TNF-α-Induced YAP/TAZ Activity Mediates Leukocyte-Endothelial Adhesion by Regulating VCAM1 Expression in Endothelial Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
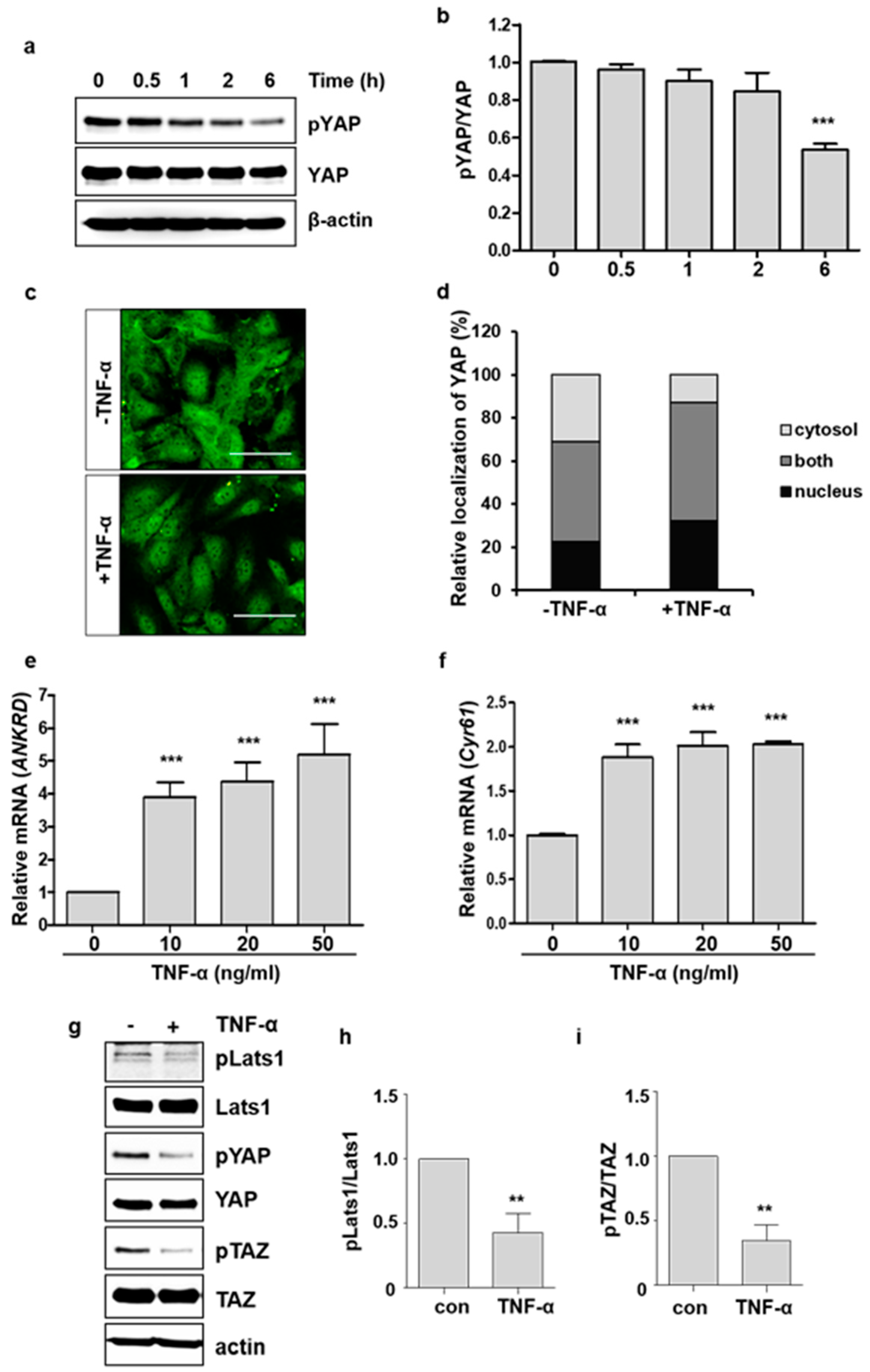
2.1. TNF-α Promotes YAP Activation
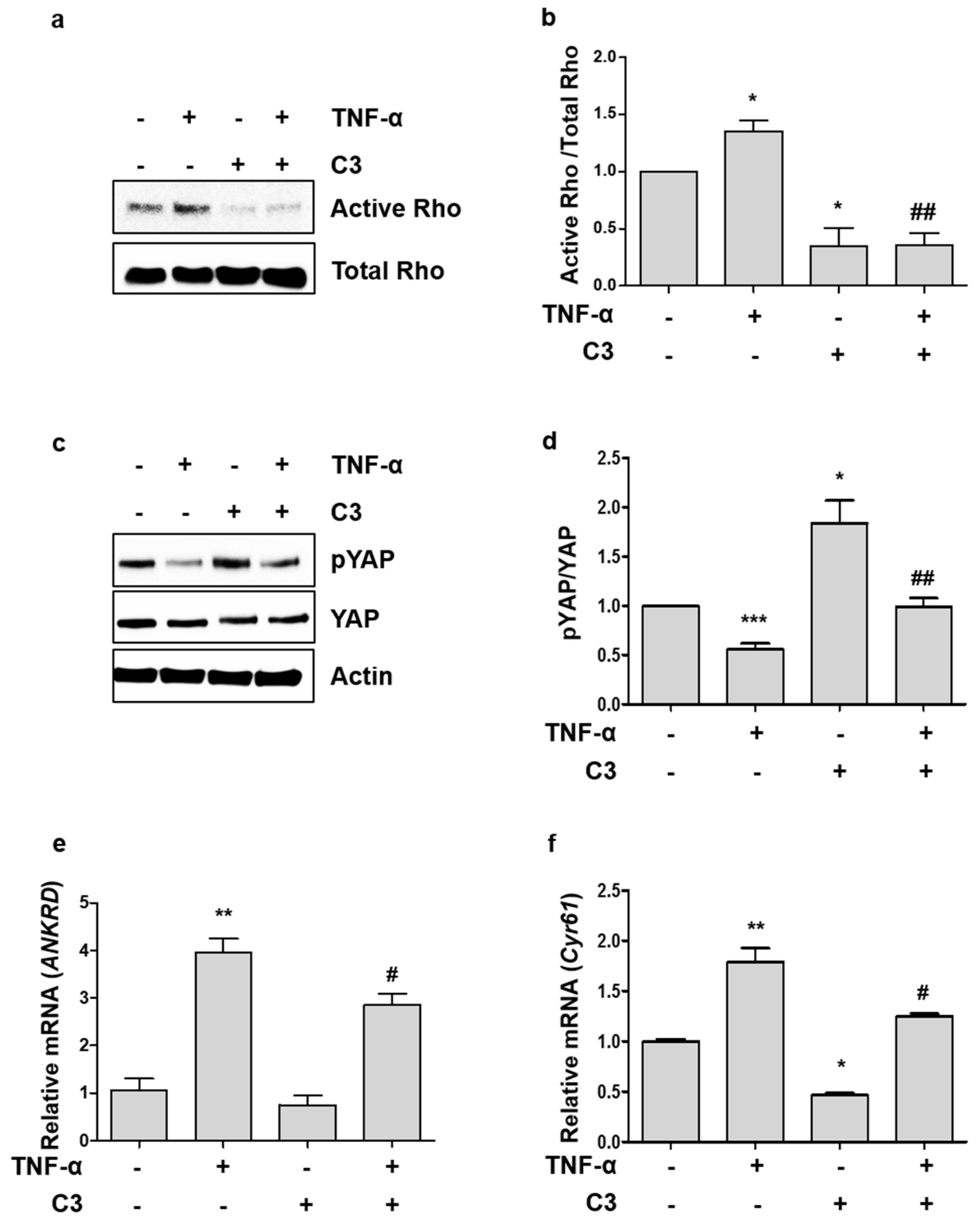
2.2. TNF-α-Induced YAP Dephosphorylation is Dependent on Activation of Rho GTPases
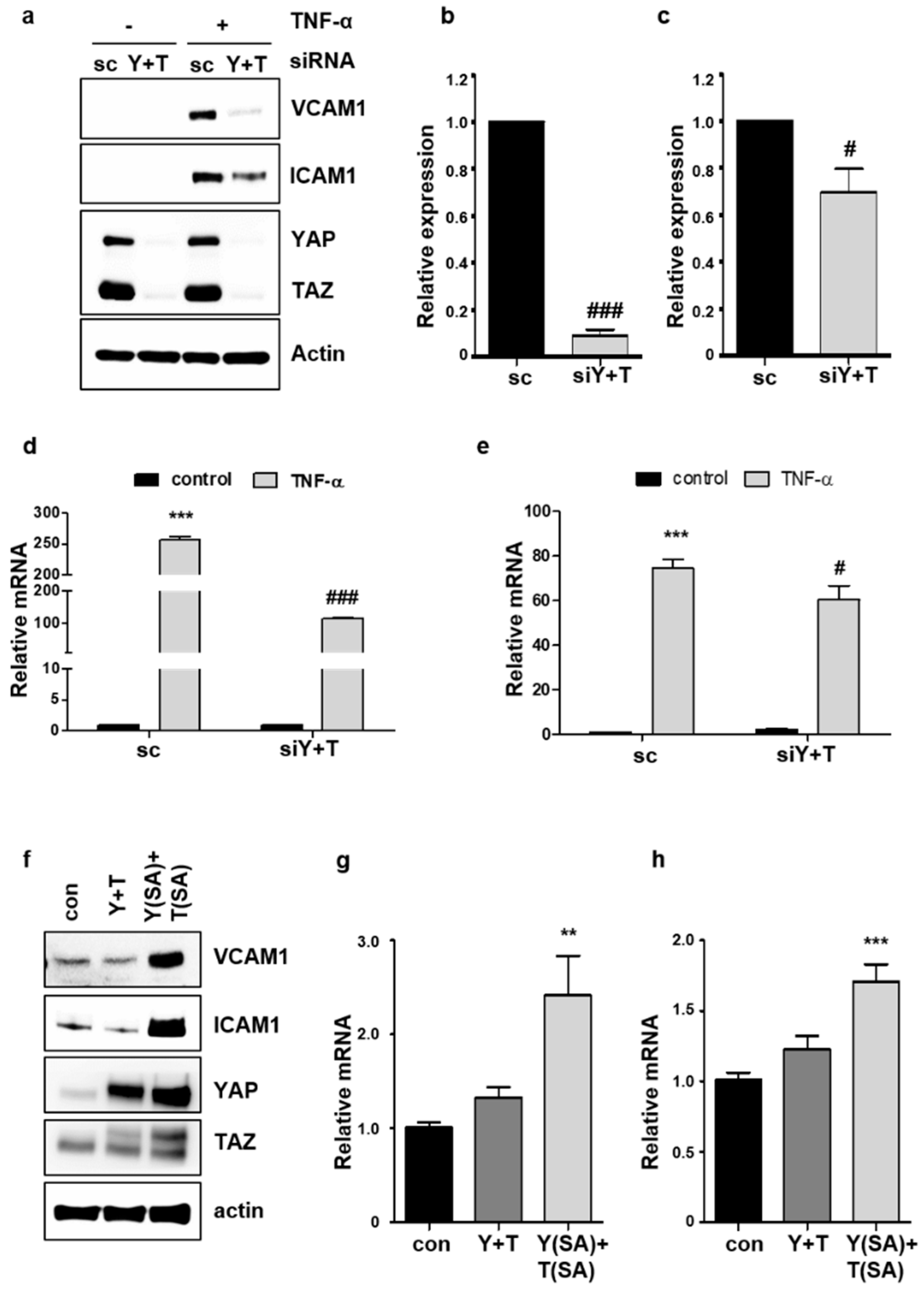
2.3. Knockdown of YAP/TAZ Inhibits TNF-α-Induced VCAM1 Expression
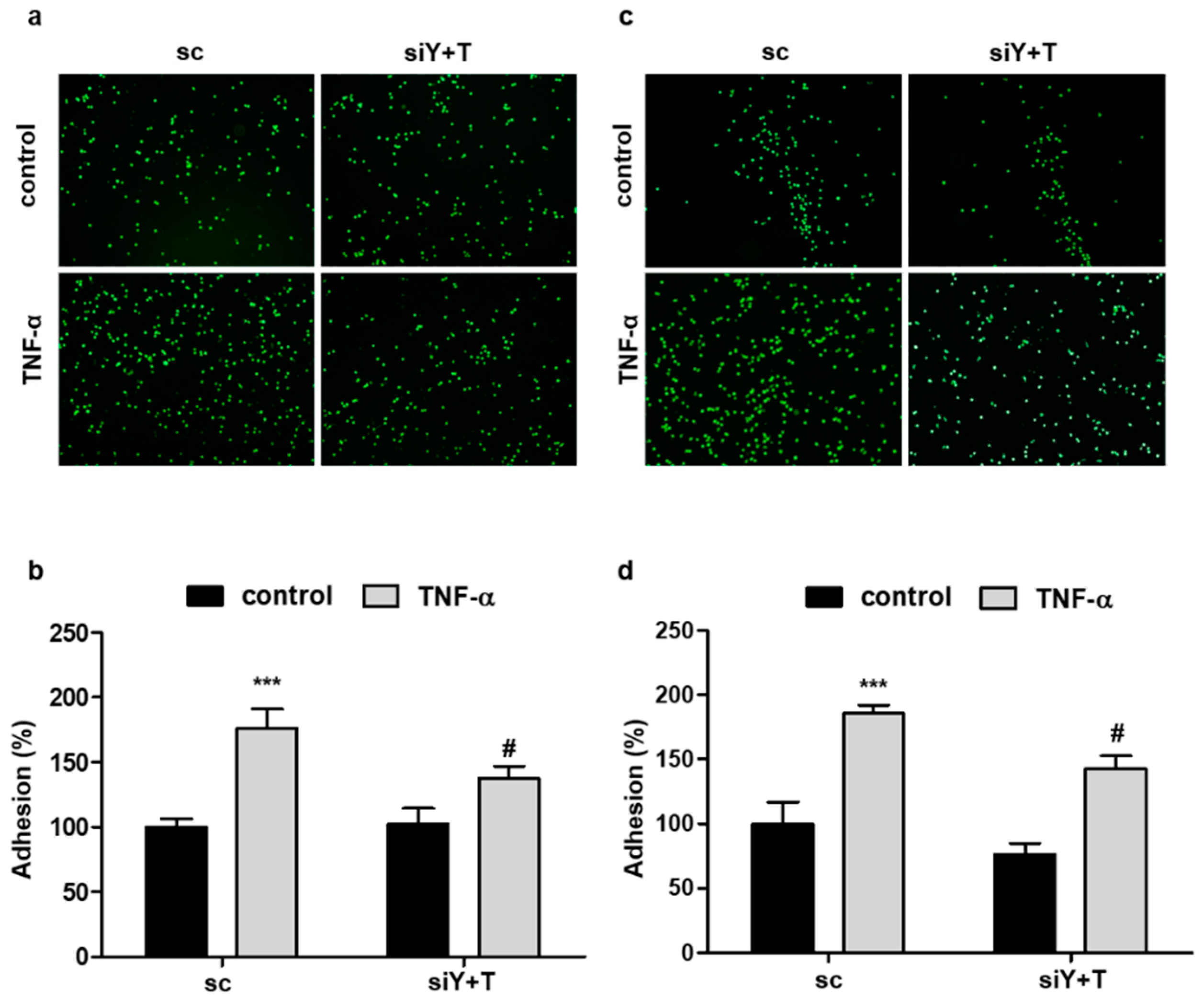
2.4. Knockdown of YAP/TAZ Reduces the Adhesion of Monocytes to Endothelial Cells
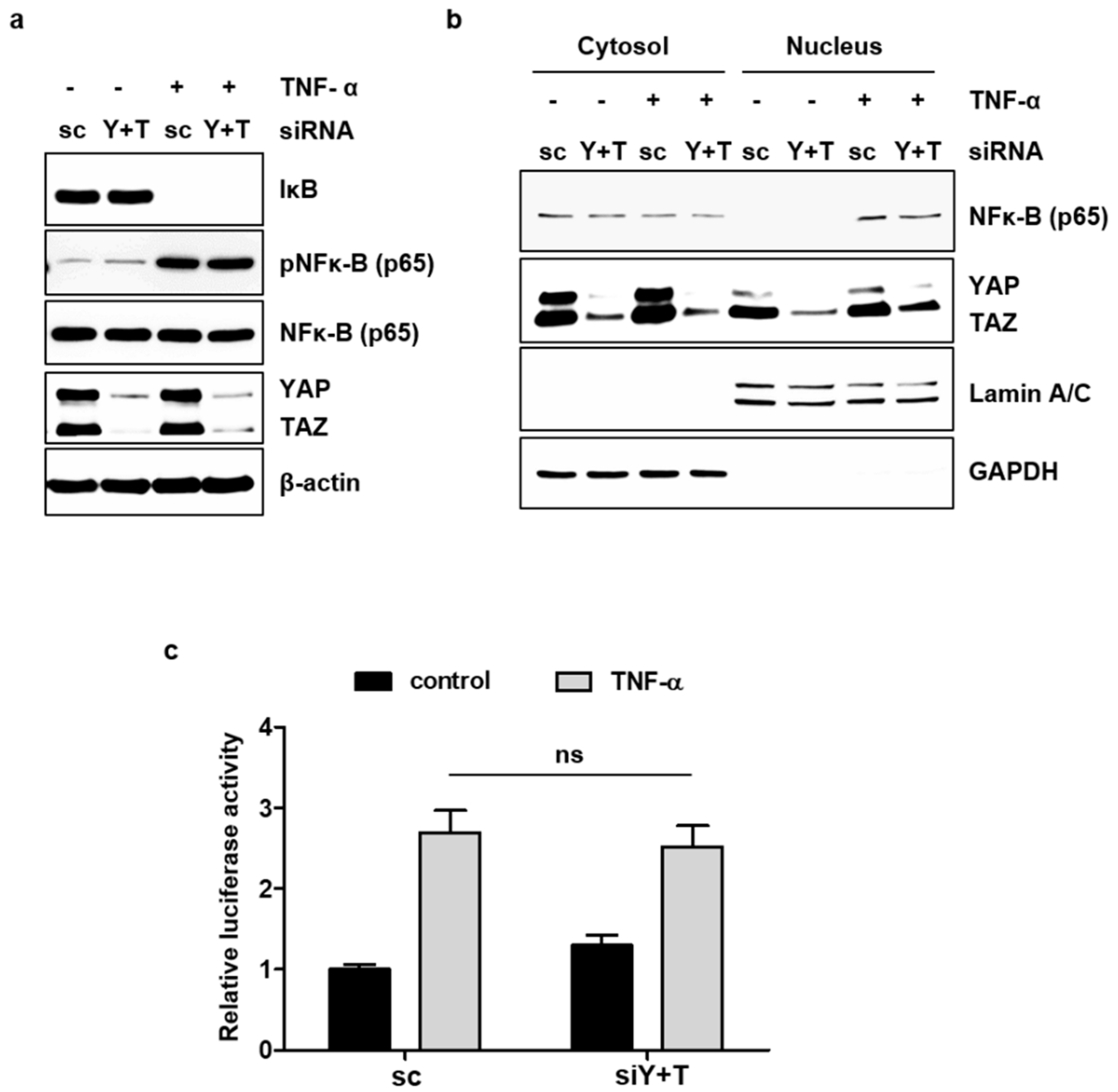
2.5. YAP/TAZ Does Not Affect TNF-α-Induced NF-κB Signaling
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Transfection with siRNA and Plasmid DNA In Vitro
4.3. Western Blot Analysis
4.4. Immunofluorescence Staining
4.5. qRT-PCR
4.6. Luciferase Assay
4.7. Rho GTPase Activation Assay
4.8. Adhesion Assay
4.9. Preparation of Nuclear and Cytoplasmic Fractions
5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef] [PubMed]
- Halder, G.; Dupont, S.; Piccolo, S. Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat. Rev. Mol. Cell Biol. 2012, 13, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Gumbiner, B.M.; Kim, N.G. The Hippo-YAP signaling pathway and contact inhibition of growth. J. Cell Sci. 2014, 127, 