Advancement of NF-κB Signaling Pathway: A Novel Target in Pancreatic Cancer



1
Department of Basic Sciences, Kentucky College of Osteopathic Medicine, University of Pikeville, Pikeville, KY 41501, USA
2
Department of Physiology, The Johns Hopkins University, School of Medicine, Baltimore, MD 21205, USA
3
Department of Biomedical Sciences, Cooper Medical School of Rowan University, Camden, NJ 08103, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(12), 3890; https://doi.org/10.3390/ijms19123890
Submission received: 25 October 2018
/
Revised: 21 November 2018
/
Accepted: 29 November 2018
/
Published: 5 December 2018
(This article belongs to the Special Issue NF-κB and Cancer)
{kind=link}
{kind=link}
Abstract
:Pancreatic ductal adenocarcinoma (PDAC) is one of the deadliest cancers and is the third highest among cancer related deaths. Despite modest success with therapy such as gemcitabine, pancreatic cancer incidence remains virtually unchanged in the past 25 years. Among the several driver mutations for PDAC, Kras mutation contributes a central role for its development, progression and therapeutic resistance. In addition, inflammation is implicated in the development of most human cancer, including pancreatic cancer. Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) is recognized as a key mediator of inflammation and has been frequently observed to be upregulated in PDAC. Several lines of evidence suggest that NF-κB pathways play a crucial role in PDAC development, progression and resistance. In this review, we focused on emphasizing the recent advancements in the involvement of NF-κB in PADC’s progression and resistance. We also highlighted the interaction of NF-κB with other signaling pathways. Lastly, we also aim to discuss how NF-κB could be an excellent target for PDAC prevention or therapy. This review could provide insight into the development of novel therapeutic strategies by considering NF-κB as a target to prevent or treat PDAC.
1. Introduction
The pancreas is a vital endocrine and exocrine organ that lies in the upper abdomen behind the stomach. The exocrine pancreas, which comprises more than 95% of the pancreatic mass, secretes digestive enzymes into the duodenum. It includes acinar and ductal cells associated with connective tissue, vessels, and nerves. The pancreatic juice produced by the exocrine pancreas flows into the common bile duct via the pancreatic duct, and empties into the duodenum to aid in digestion. The endocrine pancreas primarily consists of α and β cells which secrete glucagon, insulin, somatostatin and pancreatic polypeptide into the blood [1]. Considering the location and function of the pancreas, pancreatic cancer can have devastating effects. For example, pancreatic ductal adenocarcinoma (PDAC) arises exocrine cells. Almost 85% cases of pancreatic cancers are PDAC. The other forms of pancreatic cancers have been reported, nonetheless these are either less common or rare, such as neuroendocrine tumors, acinar carcinomas, pancreatoblastomas, colloid carcinomas, and solid-pseudopapillary neoplasms [1].
PDAC accounts for approximately 7.3% of all cancer deaths. Sadly, despite a steady increase in medium survival times for most cancers over the past few decades, survival rates of pancreatic cancer remain a formidable challenge, with the five-year survival of patients remaining as low as 8.5% in the USA [2]. According to recent cancer statistics, 55,440 new cases of pancreatic cancers will be detected and 44,330 people will die in 2018 in the USA [2]. Alarmingly, new cases of pancreatic cancer have been rising on an average of 0.5% annually over the past ten years, and it is expected to become the second leading cause of cancer-related death in the US by 2030 [3,4]. Unfortunately, the increase in mortality is not just restricted to USA; it is increasing worldwide [5].
Various factors contribute to low survival rates of PDAC. Amongst these are late stage diagnosis and an increasing prevalence in the elderly population. Almost 75% of the patients diagnosed are between 55–84 years of age and the median onset age is 71 years [6]. Additionally, metastasis also contributes in poor survival; almost 90% of patients are diagnosed with metastasis [7]. There is a genetic component and strong family history, with 10% to 15% of the PDAC cases exhibiting mutations in BRCA2, BRCA1, CDKN2A, ATM, STK11, PRSS1, MLH1 and PALB2 [7]. Furthermore, cigarette smoking is one of the major risk factors in the development of PDAC [8]. Additionally, diabetes, chronic pancreatitis, and obesity confer increased risk for PDAC [7,9,10].
Large-scale cancer genomic studies have shown a heterogeneous mutational profile in PDAC [11]. Almost 63 genetic alternations have been identified, with the most common Ras mutation. Almost 90% of PDAC are associated with Ras mutation. Inactivating mutations of TP53, CDKN2A and SMAD4 showed up in 50–80% of pancreatic cancer cases. Other genes, including ARID1A, MLL3 and TGFBR2, are mutated in ~10% of tumors [1,12].
There is a dearth of effective systemic therapies that also contributes to PDAC cancer mortality [7]. Surgery remains the only chance for cure of PDAC; however, most patients relapse and die. Also, at early diagnosis, few patients are found eligible for resection [13].
In 1997, gemcitabine (a nucleoside analogue), was approved to treat pancreatic cancer by the US FDA. Later in 2005, FDA approved a tyrosine kinase inhibitor of EGFR, erlotinib along with gemcitabine for pancreatic cancer treatment. Recently, phase 3 ACCORD-11 trial, using FOLFIRINOX regimen (oxaliplatin 85 mg/m², folinic acid [leucovorin] 400 mg/m², irinotecan 180 mg/m², bolus fluorouracil 400 mg/m², infusional fluorouracil 2400 mg/m² over 46 h, every 14 days) showed a better response, progression-free survival, and overall survival compared to gemcitabine monotherapy in patients with metastatic PDAC. Either FOLFIRINOX or the combination of combination of gemcitabine along with nanoparticle albumin–bound paclitaxel (nab-paclitaxel) is considered standard treatment for patients with PDAC. However, due to the side effects of this cocktail of chemotherapeutic drugs, only 30–40% of PDAC patients can be treated with the combination. Nonetheless, the side effect of chemotherapy remains the main obstacle in achieving quality of life of the patients [1,7,13]. As a result, there is an urgent need to extend our understanding about PDAC, in terms of pathways regulating its oncogenic role, and to develop novel therapeutic strategies in order to improve long term survival of PDAC patients.
2. NF-κB Signaling Pathways
The transcription factor, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) was first discovered in 1986 as a regulator of B-cell specific transcription factor. Since its discovery, it has been found to be involved in key processes and regulates the expression of number of genes involved in immune system, cell survival, stress responses, embryogenesis, differentiation, proliferation and cell death [14]. Because of its critical role in a variety of biological events, the dysregulation of NF-κB pathways can lead to various ailments including cancer, autoimmune diseases, neurodegenerative diseases, cardiovascular disease, and diabetes [14,15].
The NF-κB family consists of five master transcription factors, RelA/p65, RelB, c-Rel, and precursor proteins p50/p105 (NF-κB1) and p100/p52 (NF-κB2). The functional form NF-κB1 (p50) and NF-κB2 (p52) are produced by proteasome mediated processing of precursor proteins p105, and p100 respectively [16,17]. These transcription factors form various dimeric complexes and bind to κB sites, which induce or repress transcription. Out of various dimers, the well-studied dimer is p50/65. The activation of NF-κB is mediated by two pathways: classical/canonical, and non-classical/non-canonical. Any external stimulus including oxidative stress, viruses, bacteria, growth factors, lead to the activation of classical pathways where NF-κB is rapid and transiently activated. The classical pathways mainly involve p50–p65 dimers and play critical role in the regulation of gene products involved in angiogenesis, survival, metastasis, and cell proliferation. The first step in the classical activation of signaling is the activation of TGFβ activated kinase 1 (TAK1, or called as MAP3K7), followed by activation of trimeric IκB kinase (IKK) complex, which is made up of catalytic (IKKα and IKKβ) and regulatory (IKKγ, also called as NEMO) subunits. The IKK complex then phosphorylates inhibitory protein IκBα. The phosphorylation of IκBα leads to its ubiquitination and proteasomal degradation. The degradation of IκBα releases the dimer p50/65 in cytoplasm followed by nuclear translocation. Translocated complex binds with the specific sequence of DNA and regulates the gene expression [18,19]. Studies based on genetic mouse models suggest that IKKγ and IKKβ are critical in phosphorylation-dependent IκBα degradation, whereas IKKα plays a supporting role [20].
