Regulation of Leptin Methylation Not via Apoptosis by Melatonin in the Rescue of Chronic Programming Liver Steatosis

 ,
,  ,
,  , and
, and
Abstract
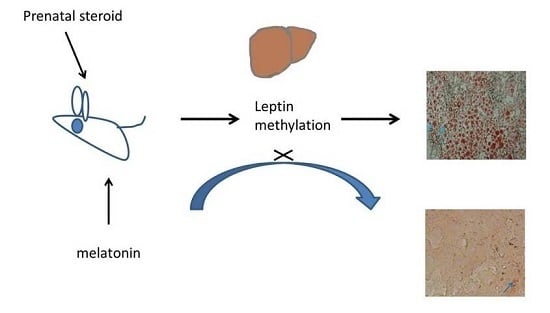
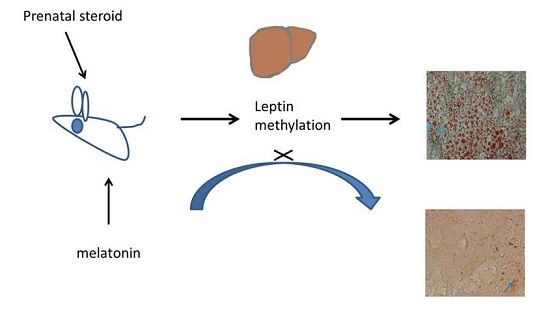
:
1. Introduction
2. Results
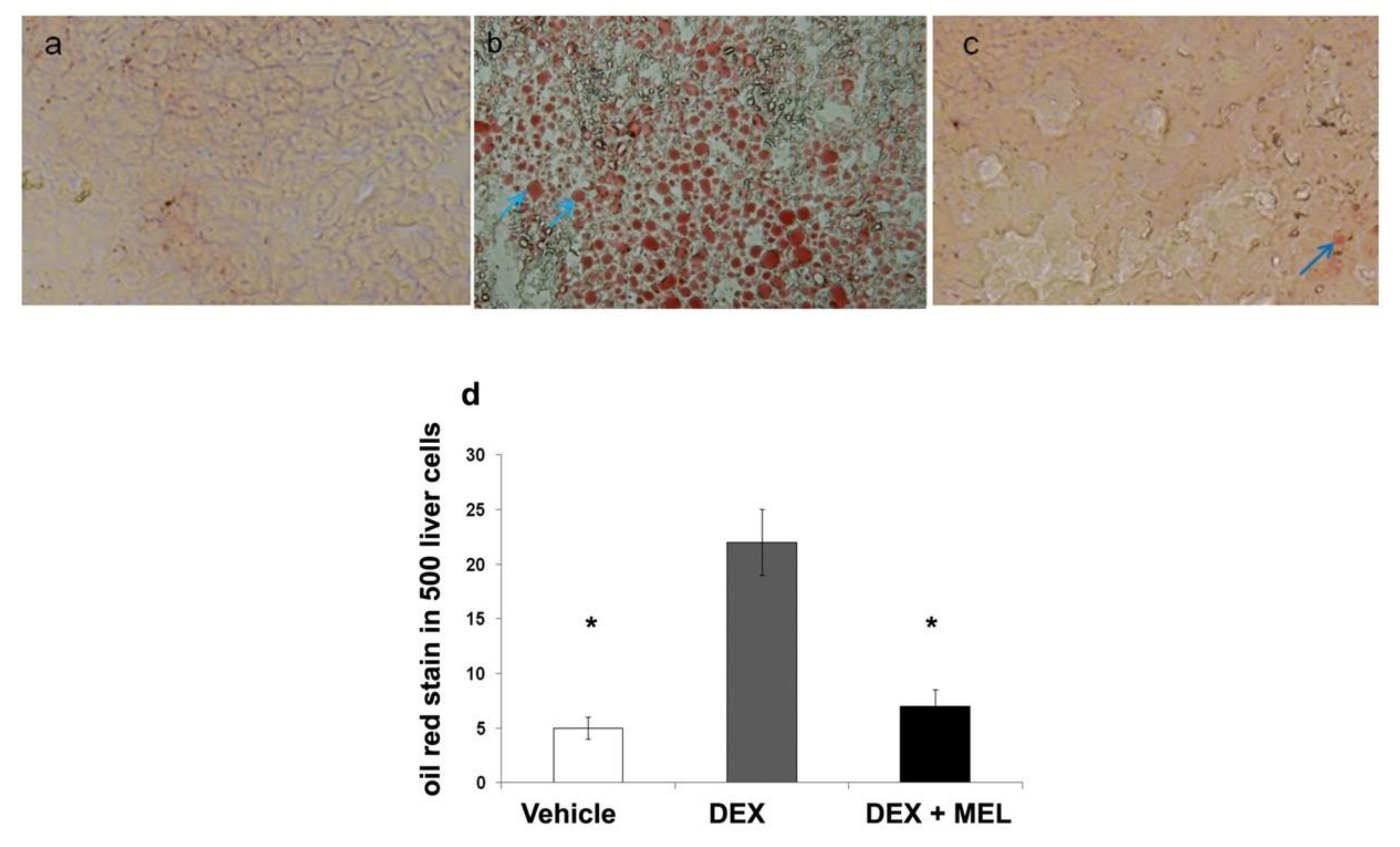
2.1. Liver Steatosis
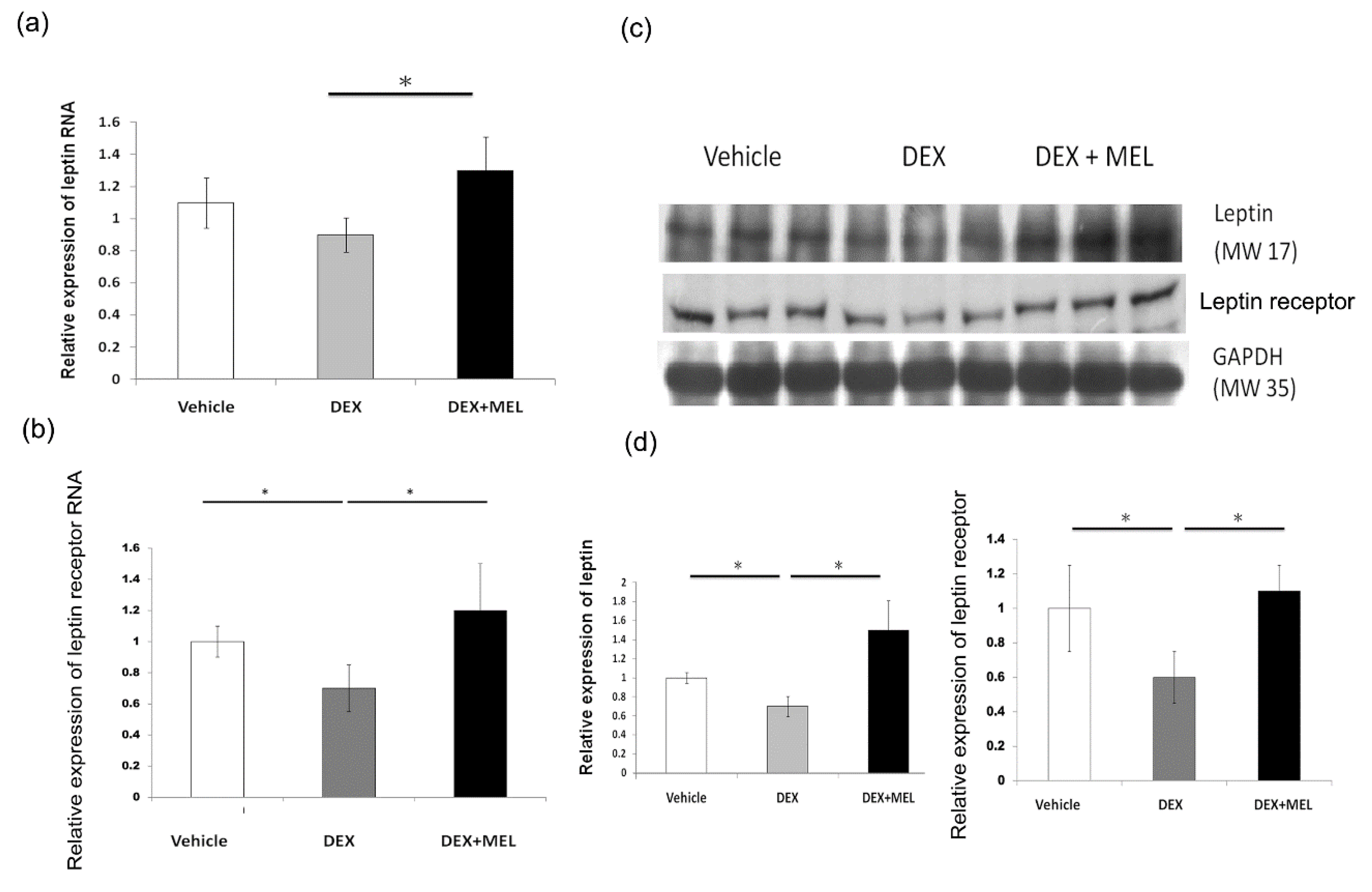
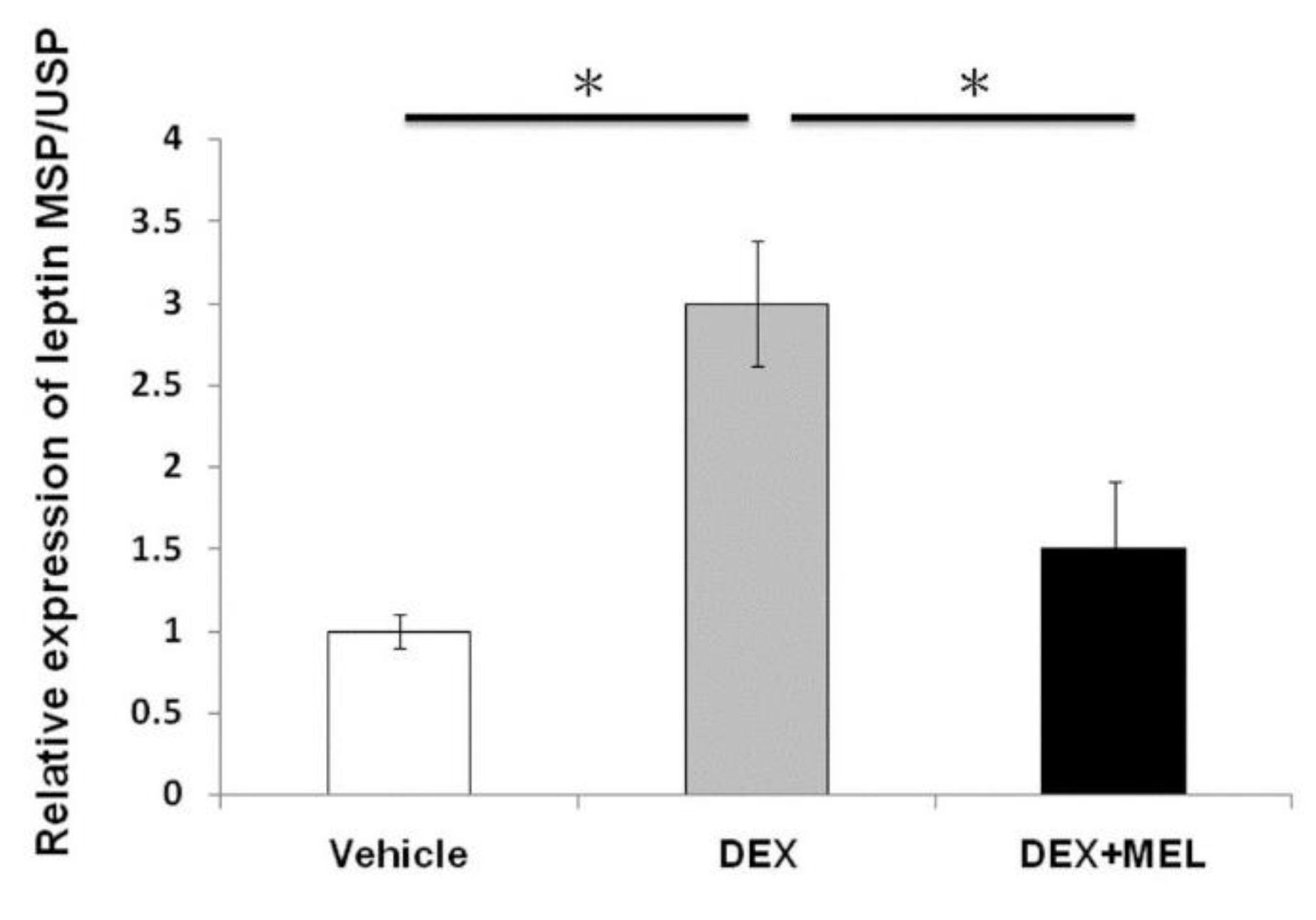
2.2. Leptin Expression and Methylation
2.3. Expression of Leptin Methylation
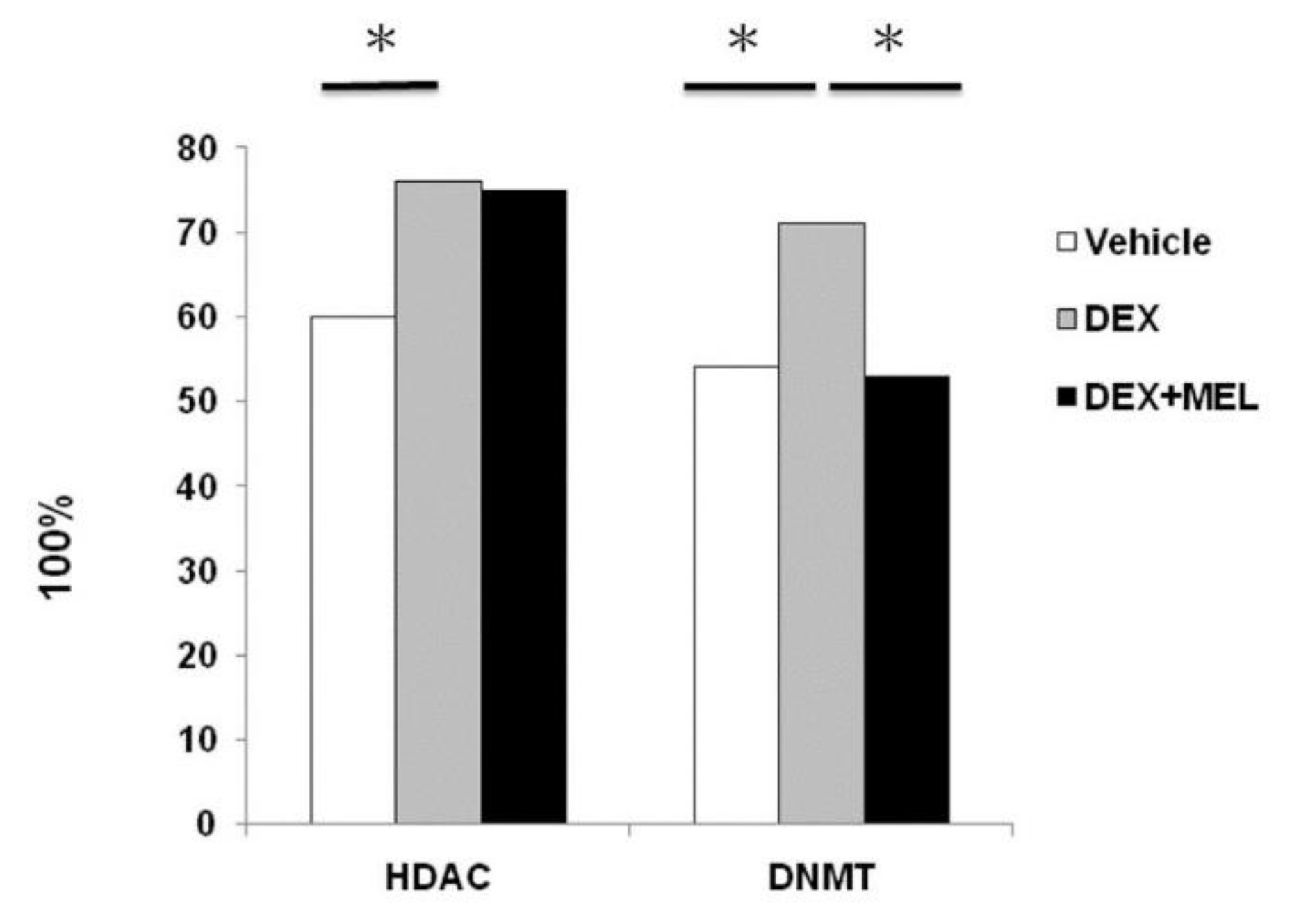
2.4. Histone Deacetylation (HDAC) and DNA Methyltransferase (DNMT) Activity in Liver
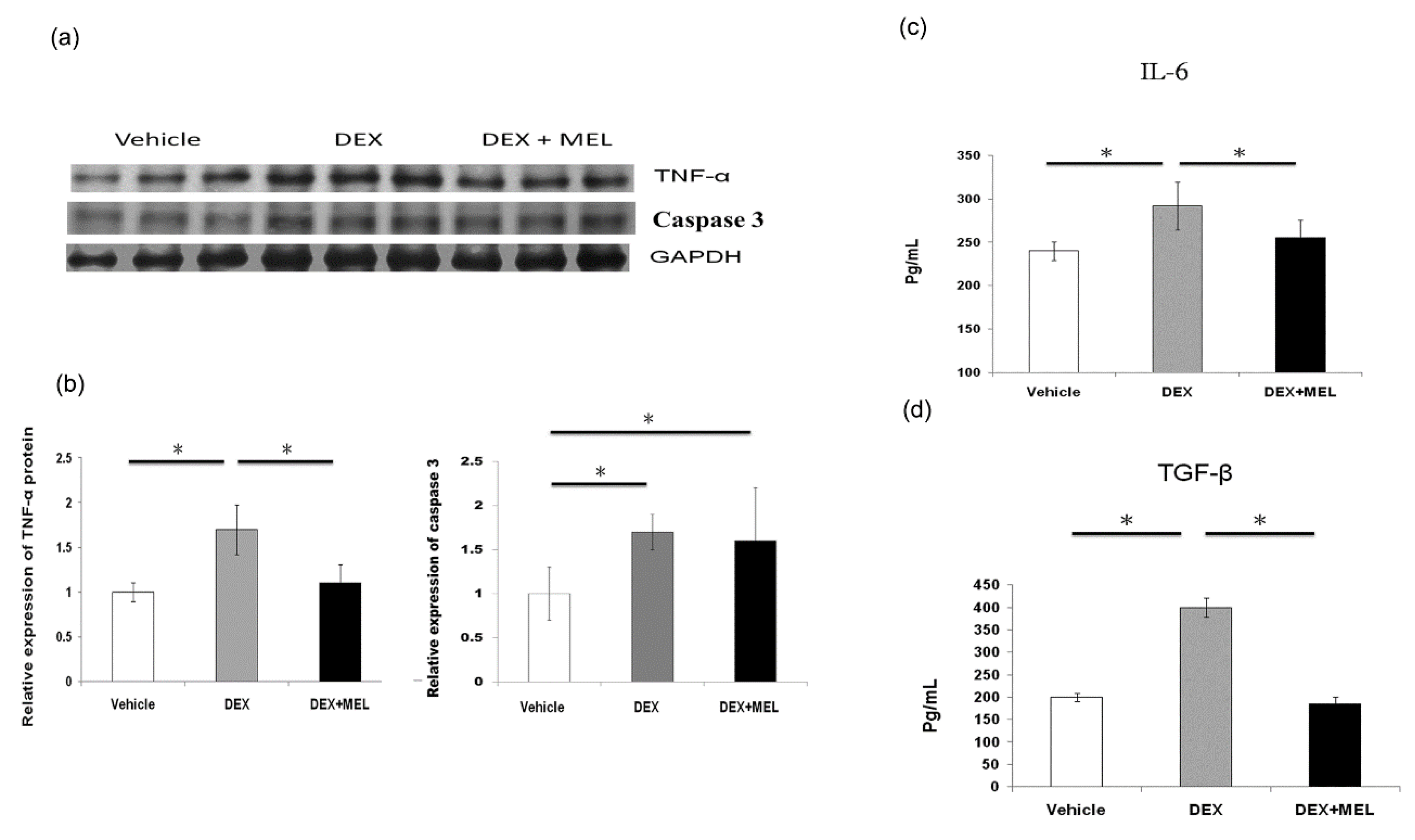
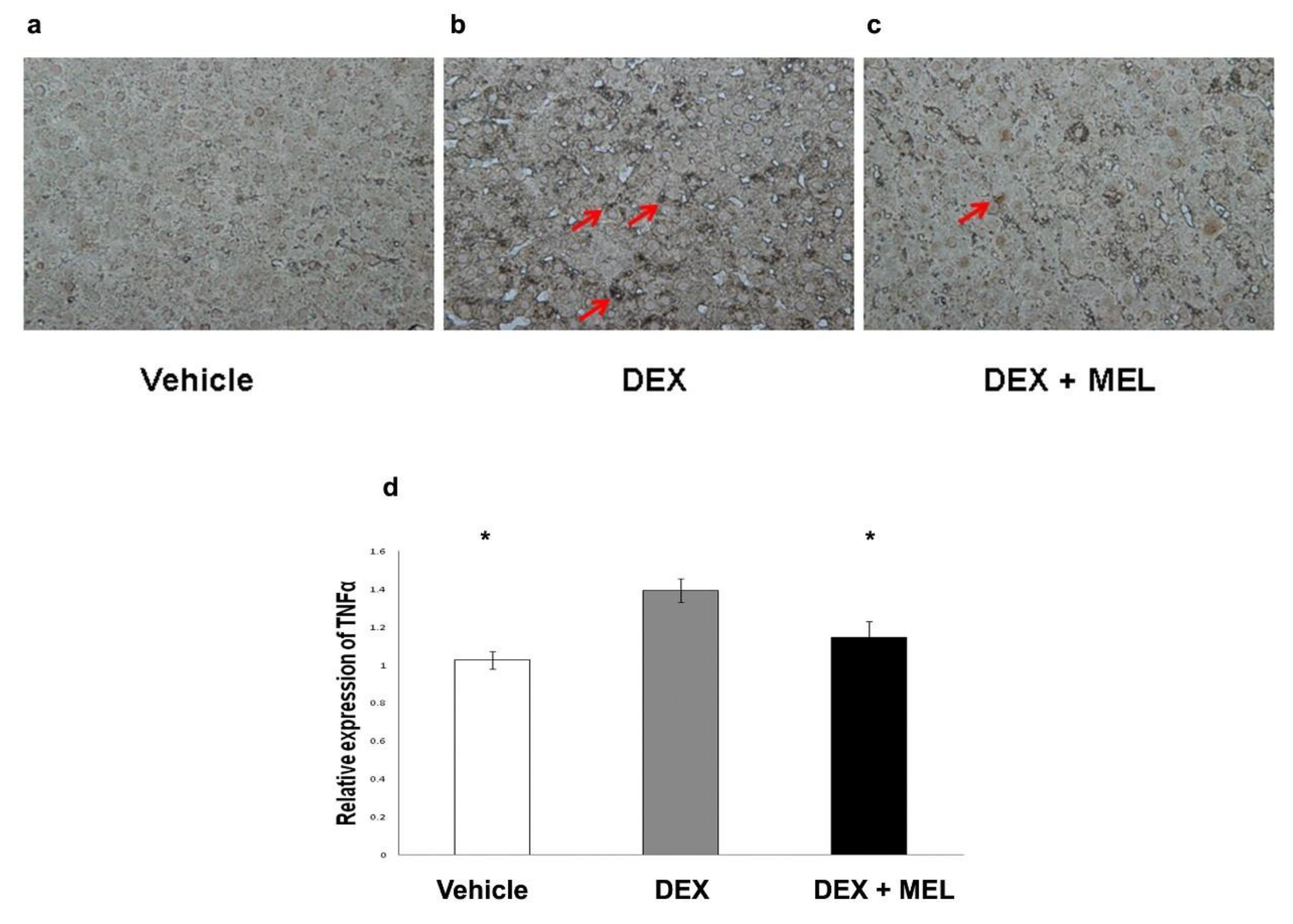
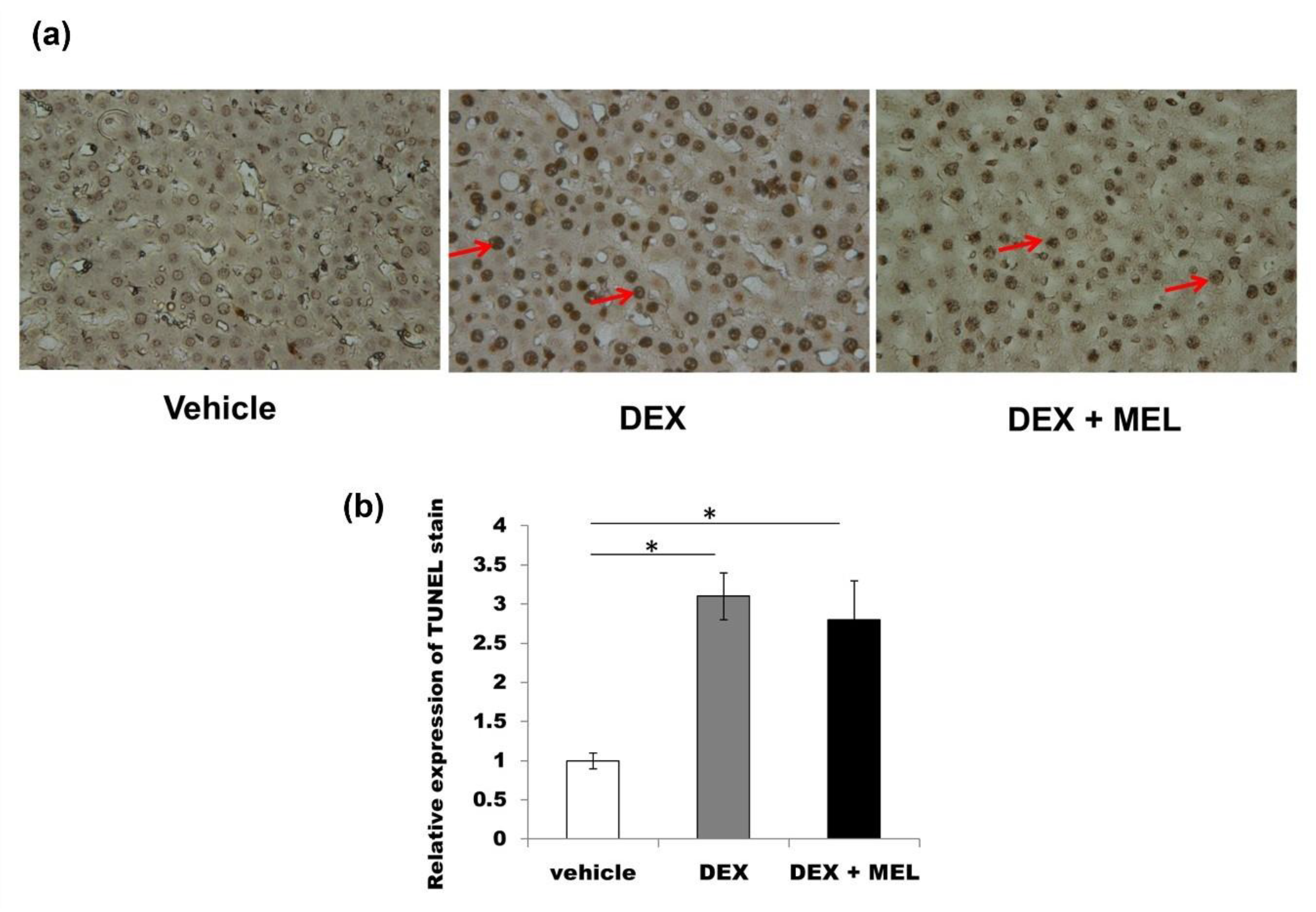
2.5. Inflammation and Apoptosis Studies
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Prenatal Dexamethasone Exposure Paradigm
4.3. Melatonin Treatment
4.4. Localization of Oil Red Stain and Analysis
4.5. RNA Isolation and Real-Time PCR
4.6. Western Blotting Analysis
4.7. Histone Extraction
4.8. HDAC/DNMT Activity Assay
4.9. Bisulfite Modification
4.10. Methylation Specific qPCR
4.11. TdT-Mediated dUTP Biotin Nick End Labeling (TUNEL)
4.12. Cytokine Secretion with Enzyme-Linked Immunosorbent Assay (ELISA)
4.13. Immunohistochemistry Stain
4.14. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALT | alanine transaminase |
| AST | aspartate transaminase |
| DEX | dexamethasone |
| DNMT | DNA methyltransferase |
| HDAC | histone deacetylases |
| MEL | melatonin |
| MSP/USP | methylation-specific PCR/unmethylation-specific PCR |
| NAFLD | steatosis in non-alcoholic fatty liver disease |
| TGF-β | transforming growth factor beta |
| TNF-α | tumor necrosis factor alpha |
References
