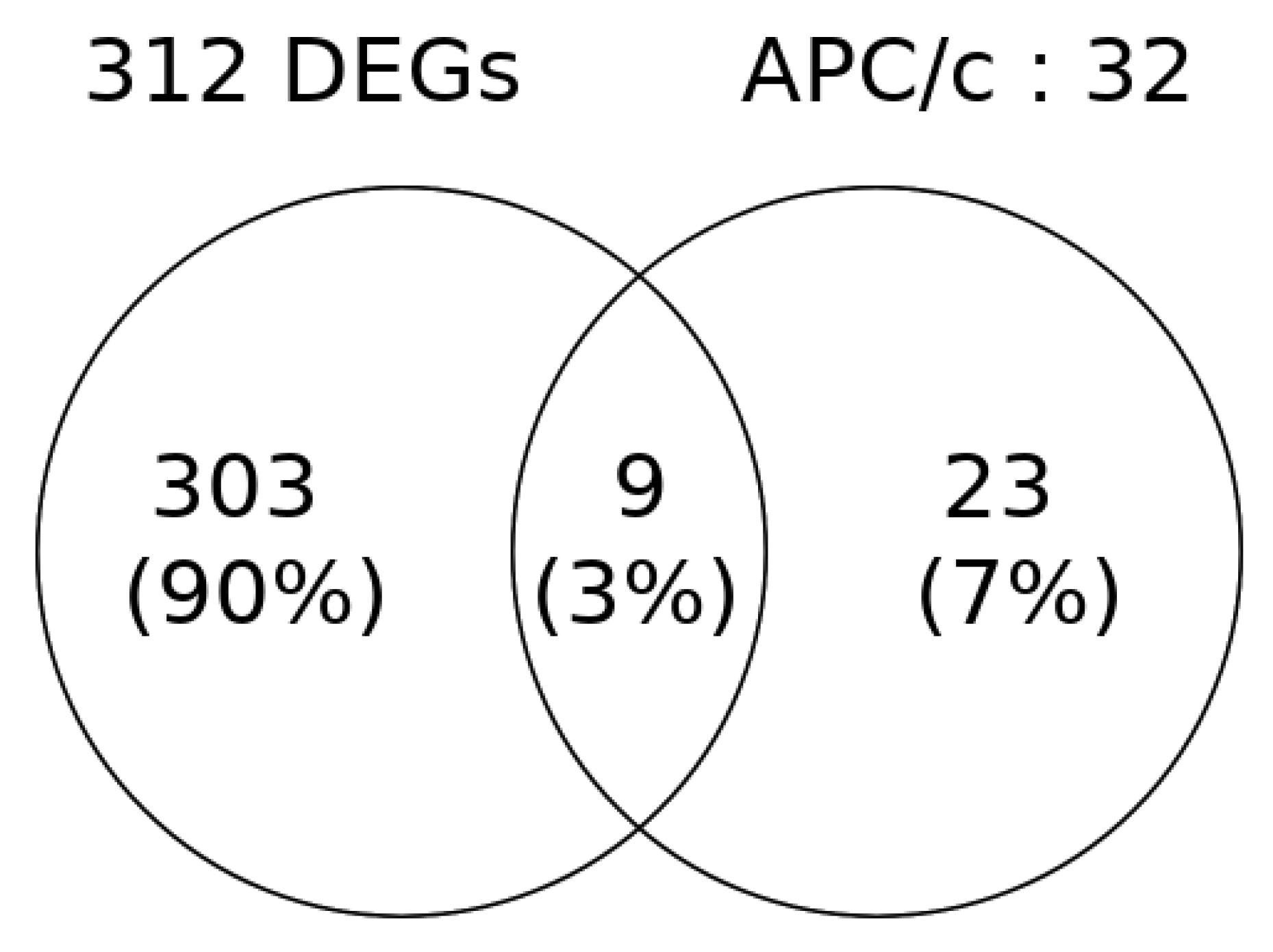
2.5. Identification of Oncogenes and Tumor Suppressor Genes
2.6. Identifying Epigenetic Modifier
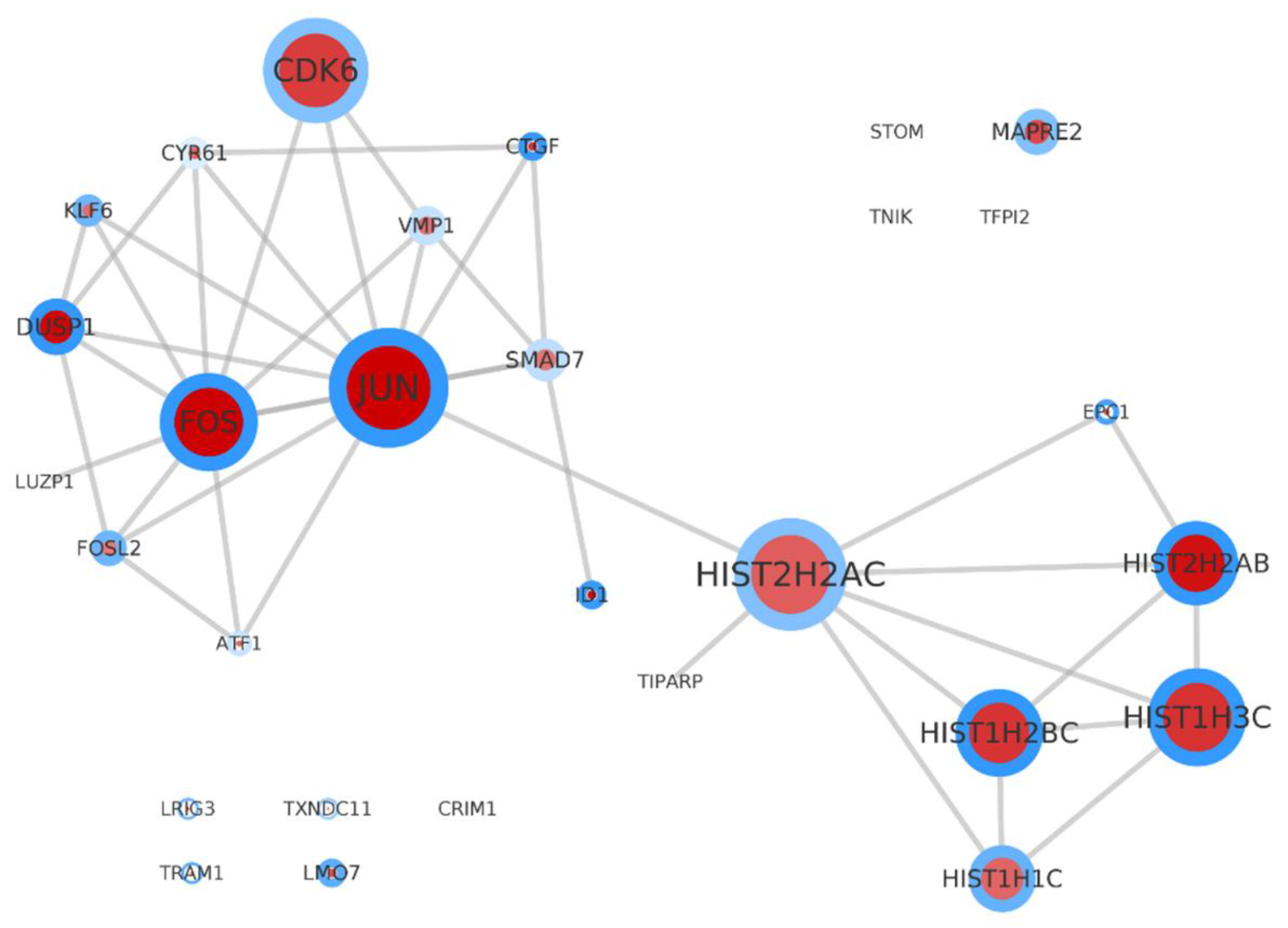
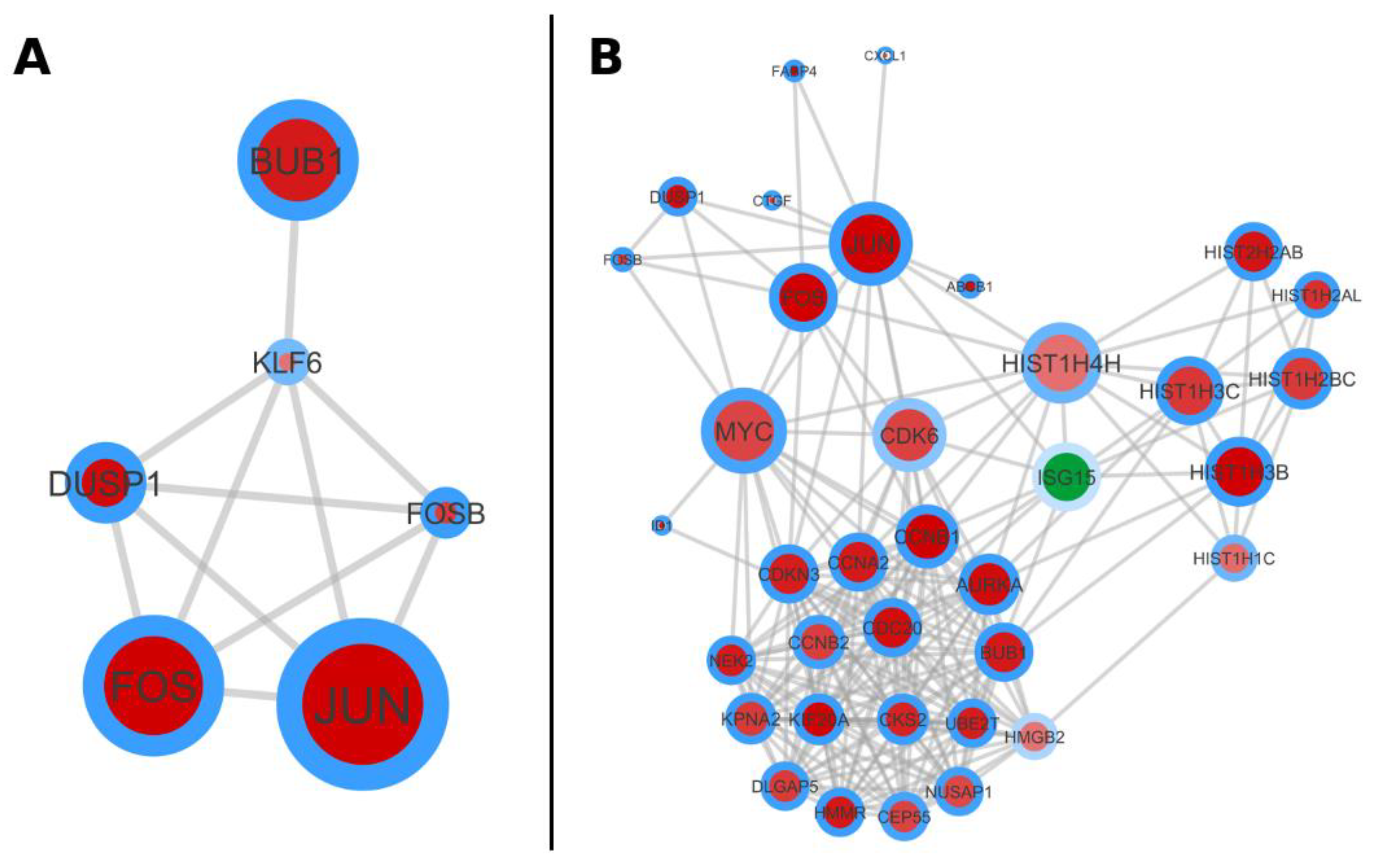
2.7. The Protein-Protein Interaction (PPI) Network Analysis Is Supporting the Annotation Derived Information
2.8. Condensing Information on Networks and Pathways by Considering on Protein Domains and Protein Complexes
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Cell Line and Cell Culture Conditions
5.2. Side Population
5.3. Cytotoxiticity Assay
5.4. Colony Assy
5.5. In Vitro Differentiation Potential
5.6. RNA Isolation and Library Preparation
5.7. Raw Data Processing/Controls
5.8. Differential Analysis/Controls
5.9. Functional and Pathway Enrichment Analysis
5.10. Identification of Tumor-Associated Genes
5.11. Construction of PPI Network and the Subnetwork Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CADO-ES1 | Ewing cell line CADO-ES1, human, female. Fusion gene: EWSR1-ERG |
| SP | Side-population, here a subfraction of Ewing sarcoma cells owning some stem cell properties |
| non-SP | The (multi clonal) main Ewing sarcoma cell population |
| MSC | Mesenchymal stem cell (here originating from bone marrow explantats) |
| CSC | Cancer stem cell, other terms and slightly different definitions are existing |
| FACS | Fluorescence activated cell sorting |
| DEG(s) | Differentially expressed gene(s) |
| FDR | False discovery rate, a multiple test correction procedure resulting in a corrected p value |
| PPI | Protein-protein interaction |
| GO | Gene Ontology |
| APC/c | The anaphase-promoting complex/cyclosome |
| DAVID | Database for Annotation, Visualization, and Integrated Discovery tool |
References
- Ewing, J. Diffuse Endothelioma of Bone. Proc. N. Y. Pathol. Soc. 1921, 21, 17–24. [Google Scholar]
- Karnuth, B.; Dedy, N.; Spieker, T.; Lawlor, E.; Gattenlöhner, S.; Ranft, A.; Dirksen, U.; Jürgens, H.; Bräuninger, A. Differentially expressed miRNAs in Ewing sarcoma compared to mesenchymal stem cells: Low miR-31 expression with effects on proliferation and invasion. PLoS ONE 2014, 9, e93067. [Google Scholar] [CrossRef] [PubMed]
- Tarazona, N.; Navarro, L.; Cejalvo, J.; Gambardella, V.; Pérez-Fidalgo, J.; Sempere, A.; Navarro, S.; Cervantes, A. Primary paraesophageal Ewing’s sarcoma: An uncommon case report and literature review. OncoTargets Ther. 2015, 8, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- Kodama, K.; Doi, O.; Higashiyama, M.; Mori, Y.; Horai, T.; Tateishi, R.; Aoki, Y.; Misawa, S. Establishment and characterization of a new Ewing’s sarcoma cell line. Cancer Genet. Cytogenet. 1991, 57, 19–30. [Google Scholar] [CrossRef]
- Delattre, O.; Zucman, J.; Melot, T.; Garau, X.S.; Zucker, J.-M.; Lenoir, G.M.; Ambros, P.F.; Sheer, D.; Turc-Carel, C.; Triche, T.J.; et al. The Ewing family of tumors—A subgroup of small-round-cell tumors defined by specific chimeric transcripts. N. Engl. J. Med. 1994, 331, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Stahl, M.; Ranft, A.; Paulussen, M.; Bölling, T.; Vieth, V.; Bielack, S.; Görtitz, I.; Braun-Munzinger, G.; Hardes, J.; Jürgens, H.; et al. Risk of recurrence and survival after relapse in patients with Ewing sarcoma. Pediatr. Blood Cancer 2011, 57, 549–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delattre, O.; Zucman, J.; Plougastel, B.; Desmaze, C.; Melot, T.; Peter, M.; Kovar, H.; Joubert, I.; de Jong, P.; Rouleau, G.; et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992, 359, 162–165. [Google Scholar] [CrossRef]
