Inhibition of Microglia-Derived Oxidative Stress by Ciliary Neurotrophic Factor Protects Dopamine Neurons In Vivo from MPP+ Neurotoxicity

Abstract
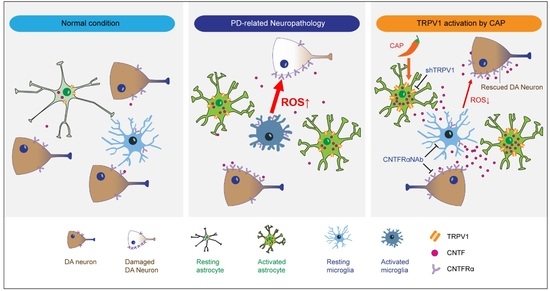
:
1. Introduction
2. Results
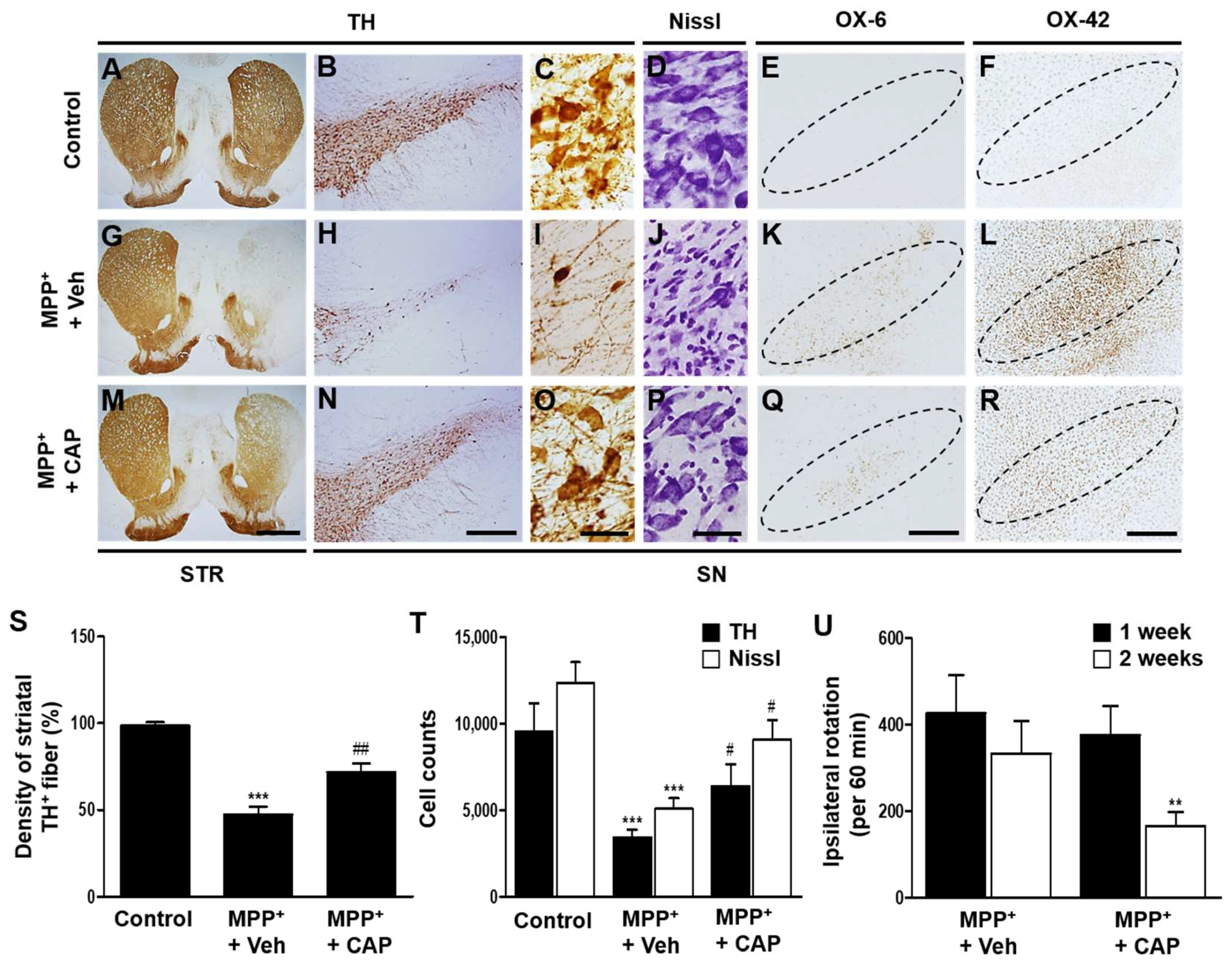
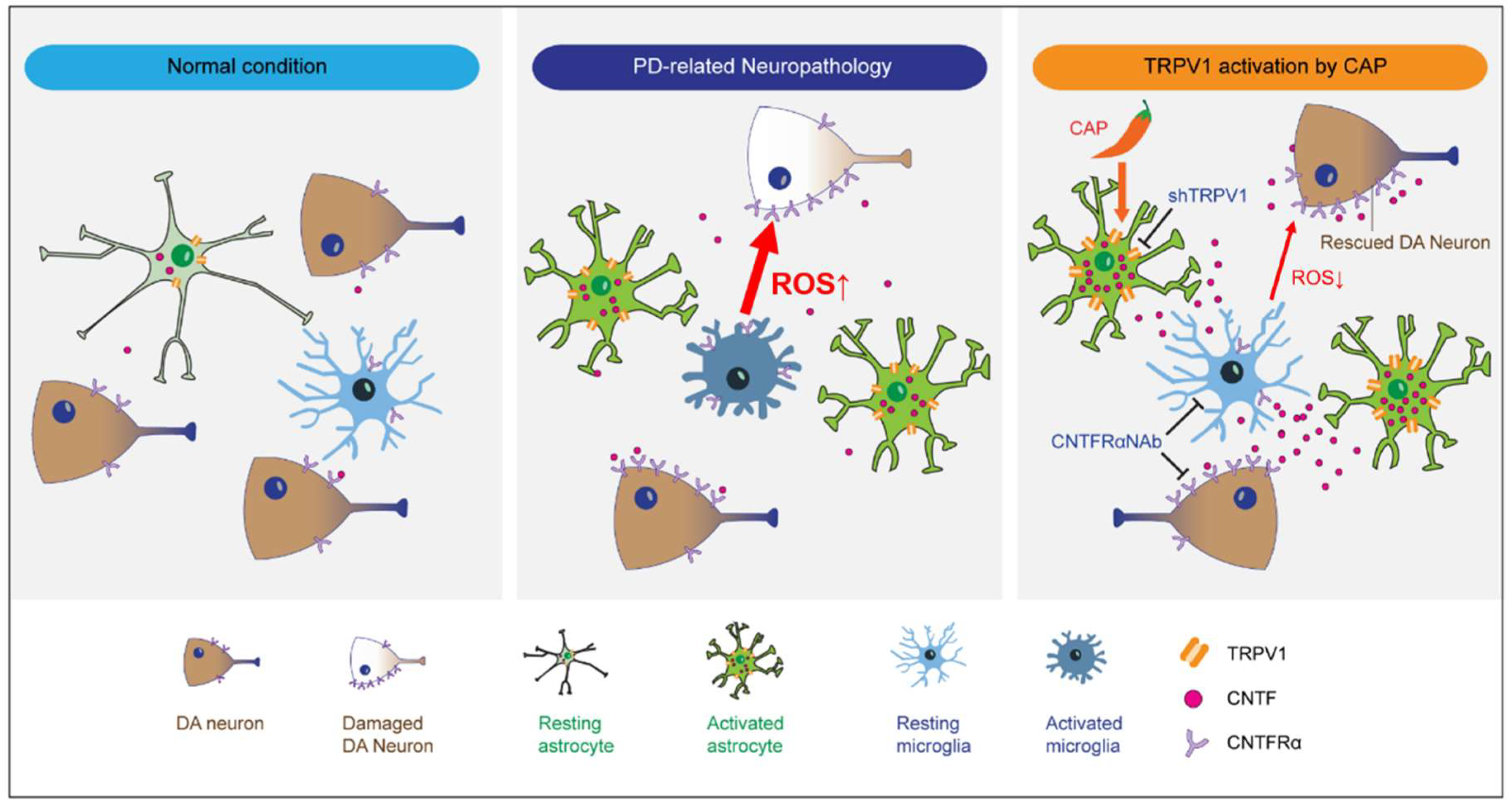
2.1. TRPV1 Activation by Capsaicin Prevents Degeneration of DA Neurons and Inhibits Microglial Activation In Vivo in MPP+-Lesioned Rat
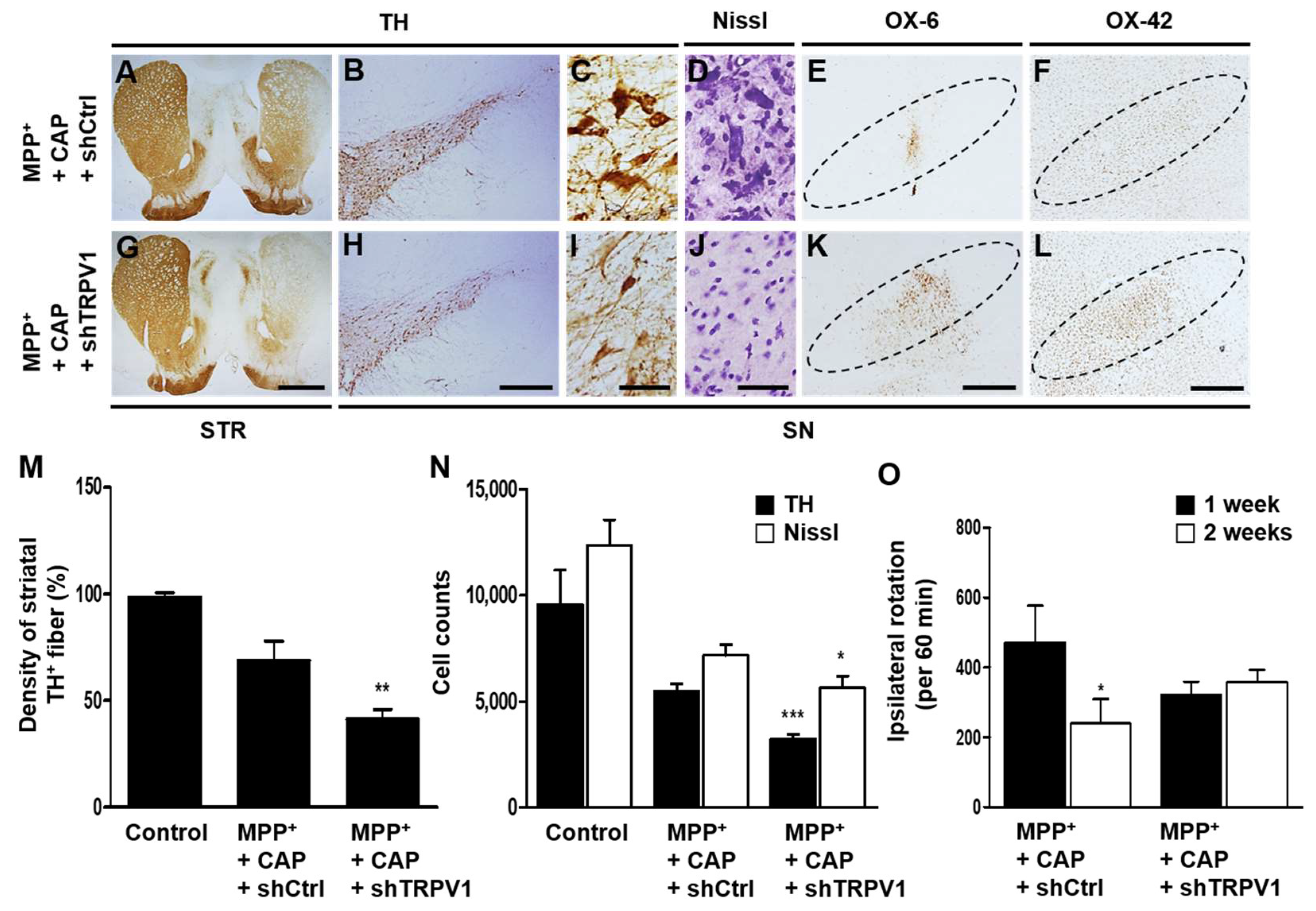
2.2. Astrocytic TRPV1 Knockdown by shRNA Alters DA Neuronal Survival and Microglial Activation In Vivo in MPP+-Lesioned Rat
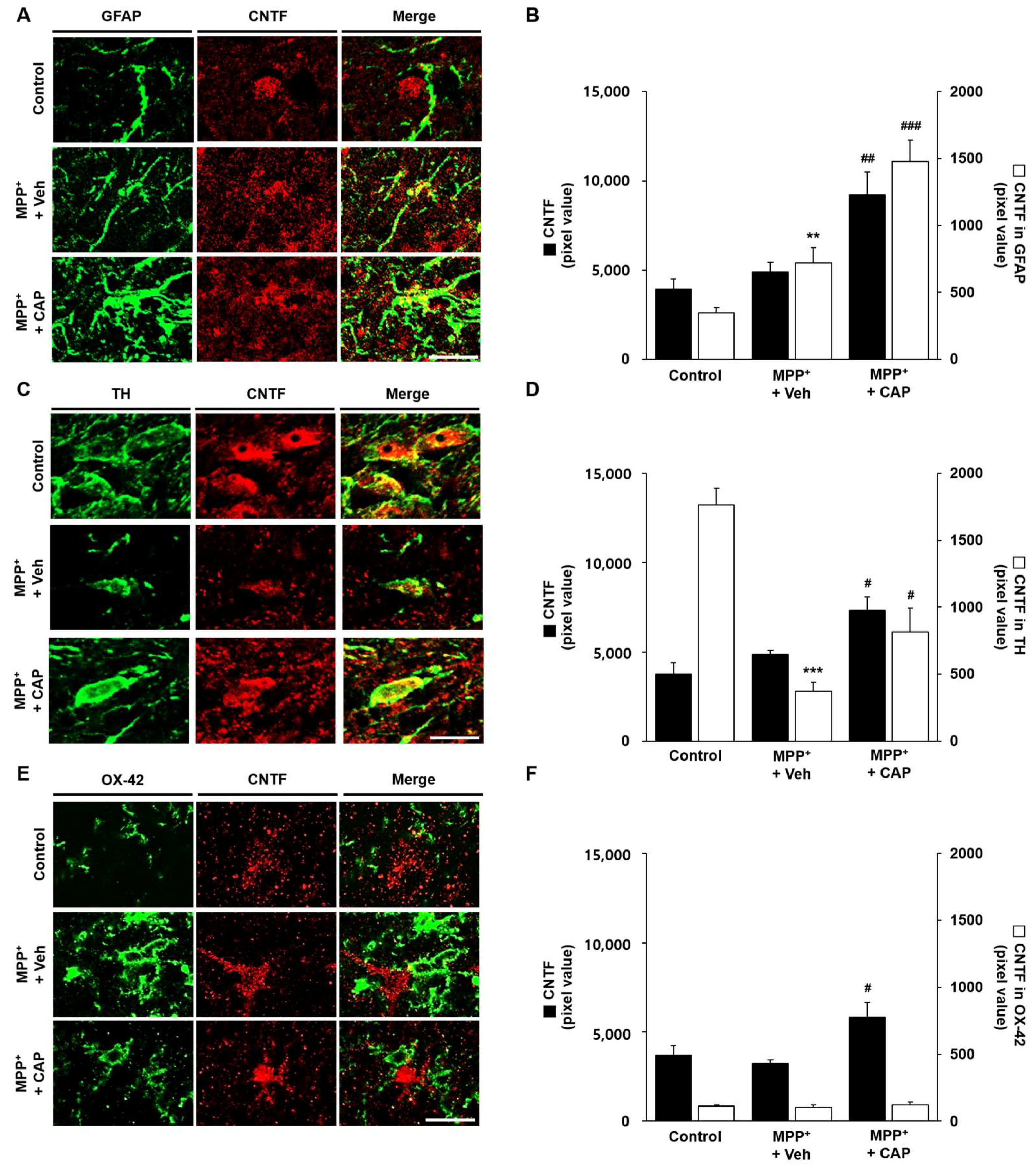
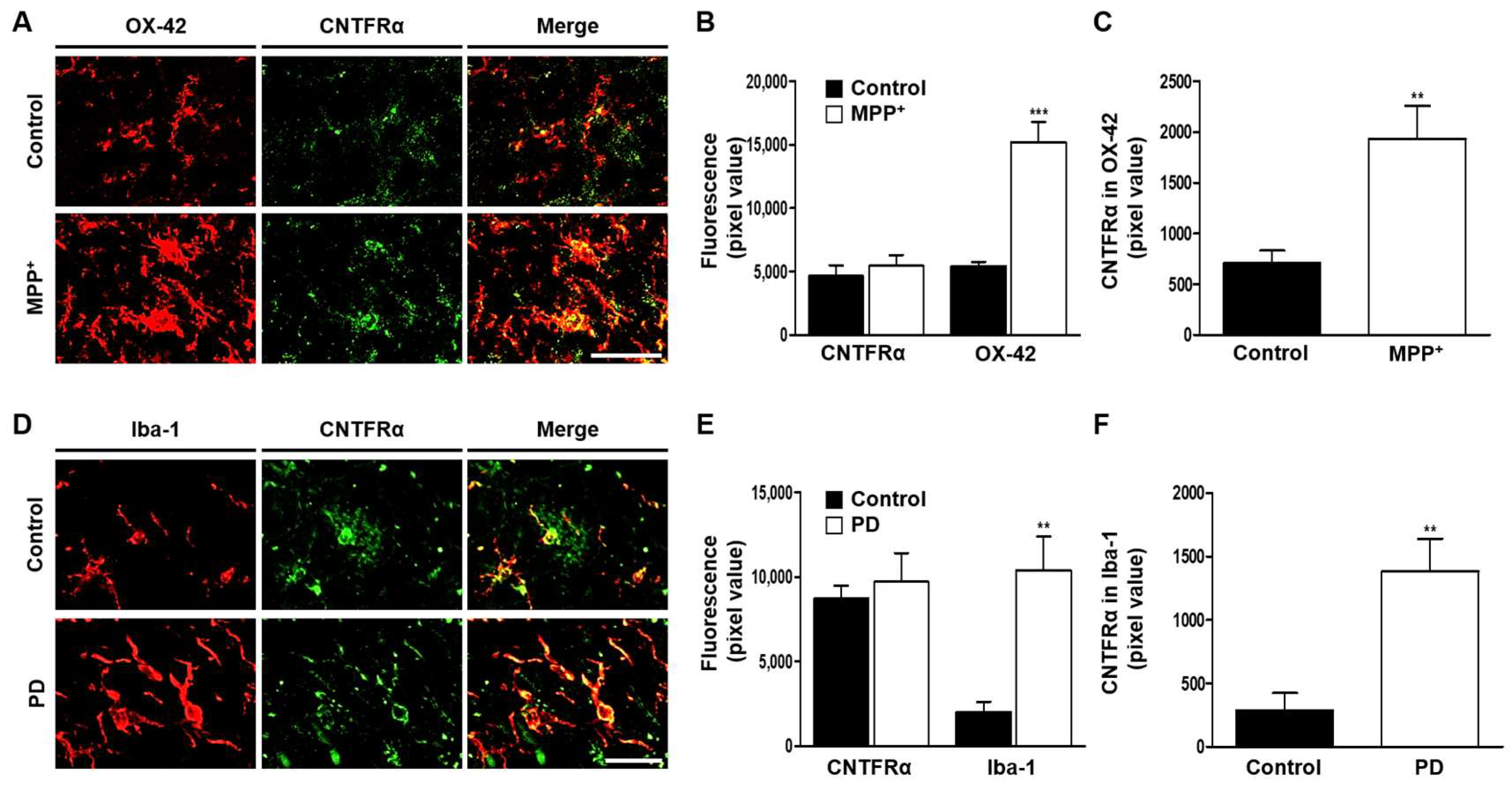
2.3. CAP Treatment Is Unable to Change Basal Levels of CNTF on Microglia in the SN In Vivo of MPP+-Lesioned Rat
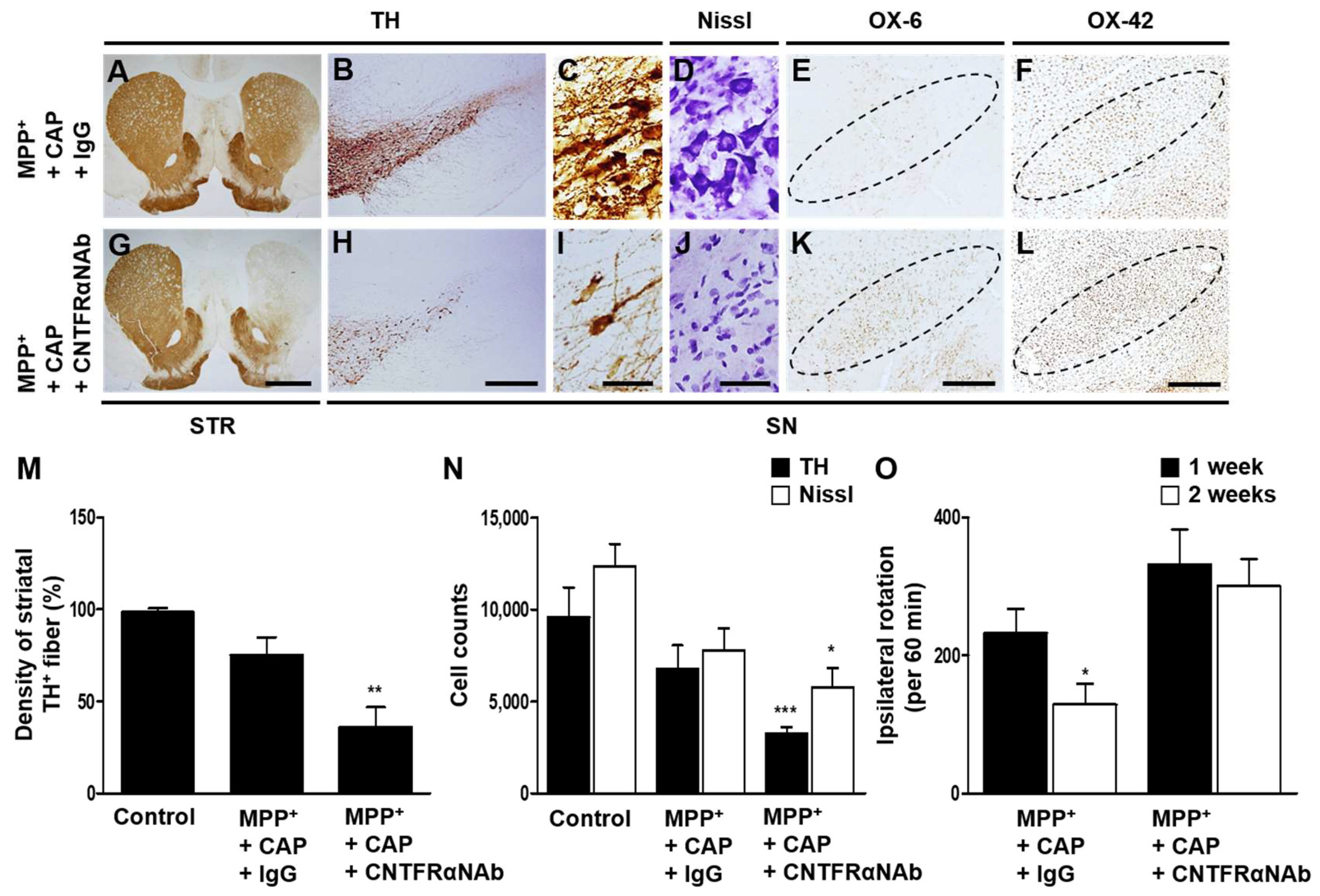
2.4. CNTFRα Neutralization Alters Neuroprotection and Microglial Activation in the SN In Vivo in MPP+-Lesioned Rat
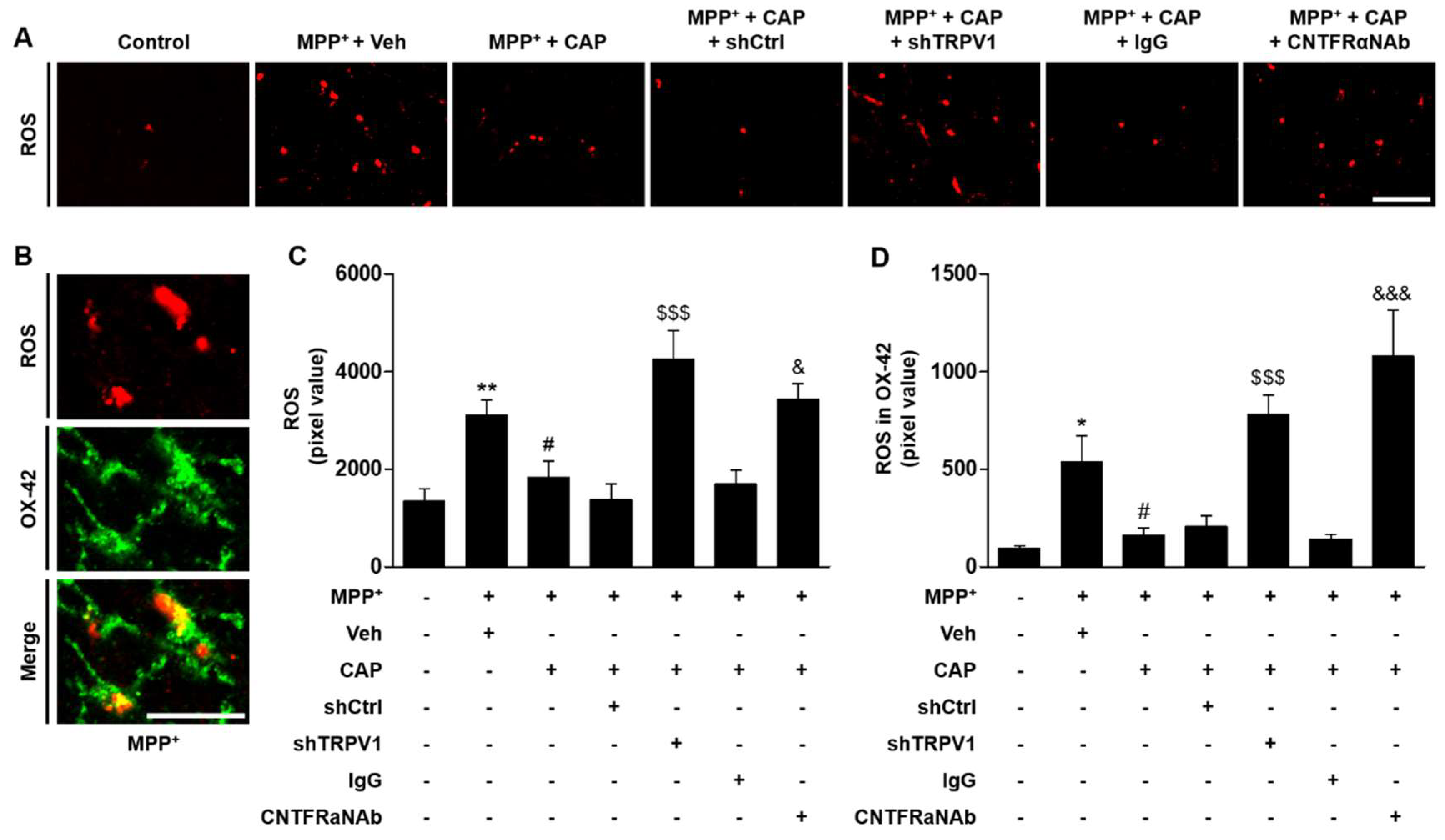
2.5. CNTF Derived from TRPV1 Activated Astrocytes Inhibits Microglial ROS Production in the SN In Vivo in MPP+-Lesioned Rat
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Human Samples
4.3. Stereotaxic Surgery and Tissue Preparation
4.4. Capsaicin (CAP) Injection
4.5. Rotational Behavior Test
4.6. Stereological Estimation
4.7. Morphological Analysis
4.8. Image J Analysis
4.9. Immunostaining
4.10. TRPV1 shRNA and Lentivirus Production
4.11. In Situ Detection of O2− and O2−-Derived Oxidants
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ginhoux, F.; Lim, S.; Hoeffel, G.; Low, D.; Huber, T. Origin and differentiation of microglia. Front. Cell. Neurosci. 2013, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Savitt, J.M.; Dawson, V.L.; Dawson, T.M. Diagnosis and treatment of Parkinson disease: Molecules to medicine. J. Clin. Investig. 2006, 116, 1744–1754. [Google Scholar] [CrossRef] [PubMed]