The alternative/non-canonical pathway depends on de novo synthesis of NF-κB-inducing kinase (NIK, also known as MAP3K14), which is activated by a sub group of cytokines such as lymphotoxin (LT), receptor activator of NF-κB ligand (RANKL/TNFSF11), CD40 ligand (CD40L), and B cell activating factor of the TNF family (BAFF/TNFSF13B). Based on genetic studies it has been established that NIK is required for non- canonical NF-κB activation [21]. NIK activates IKKα, which, mediates the processing of p100 [22]. The alternative pathway results in the activation of p52–RELB dimers and regulates the expression of genes associated with the maintenance of secondary lymphoid organs and tumor microenvironment [15,23,24].
3. Role of NF-κB Pathways in Cancer Development and Progression
Several agents including oxidative stress, pro-inflammatory cytokines, growth factors, viral and bacterial agents activate NF-κB (Figure 1). Once activated, NF-κB regulates key gene products playing critical roles in tumorigenesis. Aberrant NF-κB signaling has shown to be associated with a variety of tumors, including breast, colon, leukemia, lymphoma, lung, prostate, pancreatic, thyroid, and ovarian carcinoma [15,25].
3.1. Inflammation and Immune Modulation
The activation of canonical NF-κB signaling has been associated in inflammatory processes [26] (Figure 1). Tumor necrosis factor alpha (TNF-α) and IL-6 are two of the best-studied pro-inflammatory and pro-tumorigenic cytokines, the expression of which are elevated in many different cancers. Activated macrophages and neutrophils mainly produce TNF-α, IL-1 and IL-6 which may accelerate tumorigenesis. TNF signaling entails activation of NF-κB and mitogen-activated protein kinases (MAPKs), by activating transcription factor AP1, and promote cell survival of malignant cells. Additionally, IL-6 links NF-κB to STAT3, where this axis plays a prominent role in cancer-related inflammation and other pathologies [15,27].
Another key modulator of inflammation is reactive oxygen species (ROS), which are produced by mitochondrial and other cellular byproducts. The role of ROS has been reported in inflammatory disorders and in various stages of cancer development. Importantly, ROS can activate NF-κB by various mechanisms and paradoxically, NF-κB also regulates ROS. It has been demonstrated that NF-κB regulated genes play an important role in the detoxification of ROS. However, some gene products such as nitric oxide synthases (NOS) do perform a pro-oxidant function. ROS and reactive nitrogen species can also induce DNA damage and oncogenic mutations leading to cancer [25,28].
NF-κB signaling pathways is also critical in T and B cell development and proliferation. Thus, constitutive NF-κB activation leads to the continuous proliferation of lymphocytes which is the main culprit in leukemia and lymphoma [29]. NK cells exert anti-tumorigenic activity in leukemia and lymphomas by killing the malignant cells. NF-κB modulates the anti-tumorigenic potential of NK cells, by upregulating the expression of perforins and granzyme B, which inhibit lytic potentials of NK cells [15].
3.2. Proliferation of Cancer
NF-κB plays a major role in tumor initiation, tumor promotion and tumor progression (Figure 1). NF-κB activation promotes mutagenesis, at various stages such as in DNA strands, daughter cells, and mismatch repair genes, which eventually leads to genomic instabilities. Chronic inflammation and NF-κB can also induce chromosomal instability and aneuploidy leading to tumor initiation. NF-κB promotes cancer cell proliferation and survival, by enhancing the expression of various cyclins including cyclin D1 and cyclin D2 [15,24]. Kras and p53 mutations have been found in 20–25%, and in ~50% of all cancers, respectively. Oncogenic mutations in EGFR and PI3K also contribute to NF-κB activation and tumor proliferation [25].
3.3. Cell Death
One of the “hallmarks of cancer” is avoidance of cell death. Constitutive activation of NF-κB has been reported in variety of cancer types including pancreatic cancers, which is associated with the drug resistance [30]. NF-κB was shown to regulate pro-and anti-apoptotic genes cIAPs; TRAIL, caspase-8 and c-FLIP, BCL-2 and BCL-XL [25]. Tumor suppressor p53 activates pro-apoptotic proteins Puma and NOXA, leading to apoptosis. NF-κB and p53 antagonistically regulate each other’s activity, while IKKβ was shown to directly phosphorylate p53 and promote its polyubiquitylation and degradation [15]. In addition to p53 mediated apoptosis, studies have demonstrated that GTPase HRAS159 and c-MYC induced NF-κB activation inhibits cell death [31,32]. NF-κB and autophagy intricately regulate each other’s activity. For example, NF-κB family member p65/RelA was shown to upregulate protein expressions which are associated in autophagy such as beclin 1 and SQSTM1, whereas autophagy was shown to degrade IKK subunits and inhibit NF-κB signaling [33,34,35]. NF-κB induced chemo-resistance will be discussed in a later part of the review in the context of pancreatic cancer.
3.4. Tumor Microenvironment
Evolution of the tumor microenvironment occurs in three steps: niche construction, expansion, and maturation. It has been demonstrated that NF-κB is critical in all steps (Figure 1) [15]. Moreover, studies have further emphasized that activation non-canonical NF-κB signaling is crucial in tumor microenvironment [36]. Additionally, it has been demonstrated that cross interaction of STAT3 and NF-κB signaling pathways regulates the production of cytokines, growth factors which play an important role in the constitution of tumor microenvironment [27]. Furthermore, inhibition of NF-κB signaling in cancer associated fibroblasts (CAF’s) abolished its tumor-promoting effects, suggesting that NF-κB is critical in CAFs mediated tumor enhancing effects [15]. Additionally, NF-κB plays a key role in the constitution of tumor microenvironment in multiple myeloma. It is activated by diverse bone marrow-derived cytokines and growth factors, and also by direct physical contact between multiple myeloma cells and stromal cells [37].
3.5. Angiogenesis
The formation of new blood vessels from existing endothelial lined vessels, defined as angiogenesis, is crucial for tumor growth. The growth factors such as vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), and platelet-derived growth factor (PDGF) regulates angiogenesis. It has been demonstrated that NF-κB regulates angiogenesis. For example, inhibition of NF-κB activity inhibits the tumor growth and angiogenesis, in different cancer models, including pancreatic cancer [38,39,40,41,42]. Along these lines, κB sites have been in identified in promoter region of matrix metalloproteinases (MMPs) genes, which are involved in angiogenesis [24,43].
3.6. Metastasis
Approximately 90% of cancer deaths happen because of metastasis, which is the mobilization of cancer cells from primary tumor sites to either surrounding sites or distant organs. Epithelial–mesenchymal transition (EMT) is considered to be a hallmark of metastasis [44]. Blocking NF-κB inhibited the transcription of the EMT genes CDH2, SLUG, TWIST1 and SNAIL in different cancer cell lines (Figure 1) [19,45,46,47,48]. Furthermore, it has been demonstrated that TNF-α induced NF-κB regulates expression of TWIST1 in both normal breast epithelial and breast cancer cells leading metastatic phenotype [49]. In addition to regulating EMT genes, NF-κB can promote cell migration and invasion through other mechanisms. For example, NF-κB activation can stimulate the expression of hypoxia-inducible factor 1α (HIF1α), thereby may enhance hypoxic conditions and survival of metastasis-initiating cells [50].
