New Insight into the Chloroacetanilide Herbicide Degradation Mechanism through a Nucleophilic Attack of Hydrogen Sulfide


Abstract
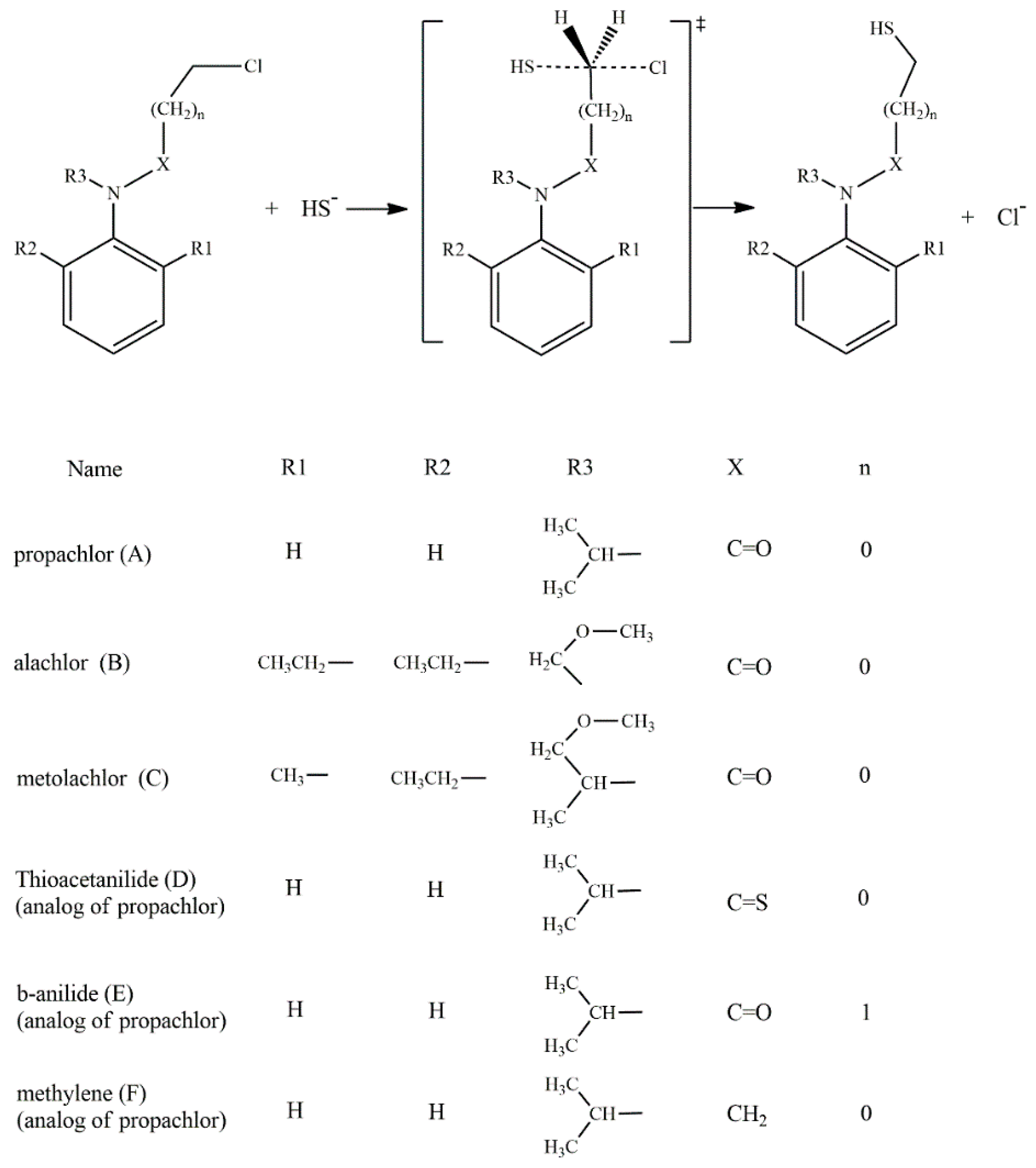
:1. Introduction
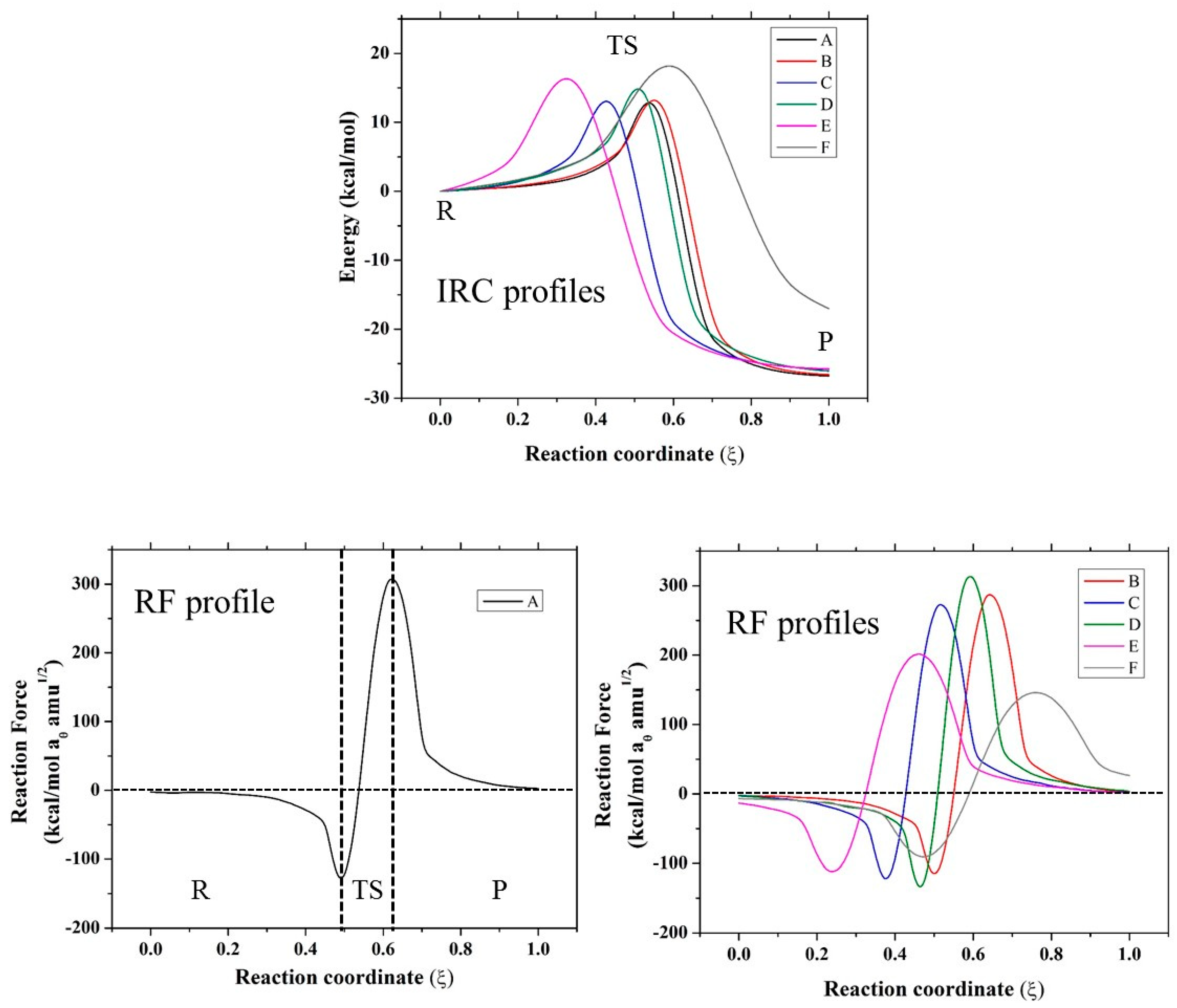
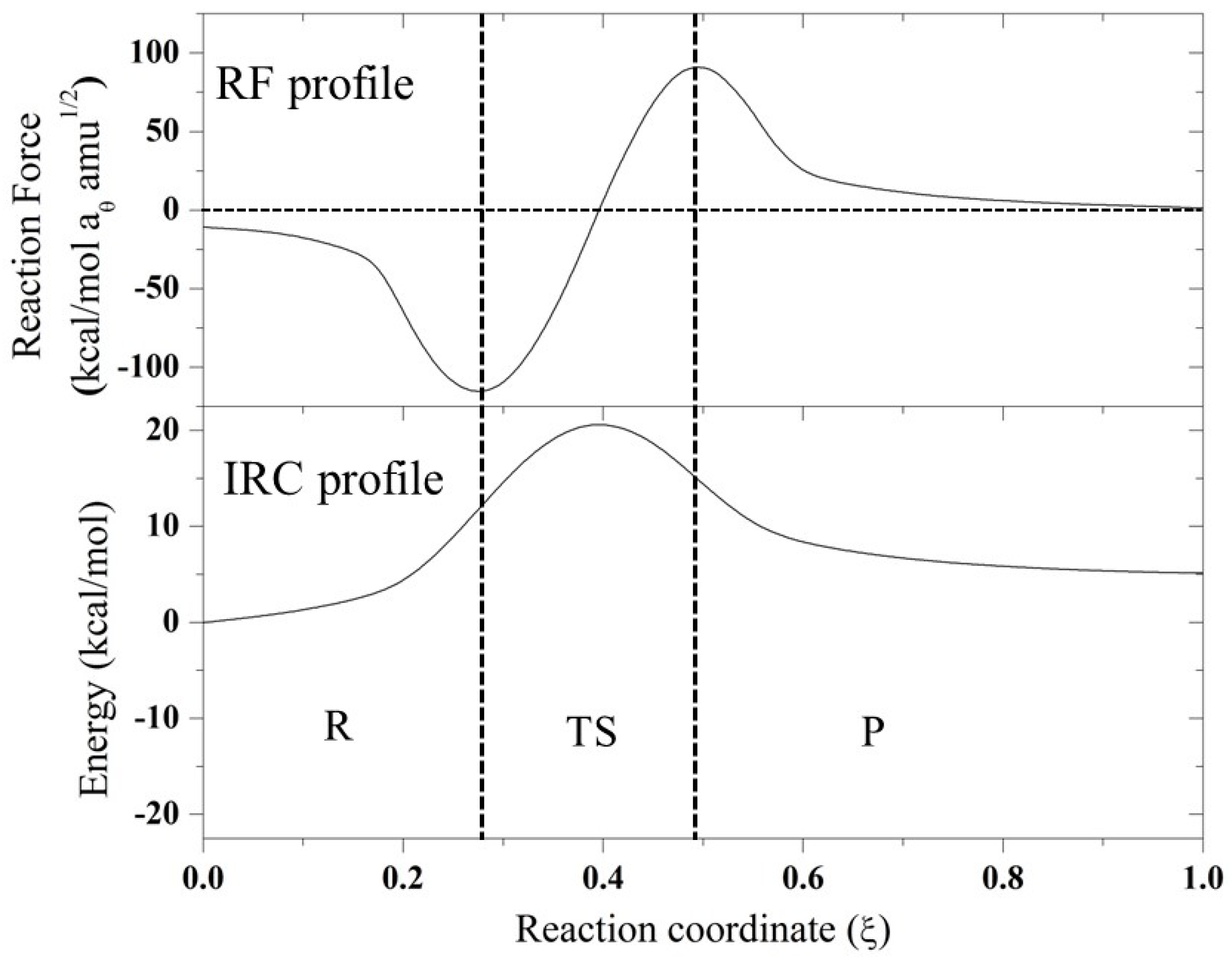
2. Results and Discussion
2.1. Thermodynamic Parameters
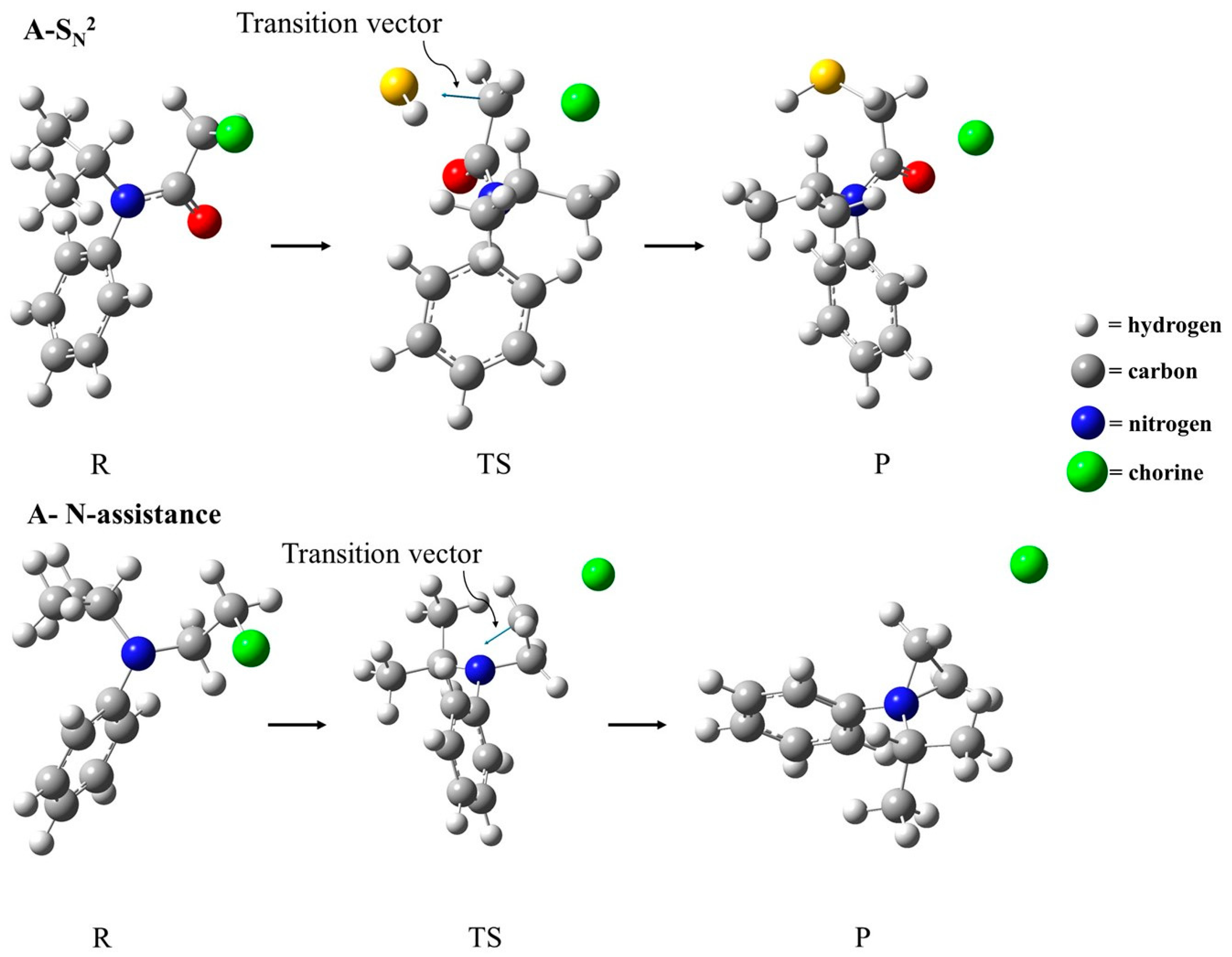
2.2. Geometric Parameters
2.3. Natural Bond Orbital (NBO) Analysis
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Vogel, J.R.; Majewski, M.S.; Capel, P.D. Pesticides in Rain in Four Agricultural Watersheds in the United States. J. Environ. Qual. 2008, 37, 1101. [Google Scholar] [CrossRef] [PubMed]
- Barbash, J.E.; Thelin, G.P.; Kolpin, D.W.; Gilliom, R.J. Major Herbicides in Ground Water: Results from the National Water-Quality Assessment Major Herbicides in Ground Water: Results from the National Water-Quality Assessment. J. Environ. Qual. 2001, 30, 831–845. [Google Scholar] [CrossRef] [PubMed]
- Dearfield, K.L.; McCarroll, N.E.; Protzel, A.; Stack, H.F.; Jackson, M.A.; Waters, M.D. A survey of EPA/OPP and open literature on selected pesticide chemicals II. Mutagenicity and carcinogenicity of selected chloroacetanilides and related compounds. Mutat. Res. 1999, 443, 183–221. [Google Scholar] [CrossRef]