- Dawson, T.M.; Dawson, V.L. Molecular pathways of neurodegeneration in Parkinson’s disease. Science 2003, 302, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Hunot, S. Neuroinflammation in Parkinson’s disease: A target for neuroprotection? Lancet Neurol. 2009, 8, 382–397. [Google Scholar] [CrossRef]
- Gaki, G.S.; Papavassiliou, A.G. Oxidative stress-induced signaling pathways implicated in the pathogenesis of Parkinson’s disease. Neuromol. Med. 2014, 16, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Park, E.S.; Kim, S.R.; Jin, B.K. Transient receptor potential vanilloid subtype 1 contributes to mesencephalic dopaminergic neuronal survival by inhibiting microglia-originated oxidative stress. Brain Res. Bull. 2012, 89, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Gunthorpe, M.J.; Szallasi, A. Peripheral TRPV1 receptors as targets for drug development: New molecules and mechanisms. Curr. Pharm. Des. 2008, 14, 32–41. [Google Scholar] [PubMed]
- Starowicz, K.; Cristino, L.; Di Marzo, V. TRPV1 receptors in the central nervous system: Potential for previously unforeseen therapeutic applications. Curr. Pharm. Des. 2008, 14, 42–54. [Google Scholar] [PubMed]
- Kauer, J.A.; Gibson, H.E. Hot flash: TRPV channels in the brain. Trends Neurosci. 2009, 32, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.H.; Park, E.S.; Won, S.Y.; Lee, Y.A.; Kim, K.I.; Jeong, J.Y.; Baek, J.Y.; Cho, E.J.; Jin, M.; Chung, Y.C.; et al. TRPV1 on astrocytes rescues nigral dopamine neurons in Parkinson’s disease via CNTF. Brain 2015, 138 Pt 12, 3610–3622. [Google Scholar] [CrossRef] [Green Version]