3.7. Cancer Stem Cells
Cancer stem cells (CSCs) are immortal cells within a tumor that can self-renew and give rise to many cell types that constitute the tumor. CSCs contribute to metastasis and drug resistance in tumor cells. NF-κB signaling is an important pathway that regulates survival, activation and differentiation of CSCs (Figure 1). In fact, both classical and alternative activation of NF-κB may promote proliferation and survival of stem cells in various cancers [51]. In pancreatic CSCs, the classical NF-κB pathway appears to increase the expression of a transcription factor SOX9, which is critical in invasion [52].
4. NF-κB and Pancreatic Cancer
Almost 90% of pancreatic cancers are driven by oncogenic mutations of Kras. Aberrant Kras signaling activates inflammatory signaling pathways that play critical roles in regulating the initiation of pancreatic intraepithelial neoplasia (PanIN) and the progression of PDAC [53]. The fact that Kras is involved in the pancreatic cancer development is established by studies of several genetic mouse models. Along these lines, several mouse models have been developed showing the direct link of IKK and Kras [54]. KRASG12D induces IL-1α expression via AP-1 activation, leading to NF-κB activation in tumor cells [54]. Activation of NF-κB drives several signaling molecules, cooperates with other signaling pathways including Notch signaling and stimulates PDAC development [55,56]. Activated Kras also regulates microRNA’s via NF-κB signaling. Many other oncogenic mutations, such as, EGFR, PI3K and p53, also contribute to NF-κB activation in PDAC [25].
Chronic inflammation can induce NF-κB activation. It has been demonstrated that during chronic pancreatitis, inflammatory cells, and macrophages enhance stroma formation, which increases the risk for PDAC development [40]. Additionally, elevated levels of cytokines and chemokines are observed in PDAC cells, which is correlated with the enhanced NF-κB signaling. For example, elevated levels of chemokine CXCL14, which promotes angiogenesis and tumor growth were found in PDAC [40].
The central region of pancreatic tumors usually contains of a hypoxic microenvironment, which increases the likelihood of angiogenesis, metastasis and drug resistance [57,58]. NF-κB regulates HIF1-α, EMT [59] and angiogenesis factors like VEGF [60] in pancreatic cancer. Fujioka S. et al. demonstrated that inhibiting constitutive NF-κB activity by expressing IκBαM (inhibitor of NF-κB phosphorylation mutant) suppressed liver metastasis in metastatic human PDAC orthotropic nude mouse model. Further, decreases in NF-κB activation reduced expression of a major proangiogenic molecule, vascular endothelial growth factor (VEGF) which shows that inhibition of NF-κB signaling can suppress the angiogenesis and metastasis of pancreatic cancer [60].
5. NF-κB in Drug Resistance
The biggest problem in the treatment of advanced pancreatic cancer is the acquired resistance against Gemcitabine [61] and NF-κB was shown to induce chemoresistance to gemcitabine in multiple ways. Studies have demonstrated that human equilibrative nucleoside transporter 1 (hENT1) and human concentrative nucleoside transporter 1 and 3 (hCNT1 and hCNT3) metabolize Gemcitabine. Furthermore, the expression of hENT1 and hCNT3 is associated with chemo sensitivity and overall worse survival in pancreatic tumors [62]. Interestingly, MUCIN 4 negatively regulates expression of hCNT1 transporter through NF-κB signaling pathways, which leads to gemcitabine resistance [63]. Also, cross talk of NF-κB with other pathways leads to gemcitabine resistance. Recently, the role of miR-1266 is explored in the chemo resistance of gemcitabine. How miR-1266 regulates chemoreistance is not clear, however it has been reported that miR-1266 negatively regulates the inhibitors of STAT3 and NF-κB pathways such as SOCS3, PTPN11, ITCH, and TNIP1, which results in constitutive activation of NF-κB and STAT3. Additionally, overexpression of miR-1255 has been linked with chemo resistance of gemcitabine in pancreatic cancer cells [64]. Downregulation of NF-κB/STAT3 signaling axis by Nexrutine1 (reduces transcriptional activity of COX-2) increased gemcitabine chemosensitivity in PDAC cells. In addition to crosstalk between pathways, NF-κB’s role in regulation of stemness also contributes to gemcitabine resistance [65]. Zhang Z et al. reported that pancreatic cancer stem cells develop chemoresistance against gemcitabine by activating NF-κB/STAT3/Nox/ROS signaling pathway [61]. These studies clearly suggest that the blocking of these signaling pathways along with gemcitabine may be a better treatment regimen [61]. Additionally, inhibition of p65, reduces NF-κB activity and consequently inhibits Bcl-2, cyclin D1 and VEGF, and activation of caspase-3. Gemcitabine works synergistically with p65 siRNA and inhibits the proliferation of pancreatic cancer cells. It also suppresses the growth and angiogenesis of pancreatic tumors in animal models [66]. Furthermore, NF-κB induces gemcitabine resistance by interacting with novel regulators, Tripartite motif containing 31 (TRIM31) which is a newly identified E3 ubiquitin-protein ligase, markedly upregulated in pancreatic cancer cell lines and tissue, that correlates to aggressive behavior and poor prognosis in pancreatic cancer patients. Interestingly, the overexpression of TRIM31 mediates gemcitabine resistance via NF-κB activation, and similarly the inhibition of TRIM31 potentiates the cytotoxic potential gemcitabine both in vitro and in vivo model of pancreatic cancer cells in NF-κB dependent manner [67]. Additionally, combined activation of NF-κB and HIF1-α, were shown to induce hypoxia-induced EMT and resistance to gemcitabine [59].
Aberrant NF-κB signaling is also involved in chemoresistance to platinum agents and topoisomerase inhibitors, part of FOLFIRINOX regime, which are standard of care for PDAC. AsPC1-R and BxPC3-R, resistant cell lines against cisplatin, were developed from the PDAC parental cell lines AsPC114 and BxPC3. Pathway Enrichment Analysis (PEA) for genes upregulated in AsPC1-R cells showed dysregulation of NF-κB signal transduction pathway [68]. Furthermore, TGF-β-activated kinase-1 (TAK1) which activates NF-κB and AP-1, has been linked to chemoresistance, because the knockdown of TAK1 in PDAC cells significantly lowered NF-κB activity. Moreover, the addition of TAK1 inhibitor LYTAK1 potentiates the cytotoxic response of oxaliplatin, SN-38, and gemcitabine in PDAC cells [69].
Taken together, the main culprit in chemo resistance is the constitutive activation of NF-κB. Thus, simultaneous targeting of NF-κB signaling pathways along with the chemotherapeutic agents may be a better option in treatment of PDAC.
6. Cross Talk of NF-κB with Other Signaling in Pancreatic Cancer
The samples of most pancreatic cancer patients (67–70%) show constitutive activation of NF-κB [40,70]. It is also reported that NF-κB signaling pathways interact with several other signaling pathways (Figure 2). For example, it has been shown that KrasG12D mutation, which is seen in almost all patients, is the main driver of constitutive NF-κB expression. How Kras mutation drives the constitutive expression of NF-κB has been explored, and it has been shown that cytokine interleukin-1α (IL-1α), mediates this process by activating transcription factor AP-1 [71]. Furthermore, recent studies suggest that cross talk of Notch and NF-κB signaling regulates inflammatory processes in transformed cells [56].