- Coates, J.D.; Bruce, R.A.; Haddock, J.D. Anoxic bioremediation of hydrocarbons. Nature 1998, 396, 730. [Google Scholar] [CrossRef] [PubMed]
- Adrian, L.; Szewzyk, U.; Wecke, J.; Görisch, H. Bacterial dehalorespiration with chlorinated benzenes. Nature 2000, 408, 580–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwillich, T. A tentative comeback for bioremediation. Science 2000, 289, 2266–2267. [Google Scholar] [CrossRef] [PubMed]
- Lovley, D.R. Bioremediation: Anaerobes to the rescue. Science 2001, 293, 1444–1446. [Google Scholar] [CrossRef] [PubMed]
- Fuerst, E.P. Understanding the Mode of Action of the Chloroacetamide and Thiocarbamate Herbicides. Weed Technol. 1987, 1, 270–277. [Google Scholar] [CrossRef]
- Gan, J.; Wang, Q.; Yates, S.R.; Koskinen, W.C.; Jury, W.A. Dechlorination of chloroacetanilide herbicides by thiosulfate salts. Proc. Natl. Acad. Sci. USA 2002, 99, 5189–5194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loch, A.R.; Lippa, K.A.; Carlson, D.L.; Chin, Y.P.; Traina, S.J.; Roberts, A.L. Nucleophilic aliphatic substitution reactions of propachlor, alachlor, and metolachlor with bisulfide (HS−) and polysulfides (Sn2−). Environ. Sci. Technol. 2002, 36, 4065–4073. [Google Scholar] [CrossRef] [PubMed]