- Shang, K.; Amna, T.; Amina, M.; Al-Musayeib, N.M.; Al-Deyab, S.S.; Hwang, I. Influence of Capsaicin on Inflammatory Cytokines Induced by Lipopolysaccharide in Myoblast Cells under In vitro Environment. Pharmacogn. Mag. 2017, 13 (Suppl. 1), S26–S32. [Google Scholar] [PubMed]
- Guo, S.Y.; Yang, G.P.; Jiang, D.J.; Wang, F.; Song, T.; Tan, X.H.; Sun, Z.Q. Protection of capsaicin against hypoxia-reoxygenation-induced apoptosis of rat hippocampal neurons. Can. J. Physiol. Pharmacol. 2008, 86, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, H.S.; Roelofs, N.; Muñoz, E.; Fiebich, B.L. Alleviation of Microglial Activation Induced by p38 MAPK/MK2/PGE 2 Axis by Capsaicin: Potential Involvement of other than TRPV1 Mechanism/s. Sci. Rep. 2017, 7, 116. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.W.; Lee, S.T.; Wu, W.T.; Fu, W.M.; Ho, F.M.; Lin, W.W. Signal transduction for inhibition of inducible nitric oxide synthase and cyclooxygenase-2 induction by capsaicin and related analogs in macrophages. Br. J. Pharmacol. 2003, 140, 1077–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.-S.; Kawada, T.; Kim, B.-S.; Han, I.-S.; Choe, S.-Y.; Kurata, T.; Yu, R. Capsaicin exhibits anti-inflammatory property by inhibiting IkB-a degradation in LPS-stimulated peritoneal macrophages. Cell. Signal. 2003, 15, 299–306. [Google Scholar] [CrossRef]
- Stöckli, K.; Lillien, L.; Näher-Noé, M.; Breitfeld, G.; Hughes, R.A.; Raff, M.; Thoenen, H.; Sendtner, M. Regional distribution, developmental changes, and cellular localization of CNTF-mRNA and protein in the rat brain. J. Cell Biol. 1991, 115, 447–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallner, C.; Woods, A.G.; Deller, T.; Kirsch, M.; Hofmann, H.D. CNTF and CNTF receptor alpha are constitutively expressed by astrocytes in the mouse brain. Glia 2002, 37, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Paintlia, M.K.; Paintlia, A.S.; Singh, A.K.; Singh, I. S-nitrosoglutathione induces ciliary neurotrophic factor expression in astrocytes, which has implications to protect the central nervous system under pathological conditions. J. Biol. Chem. 2013, 288, 3831–3843. [Google Scholar] [CrossRef] [PubMed]