NF-κB and Notch signaling pathways are activated in many cancers, including pancreatic cancer [72,73,74,75]. Activation of Notch signaling leads to proteolytic cleavage of Notch and the release of Notch intracellular domain (NICD), which can translocate and interacts to Rbp-j a DNA binding protein. The nuclear interaction of NICD to Rbp-j regulates gene expression of transcriptional repressors Hes and Hey [76], which are upregulated in PanINs and PDAC. Furthermore, Hes1 regulates expression of nuclear receptor PPARγ. Studies of Maniati et al., suggested that in pre-malignant pancreatic epithelial cells PPARγ is suppressed by the complex of Tnf-α/Hes1. Thus, coordinated activities of NF-κB, Notch and PPARγ regulates key events which play critical roles in the etiology of pancreatic cancers. Along these lines, it has been demonstrated that y-secretase inhibitor which is inhibitor of NF-κB and Notch pathways, suppressed inflammatory genes expression in transformed cells [56]. These findings clearly demonstrate that cross talk of NF-κB, Kras, and Notch signaling pathways governs the chronic inflammatory pathway and eventually promote tumorigenesis [40].
The activation of transforming growth factor- β (TGF-β) exhibits its growth inhibitory effects in normal epithelial cells, including pancreatic cells, yet it exhibits its growth promoting effect in many neoplastic transformed cells, including pancreatic cancer cells. Importantly, it has been established that TGF-β activates NF-κB and plays a key role in the development of PDAC. For example, NF-κB activated by TGF-β suppresses expression of PTEN in pancreatic cancer cells [72]. Suppression of PTEN by TGF-β could enhance pancreatic cancer molality and facilitate the metastasis. Therefore, activation of NF-κB mediate both TGF-β induced cell motility and suppression of PTEN expression. In conclusion, blocking NF-κB by small molecules would elevate the PTEN expression levels and reduce the pancreatic cancer burden.
Varieties of anti-inflammatory and anti-carcinogenic phytochemicals activate Nrf-2 by inducing oxidative stress and lead to the inhibition of NF-κB signaling [73,75]. Curcumin, a PDAC chemosensitizer, regulates NF-κB and Nrf-2. Recent evidences suggested that epigallocatechine-3-gallate exhibits its anti-inflammatory activity through NrF-2 mediated NF-κB inhibition [71]. Further, several Nrf-2 activators include Phenethyl isocyanate (PEITC), SFN and curcumin attenuate liposaccharide induced NF-κB activation [74]. Furthermore, PEITC, and SFN treatment inhibit the phosphorylation of IKK/IκB, nuclear translocation of p65 NF-κB, and inhibit NF-κB signaling. On the other hand, it was reported that NF-κB directly repress the Nrf-2 signaling at the transcriptional level, suggesting the potential cross talk between Nrf-2 and NF-κB [71]. However, it is also unclear whether the failure of clinical trials in the PDACs is because of the Nrf-2 activation by many of these molecules. Therefore, it would be interesting to further evaluate whether using an Nrf-2 inhibitor can improve the outcome in NF-κB inhibiting strategy.
7. NF-κB, An Excellent Target for Pancreatic Cancer Prevention and Therapy
A mammoth amount of information suggests that both classical and non-classical NF-κB signaling plays an important role in pancreatic cancer [40]. Studies have demonstrated that the constitutive activation of NF-κB leads to the promotion of cell proliferation, angiogenesis, invasion and metastasis [41,60,76]. Thus, targeting of NF-κB is important in order to prevent pancreatic cancer [40]. Along these lines, several attempts have been made to inhibit NF-κB. Below are the main agents that have been used to inhibit pancreatic cancer.
7.1. Cyclooxygenase Inhibitors
A variety of COX inhibitors have been used to suppress pancreatic cancer growth [77]. One of the popular COX inhibitors, aspirin, blocks adenosine triphosphate binding to IKKβ, thus, NF-κB activation [78]. Celecoxib also has some effect on NF-κB, however it has a differential response on pancreatic cancer cells. For example, when celecoxib was used in combination with gemcitabine it reduced the activation of NF-κB in BxPC-3 cells but had no effect on Paca-2 cells [79]. Another COX inhibitor, sulindac, inhibited NF-κB and pancreatic cell growth when used in combination with parthenolide [80]. Sclabas et al. demonstrated that aspirin could be used as a chemo-preventive agent [81]. These studies suggest that NF-κB is a potential target for cancer prevention and COX inhibitors could be utilized as chemo-preventive agent.
7.2. Curcumin
Curcumin is a natural yellow compound found in turmeric. Almost 4000 articles have been published on Curcumin, and some of the studies have named this molecule as a “magic bullet” [82,83,84]. Curcumin exerts its anti-cancer activities through a variety of mechanisms including inhibition of NF-κB [85,86]. Several studies have demonstrated that Curcumin shows its anti-cancer and anti-inflammatory activities by inhibiting both IKK and NF-κB activity in a variety of cancer cells, including pancreatic cancer [74,87,88]. A significant amount of studies has demonstrated that Curcumin inhibits NF-κB and promotes apoptosis in a variety of pancreatic cancer cells and orthotropic model [74,88]. Recently, a phase 2 clinical trial was completed at MD Anderson Cancer Center, and the study concluded that oral curcumin is well tolerated and exerts biological activities in pancreatic cancer patients [89].
7.3. Other Agents
Several other NF-κB inhibitors have shown great potential to inhibit pancreatic cancer but have limited studies. Some of these agents are protein kinase C inhibitors, parthenolide, polyphenols, protein tyrosine kinase inhibitor, GSK-3 kinase inhibitor, proteasome inhibitors, and IκBα mutants [77]. As drug discovery and cancer research progresses, we anticipate the discovery of more exciting inhibitors of NF-κB in order to combat pancreatic cancer.
8. Conclusions
Several lines of evidence suggested that both canonical and non-canonical NF-κB activation is responsible for PDAC progression and metastasis. Further, the cross-talk of NF-κB with other signaling pathways indicates that it is the cellular wiring which is critical in development of PDAC. Targeting of this signaling pathway is important in order to prevent PDAC. As described above, there are a variety of NF-κB inhibitors which show great promise and there are many more to be discovered. However, despite the effectiveness of these molecules in laboratory studies, only few molecules are in clinical trials, thus more studies on patient samples are needed. In addition, several studies have demonstrated that combination therapy could be beneficial in order to treat pancreatic cancer or prevent the chemotherapy resistance to pancreatic cancer.
Overall, the mammoth studies of NF-κB and its relationship with pancreatic cancer have laid a solid foundation for drug development. It is an opportune time for the development of novel agents that target these signaling pathways. Several agents have been discovered already and many more are in the pipeline. However, extensive studies on animal models followed by clinical trials are needed to fully understand the mechanism of these drugs.
Author Contributions
K.C.P. conceived, designed, wrote, analyzed data & revised the manuscript. M.K.P. designed, wrote & revised the manuscript. M.R.M. wrote the part of manuscript. K.B. has also wrote the part of manuscript.
Funding
This work was supported by University of Pikeville, Kentucky College of Osteopathic Medicine (KYCOM)’s start-up fund.