- Kapoor, A.; Petropoulos, S.; Matthews, S.G. Fetal programming of hypothalamic-pituitary-adrenal (HPA) axis function and behavior by synthetic glucocorticoids. Brain Res. Rev. 2008, 57, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Tiao, M.M.; Huang, L.T.; Chen, C.J.; Sheen, J.M.; Tain, Y.L.; Chen, C.C.; Kuo, H.C.; Huang, Y.H.; Tang, K.S.; Chu, E.W.; et al. Melatonin in the regulation of liver steatosis following prenatal glucocorticoid exposure. BioMed Res. Int. 2014, 2014, 942172. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Chen, C.C.; Sheen, J.M.; Yu, H.R.; Tiao, M.M.; Kuo, H.C.; Huang, L.T. Melatonin attenuates prenatal dexamethasone-induced blood pressure increase in a rat model. J. Am. Soc. Hypertens. 2014, 8, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Cuevas Guaman, M.; Sbrana, E.; Shope, C.; Showalter, L.; Hu, M.; Meloche, S.; Huang, L.T. Administration of antenatal glucocorticoids and postnatal surfactant ameliorates respiratory distress syndrome-associated neonatal lethality in Erk3(−/−) mouse pups. Pediatr. Res. 2014, 76, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Entringer, S.; Buss, C.; Wadhwa, P.D. Prenatal stress and developmental programming of human health and disease risk: Concepts and integration of empirical findings. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Varcoe, T.J.; Boden, M.J.; Voultsios, A.; Salkeld, M.D.; Rattanatray, L.; Kennaway, D.J. Characterisation of the Maternal Response to Chronic Phase Shifts during Gestation in the Rat: Implications for Fetal Metabolic Programming. PLoS ONE 2013, 8, e53800. [Google Scholar] [CrossRef] [PubMed]
- Thornburg, K.L.; Shannon, J.; Thuillier, P.; Turker, M.S. In utero life and epigenetic predisposition for disease. Adv. Genet. 2010, 71, 57–78. [Google Scholar] [PubMed]