- Bordwell, F.G.; Brannen, W.T. The Effect of the Carbonyl and Related Groups on the Reactivity of Halides in SN2 Reactions. J. Am. Chem. Soc. 1964, 86, 4645–4650. [Google Scholar] [CrossRef]
- Bach, R.D.; Coddens, B.A.; Wolber, G.J. Origin of the Reactivity of Allyl Chloride and a-Chloroacetaldehyde in SN2 Nucleophilic Substitution Reactions: A Theoretical Comparison. J. Org. Chem. 1986, 51, 1030–1033. [Google Scholar] [CrossRef]
- Lippa, K.A.; Roberts, A.L. Correlation analyses for bimolecular nucleophilic substitution reactions of chloroacetanilide herbicides and their structural analogs with environmentally relevant nucleophiles. Environ. Toxicol. Chem. 2005, 24, 2401–2409. [Google Scholar] [CrossRef] [PubMed]
- Arcelli, A.; Papa, M.; Porzi, G.; Sandri, S. Participation of neighbouring amide group in the competitive acid catalysed hydrolysis of ether linkage and an intramolecular S(N)2 reactions. Tetrahedron 1997, 53, 10513–10516. [Google Scholar] [CrossRef]
- Lippa, K.A.; Demel, S.; Lau, I.H.; Roberts, A.L. Kinetics and mechanism of the nucleophilic displacement reactions of chloroacetanilide herbicides: Investigation of alpha-substituent effects. J. Agric. Food Chem. 2004, 52, 3010–3021. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A. 03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skara, G.; Pinter, B.; Top, J.; Geerlings, P.; De Proft, F.; De Vleeschouwer, F. Conceptual Quantum Chemical Analysis of Bonding and Noncovalent Interactions in the Formation of Frustrated Lewis Pairs. Chem. A Eur. J. 2015, 21, 5510–5519. [Google Scholar] [CrossRef] [PubMed]
- Villegas-Escobar, N.; Toro-Labbé, A.; Becerra, M.; Real-Enriquez, M.; Mora, J.R.; Rincon, L. A DFT study of hydrogen and methane activation by B(C6F5)3/P(t-Bu)3 and Al(C6F5)3/P(t-Bu)3 frustrated Lewis pairs. J. Mol. Model. 2017, 23, 234. [Google Scholar] [CrossRef] [PubMed]
- Papajak, E.; Zheng, J.; Xu, X.; Leverentz, H.R.; Truhlar, D. Perspectives on Basis Sets Beautiful: Seasonal Plantings of Diffuse Basis Functions. J. Chem. Theory Comput. 2011, 7, 3027–3034. [Google Scholar] [CrossRef] [PubMed]
- Julio, L.L.; Mora, J.R.; Maldonado, A.; Chuchani, G. Gas-phase elimination kinetics of selected aliphatic α,β-unsaturated aldehydes catalyzed by hydrogen chloride. J. Phys. Org. Chem. 2015, 28, 261–265. [Google Scholar] [CrossRef]
- Ochterski, J.W. Thermochemistry in Gaussian; Gaussian Inc.: Pittsburgh, PA, USA, 2000; Volume 264, pp. 1–19. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Hratchian, H.P.; Schlegel, H.B. Accurate reaction paths using a Hessian based predictor-corrector integrator. J. Chem. Phys. 2004, 120, 9918–9924. [Google Scholar] [CrossRef] [PubMed]