Acknowledgments
We would like to thank Daniel Atchley and Steven R. Harris for the editing of this review.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| PDAC | Pancreatic ductal adenocarcinoma |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NEMO | Regulatory subunit IKKγ |
| EMT | Epithelial-to-mesenchymal transition |
| TNF-α | Tumor necrosis factor alpha |
| CAF | cancer associated fibroblasts |
| EGFR | Epidermal growth factor receptor |
| BRCA | Breast cancer |
| CDKN2A | cyclin-dependent kinase Inhibitor 2A |
| ATM | Ataxia-Telangiectasia mutated |
| STK | Serine/threonine kinase |
| PRSS | serine protease |
| MLH | MutL homolog |
| PALB2 | Partner and localizer of BRCA2 |
| TP53 | Tumor protein p53 |
| ARID1A | AT-rich interactive domain-containing protein 1A |
| MLL | mixed-lineage leukemia protein |
| TGFBR | TGF-β receptor type |
| Rel | Reticuloendotheliosis |
| IKK | Inhibitor of κB kinase |
| MAPKK | Mitogen-activated protein kinase kinase kinase |
| RANKL | Receptor activator of nuclear factor kappa-Β ligand |
| BAFF | B-cell activating factor |
| IL | interleukin |
| STAT | Signal transducer and activator of transcription |
| PI3K | phosphoinositide 3-kinase |
| NK | Natural killer |
| TRAIL | TNF-related apoptosis-inducing ligand |
| c-FLIP | Cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein |
| BCL | B-cell lymphoma |
| BCL-XL | B-cell lymphoma-extra large |
| Puma | p53 upregulated modulator of apoptosis |
| CXCL | chemokine (C-X-C motif) ligand |
| SOCS | Suppressor of cytokine signaling |
| PTPN | Tyrosine-protein phosphatase non-receptor type |
| ADAM | Disintegrin and metalloproteinase domain-containing protein |
| MMP | Matrix Metallopeptidase |
| SFN | Sulforaphane |
| SMAD | Single mothers against decapentaplegic |
| US FDA | United State food and drug administration |
| ACCORD | Action to Control Cardiovascular Risk in Diabetes |
| FOLFIRINOX | fluorouracil [5-FU], leucovorin, irinotecan, oxaliplatin |
| TGF | Transforming growth factor |
| NIK | NF-kappa-B-inducing kinase |
| SQSMT1 | sequestosome-1 |
| SOX9 | Sry-related HMG box 9 |
| CDH2 | Cadherin-2 |
| SLUG | SNAI2 |
| TWIST | Twist-related protein |
| SNAIL-1 | Snail Family Transcriptional Repressor 1 |
| AP1 | Activator protein 1 |
| TNIP1 | TNFAIP3-interacting protein 1 |
| PPAR | peroxisome proliferator-activated receptors |
| PTEN | Phosphatase and tensin homolog |
| ROS | Reactive oxygen species |
| VEGF | Vascular endothelial growth factor |
| COX-2 | Cyclooxygenase |
| HIF1α | Hypoxia-inducible factor 1-alpha |
| GSK-3 | Glycogen synthase kinase 3 |
| Nrf-2 | Nuclear respiratory factor 2 |
References
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Saad, A.M.; Turk, T.; Al-Husseini, M.J.; Abdel-Rahman, O. Trends in pancreatic adenocarcinoma incidence and mortality in the united states in the last four decades; a seer-based study. BMC Cancer 2018, 18, 688. [Google Scholar] [CrossRef] [PubMed]
- Ilic, M.; Ilic, I. Epidemiology of pancreatic cancer. World J. Gastroenterol. 2016, 22, 9694–9705. [Google Scholar] [CrossRef] [PubMed]
- White, M.C.; Holman, D.M.; Boehm, J.E.; Peipins, L.A.; Grossman, M.; Henley, S.J. Age and cancer risk: A potentially modifiable relationship. Am. J. Prev. Med. 2014, 46, S7–S15. [Google Scholar] [CrossRef] [PubMed]
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85. [Google Scholar] [CrossRef]
- Iodice, S.; Gandini, S.; Maisonneuve, P.; Lowenfels, A.B. Tobacco and the risk of pancreatic cancer: A review and meta-analysis. Langenbecks Arch. Surg. 2008, 393, 535–545. [Google Scholar] [CrossRef]
- Arslan, A.A.; Helzlsouer, K.J.; Kooperberg, C.; Shu, X.O.; Steplowski, E.; Bueno-de-Mesquita, H.B.; Fuchs, C.S.; Gross, M.D.; Jacobs, E.J.; Lacroix, A.Z.; et al. Anthropometric measures, body mass index, and pancreatic cancer: A pooled analysis from the pancreatic cancer cohort consortium (panscan). Arch. Intern. Med. 2010, 170, 791–802. [Google Scholar] [CrossRef]
- Bosetti, C.; Rosato, V.; Li, D.; Silverman, D.; Petersen, G.M.; Bracci, P.M.; Neale, R.E.; Muscat, J.; Anderson, K.; Gallinger, S.; et al. Diabetes, antidiabetic medications, and pancreatic cancer risk: An analysis from the international pancreatic cancer case-control consortium. Ann. Oncol. 2014, 25, 2065–2072. [Google Scholar] [CrossRef]
- Biankin, A.V.; Maitra, A. Subtyping pancreatic cancer. Cancer Cell 2015, 28, 411–413. [Google Scholar] [CrossRef] [PubMed]
- Falasca, M.; Kim, M.; Casari, I. Pancreatic cancer: Current research and future directions. Biochim. Biophys. Acta 2016, 1865, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Neoptolemos, J.P.; Kleeff, J.; Michl, P.; Costello, E.; Greenhalf, W.; Palmer, D.H. Therapeutic developments in pancreatic cancer: Current and future perspectives. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 years of NF-κB: A blossoming of relevance to human pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; DeMartino, G.N.; Greene, W.C. Cotranslational biogenesis of NF-κB p50 by the 26s proteasome. Cell 1998, 92, 819–828. [Google Scholar] [CrossRef]
- Sun, S.C.; Ley, S.C. New insights into NF-κB regulation and function. Trends Immunol. 2008, 29, 469–478. [Google Scholar] [CrossRef] [Green Version]
- Ijichi, H. Genetically-engineered mouse models for pancreatic cancer: Advances and current limitations. World J. Clin. Oncol. 2011, 2, 195–202. [Google Scholar] [CrossRef]
- Qin, Y.; Zhao, D.; Zhou, H.G.; Wang, X.H.; Zhong, W.L.; Chen, S.; Gu, W.G.; Wang, W.; Zhang, C.H.; Liu, Y.R.; et al. Apigenin inhibits NF-κB and snail signaling, emt and metastasis in human hepatocellular carcinoma. Oncotarget 2016, 7, 41421–41431. [Google Scholar] [CrossRef]