- Drake, A.J.; Raubenheimer, P.J.; Kerrigan, D.; McInnes, K.J.; Seckl, J.R.; Walker, B.R. Prenatal dexamethasone programs expression of genes in liver and adipose tissue and increased hepatic lipid accumulation but not obesity on a high-fat diet. Endocrinology 2010, 151, 1581–1587. [Google Scholar] [CrossRef] [PubMed]
- Pogribny, I.P.; Tryndyak, V.P.; Bagnyukova, T.V.; Melnyk, S.; Montgomery, B.; Ross, S.A.; Latendresse, J.R.; Rusyn, I.; Beland, F.A. Hepatic epigenetic phenotype predetermines individual susceptibility to hepatic steatosis in mice fed a lipogenic methyl-deficient diet. J. Hepatol. 2009, 51, 176–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.R.; Kuo, H.C.; Chen, C.C.; Sheen, J.M.; Tiao, M.M.; Chen, Y.C.; Chang, K.A.; Tain, Y.L.; Huang, L.T. Prenatal dexamethasone exposure in rats results in long-term epigenetic histone modifications and tumor necrosis factor-alpha production decrease. Immunology 2014, 143, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Ducroc, R.; Sakar, Y.; Fanjul, C.; Barber, A.; Bado, A.; Lostao, M.P. Luminal leptin inhibits L-glutamine transport in rat small intestine: Involvement of ASCT2 and B0AT1. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G179–G185. [Google Scholar] [CrossRef] [PubMed]
- Gautron, L.; Elmquist, J.K. Sixteen years and counting: An update on leptin in energy balance. J. Clin. Investig. 2011, 121, 2087–2093. [Google Scholar] [CrossRef] [PubMed]
- Ravussin, E.; Smith, S.R.; Mitchell, J.A.; Shringarpure, R.; Shan, K.; Maier, H.; Koda, J.E.; Weyer, C. Enhanced weight loss with pramlintide/metreleptin: An integrated neurohormonal approach to obesity pharmacotherapy. Obesity 2009, 17, 1736–1743. [Google Scholar] [CrossRef] [PubMed]