- Sorensen, P.; Lessnick, S.; Lopez-Terrada, D.; Liu, X.; Triche, T.; Denny, C. A second Ewing’s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat. Genet. 1994, 6, 146–151. [Google Scholar] [CrossRef]
- Le Deley, M.C.; Delattre, O.; Schaefer, K.L.; Burchill, S.A.; Koehler, G.; Hogendoorn, P.C.; Lion, T.; Poremba, C.; Marandet, J.; Ballet, S.; et al. Impact of EWS-ETS fusion type on disease progression in Ewing’s sarcoma/peripheral primitive neuroectodermal tumor: Prospective results from the cooperative Euro-E.W.I.N.G. 99 trial. J. Clin. Oncol. 2010, 28, 1982–1988. [Google Scholar] [CrossRef]
- Sankar, S.; Lessnick, S. Promiscuous partnerships in Ewing’s sarcoma. Cancer Genet. 2011, 204, 351–365. [Google Scholar] [CrossRef]
- Potratz, J.; Dirksen, U.; Jürgens, H.; Craft, A. Ewing sarcoma: Clinical state-of-the-art. Pediatr. Hematol. Oncol. 2012, 29, 1–11. [Google Scholar] [CrossRef]
- Notta, F.; Mullighan, C.G.; Wang, J.C.Y.; Poeppl, A.; Doulatov, S.; Phillips, L.A.; Ma, J.; Minden, M.D.; Downing, J.R.; Dick, J.E. Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature 2011, 469, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Tirode, F.; Laud-Duval, K.; Prieur, A.; Delorme, B.; Charbord, P.; Delattre, O. Mesenchymal stem cell features of Ewing tumors. Cancer Cell 2007, 11, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Riggi, N.; Suvà, M.L.; De Vito, C.; Provero, P.; Stehle, J.C.; Baumer, K.; Cironi, L.; Janiszewska, M.; Petricevic, T.; Suvà, D.; et al. EWS-FLI-1 modulates miRNA145 and SOX2 expression to initiate mesenchymal stem cell reprogramming toward Ewing sarcoma cancer stem cells. Genes Dev. 2010, 24, 916–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minguell, J.; Erices, A.; Conget, P. Mesenchymal Stem Cells. Exp. Boil. Med. 2001, 226, 507–520. [Google Scholar] [CrossRef]
- Riggi, N.; Cironi, L.; Provero, P.; Suvà, M.; Kaloulis, K.; Garcia-Echeverria, C.; Hoffmann, F.; Trumpp, A.; Stamenkovic, I. Development of Ewing’s sarcoma from primary bone marrow-derived mesenchymal progenitor cells. Cancer Res. 2005, 65, 11459–11468. [Google Scholar] [CrossRef]
- Riggi, N.; Suva, M.-L.; Suva, D.; Cironi, L.; Provero, P.; Tercier, S.; Joseph, J.-M.; Stehle, J.-C.; Baumer, K.; Kindler, V.; et al. EWS-FLI-1 expression triggers a Ewing’s sarcoma initiation program in primary human mesenchymal stem cells. Cancer Res. 2008, 68, 2176–2185. [Google Scholar] [CrossRef]
- Von Levetzow, C.; Jiang, X.; Gwye, Y.; von Levetzow, G.; Hung, L.; Cooper, A.; Hsu, J.; Lawlor, E. Modeling initiation of Ewing sarcoma in human neural crest cells. PLoS ONE 2011, 6, e19305. [Google Scholar] [CrossRef]
- Hirschmann-Jax, C.; Foster, A.; Wulf, G.; Nuchtern, J.; Jax, T.; Gobel, U.; Goodell, M.; Brenner, M. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc. Natl. Acad. Sci. USA 2004, 101, 14228–14233. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Maley, C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.S.K.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodell, M. Stem cell identification and sorting using the Hoechst 33342 side population (SP). Curr. Protoc. Cytom. 2005. [Google Scholar] [CrossRef]