- Hratchian, H.P.; Schlegel, H.B. Using Hessian updating to increase the efficiency of a Hessian based predictor-corrector reaction path following method. J. Chem. Theory Comput. 2005, 1, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.; Bernhard Schlegel, H. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- Toro-Labbe, A. Characterization of chemical reactions from the profiles of energy, chemical potential and hardness. J. Phys. Chem. A 1999, 103, 4398–4403. [Google Scholar] [CrossRef]
- Herrera, B.; Toro-Labbé, A. The role of reaction force and chemical potential in characterizing the mechanism of double proton transfer in the adenine-uracil complex. J. Phys. Chem. A 2007, 111, 5921–5926. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.; Toro-Labbé, A. The reaction force. A scalar property to characterize reaction mechanisms. J. Math. Chem. 2009, 45, 911–927. [Google Scholar] [CrossRef]
- Pérez, P.; Toro-Labbé, A. Theoretical analysis of some substituted imine-enamine tautomerism. Theor. Chem. Acc. 2001, 105, 422–430. [Google Scholar] [CrossRef]
- Moyano, A.; Pericàs, M.A.; Valentí, E. A Theoretical Study on the Mechanism of the Thermal and the Acid-Catalyzed Decarboxylation of 2-Oxetanones (β-Lactones). J. Org. Chem. 1989, 54, 573–582. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