- Israel, A. The ikk complex, a central regulator of NF-κB activation. Cold Spring Harb. Perspect. Biol. 2010, 2, a000158. [Google Scholar] [CrossRef]
- Xiao, G.; Harhaj, E.W.; Sun, S.C. NF-κB-inducing kinase regulates the processing of NF-κB2 p100. Mol. Cell 2001, 7, 401–409. [Google Scholar] [CrossRef]
- Senftleben, U.; Cao, Y.; Xiao, G.; Greten, F.R.; Krahn, G.; Bonizzi, G.; Chen, Y.; Hu, Y.; Fong, A.; Sun, S.C.; et al. Activation by ikkalpha of a second, evolutionary conserved, NF-κB signaling pathway. Science 2001, 293, 1495–1499. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, M.M.; Sung, B.; Yadav, V.R.; Kannappan, R.; Aggarwal, B.B. NF-κB addiction and its role in cancer: ‘One size does not fit all’. Oncogene 2011, 30, 1615–1630. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Hong, J.T. Roles of NF-κB in cancer and inflammatory diseases and their therapeutic approaches. Cells 2016, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Shen, S.; Verma, I.M. NF-κB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Karin, M. Dangerous liaisons: Stat3 and NF-κB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010, 21, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Jost, P.J.; Ruland, J. Aberrant NF-κB signaling in lymphoma: Mechanisms, consequences, and therapeutic implications. Blood 2007, 109, 2700–2707. [Google Scholar]
- Godwin, P.; Baird, A.M.; Heavey, S.; Barr, M.P.; O’Byrne, K.J.; Gately, K. Targeting nuclear factor-kappa B to overcome resistance to chemotherapy. Front. Oncol. 2013, 3, 120. [Google Scholar] [CrossRef] [Green Version]
- Joneson, T.; Bar-Sagi, D. Suppression of ras-induced apoptosis by the rac gtpase. Mol. Cell. Biol. 1999, 19, 5892–5901. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Madrid, L.V.; Saims, D.; Sedivy, J.; Wang, C.Y. C-myc sensitizes cells to tumor necrosis factor-mediated apoptosis by inhibiting nuclear factor κB transactivation. J. Biol. Chem. 2002, 277, 36671–36677. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.S. Regulation of cell death and autophagy by ikk and NF-κB: Critical mechanisms in immune function and cancer. Immunol. Rev. 2012, 246, 327–345. [Google Scholar] [CrossRef] [PubMed]
- Copetti, T.; Bertoli, C.; Dalla, E.; Demarchi, F.; Schneider, C. P65/rela modulates becn1 transcription and autophagy. Mol. Cell. Biol. 2009, 29, 2594–2608. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Hyttinen, J.M.; Kauppinen, A.; Kaarniranta, K. Context-dependent regulation of autophagy by ikk-NF-κB signaling: Impact on the aging process. Int. J. Cell Biol. 2012, 2012, 849541. [Google Scholar] [CrossRef]
- Wharry, C.E.; Haines, K.M.; Carroll, R.G.; May, M.J. Constitutive non-canonical nfkappab signaling in pancreatic cancer cells. Cancer Biol. Ther. 2009, 8, 1567–1576. [Google Scholar] [CrossRef]
- Matthews, G.M.; de Matos Simoes, R.; Dhimolea, E.; Sheffer, M.; Gandolfi, S.; Dashevsky, O.; Sorrell, J.D.; Mitsiades, C.S. NF-κB dysregulation in multiple myeloma. Semin. Cancer Biol. 2016, 39, 68–76. [Google Scholar] [CrossRef]
- Chen, W.; Li, Z.; Bai, L.; Lin, Y. NF-κB in lung cancer, a carcinogenesis mediator and a prevention and therapy target. Front. Biosci. (Landmark Ed.) 2011, 16, 1172–1185. [Google Scholar] [CrossRef]
- Meteoglu, I.; Erdogdu, I.H.; Meydan, N.; Erkus, M.; Barutca, S. NF-κB expression correlates with apoptosis and angiogenesis in clear cell renal cell carcinoma tissues. J. Exp. Clin. Cancer Res. 2008, 27, 53. [Google Scholar] [CrossRef]
- Prabhu, L.; Mundade, R.; Korc, M.; Loehrer, P.J.; Lu, T. Critical role of NF-κB in pancreatic cancer. Oncotarget 2014, 5, 10969–10975. [Google Scholar] [CrossRef] [Green Version]
- Shamoto, T.; Matsuo, Y.; Shibata, T.; Tsuboi, K.; Nagasaki, T.; Takahashi, H.; Funahashi, H.; Okada, Y.; Takeyama, H. Zerumbone inhibits angiogenesis by blocking NF-κB activity in pancreatic cancer. Pancreas 2014, 43, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.X.; Xia, Z.; Zhang, N.; Gong, W.; Huang, S. Constitutive NF-κB activity regulates the expression of vegf and il-8 and tumor angiogenesis of human glioblastoma. Oncol. Rep. 2010, 23, 725–732. [Google Scholar] [PubMed]
- Soubannier, V.; Stifani, S. NF-κB signalling in glioblastoma. Biomedicines 2017, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.N.; Mei, Y.; Zhang, J. Cancer metastasis: Issues and challenges. Chin. J. Cancer 2017, 36, 38. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Liu, J.; Zhang, L.; Xiao, X.; Li, W. Curcumin inhibits h2o2-induced invasion and migration of human pancreatic cancer via suppression of the erk/NF-κB pathway. Oncol. Rep. 2016, 36, 2245–2251. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.A.; Azoitei, N.; Baumann, B.; Grunert, S.; Sommer, A.; Pehamberger, H.; Kraut, N.; Beug, H.; Wirth, T. NF-κB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J. Clin. Investig. 2004, 114, 569–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, M.; Allison, D.F.; Baranova, N.N.; Wamsley, J.J.; Katz, A.J.; Bekiranov, S.; Jones, D.R.; Mayo, M.W. NF-κB regulates mesenchymal transition for the induction of non-small cell lung cancer initiating cells. PLoS ONE 2013, 8, e68597. [Google Scholar] [CrossRef] [PubMed]
- Pires, B.R.; Mencalha, A.L.; Ferreira, G.M.; de Souza, W.F.; Morgado-Diaz, J.A.; Maia, A.M.; Correa, S.; Abdelhay, E.S. NF-κB is involved in the regulation of emt genes in breast cancer cells. PLoS ONE 2017, 12, e0169622. [Google Scholar] [CrossRef] [PubMed]
- Li, C.W.; Xia, W.; Huo, L.; Lim, S.O.; Wu, Y.; Hsu, J.L.; Chao, C.H.; Yamaguchi, H.; Yang, N.K.; Ding, Q.; et al. Epithelial-mesenchymal transition induced by tnf-α requires NF-κB-mediated transcriptional upregulation of twist1. Cancer Res. 2012, 72, 1290–1300. [Google Scholar] [CrossRef]