- Singh Nanda, J.; Kumar, R.; Raghava, G. dbEM: A database of epigenetic modifiers curated from cancerous and normal genomes. Sci. Rep. 2016, 6, 19340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korsching Group. Available online: http://complex-systems.uni-muenster.de/cy_ew1/index.html (accessed on 23 November 2017).
- Ideker, T.; Ozier, O.; Schwikowski, B.; Siegel, A. Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics 2002, 18 (Suppl. 1), S233–S240. [Google Scholar] [CrossRef]
- Cironi, L.; Riggi, N.; Provero, P.; Wolf, N.; Suvà, M.; Suvà, D.; Kindler, V.; Stamenkovic, I. IGF1 is a common target gene of Ewing’s sarcoma fusion proteins in mesenchymal progenitor cells. PLoS ONE 2008, 3, e2634. [Google Scholar] [CrossRef]
- Yates, L.; Campbell, P. Evolution of the cancer genome. Nat. Rev. Genet. 2012, 13, 795–806. [Google Scholar] [CrossRef] [Green Version]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [Green Version]
- Todorova, R. Ewing’s sarcoma cancer stem cell targeted therapy. Curr. Stem Cell Res. Ther. 2014, 9, 46–62. [Google Scholar] [CrossRef]
- Ponti, D.; Costa, A.; Zaffaroni, N.; Pratesi, G.; Petrangolini, G.; Coradini, D.; Pilotti, S.; Pierotti, M.; Daidone, M. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005, 65, 5506–5511. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhang, H.; Hu, Y.; Xia, Q.; Liu, C.; Li, Y.; Yu, B.; Gu, T.; Zhang, X.; Yu, X.; Kong, W. Sphere formation assay is not an effective method for cancer stem cell derivation and characterization from the Caco-2 colorectal cell line. Curr. Stem Cell Res. Ther. 2014, 9, 82–88. [Google Scholar] [CrossRef]
- Szakács, G.; Váradi, A.; Ozvegy-Laczka, C.; Sarkadi, B. The role of ABC transporters in drug absorption, distribution, metabolism, excretion and toxicity (ADME-Tox). Drug Discov. Today 2008, 13, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Camarda, R.; Odegaard, J.; Paik, H.; Oskotsky, B.; Krings, G.; Goga, A.; Sirota, M.; Butte, A. Comprehensive analysis of normal adjacent to tumor transcriptomes. Nat. Commun. 2017, 8, 1077. [Google Scholar] [CrossRef]
- Eferl, R.; Wagner, E. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Papoudou-Bai, A.; Hatzimichael, E.; Barbouti, A.; Kanavaros, P. Expression patterns of the activator protein-1 (AP-1) family members in lymphoid neoplasms. Clin. Exp. Med. 2017, 17, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Garces de Los Fayos Alonso, I.; Liang, H.; Turner, S.; Lagger, S.; Merkel, O.; Kenner, L. The Role of Activator Protein-1 (AP-1) Family Members in CD30-Positive Lymphomas. Cancers 2018, 10, 93. [Google Scholar] [CrossRef]
- Dominguez, D.; Tsai, Y.; Gomez, N.; Jha, D.; Davis, I.; Wang, Z. A high-resolution transcriptome map of cell cycle reveals novel connections between periodic genes and cancer. Cell Res. 2016, 26, 946–962. [Google Scholar] [CrossRef]
- Squire, L.; Berg, D.; Bloom, F.; du Lac, S.; Ghosh, A.; Spitzer, N. Fundamental Neuroscience; Elsevier: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Clark, D.; Pazdernik, N. Biotechnology, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Ameyar, M.; Wisniewska, M.; Weitzman, J. A role for AP-1 in apoptosis: The case for and against. Biochimie 2003, 85, 747–752. [Google Scholar] [CrossRef]