709–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, Y.H.; Kim, J.; Park, D.Y.; Bae, H.; Lee, D.H.; Kim, K.H.; Hong, S.P.; Jang, S.P.; Kubota, Y.; et al. YAP/TAZ regulates sprouting angiogenesis and vascular barrier maturation. J. Clin. Investig. 2017, 127, 3441–3461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Bao, Q.; Zhang, Y.; Liu, M.; Lv, H.; Liu, Y.; Yao, L.; Li, B.; Zhang, C.; He, S.; et al. Yes-Associated Protein Promotes Angiogenesis via Signal Transducer and Activator of Transcription 3 in Endothelial Cells. Circ. Res. 2018, 122, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Freire Valls, A.; Schermann, G.; Shen, Y.; Moya, I.M.; Castro, L.; Urban, S.; Solecki, G.M.; Winkler, F.; Riedemann, L.; et al. YAP/TAZ Orchestrate VEGF Signaling during Developmental Angiogenesis. Dev. Cell 2017, 42, 462–478. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Kwon, Y.G. Roles of YAP in mediating endothelial cell junctional stability and vascular remodeling. BMB Rep. 2015, 48, 429–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.J.; Zhang, H.; Park, H.; Choi, K.S.; Lee, H.W.; Agrawal, V.; Kim, Y.M.; Kwon, Y.G. Yes-associated protein regulates endothelial cell contact-mediated expression of angiopoietin-2. Nat. Commu. 2015, 6, 6943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.K.; Thiagarajan, P. Role of endothelium in thrombosis and hemostasis. Annu. Rev. Med. 1996, 47, 315–331. [Google Scholar] [PubMed]