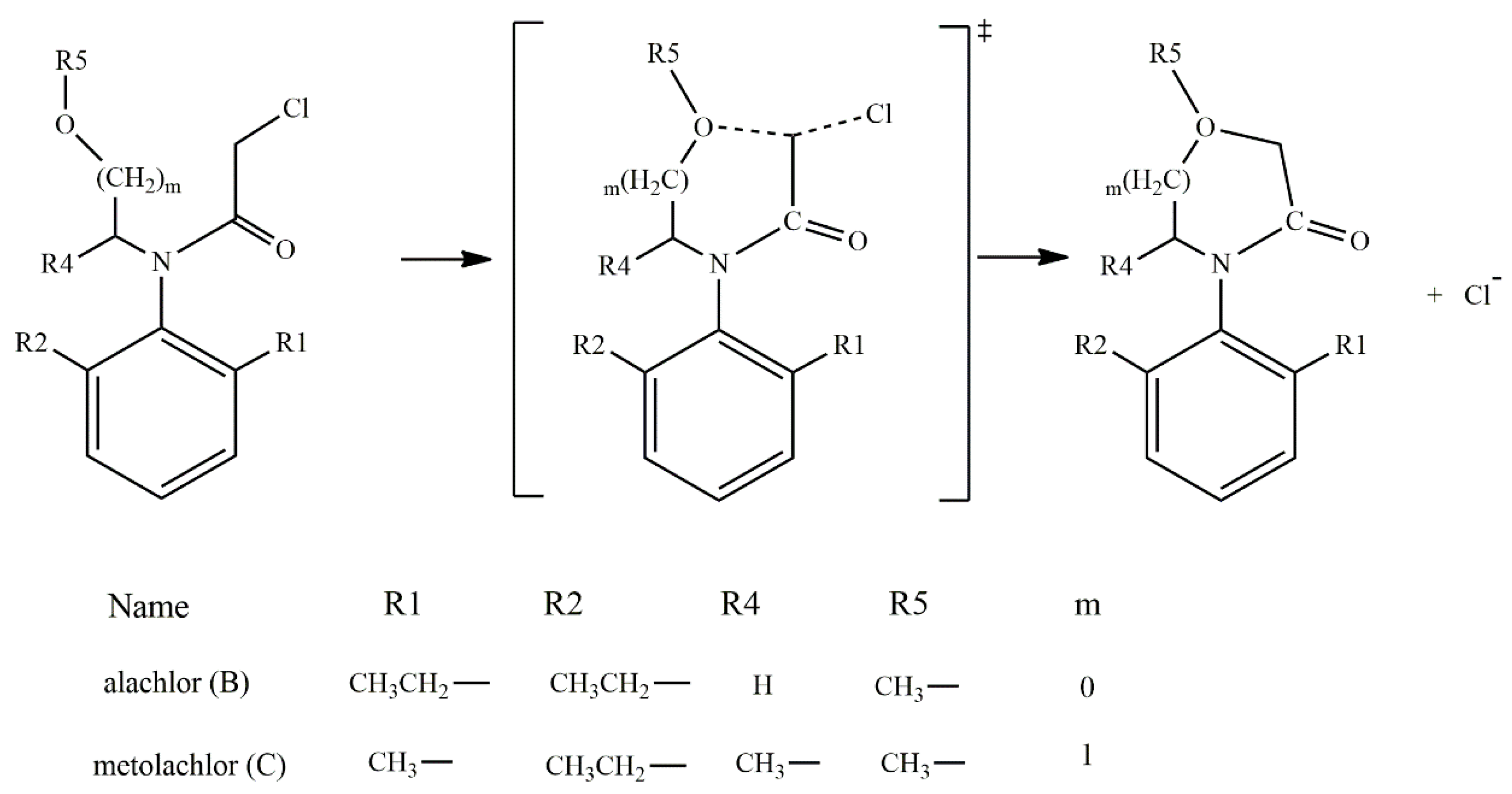{kind=link}
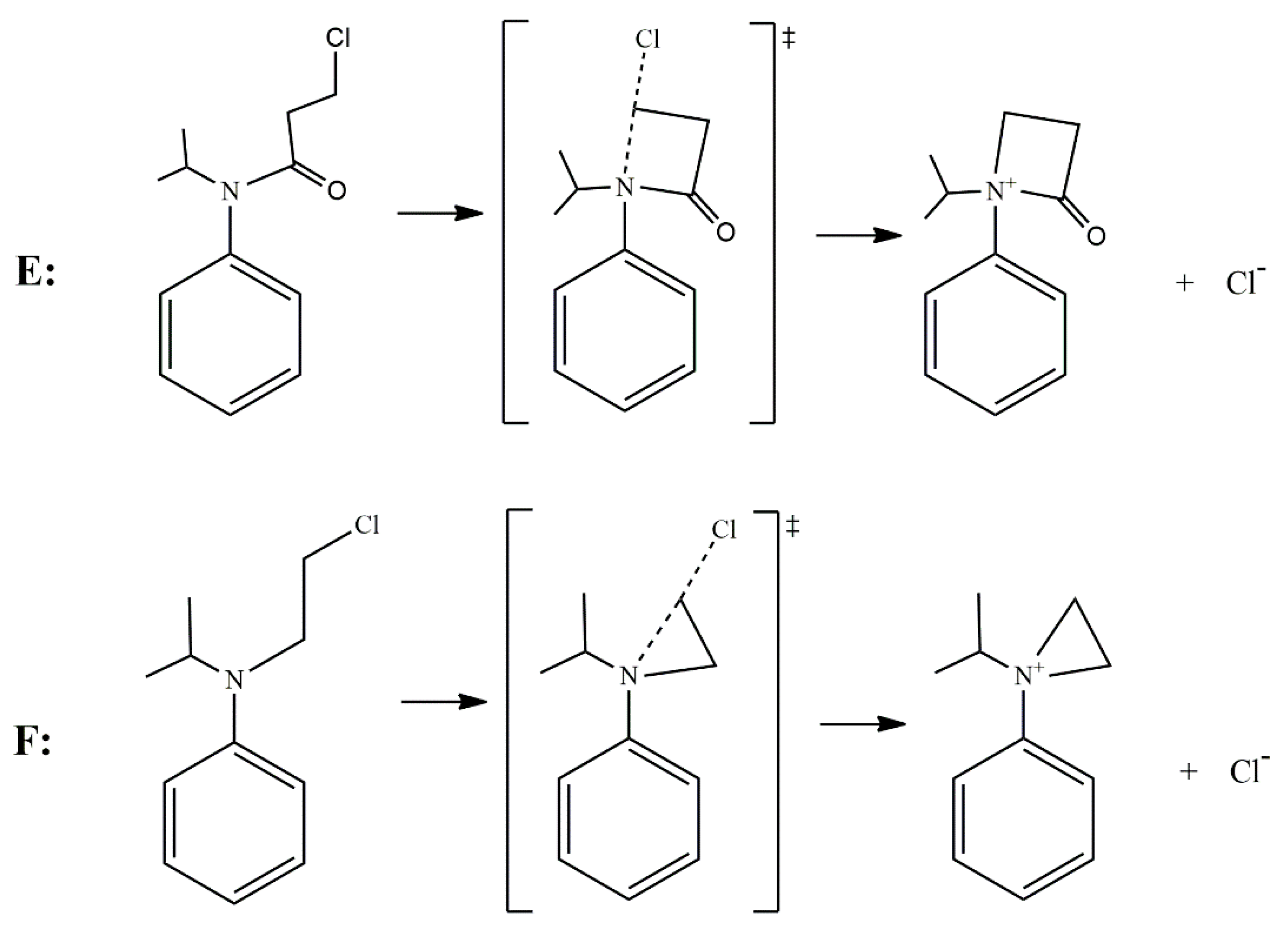{kind=link}
{kind=link}
| Compound | Mechanism | Theory | Experimental | ||||
|---|---|---|---|---|---|---|---|
| ∆H‡ | ∆S‡ | ∆G‡ | ∆H‡ | ∆S‡ | ∆G‡ | ||
| kcal/mol | cal/molK | kcal/mol | kcal/mol | cal/molK | kcal/mol | ||
| A | SN2 | 11.45 | −23.64 | 18.50 | 13.79 | −18.24 | 19.22 |
| B | SN2 | 11.50 | −25.43 | 19.08 | 13.84 | −19.98 | 19.79 |
| O-assistance | 33.02 | −2.17 | 33.67 | ||||
| C | SN2 | 11.91 | −23.67 | 18.97 | 15.25 | −19.29 | 20.98 |
| O-assistance | 26.60 | −6.36 | 28.50 | ||||
| D | SN2 | 13.44 | −18.24 | 18.87 | 11.57 | −23.80 | 18.64 |
| E | SN2 | 16.89 | −21.36 | 23.26 | 25.33 | 4.97 | 23.85 |
| N-assistance | 41.38 | −0.22 | 41.45 | ||||
| F | SN2 | 17.66 | −21.05 | 23.94 | --- | --- | --- |
| N-assistance | 23.45 | −0.23 | 23.52 | --- | --- | --- | |
| Mechanism | Compound | W1 | W2 | W3 | W4 | Er |
|---|---|---|---|---|---|---|
| SN2 | A | 9.19 | 3.80 | −16.01 | −23.59 | −26.61 |
| B | 9.41 | 3.78 | −16.36 | −23.84 | −27.01 | |
| C | 8.99 | 4.04 | −15.21 | −24.03 | −26.21 | |
| D | 11.10 | 3.89 | −17.03 | −23.87 | −25.91 | |
| E | 9.91 | 6.40 | −18.05 | −24.01 | −25.75 | |