- Huynh, F.K.; Neumann, U.H.; Wang, Y.; Rodrigues, B.; Kieffer, T.J.; Covey, S.D. A role for hepatic leptin signaling in lipid metabolism via altered very low density lipoprotein composition and liver lipase activity in mice. Hepatology 2013, 57, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Terron, M.P.; Flores, L.J.; Czarnocki, Z. Melatonin and its metabolites: New findings regarding their production and their radical scavenging actions. Acta Biochim. Pol. 2007, 54, 1–9. [Google Scholar] [PubMed]
- Zhong, M.; Xue, Z.G.; Cang, J.; Wu, W.; Huang, J.F.; Zhu, D.M. Protective effect of melatonin on reexpansion pulmonary edema in rats model. Chin. Med. J. 2013, 126, 500–504. [Google Scholar] [PubMed]
- Zaitone, S.; Hassan, N.; El-Orabi, N.; El-Awady el, S. Pentoxifylline and melatonin in combination with pioglitazone ameliorate experimental non-alcoholic fatty liver disease. Eur. J. Pharmacol. 2011, 662, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Perfield, J.W., 2nd; Ortinau, L.C.; Pickering, R.T.; Ruebel, M.L.; Meers, G.M.; Rector, R.S. Altered hepatic lipid metabolism contributes to nonalcoholic Fatty liver disease in leptin-deficient ob/ob mice. J. Obes. 2013, 2013, 296537. [Google Scholar] [CrossRef] [PubMed]
- Shimokobe, H.; Sumida, Y.; Tanaka, S.; Mori, K.; Kitamura, Y.; Fukumoto, K.; Kakutani, A.; Ohno, T.; Kanemasa, K.; Imai, S.; et al. Human placental extract treatment for nonalcoholic steatohepatitis nonresponsive to lifestyle intervention: A pilot study. Hepatol. Res. 2015, 45, 1034–1040. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Nikolopoulos, P.; Stogianni, A.; Romiopoulos, I.; Katsinelos, P.; Kountouras, J. Effect of helicobacter pylori eradication on hepatic steatosis, NAFLD fibrosis score and HSENSI in patients with nonalcoholic steatohepatitis: A MR imaging-based pilot open-label study. Arq. Gastroenterol. 2014, 51, 261–268. [Google Scholar] [CrossRef] [PubMed]
- DePaoli, A. Leptin in common obesity and associated disorders of metabolism. J. Endocrinol. 2014, 223, T71–T81. [Google Scholar] [CrossRef] [PubMed]