- McKellar, G.E.; McCarey, D.W.; Sattar, N.; McInnes, I.B. Role for TNF in atherosclerosis? Lessons from autoimmune disease. Nat. Rev. Cardiol. 2009, 6, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Park, Y.; Wu, J.; Chen, X.; Lee, S.; Yang, J.; Dellsperger, K.C.; Zhang, C. Role of TNF-alpha in vascular dysfunction. Clin. Sci. 2009, 116, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Wu, L.W.; Grivennikov, S.I.; de Jong, P.R.; Lian, I.; Yu, F.X.; Wang, K.; Ho, S.B.; Boland, B.S.; Chang, J.T.; et al. A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature 2015, 519, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wang, Y.; Hu, G.; Zhou, J.; Mei, L.; Xiong, W.C. YAP Is a Critical Inducer of SOCS3, Preventing Reactive Astrogliosis. Cereb. Cortex 2016, 26, 2299–2310. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.S.; Yu, F.X.; Gong, R.; Brown, J.H.; Guan, K.L. Regulation of the Hippo-YAP pathway by protease-activated receptors (PARs). Genes Dev. 2012, 26, 2138–2143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pober, J.S.; Cotran, R.S. Cytokines and endothelial cell biology. Physiol. Rev. 1990, 70, 427–451. [Google Scholar] [CrossRef] [PubMed]