- Van Dam, H.; Castellazzi, M. Distinct roles of Jun: Fos and Jun: ATF dimers in oncogenesis. Oncogene 2001, 20, 2453–2464. [Google Scholar] [CrossRef]
- Yamashita, J.; McCauley, L. The activating protein-1 transcriptional complex—Essential and multifaceted roles in bone. Clin. Rev. Bone Miner. Metab. 2006, 4, 107–122. [Google Scholar] [CrossRef]
- Hoeller, D.; Hecker, C.; Dikic, I. Ubiquitin and ubiquitin-like proteins in cancer pathogenesis. Nat. Rev. Cancer 2006, 6, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Frescas, D.; Pagano, M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: Tipping the scales of cancer. Nat. Rev. Cancer 2008, 8, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wan, L.; Zhong, J.; Inuzuka, H.; Liu, P.; Sarkar, F.; Wei, W. Cdc20: A potential novel therapeutic target for cancer treatment. Curr. Pharm. Des. 2013, 19, 3210–3214. [Google Scholar] [CrossRef] [PubMed]
- McLean, J.; Chaix, D.; Ohi, M.; Gould, K. State of the APC/C: Organization, function, and structure. Crit. Rev. Biochem. Mol. Boil. 2011, 46, 118–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolders, L.; Teodoro, J. Targeting the anaphase promoting complex: Common pathways for viral infection and cancer therapy. Expert Opin. Ther. Targets 2011, 15, 767–780. [Google Scholar] [CrossRef]
- Clarke, D.; Segal, M.; Andrews, C.; Rudyak, S.; Jensen, S.; Smith, K.; Reed, S. S-phase checkpoint controls mitosis via an APC-independent Cdc20p function. Nat. Cell Boil. 2003, 5, 928–935. [Google Scholar] [CrossRef]
- Kidokoro, T.; Tanikawa, C.; Furukawa, Y.; Katagiri, T.; Nakamura, Y.; Matsuda, K. CDC20, a potential cancer therapeutic target, is negatively regulated by p53. Oncogene 2008, 27, 1562–1571. [Google Scholar] [CrossRef]
- Labit, H.; Fujimitsu, K.; Bayin, N.; Takaki, T.; Gannon, J.; Yamano, H. Dephosphorylation of Cdc20 is required for its C-box-dependent activation of the APC/C. EMBO J. 2012, 31, 3351–3362. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Jedinak, A.; Sliva, D. Ganodermanontriol (GDNT) exerts its effect on growth and invasiveness of breast cancer cells through the down-regulation of CDC20 and uPA. Biochem. Biophys. Res. Commun. 2011, 415, 325–329. [Google Scholar] [CrossRef]
- Rajkumar, T.; Sabitha, K.; Vijayalakshmi, N.; Shirley, S.; Bose, M.; Gopal, G.; Selvaluxmy, G. Identification and validation of genes involved in cervical tumourigenesis. BMC Cancer 2011, 11, 80. [Google Scholar] [CrossRef] [PubMed]
- Marucci, G.; Morandi, L.; Magrini, E.; Farnedi, A.; Franceschi, E.; Miglio, R.; Calò, D.; Pession, A.; Foschini, M.P.; Eusebi, V. Gene expression profiling in glioblastoma and immunohistochemical evaluation of IGFBP-2 and CDC20. Virchows Arch. Int. J. Pathol. 2008, 453, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Ouellet, V.; Guyot, M.; Le Page, C.; Filali-Mouhim, A.; Lussier, C.; Tonin, P.; Provencher, D.; Mes-Masson, A. Tissue array analysis of expression microarray candidates identifies markers associated with tumor grade and outcome in serous epithelial ovarian cancer. Int. J. Cancer 2006, 119, 599–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, T.; Daigo, Y.; Aragaki, M.; Ishikawa, K.; Sato, M.; Kaji, M. Overexpression of CDC20 predicts poor prognosis in primary non-small cell lung cancer patients. J. Surg. Oncol. 