- Tsochatzis, E.; Papatheodoridis, G.V.; Archimandritis, A.J. The evolving role of leptin and adiponectin in chronic liver diseases. Am. J. Gastroenterol. 2006, 101, 2629–2640. [Google Scholar] [CrossRef] [PubMed]
- Oral, E.A.; Simha, V.; Ruiz, E.; Andewelt, A.; Premkumar, A.; Snell, P.; Wagner, A.J.; DePaoli, A.M.; Reitman, M.L.; Taylor, S.I.; et al. Leptin-replacement therapy for lipodystrophy. N. Engl. J. Med. 2002, 346, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Ikejima, K.; Okumura, K.; Lang, T.; Honda, H.; Abe, W.; Yamashina, S.; Enomoto, N.; Takei, Y.; Sato, N. The role of leptin in progression of non-alcoholic fatty liver disease. Hepatol. Res. 2005, 33, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Stehle, J.H.; Saade, A.; Rawashdeh, O.; Ackermann, K.; Jilg, A.; Sebesteny, T.; Maronde, E. A survey of molecular details in the human pineal gland in the light of phylogeny, structure, function and chronobiological diseases. J. Pineal Res. 2011, 51, 17–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatzis, G.; Ziakas, P.; Kavantzas, N.; Triantafyllou, A.; Sigalas, P.; Andreadou, I.; Ioannidis, K.; Chatzis, S.; Filis, K.; Papalampros, A.; et al. Melatonin attenuates high fat diet-induced fatty liver disease in rats. World J. Hepatol. 2013, 5, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.S.; Amaral, F.G.; Mesquita, C.C.; Barbosa, A.P.; Lellis-Santos, C.; Turati, A.O.; Santos, L.R.; Sollon, C.S.; Gomes, P.R.; Faria, J.A.; et al. Maternal melatonin programs the daily pattern of energy metabolism in adult offspring. PLoS ONE 2012, 7, e38795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonciarz, M.; Bielanski, W.; Partyka, R.; Brzozowski, T.; Konturek, P.C.; Eszyk, J.; Celiński, K.; Reiter, R.J.; Konturek, S.J. Plasma insulin, leptin, adiponectin, resistin, ghrelin, and