- Collins, T.; Read, M.A.; Neish, A.S.; Whitley, M.Z.; Thanos, D.; Maniatis, T. Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. FASEB J. 1995, 9, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.K. Linking endothelial dysfunction with endothelial cell activation. J.Clin. Investig. 2013, 123, 540–541. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Luo, J.Y.; Li, B.; Tian, X.Y.; Chen, L.J.; Huang, Y.; Liu, J.; Deng, D.; Lau, C.W.; Wan, S.; et al. Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature 2016, 540, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Koroleva, M.; Yin, M.; Jin, Z.G. Atheroprotective laminar flow inhibits Hippo pathway effector YAP in endothelial cells. Transl. Res. 2016, 176, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Huang, X.; Guo, F.; Zhou, Z.; Dou, Y.; Huan, J. Yes-associated protein (YAP) signaling regulates lipopolysaccharide-induced tissue factor expression in human endothelial cells. Surgery 2016, 159, 1436–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohgushi, M.; Minaguchi, M.; Sasai, Y. Rho-Signaling-Directed YAP/TAZ Activity Underlies the Long-Term Survival and Expansion of Human Embryonic Stem Cells. Cell Stem Cell 2015, 17, 448–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Chiu, J.J.; Lee, P.L.; Chen, C.N.; Lee, C.I.; Chang, S.F.; Chen, L.J.; Lien, S.C.; Ko, Y.C.; Usami, S.; Chien, S. Shear stress increases ICAM-1 and decreases VCAM-1 and E-selectin expressions induced by tumor necrosis factor-[alpha] in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Nagel, T.; Resnick, N.; Atkinson, W.J.; Dewey, C.F., Jr.; Gimbrone, M.A., Jr. Shear stress selectively upregulates intercellular adhesion molecule-1 expression in cultured human vascular endothelial cells. J. Clin. Investig. 