2012, 106, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Shuck, R.; Kurenbekova, L.; Allen-Rhoades, W.; Edwards, D.; Huang, S.; Rajapakshe, K.; Coarfa, C.; Donehower, L.A.; Yustein, J.T. miR-130b directly targets ARHGAP1 to drive activation of a metastatic CDC42-PAK1-AP1 positive feedback loop in Ewing sarcoma. Int. J. Cancer 2017, 141, 2062–2075. [Google Scholar] [CrossRef]
- Whitehurst, A.; Ram, R.; Shivakumar, L.; Gao, B.; Minna, J.; White, M. The RASSF1A tumor suppressor restrains anaphase-promoting complex/cyclosome activity during the G1/S phase transition to promote cell cycle progression in human epithelial cells. Mol. Cell. Boil. 2008, 28, 3190–3197. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Xiao, B.; Jha, H.; Lu, J.; Banerjee, S.; Robertson, E. Kaposi’s sarcoma-associated herpesvirus-encoded LANA can induce chromosomal instability through targeted degradation of the mitotic checkpoint kinase Bub1. J. Virol. 2014, 88, 7367–7378. [Google Scholar] [CrossRef]
- Tulchinsky, E. Fos family members: Regulation, structure and role in oncogenic transformation. Histol. Histopathol. 2000, 15, 921–928. [Google Scholar] [CrossRef]
- Milde-Langosch, K. The Fos family of transcription factors and their role in tumourigenesis. Eur. J. Cancer 2005, 41, 2449–2461. [Google Scholar] [CrossRef]
- Gazon, H.; Barbeau, B.; Mesnard, J.; Peloponese, J. Hijacking of the AP-1 Signaling Pathway during Development of ATL. Front. Microbiol. 2017, 8, 2686. [Google Scholar] [CrossRef]
- Han, R.; Li, L.; Ugalde, A.; Tal, A.; Manber, Z.; Barbera, E.; Chiara, V.; Elkon, R.; Agami, R. Functional CRISPR screen identifies AP1-associated enhancer regulating FOXF1 to modulate oncogene-induced senescence. Genome Boil. 2018, 19, 118. [Google Scholar] [CrossRef]
- Lopez-Bergami, P.; Lau, E.; Ronai, Z. Emerging roles of ATF2 and the dynamic AP1 network in cancer. Nat. Rev. Cancer 2010, 10, 65–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newton, K.; Dixit, V. Signaling in innate immunity and inflammation. Cold Spring Harb. Perspect. Boil. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, C.; Zhang, S.; Barford, D. Visualizing the complex functions and mechanisms of the anaphase promoting complex/cyclosome (APC/C). Open Boil. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; He, M.; Shah, A.; Wan, Y. Insights into APC/C: From cellular function to diseases and therapeutics. Cell Div. 2016, 11, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harkness, T. Activating the Anaphase Promoting Complex to Enhance Genomic Stability and Prolong Lifespan. Int. J. Mol. Sci. 2018, 19, 1888. [Google Scholar] [CrossRef] [PubMed]
- Nicolau-Neto, P.; Palumbo, A.; De Martino, M.; Esposito, F.; de Almeida Simão, T.; Fusco, A.; Nasciutti, L.; Meireles Da Costa, N.; Ribeiro Pinto, L. UBE2C Is a Transcriptional Target of the Cell Cycle Regulator FOXM1. Genes 2018, 9, 188. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Li, B.; Yu, H. The Bub1-Plk1 kinase complex promotes spindle checkpoint signalling through Cdc20 phosphorylation. Nat. Commun. 2016, 7, 10818. [Google Scholar] [CrossRef]