| F | 11.59 | 6.58 | −15.70 | −20.11 | −17.64 | |
| N-assistance | F | 11.89 | 8.72 | −5.78 | −9.67 | 5.16 |
| Mechanism | Compound | Stationary Point | S–C (Å) | C–Cl (Å) | Angle S–C–Cl | Imaginary Frequency (cm−1) |
| SN2 | A | R | 3.680 | 1.807 | 174.43 | −545.06 |
| TS | 2.551 | 2.197 | ||||
| P | 1.829 | 3.557 | ||||
| B | R | 3.711 | 1.808 | 172.19 | −543.78 | |
| TS | 2.554 | 2.187 | ||||
| P | 1.829 | 3.421 | ||||
| C | R | 3.642 | 1.811 | 172.06 | −542.17 | |
| TS | 2.553 | 2.205 | ||||
| P | 1.827 | 3.481 | ||||
| D | R | 3.769 | 1.811 | 170.03 | −530.77 | |
| TS | 2.565 | 2.208 | ||||
| P | 1.831 | 3.614 | ||||
| E | R | 3.372 | 1.822 | 163.54 | −534.96 | |
| TS | 2.604 | 2.296 | ||||
| P | 1.830 | 3.723 | ||||
| F | R | 3.531 | 1.823 | 158.66 | −525.35 | |
| TS | 2.612 | 2.326 | ||||
| P | 1.849 | 3.119 | ||||
| Stationary Point | N–C (Å) | C–Cl (Å) | Angle N–C–Cl | Imaginary Frequency (cm−1) | ||
| N-assistance | F | R | 2.384 | 1.815 | 155.99 | −526.43 |
| TS | 1.895 | 2.381 | ||||
| P | 1.490 | 3.423 |
| Mechanims | Compound | δQS | δQC | δQCl |
| SN2 | A | 0.363 | 0.043 | −0.378 |
| B | 0.374 | 0.092 | −0.388 | |
| C | 0.361 | 0.097 | −0.386 | |
| D | 0.382 | 0.108 | −0.384 | |
| E | 0.44 | 0.161 | −0.581 | |
| F | 0.316 | 0.164 | −0.447 | |
| δQN | δQC | δQCl | ||