1994, 94, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Marui, N.; Offermann, M.K.; Swerlick, R.; Kunsch, C.; Rosen, C.A.; Ahmad, M.; Alexander, R.W.; Medford, R.M. Vascular cell adhesion molecule-1 (VCAM-1) gene transcription and expression are regulated through an antioxidant-sensitive mechanism in human vascular endothelial cells. J. Clin. Investig. 1993, 92, 1866–1874. [Google Scholar] [CrossRef] [PubMed]
- Cook-Mills, J.M.; Marchese, M.E.; Abdala-Valencia, H. Vascular cell adhesion molecule-1 expression and signaling during disease: Regulation by reactive oxygen species and antioxidants. Antioxid. Redox Signal. 2011, 15, 1607–1638. [Google Scholar] [CrossRef] [PubMed]
- Iiyama, K.; Hajra, L.; Iiyama, M.; Li, H.; DiChiara, M.; Medoff, B.D.; Cybulsky, M.I. Patterns of vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 expression in rabbit and mouse atherosclerotic lesions and at sites predisposed to lesion formation. Circ. Res. 1999, 85, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Cybulsky, M.I.; Iiyama, K.; Li, H.; Zhu, S.; Chen, M.; Iiyama, M.; Davis, V.; Gutierrez-Ramos, J.C.; Connelly, P.W.; Milstone, D.S. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J. Clin. Investig. 