- D’Ignazio, L.; Bandarra, D.; Rocha, S. NF-κB and hif crosstalk in immune responses. FEBS J. 2016, 283, 413–424. [Google Scholar] [CrossRef]
- Matsui, W.H. Cancer stem cell signaling pathways. Medicine 2016, 95, S8–S19. [Google Scholar] [CrossRef]
- Sun, L.; Mathews, L.A.; Cabarcas, S.M.; Zhang, X.; Yang, A.; Zhang, Y.; Young, M.R.; Klarmann, K.D.; Keller, J.R.; Farrar, W.L. Epigenetic regulation of sox9 by the NF-κB signaling pathway in pancreatic cancer stem cells. Stem Cells 2013, 31, 1454–1466. [Google Scholar] [CrossRef]
- Yuan, P.; He, X.H.; Rong, Y.F.; Cao, J.; Li, Y.; Hu, Y.P.; Liu, Y.; Li, D.; Lou, W.; Liu, M.F. Kras/NF-κB/yy1/mir-489 signaling axis controls pancreatic cancer metastasis. Cancer Res. 2017, 77, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Ling, J.; Kang, Y.; Zhao, R.; Xia, Q.; Lee, D.F.; Chang, Z.; Li, J.; Peng, B.; Fleming, J.B.; Wang, H.; et al. Krasg12d-induced ikk2/β/NF-κB activation by il-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 105–120. [Google Scholar] [CrossRef]
- Bang, D.; Wilson, W.; Ryan, M.; Yeh, J.J.; Baldwin, A.S. Gsk-3α promotes oncogenic kras function in pancreatic cancer via tak1-tab stabilization and regulation of noncanonical NF-κB. Cancer Discov. 2013, 3, 690–703. [Google Scholar] [CrossRef] [PubMed]
- Maniati, E.; Bossard, M.; Cook, N.; Candido, J.B.; Emami-Shahri, N.; Nedospasov, S.A.; Balkwill, F.R.; Tuveson, D.A.; Hagemann, T. Crosstalk between the canonical NF-κB and notch signaling pathways inhibits ppargamma expression and promotes pancreatic cancer progression in mice. J. Clin. Investig. 2011, 121, 4685–4699. [Google Scholar] [CrossRef] [PubMed]
- Lohse, I.; Rasowski, J.; Cao, P.; Pintilie, M.; Do, T.; Tsao, M.S.; Hill, R.P.; Hedley, D.W. Targeting hypoxic microenvironment of pancreatic xenografts with the hypoxia-activated prodrug th-302. Oncotarget 2016, 7, 33571–33580. [Google Scholar] [CrossRef]
- Yuen, A.; Diaz, B. The impact of hypoxia in pancreatic cancer invasion and metastasis. Hypoxia 2014, 2, 91–106. [Google Scholar] [PubMed]
- Cheng, Z.X.; Wang, D.W.; Liu, T.; Liu, W.X.; Xia, W.B.; Xu, J.; Zhang, Y.H.; Qu, Y.K.; Guo, L.Q.; Ding, L.; et al. Effects of the hif-1α and NF-κB loop on epithelial-mesenchymal transition and chemoresistance induced by hypoxia in pancreatic cancer cells. Oncol. Rep. 2014, 31, 1891–1898. [Google Scholar] [CrossRef]
- Fujioka, S.; Sclabas, G.M.; Schmidt, C.; Frederick, W.A.; Dong, Q.G.; Abbruzzese, J.L.; Evans, D.B.; Baker, C.; Chiao, P.J. Function of nuclear factor κB in pancreatic cancer metastasis. Clin. Cancer Res. 2003, 9, 346–354. [Google Scholar]
- Zhang, Z.; Duan, Q.; Zhao, H.; Liu, T.; Wu, H.; Shen, Q.; Wang, C.; Yin, T. Gemcitabine treatment promotes pancreatic cancer stemness through the nox/ros/NF-κB/stat3 signaling cascade. Cancer Lett. 2016, 382, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Amrutkar, M.; Gladhaug, I.P. Pancreatic cancer chemoresistance to gemcitabine. Cancers 2017, 9, 157. [Google Scholar] [CrossRef] [PubMed]
- Skrypek, N.; Duchene, B.; Hebbar, M.; Leteurtre, E.; van Seuningen, I.; Jonckheere, N. The muc4 mucin mediates gemcitabine resistance of human pancreatic cancer cells via the concentrative nucleoside transporter family. Oncogene 2013, 32, 1714–1723. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ren, D.; Wu, X.; Lin, X.; Ye, L.; Lin, C.; Wu, S.; Zhu, J.; Peng, X.; Song, L. Mir-1266 contributes to pancreatic cancer progression and chemoresistance by the stat3 and NF-κB signaling pathways. Mol. Ther. Nucleic Acids 2018, 11, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Munoz, A.R.; Pingali, S.; Payton-Stewart, F.; Chan, D.E.; Freeman, J.W.; Ghosh, R.; Kumar, A.P. Downregulation of stat3/NF-κB potentiates gemcitabine activity in pancreatic cancer cells. Mol. Carcinog. 2017, 56, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Arumugam, T.; Yamamoto, T.; Levin, P.A.; Ramachandran, V.; Ji, B.; Lopez-Berestein, G.; Vivas-Mejia, P.E.; Sood, A.K.; McConkey, D.J.; et al. Nuclear factor-κB p65/rela silencing induces apoptosis and increases gemcitabine effectiveness in a subset of pancreatic cancer cells. Clin. Cancer Res. 2008, 14, 8143–8151. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Chen, S.; Guo, Y.; Sun, C. Oncogenic trim31 confers gemcitabine resistance in pancreatic cancer via activating the NF-κB signaling pathway. Theranostics 2018, 8, 3224–3236. [Google Scholar] [CrossRef]
- Mezencev, R.; Matyunina, L.V.; Wagner, G.T.; McDonald, J.F. Acquired resistance of pancreatic cancer cells to cisplatin is multifactorial with cell context-dependent involvement of resistance genes. Cancer Gene Ther. 2016, 23, 446–453. [Google Scholar] [CrossRef] [Green Version]
- Melisi, D.; Xia, Q.; Paradiso, G.; Ling, J.; Moccia, T.; Carbone, C.; Budillon, A.; Abbruzzese, J.L.; Chiao, P.J. Modulation of pancreatic cancer chemoresistance by inhibition of tak1. J. Natl. Cancer Inst. 2011, 103, 1190–1204. [Google Scholar] [CrossRef]
- Wang, W.; Abbruzzese, J.L.; Evans, D.B.; Larry, L.; Cleary, K.R.; Chiao, P.J. The nuclear factor-κB rela transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin. Cancer Res. 1999, 5, 119–127. [Google Scholar]
- Jiang, J.; Mo, Z.C.; Yin, K.; Zhao, G.J.; Lv, Y.C.; Ouyang, X.P.; Jiang, Z.S.; Fu, Y.; Tang, C.K. Epigallocatechin-3-gallate prevents tnf-α-induced NF-κB activation thereby upregulating abca1 via the nrf2/keap1 pathway in macrophage foam cells. Int. J. Mol. Med. 2012, 29, 946–956. [Google Scholar] [PubMed]