- Human Protein Atlas—Pathology. Available online: https://www.proteinatlas.org/ENSG00000048052-HDAC9/pathology (accessed on 1 July 2018).
- Milde, T.; Oehme, I.; Korshunov, A.; Kopp-Schneider, A.; Remke, M.; Northcott, P.; Deubzer, H.E.; Lodrini, M.; Taylor, M.D.; von Deimling, A.; et al. HDAC5 and HDAC9 in medulloblastoma: Novel markers for risk stratification and role in tumor cell growth. Clin. Cancer Res. 2010, 16, 3240–3252. [Google Scholar] [CrossRef]
- Fleming, J.L.; Dworkin, A.M.; Allain, D.C.; Fernandez, S.; Wei, L.; Peters, S.B.; Iwenofu, O.H.; Ridd, K.; Bastian, B.C.; Toland, A.E. Allele-specific imbalance mapping identifies HDAC9 as a candidate gene for cutaneous squamous cell carcinoma. Int. J. Cancer 2014, 134, 244–248. [Google Scholar] [CrossRef]
- Chen, Y.; Chung, C.; Liu, Y.; Yeh, S.; Hsu, J.; Hung, M.; Su, H.; Li, L. Enhancer of Zeste Homolog 2 and Histone Deacetylase 9c Regulate Age-Dependent Mesenchymal Stem Cell Differentiation into Osteoblasts and Adipocytes. Stem Cells 2016, 34, 2183–2193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Park, J.-R.; Seo, M.-S.; Roh, K.-H.; Park, S.-B.; Hwang, J.-W.; Sun, B.; Seo, K.; Lee, Y.-S.; Kang, S.-K. Histone deacetylase inhibitors decrease proliferation potential and multilineage differentiation capability of human mesenchymal stem cells. Cell Prolif. 2009, 42, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, M.; Crompot, E.; Meuleman, N.; Mineur, P.; Dessars, B.; El Housni, H.; Bron, D.; Lagneaux, L.; Stamatopoulos, B. Global histone deacetylase enzymatic activity is an independent prognostic marker associated with a shorter overall survival in chronic lymphocytic leukemia patients. Epigenetics 2014, 9, 1374–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Array Express. Available online: https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-6067/ (accessed on 23 February 2018).
- Trapnell, C.; Pachter, L.; Salzberg, S. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, B. A comprehensive evaluation of ensembl, RefSeq, and UCSC annotations in the context of RNA-seq read mapping and gene quantification. BMC Genom. 2015, 16, 97. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Boil. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Harris, M.A.; Clark, J.; Ireland, A.; Lomax, J.; Ashburner, M.; Foulger, R.; Eilbeck, K.; Lewis, S.; Marshall, B.; Mungall, C.; et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.; Wang, J.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef] [PubMed]
- Vastrik, I.; D’Eustachio, P.; Schmidt, E.; Joshi-Tope, G.; Gopinath, G.; Croft, D.; de Bono, B.; Gillespie, M.; Jassal, B.; Lewis, S.; et al. Reactome: A knowledge base of biologic pathways and processes. Genome Boil. 2007, 8, R39. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar]
- Zhao, M.; Kim, P.; Mitra, R.; Zhao, J.; Zhao, Z. TSGene 2.0: An updated literature-based knowledgebase for tumor suppressor genes. Nucleic Acids Res. 2016, 44, D1023–D1031. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sun, J.; Zhao, M. ONGene: A literature-based database for human oncogenes. J. Genet. Genom. 2017, 44, 119–121. [Google Scholar] [CrossRef] [PubMed]
- TSGene. Available online: https://bioinfo.uth.edu/TSGene/Human_TSGs.txt (accessed on 13 July 2018).