| N-assistance | F | 0.101 | 0.231 | −0.486 |
| Mechanism | Compound | δBi(S–C) % | δBi(C–Cl) % | δBav % | Sy |
| SN2 | A | 38.13 | 45.16 | 41.64 | 0.916 |
| B | 39.97 | 46.55 | 43.26 | 0.924 | |
| C | 38.42 | 45.83 | 42.12 | 0.912 | |
| D | 37.22 | 45.87 | 41.55 | 0.896 | |
| E | 38.22 | 47.46 | 42.84 | 0.892 | |
| F | 39.92 | 49.93 | 44.93 | 0.889 | |
| δBi(N–C) % | δBi(C–Cl) % | δBav % | Sy | ||
| N-assistance | F | 38.12 | 48.11 | 43.12 | 0.884 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mora, J.R.; Cervantes, C.; Marquez, E. New Insight into the Chloroacetanilide Herbicide Degradation Mechanism through a Nucleophilic Attack of Hydrogen Sulfide. Int. J. Mol. Sci. 2018, 19, 2864. https://doi.org/10.3390/ijms19102864
Mora JR, Cervantes C, Marquez E. New Insight into the Chloroacetanilide Herbicide Degradation Mechanism through a Nucleophilic Attack of Hydrogen Sulfide. International Journal of Molecular Sciences. 2018; 19(10):2864. https://doi.org/10.3390/ijms19102864
Chicago/Turabian StyleMora, José R., Cristian Cervantes, and Edgar Marquez. 2018. "New Insight into the Chloroacetanilide Herbicide Degradation Mechanism through a Nucleophilic Attack of Hydrogen Sulfide" International Journal of Molecular Sciences 19, no. 10: 2864. https://doi.org/10.3390/ijms19102864