- Chow, J.Y.; Ban, M.; Wu, H.L.; Nguyen, F.; Huang, M.; Chung, H.; Dong, H.; Carethers, J.M. TGF-β downregulates pten via activation of NF-κB in pancreatic cancer cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G275–G282. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Chiao, P.; Zhang, X.; Zhang, X.; Lazar, M.A.; Seto, E.; Young, H.A.; Ye, J. Coactivators and corepressors of NF-κB in IκBα gene promoter. J. Biol. Chem. 2005, 280, 21091–21098. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.S.; Kim, I.W.; Hu, R.; Kong, A.N. Modulatory properties of various natural chemopreventive agents on the activation of NF-κB signaling pathway. Pharm. Res. 2004, 21, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Khor, T.O.; Xu, C.; Shen, G.; Jeong, W.S.; Yu, S.; Kong, A.N. Activation of nrf2-antioxidant signaling attenuates NFκB-inflammatory response and elicits apoptosis. Biochem. Pharmacol. 2008, 76, 1485–1489. [Google Scholar] [CrossRef] [PubMed]
- Tao, M.; Liu, L.; Shen, M.; Zhi, Q.; Gong, F.R.; Zhou, B.P.; Wu, Y.; Liu, H.; Chen, K.; Shen, B.; et al. Inflammatory stimuli promote growth and invasion of pancreatic cancer cells through NF-κB pathway dependent repression of pp2ac. Cell Cycle 2016, 15, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Holcomb, B.; Yip-Schneider, M.; Schmidt, C.M. The role of nuclear factor kappab in pancreatic cancer and the clinical applications of targeted therapy. Pancreas 2008, 36, 225–235. [Google Scholar] [CrossRef]
- Yin, M.J.; Yamamoto, Y.; Gaynor, R.B. The anti-inflammatory agents aspirin and salicylate inhibit the activity of iκB kinase-β. Nature 1998, 396, 77–80. [Google Scholar] [CrossRef]
- El-Rayes, B.F.; Ali, S.; Sarkar, F.H.; Philip, P.A. Cyclooxygenase-2-dependent and -independent effects of celecoxib in pancreatic cancer cell lines. Mol. Cancer Ther. 2004, 3, 1421–1426. [Google Scholar]
- Yip-Schneider, M.T.; Nakshatri, H.; Sweeney, C.J.; Marshall, M.S.; Wiebke, E.A.; Schmidt, C.M. Parthenolide and sulindac cooperate to mediate growth suppression and inhibit the nuclear factor-κB pathway in pancreatic carcinoma cells. Mol. Cancer Ther. 2005, 4, 587–594. [Google Scholar] [CrossRef]
- Sclabas, G.M.; Uwagawa, T.; Schmidt, C.; Hess, K.R.; Evans, D.B.; Abbruzzese, J.L.; Chiao, P.J. Nuclear factor kappa B activation is a potential target for preventing pancreatic carcinoma by aspirin. Cancer 2005, 103, 2485–2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bimonte, S.; Barbieri, A.; Leongito, M.; Piccirillo, M.; Giudice, A.; Pivonello, C.; de Angelis, C.; Granata, V.; Palaia, R.; Izzo, F. Curcumin anticancer studies in pancreatic cancer. Nutrients 2016, 8, 433. [Google Scholar] [CrossRef] [PubMed]
- Boreddy, S.R.; Srivastava, S.K. Pancreatic cancer chemoprevention by phytochemicals. Cancer Lett. 2013, 334, 86–94. [Google Scholar] [CrossRef] [Green Version]
- Devassy, J.G.; Nwachukwu, I.D.; Jones, P.J. Curcumin and cancer: Barriers to obtaining a health claim. Nutr. Rev. 2015, 73, 155–165. [Google Scholar] [CrossRef]
- Surh, Y.J.; Chun, K.S.; Cha, H.H.; Han, S.S.; Keum, Y.S.; Park, K.K.; Lee, S.S. Molecular mechanisms underlying chemopreventive activities of anti-inflammatory phytochemicals: Down-regulation of cox-2 and inos through suppression of NF-κB activation. Mutat. Res. 2001, 480, 243–268. [Google Scholar] [CrossRef]
- Vallianou, N.G.; Evangelopoulos, A.; Schizas, N.; Kazazis, C. Potential anticancer properties and mechanisms of action of curcumin. Anticancer Res. 2015, 35, 645–651. [Google Scholar]
- Bharti, A.C.; Donato, N.; Singh, S.; Aggarwal, B.B. Curcumin (diferuloylmethane) down-regulates the constitutive activation of nuclear factor-κB and IκBα kinase in human multiple myeloma cells, leading to suppression of proliferation and induction of apoptosis. Blood 2003, 101, 1053–1062. [Google Scholar] [CrossRef]
- Kunnumakkara, A.B.; Guha, S.; Krishnan, S.; Diagaradjane, P.; Gelovani, J.; Aggarwal, B.B. Curcumin potentiates antitumor activity of gemcitabine in an orthotopic model of pancreatic cancer through suppression of proliferation, angiogenesis, and inhibition of nuclear factor-κB-regulated gene products. Cancer Res. 2007, 67, 3853–3861. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, N.; Aggarwal, B.B.; Newman, R.A.; Wolff, R.A.; Kunnumakkara, A.B.; Abbruzzese, J.L.; Ng, C.S.; Badmaev, V.; Kurzrock, R. Phase ii trial of curcumin in patients with advanced pancreatic cancer. Clin. Cancer Res. 2008, 14, 4491–4499. [Google Scholar] [CrossRef]
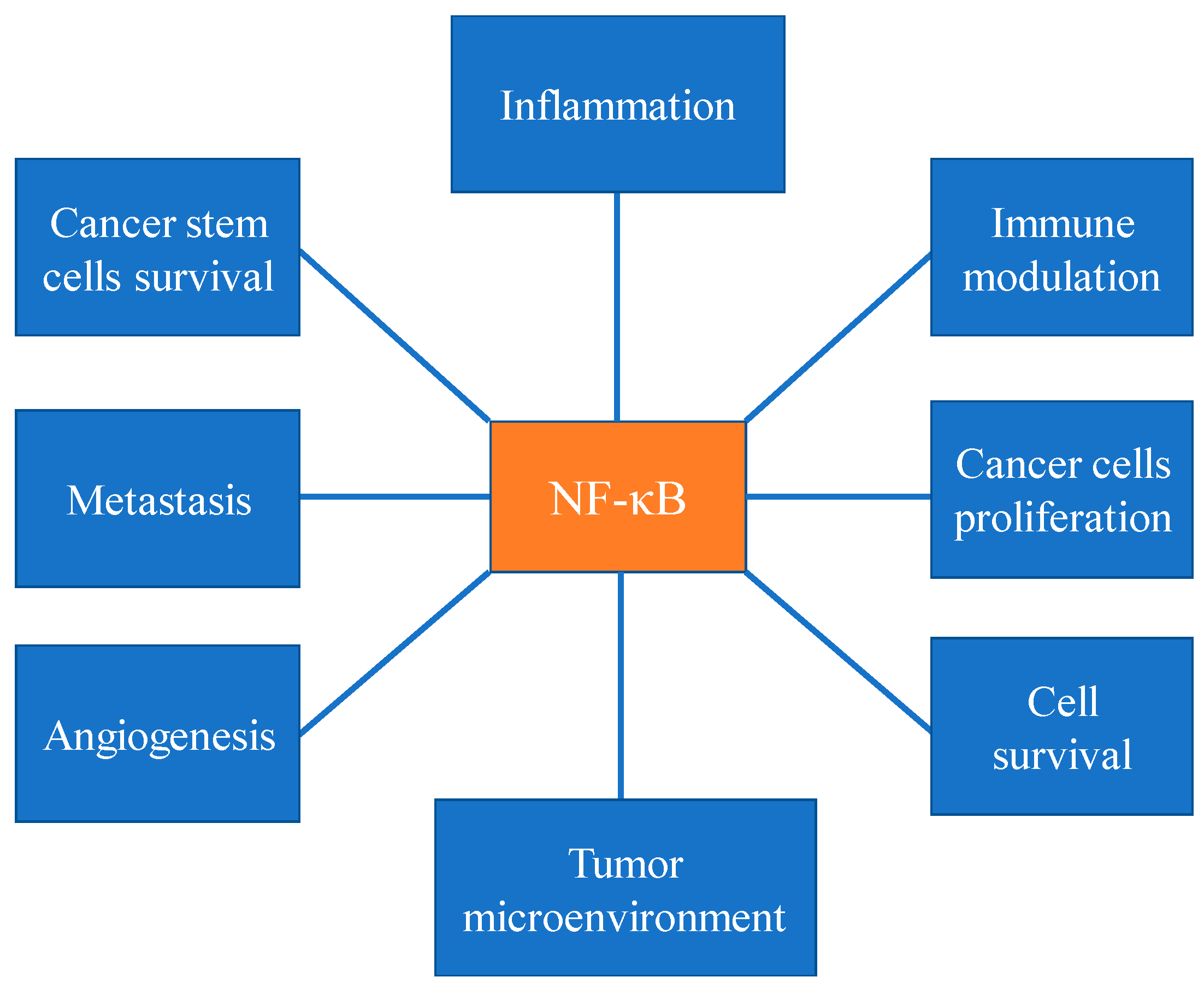
Figure 1.
Different cellular events, including inflammatory response, immune cells modulation, cancer cell proliferation, cancer cell survival, tumor microenvironment, angiogenesis, metastasis, stem cell survival activates NF-κB pathway.
Figure 1.
Different cellular events, including inflammatory response, immune cells modulation, cancer cell proliferation, cancer cell survival, tumor microenvironment, angiogenesis, metastasis, stem cell survival activates NF-κB pathway.

Figure 2.
Cross talk of NF-κB signaling with others signaling in pancreatic cancer.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pramanik, K.C.; Makena, M.R.; Bhowmick, K.; Pandey, M.K. Advancement of NF-κB Signaling Pathway: A Novel Target in Pancreatic Cancer. Int. J. Mol. Sci. 2018, 19, 3890. https://doi.org/10.3390/ijms19123890
AMA Style
Pramanik KC, Makena MR, Bhowmick K, Pandey MK. Advancement of NF-κB Signaling Pathway: A Novel Target in Pancreatic Cancer. International Journal of Molecular Sciences. 2018; 19(12):3890. https://doi.org/10.3390/ijms19123890
Chicago/Turabian StylePramanik, Kartick C., Monish Ram Makena, Kuntal Bhowmick, and Manoj K. Pandey. 2018. "Advancement of NF-κB Signaling Pathway: A Novel Target in Pancreatic Cancer" International Journal of Molecular Sciences 19, no. 12: 3890. https://doi.org/10.3390/ijms19123890
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.