- ONgene. Available online: http://ongene.bioinfo-minzhao.org/index.html (accessed on 13 July 2018).
- Snel, B.; Lehmann, G.; Bork, P.; Huynen, M. STRING: A web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 2000, 28, 3442–3444. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
- Mitra, K.; Carvunis, A.; Ramesh, S.; Ideker, T. Integrative approaches for finding modular structure in biological networks. Nat. Rev. Genet. 2013, 14, 719–732. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Lin, D.; Zhang, J.; Wang, Y.; Deng, H. Comparison of statistical methods for subnetwork detection in the integration of gene expression and protein interaction network. BMC Bioinform. 2017, 18, 149. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GO-ID | GO Name | Gene Counts | p values (FDR) | Candidates Also Part of Enriched Pathways |
|---|---|---|---|---|
| 22403 | cell cycle phase | 48 | 2.3 × 10−31 | 36 (75%) |
| 279 | M phase | 44 | 3.1 × 10−31 | 33 (75%) |
| 6996 | organelle organization | 75 | 3.1 × 10−31 | 65 (87%) |
| 22402 | cell cycle process | 52 | 2.3 × 10−30 | 39 (75%) |
| 278 | mitotic cell cycle | 44 | 5.6 × 10−30 | 34 (77%) |
| 87 | M phase of mitotic cell cycle | 36 | 3.5 × 10−28 | 27 (75%) |
| 280 | nuclear division | 35 | 1.8 × 10−27 | 26 (74%) |
| 7067 | mitosis | 35 | 1.8 × 10−27 | 26 (74%) |
| 7049 | cell cycle | 55 | 5.1 × 10−27 | 42 (76%) |
| 48285 | organelle fission | 35 | 5.5 × 10−27 | 26 (74%) |
| Pathway Name | Set Size | Candidates Contained | p Value | q Value | Source |
|---|---|---|---|---|---|
| SET-1 | |||||
| Cell Cycle, Mitotic | 468 | 53 (11%) | 1.7 × 10−39 | 1.0 × 10−36 | Reactome |
| Cell Cycle | 551 | 55 (10%) | 4.0 × 10−38 | 1.2 × 10−35 | Reactome |
| M Phase | 267 | 40 (15%) | 3.0 × 10−34 | 6.1 × 10−32 | Reactome |
| RHO GTPase Effectors | 299 | 32 (11%) | 1.2 × 10−22 | 1.9 × 10−20 | Reactome |
| Condensation of Prophase Chromosomes | 77 | 20 (26%) | 1.9 × 10−22 | 2.3 × 10−20 | Reactome |
| Mitotic Prophase | 143 | 24 (17%) | 7.9 × 10−22 | 8.0 × 10−20 | Reactome |
| Senescence-Associated Secretory Phenotype (SASP) | 113 | 22 (20%) | 1.4 × 10−21 | 1.2 × 10−19 | Reactome |
| Cellular Senescence | 192 | 25 (13%) | 6.9 × 10−20 | 5.2 × 10−18 | Reactome |
| HATs acetylate histones | 143 | 22 (15%) | 3.8 × 10−19 | 2.6 × 10−17 | Reactome |
| HDACs deacetylate histones | 94 | 19 (20%) | 5.3 × 10−19 | 3.2 × 10−17 | Reactome |
| SET-2 | |||||
| Cellular Senescence | 192 | 9 (5%) | 1.8 × 10−10 | 4.2 × 10−08 | Reactome |
| Oxidative Stress-Induced Senescence | 129 | 7 (6%) | 9.1 × 10−09 | 1.1 × 10−06 | Reactome |
| Cellular responses to stress | 393 | 9 (2%) | 9.6 × 10−08 | 7.4 × 10−06 | Reactome |
| Senescence-Associated Secretory Phenotype (SASP) | 113 | 6 (5%) | 1.4 × 10−07 | 7.8 × 10−06 | Reactome |
| AP-1 transcription factor network | 71 | 5 (7%) | 4.3 × 10−07 | 2.0 × 10−05 | PID |
| HATs acetylate histones | 143 | 5 (4%) | 1.4 × 10−05 | 5.3 × 10−04 | Reactome |
| MAPK targets/Nuclear events mediated by MAP kinases | 31 | 3 (10%) | 4.2 × 10−05 | 1.4 × 10−03 | Reactome |
| HDACs deacetylate histones | 94 | 4 (4%) | 5.1 × 10−05 | 1.5 × 10−03 | Reactome |
| ErbB1 downstream signaling | 107 | 4 (4%) | 8.2 × 10−05 | 2.1 × 10−03 | PID |
| Hfe effect on hepcidin production | 7 | 2 (27%) | 9.9 × 10−05 | 2.1 × 10−03 | Wiki-pathways |
| Term | Protein Domains | Candidates Contained | p Value (FDR) |
|---|---|---|---|
| SET-1 | set size 204 | ||
| PF00125 | Core histone H2A/H2B/H3/H4 | 18 (9%) | 9.8 × 10−18 |
| PF00225 | Kinesin motor domain | 6 (3%) | 6.1 × 10−03 |