- Harada, T.; Harada, C.; Kohsaka, S.; Wada, E.; Yoshida, K.; Ohno, S.; Mamada, H.; Tanaka, K.; Parada, L.F.; Wada, K. Microglia-Muller glia cell interactions control neurotrophic factor production during light-induced retinal degeneration. J. Neurosci. 2002, 22, 9228–9236. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Murakami, K.; Bando, Y.; Yoshida, S. Minocycline reduces remyelination by suppressing ciliary neurotrophic factor expression after cuprizone-induced demyelination. J. Neurochem. 2013, 127, 259–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albrecht, P.J.; Dahl, J.P.; Stoltzfus, O.K.; Levenson, R.; Levison, S.W. Ciliary neurotrophic factor activates spinal cord astrocytes, stimulating their production and release of fibroblast growth factor-2, to increase motor neuron survival. Exp. Neurol. 2002, 173, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Krady, J.K.; Lin, H.W.; Liberto, C.M.; Basu, A.; Kremlev, S.G.; Levison, S.W. Ciliary neurotrophic factor and interleukin-6 differentially activate microglia. J. Neurosci. Res. 2008, 86, 1538–1547. [Google Scholar] [CrossRef] [PubMed]
- Oppenheim, R.W.; Prevette, D.; Yin, Q.; Collins, F.; MacDonald, J. Control of embryonic motoneuron survival in vivo by ciliary neurotrophic factor. Science 1991, 251, 1616–1618. [Google Scholar] [CrossRef] [PubMed]
- Mitsumoto, H.; Ikeda, K.; Klinkosz, B.; Cedarbaum, J.M.; Wong, V.; Lindsay, R.M. Arrest of motor neuron disease in wobbler mice cotreated with CNTF and BDNF. Science 1994, 265, 1107–1110. [Google Scholar] [CrossRef] [PubMed]