melatonin in nonalcoholic steatohepatitis patients treated with melatonin. J. Pineal Res. 2013, 54, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Milagro, F.I.; Campion, J.; Garcia-Diaz, D.F.; Goyenechea, E.; Paternain, L.; Martinez, J.A. High fat diet-induced obesity modifies the methylation pattern of leptin promoter in rats. J. Physiol. Biochem. 2009, 65, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ding, R.B.; Bao, J.; Deng, C.X. Emerging roles of SIRT1 in fatty liver diseases. Int. J. Biol. Sci. 2017, 13, 852–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiao, M.M.; Lin, Y.J.; Yu, H.R.; Sheen, J.M.; Lin, I.C.; Lai, Y.J.; Tain, Y.L.; Huang, L.T.; Tsai, C.C. Resveratrol ameliorates maternal and post-weaning high-fat diet-induced nonalcoholic fatty liver disease via renin-angiotensin system. Lipids Health Dis. 2018, 17, 178. [Google Scholar] [CrossRef] [PubMed]
- Peredo-Escárcega, A.E.; Guarner-Lans, V.; Pérez-Torres, I.; Ortega-Ocampo, S.; Carreón-Torres, E.; Castrejón-Tellez, V.; Díaz-Díaz, E.; Rubio-Ruiz, M.E. The Combination of Resveratrol and Quercetin Attenuates Metabolic Syndrome in Rats by Modifying the Serum Fatty Acid Composition and by Upregulating SIRT 1 and SIRT 2 Expression in White Adipose Tissue. Evid.-Based Complement. Altern. Med. 2015, 474032. [Google Scholar] [CrossRef] [Green Version]
- Thompson, C.B. Apoptosis in the pathogenesis and treatment of disease. Science 1995, 267, 1456–1462. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Luedde, T.; Trautwein, C. Inflammatory pathways in liver homeostasis and liver injury. Clin. Rev. Allergy Immunol. 2009, 36, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Ferguson-Smith, A.C.; Chen, Y.F.; Newman, M.S.; May, L.T.; Sehgal, P.B.; Ruddle, F.H. Regional localization of the interferon-β2/B-cell stimulatory factor 2/hepatocyte stimulating factor gene to human chromosome 7. Genomics 1988, 2, 203–208. [Google Scholar] [CrossRef]