2001, 107, 1255–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barringhaus, K.G.; Phillips, J.W.; Thatte, J.S.; Sanders, J.M.; Czarnik, A.C.; Bennett, D.K.; Ley, K.F.; Sarembock, I.J. Alpha4beta1 integrin (VLA-4) blockade attenuates both early and late leukocyte recruitment and neointimal growth following carotid injury in apolipoprotein E (-/-) mice. J. Vasc. Res. 2004, 41, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Shi, X.; Zhang, H.; Sun, W.; Han, S.; Yu, C.; Li, J. VCAM-1 siRNA reduces neointimal formation after surgical mechanical injury of the rat carotid artery. J. Vasc. Surg. 2009, 50, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Yeh, Y.T.; Nguyen, P.; Limqueco, E.; Lopez, J.; Thorossian, S.; Guan, K.L.; Li, Y.J.; Chien, S. Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc. Natl. Acad. Sci. USA 2016, 113, 11525–11530. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Zhang, Z.; Zou, C.; Tian, Q.; Chen, X.; Hong, M.; Liu, X.; Chen, Q.; Xu, Z.; Li, M.; et al. Hippo/YAP signaling pathway mitigates blood-brain barrier disruption after cerebral ischemia/reperfusion injury. Behav. Brain Res. 2019, 356, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Kim, K.; Sheng, Y.; Cho, J.; Qian, Z.; Zhao, Y.Y.; Hu, G.; Pan, D.; Malik, A.B.; Hu, G. YAP Controls Endothelial Activation and Vascular Inflammation Through TRAF6. Circ. Res. 2018, 123, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Finch-Edmondson, M.L.; Strauss, R.P.; Passman, A.M.; Sudol, M.; Yeoh, G.C.; Callus, B.A. TAZ Protein Accumulation Is Negatively Regulated by YAP Abundance in Mammalian Cells. J. Biol. Chem. 2015, 290, 27928–27938. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.S.; Park, J.A.; Kim, J.; Rho, S.S.; Park, H.; Kim, Y.M.; Kwon, Y.G. Nuclear IL-33 is a transcriptional regulator of NF-kappaB p65 and induces endothelial cell activation. Biochem. Biophys. Res Commun. 2012, 421, 305–311. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, H.-J.; Kim, N.-E.; Kim, B.M.; Seo, M.; Heo, J.H. TNF-α-Induced YAP/TAZ Activity Mediates Leukocyte-Endothelial Adhesion by Regulating VCAM1 Expression in Endothelial Cells. Int. J. Mol. Sci. 2018, 19, 3428. https://doi.org/10.3390/ijms19113428
Choi H-J, Kim N-E, Kim BM, Seo M, Heo JH. TNF-α-Induced YAP/TAZ Activity Mediates Leukocyte-Endothelial Adhesion by Regulating VCAM1 Expression in Endothelial Cells. International Journal of Molecular Sciences. 2018; 19(11):3428. https://doi.org/10.3390/ijms19113428
Chicago/Turabian StyleChoi, Hyun-Jung, Na-Eun Kim, Byeong Mo Kim, Miran Seo, and Ji Hoe Heo. 2018. "TNF-α-Induced YAP/TAZ Activity Mediates Leukocyte-Endothelial Adhesion by Regulating VCAM1 Expression in Endothelial Cells" International Journal of Molecular Sciences 19, no. 11: 3428. https://doi.org/10.3390/ijms19113428