- Sagot, Y.; Tan, S.; Baetge, E.; Schmalbruch, H.; Kato, A.; Aebischer, P. Polymer encapsulated cell lines genetically engineered to release ciliary neurotrophic factor can slow down progressive motor neuronopathy in the mouse. Eur. J. Neurosci. 1995, 7, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Hofmann, H.D.; Kirsch, M. Glial reactivity in ciliary neurotrophic factor-deficient mice after optic nerve lesion. J. Neurosci. 2003, 23, 5416–5424. [Google Scholar] [CrossRef] [PubMed]
- Pasquin, S.; Sharma, M.; Gauchat, J.F. Ciliary neurotrophic factor (CNTF): New facets of an old molecule for treating neurodegenerative and metabolic syndrome pathologies. Cytokine Growth Factor Rev. 2015, 26, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Rudge, J.S.; Li, Y.; Pasnikowski, E.M.; Mattsson, K.; Pan, L.; Yancopoulos, G.D.; Wiegand, S.J.; Lindsay, R.M.; Ip, N.Y. Neurotrophic factor receptors and their signal transduction capabilities in rat astrocytes. Eur. J. Neurosci. 1994, 6, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-Y.; Deller, T.; Kirsch, M.; Frotscher, M.; Hofmann, H.-D. Differential regulation of ciliary neurotrophic factor (CNTF) and CNTF receptor α expression in astrocytes and neurons of the fascia dentata after entorhinal cortex lesion. J. Neurosci. 1997, 17, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.W.; Jain, M.R.; Li, H.; Levison, S.W. Ciliary neurotrophic factor (CNTF) plus soluble CNTF receptor alpha increases cyclooxygenase-2 expression, PGE2 release and interferon-gamma-induced CD40 in murine microglia. J. Neuroinflamm. 2009, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Kahn, M.A.; Ellison, J.A.; Speight, G.J.; de Vellis, J. CNTF regulation of astrogliosis and the activation of microglia in the developing rat central nervous system. Brain Res. 1995, 685, 55–67. [Google Scholar] [CrossRef]