- Diehl, A.M. Nonalcoholic steatosis and steatohepatitis IV. Nonalcoholic fatty liver disease abnormalities in macrophage function and cytokines. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G1–G5. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar] [PubMed]
- Gabele, E.; Dostert, K.; Hofmann, C.; Wiest, R.; Scholmerich, J.; Hellerbrand, C.; Obermeier, F. DSS induced colitis increases portal LPS levels and enhances hepatic inflammation and fibrogenesis in experimental NASH. J. Hepatol. 2011, 55, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Canbakan, B.; Senturk, H.; Canbakan, M.; Toptas, T.; Tabak, O.; Balci, H.; Olgac, V.; Ozbay, G. Is alanine aminotransferase level a surrogate biomarker of hepatic apoptosis in nonalcoholic fatty liver disease? Biomark. Med. 2010, 4, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Hauser, J.; Feldon, J.; Pryce, C.R. Direct and dam-mediated effects of prenatal dexamethasone on emotionality, cognition and HPA axis in adult Wistar rats. Horm. Behav. 2009, 56, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.K.; Huang, L.T.; Huang, Y.H.; Tiao, M.M.; Tang, K.S.; Liou, C.W. The effect of the red wine polyphenol resveratrol on a rat model of biliary obstructed cholestasis: Involvement of anti-apoptotic signalling, mitochondrial biogenesis and the induction of autophagy. Apoptosis 2012, 17, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Tiao, M.M.; Wang, F.S.; Huang, L.T.; Chuang, J.H.; Kuo, H.C.; Yang, Y.L.; Huang, Y.H. MicroRNA-29a protects against acute liver injury in a mouse model of obstructive jaundice via inhibition of the extrinsic apoptosis pathway. Apoptosis 2014, 19, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Ling, X.; Cao, S.; Cheng, Q.; Keefe, J.T.; Rustum, Y.M.; Li, F. A novel small molecule FL118 that selectively inhibits survivin, Mcl-1, XIAP and cIAP2 in a p53-independent manner, shows superior antitumor activity. PLoS ONE 2012, 7, e45571. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vehicle | DEX | DEX+MEL | |
|---|---|---|---|
| body weight (gm) | 527.08 ± 8.77 | 521.50 ± 9.93 | 479.56 ± 15.86 * |
| liver weight (gm) | 16.98 ± 0.77 * | 17.94 ± 0.87 | 16.96 ± 0.80 * |
| liver/BW (%) | 3.19 ± 0.11 | 3.49 ± 0.12 | 3.53 ± 0.10 |
| triglyceride (mg/dL) | 74.85 ± 2.15 | 74.78 ± 3.39 | 64.13 ± 3.54 * |
| cholesterol (mg/dL) | 123.15 ± 9.85 | 126.33 ± 11.78 | 84.25 ± 5.89 * † |
| AST (U/L) | 81.0 ± 5.13 | 89.44 ± 7.57 | 87.50 ± 6.03 |
| ALT (U/L) | 44.85 ± 2.69 | 47.63 ± 5.36 | 48.25 ± 5.89 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsai, C.-C.; Lin, Y.-J.; Yu, H.-R.; Sheen, J.-M.; Lin, I.-C.; Lai, Y.-J.; Tain, Y.-L.; Huang, L.-T.; Tiao, M.-M. Regulation of Leptin Methylation Not via Apoptosis by Melatonin in the Rescue of Chronic Programming Liver Steatosis. Int. J. Mol. Sci. 2018, 19, 3565. https://doi.org/10.3390/ijms19113565
Tsai C-C, Lin Y-J, Yu H-R, Sheen J-M, Lin I-C, Lai Y-J, Tain Y-L, Huang L-T, Tiao M-M. Regulation of Leptin Methylation Not via Apoptosis by Melatonin in the Rescue of Chronic Programming Liver Steatosis. International Journal of Molecular Sciences. 2018; 19(11):3565. https://doi.org/10.3390/ijms19113565
Chicago/Turabian StyleTsai, Ching-Chou, Yu-Ju Lin, Hong-Ren Yu, Jiunn-Ming Sheen, I-Chun Lin, Yun-Ju Lai, You-Lin Tain, Li-Tung Huang, and Mao-Meng Tiao. 2018. "Regulation of Leptin Methylation Not via Apoptosis by Melatonin in the Rescue of Chronic Programming Liver Steatosis" International Journal of Molecular Sciences 19, no. 11: 3565. https://doi.org/10.3390/ijms19113565