| PF00170 | bZIP transcription factor | 5 (2%) | 8.4 × 10−03 |
| PF00069 | Protein kinase domain | 11 (5%) | 9.9 × 10−02 |
| PF02984 | Cyclin, C-terminal domain | 3 (1%) | 3.8 × 10−01 |
| PF00219 | Insulin-like growth factor-binding protein | 3 (1%) | 3.8 × 10−01 |
| PF08311 | Mad3/BUB1 homology region 1 | 2 (1%) | 4.0 × 10−01 |
| PF00307 | Calponin homology (CH) domain | 4 (2%) | 4.6 × 10−01 |
| PF00093 | von Willebrand factor type C domain | 3 (1%) | 4.6 × 10−01 |
| PF00134 | Cyclin, N-terminal domain | 3 (1%) | 5.5 × 10−01 |
| SET-2 | set size 41 | ||
| PF00170 | bZIP transcription factor | 4 (10%) | 8.1 × 10−04 |
| PF00125 | Core histone H2A/H2B/H3/H4 | 4 (10%) | 5.9 × 10−03 |
| PF00219 | Insulin-like growth factor binding protein | 3 (7%) | 9.2 × 10−03 |
| PF00093 | von Willebrand factor type C domain | 3 (7%) | 1.4 × 10−02 |
| PF00007 | Cystine-knot domain | 2 (5%) | 2.4 × 10−01 |
| Name of Protein Complex | Set Size | Candidates Contained | p Value | q Value | Source |
|---|---|---|---|---|---|
| SET-1 | |||||
| AP-1 | 5 | 4 (80%) | 1.4 × 10−07 | 1.5 × 10−05 | INOH |
| CycB-Cdc2.complex | 3 | 3 (100%) | 2.2 × 10−06 | 1.2 × 10−04 | Spike |
| Centrosome:AURKA:TPX2:HMMR | 75 | 8 (11%) | 4.8 × 10−06 | 1.3 × 10−04 | Reactome |
| MASH1 promoter-coactivator complex | 37 | 6 (16%) | 7.2 × 10−06 | 1.3 × 10−04 | CORUM |
| Nek2A:MCC:APC/C complex | 22 | 5 (23%) | 7.6 × 10−06 | 1.3 × 10−04 | Reactome |
| H3.1 com | 38 | 6 (16%) | 8.5 × 10−06 | 1.3 × 10−04 | PINdb |
| hBUBR1:hBUB3:MAD2*:CDC20 complex | 4 | 3 (75%) | 8.6 × 10−06 | 1.3 × 10−04 | Reactome |
| Cell cycle kinase complex CDC2 | 6 | 3 (50%) | 4.2 × 10−05 | 5.7 × 10−04 | CORUM |
| Histone H3.1 complex | 32 | 5 (16%) | 5.3 × 10−05 | 6.4 × 10−04 | CORUM |
| Emerin regulatory complex | 18 | 4 (22%) | 7.3 × 10−05 | 7.3 × 10−04 | CORUM |
| SET-2 | |||||
| AP-1 | 5 | 3 (60%) | 1.4 × 10−07 | 2.7 × 10−06 | INOH |
| p-2S-cJUN:p-2S,2T-cFOS | 2 | 2 (100%) | 6.0 × 10−06 | 2.4 × 10−05 | Reactome |
| Fra2/JUN | 2 | 2 (100%) | 6.0 × 10−06 | 2.4 × 10−05 | PID |
| c-FOS/c-JUN/CREB/CREB | 3 | 2 (67%) | 1.8 × 10−05 | 2.4 × 10−05 | BioCarta |
| ERG-JUN-FOS DNA-protein complex | 3 | 2 (67%) | 1.8 × 10−05 | 2.4 × 10−05 | CORUM |
| JUN/FOS/ER alpha | 3 | 2 (67%) | 1.8 × 10−05 | 2.4 × 10−05 | PID |
| ETS2-FOS-JUN complex | 3 | 2 (67%) | 1.8 × 10−05 | 2.4 × 10−05 | CORUM |
| JUN/FOS/GATA2 | 3 | 2 (67%) | 1.8 × 10−05 | 2.4 × 10−05 | PID |
| cortisol/GR alpha (monomer)/JUN/FOS | 3 | 2 (67%) | 1.8 × 10−05 | 2.4 × 10−05 | PID |
| p-2S-JUN:p-2S,2T-FOS:IGFBP7 Gene | 3 | 2 (67%) | 1.8 × 10−05 | 2.4 × 10−05 | Reactom |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hotfilder, M.; Mallela, N.; Seggewiß, J.; Dirksen, U.; Korsching, E. Defining a Characteristic Gene Expression Set Responsible for Cancer Stem Cell-Like Features in a Sub-Population of Ewing Sarcoma Cells CADO-ES1. Int. J. Mol. Sci. 2018, 19, 3908. https://doi.org/10.3390/ijms19123908
Hotfilder M, Mallela N, Seggewiß J, Dirksen U, Korsching E. Defining a Characteristic Gene Expression Set Responsible for Cancer Stem Cell-Like Features in a Sub-Population of Ewing Sarcoma Cells CADO-ES1. International Journal of Molecular Sciences. 2018; 19(12):3908. https://doi.org/10.3390/ijms19123908
Chicago/Turabian StyleHotfilder, Marc, Nikhil Mallela, Jochen Seggewiß, Uta Dirksen, and Eberhard Korsching. 2018. "Defining a Characteristic Gene Expression Set Responsible for Cancer Stem Cell-Like Features in a Sub-Population of Ewing Sarcoma Cells CADO-ES1" International Journal of Molecular Sciences 19, no. 12: 3908. https://doi.org/10.3390/ijms19123908