- Cen, L.P.; Luo, J.M.; Zhang, C.W.; Fan, Y.M.; Song, Y.; So, K.F.; van Rooijen, N.; Pang, C.P.; Lam, D.S.; Cui, Q. Chemotactic effect of ciliary neurotrophic factor on macrophages in retinal ganglion cell survival and axonal regeneration. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4257–4266. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.I.; Yang, C.S.; Fang, K.M.; Tzeng, S.F. Role of ciliary neurotrophic factor in microglial phagocytosis. Neurochem. Res. 2009, 34, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Keasey, M.P.; Cai, J.; Hagg, T. Loss of neuron-astroglial interaction rapidly induces protective CNTF expression after stroke in mice. J. Neurosci. 2012, 32, 9277–9287. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.-C.; Ko, H.-W.; Bok, E.-G.; Park, E.-S.; Huh, S.-H.; Nam, J.-H.; Jin, B.-K. The role of neuroinflammation on the pathogenesis of Parkinsons disease. BMB Rep. 2010, 43, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.J.; Flood, P.M. Oxidative stress and microglial cells in Parkinson’s disease. Mediat. Inflamm. 2012. [Google Scholar] [CrossRef] [PubMed]
- Berndt, C.; Lillig, C.H. Glutathione, Glutaredoxins, and Iron. Antioxid. Redox Signal. 2017. [Google Scholar] [CrossRef] [PubMed]
- Venkateshappa, C.; Harish, G.; Mythri, R.B.; Mahadevan, A.; Bharath, M.M.; Shankar, S.K. Increased oxidative damage and decreased antioxidant function in aging human substantia nigra compared to striatum: Implications for Parkinson’s disease. Neurochem. Res. 2012, 37, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Mischley, L.K.; Allen, J.; Bradley, R. Coenzyme Q10 deficiency in patients with Parkinson’s disease. J. Neurol. Sci. 2012, 318, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Kim, C.J.; Shin, S.L.; Moon, S.H.; Chun, M.H. Increased ciliary neurotrophic factor expression in reactive astrocytes following spinal cord injury in the rat. Neurosci. Lett. 1998, 255, 79–82. [Google Scholar] [CrossRef]
- Park, C.K.; Ju, W.K.; Hofmann, H.D.; Kirsch, M.; Ki Kang, J.; Chun, M.H.; Lee, M.Y. Differential regulation of ciliary neurotrophic factor and its receptor in the rat hippocampus following transient global ischemia. Brain Res. 2000, 861, 345–353. [Google Scholar] [CrossRef]
- Kong, W.L.; Peng, Y.Y.; Peng, B.W. Modulation of neuroinflammation: Role and therapeutic potential of TRPV1 in the neuro-immune axis. Brain Behav. Immun. 2017, 64, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 6th ed.; Academic Press/Elsevier: Amsterdam, The Netherlands; Boston, MA, USA, 2007. [Google Scholar]
- Choi, S.H.; Joe, E.H.; Kim, S.U.; Jin, B.K. Thrombin-induced microglial activation produces degeneration of nigral dopaminergic neurons in vivo. J. Neurosci. 2003, 23, 5877–5886. [Google Scholar] [CrossRef] [PubMed]
- Veldhuis, W.B.; van der Stelt, M.; Wadman, M.W.; van Zadelhoff, G.; Maccarrone, M.; Fezza, F.; Veldink, G.A.; Vliegenthart, J.F.; Bar, P.R.; Nicolay, K.; et al. Neuroprotection by the endogenous cannabinoid anandamide and arvanil against in vivo excitotoxicity in the rat: Role of vanilloid receptors and lipoxygenases. J. Neurosci. 2003, 23, 4127–4133. [Google Scholar] [CrossRef] [PubMed]
- Christoph, T.; Bahrenberg, G.; De Vry, J.; Englberger, W.; Erdmann, V.A.; Frech, M.; Kogel, B.; Rohl, T.; Schiene, K.; Schroder, W.; et al. Investigation of TRPV1 loss-of-function phenotypes in transgenic shRNA expressing and knockout mice. Mol. Cell. Neurosci. 2008, 37, 579–589. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample No. | Final Diagnosis | Age | Sex | PMD | Staining | Tissue |
|---|---|---|---|---|---|---|
| 04-424 | Control | 75.1 | F | 22.5 | Iba-1 + CNTFRα | SN |
| 08-026 | 67.3 | F | 24 | |||
| 07-787 | 66.5 | M | 19 | |||
| 09-260 | PD | 66.8 | F | 20 | ||
| V11-042 | 72.1 | M | 25 | |||
| V11-007 | 74 | F | 14 |
| Capsaicin Dose (mg/kg) | Lethality | Pain Degree | Behavioral Symptoms (Paroxysm, Spasticity, Couching) | ||
|---|---|---|---|---|---|
| Dead | Total | % | |||
| 1 mg/kg | 0 | 18 | 0 | + | Last for 1–2 min after capsaicin injection only at 1st day |
| 5 mg/kg | 1 | 15 | 6.6 | ++ | Last for 5–10 min after capsaicin injection for 7 days |
| 10 mg/kg | 9 | 15 | 60 | +++ | Last for more than 10 min after capsaicin injection for 7 days Some of rat (N = 9) was dead in less than 1 min |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baek, J.Y.; Jeong, J.Y.; Kim, K.I.; Won, S.-Y.; Chung, Y.C.; Nam, J.H.; Cho, E.J.; Ahn, T.-B.; Bok, E.; Shin, W.-H.; et al. Inhibition of Microglia-Derived Oxidative Stress by Ciliary Neurotrophic Factor Protects Dopamine Neurons In Vivo from MPP+ Neurotoxicity. Int. J. Mol. Sci. 2018, 19, 3543. https://doi.org/10.3390/ijms19113543
Baek JY, Jeong JY, Kim KI, Won S-Y, Chung YC, Nam JH, Cho EJ, Ahn T-B, Bok E, Shin W-H, et al. Inhibition of Microglia-Derived Oxidative Stress by Ciliary Neurotrophic Factor Protects Dopamine Neurons In Vivo from MPP+ Neurotoxicity. International Journal of Molecular Sciences. 2018; 19(11):3543. https://doi.org/10.3390/ijms19113543
Chicago/Turabian StyleBaek, Jeong Yeob, Jae Yeong Jeong, Kyoung In Kim, So-Yoon Won, Young Cheul Chung, Jin Han Nam, Eun Ju Cho, Tae-Beom Ahn, Eugene Bok, Won-Ho Shin, and et al. 2018. "Inhibition of Microglia-Derived Oxidative Stress by Ciliary Neurotrophic Factor Protects Dopamine Neurons In Vivo from MPP+ Neurotoxicity" International Journal of Molecular Sciences 19, no. 11: 3543. https://doi.org/10.3390/ijms19113543