Novel Methylselenoesters Induce Programed Cell Death via Entosis in Pancreatic Cancer Cells

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
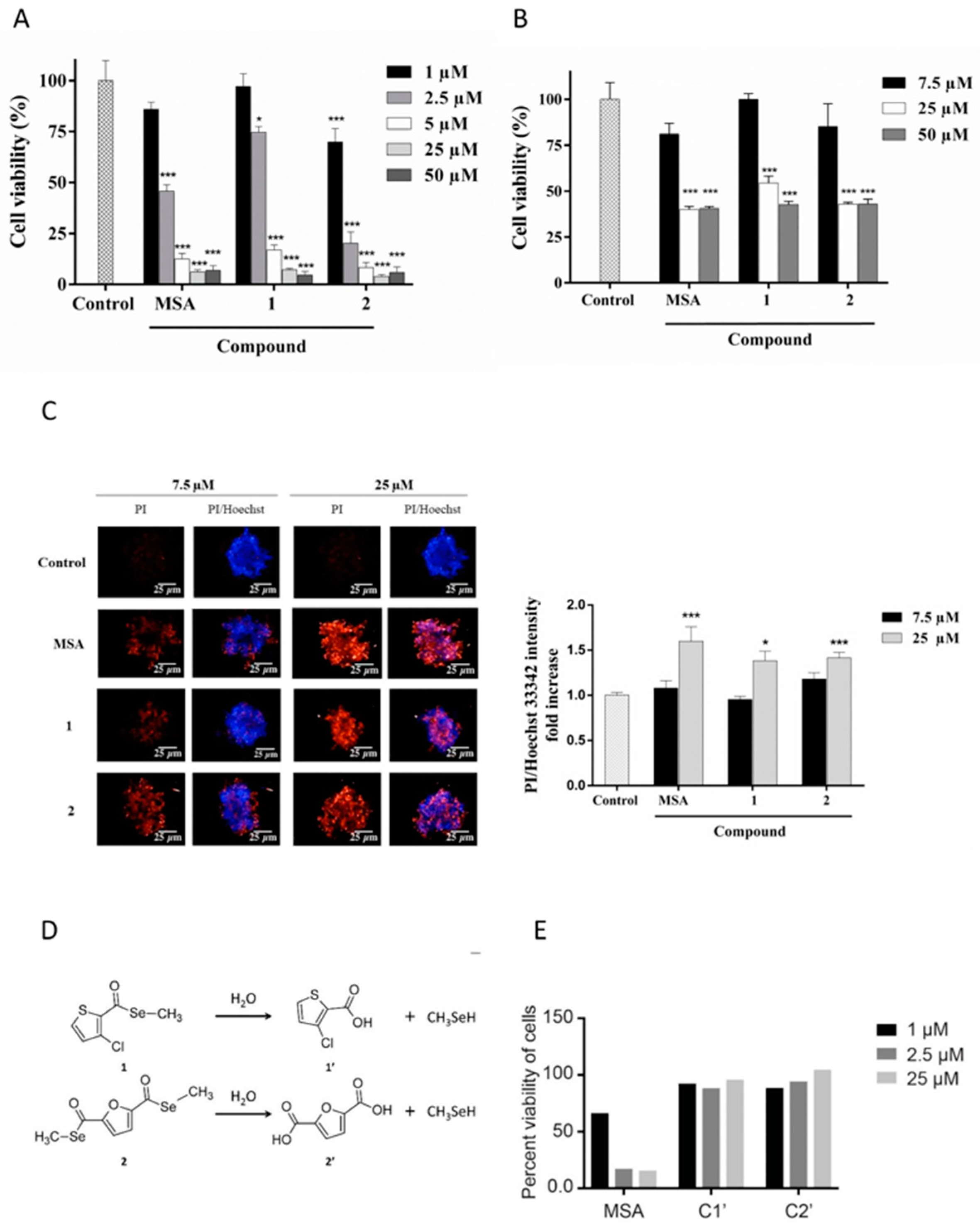
2.1. MSA, and Compounds 1 and 2 Reduce Panc-1 Cell Viability Both in 2D and 3D Cultures
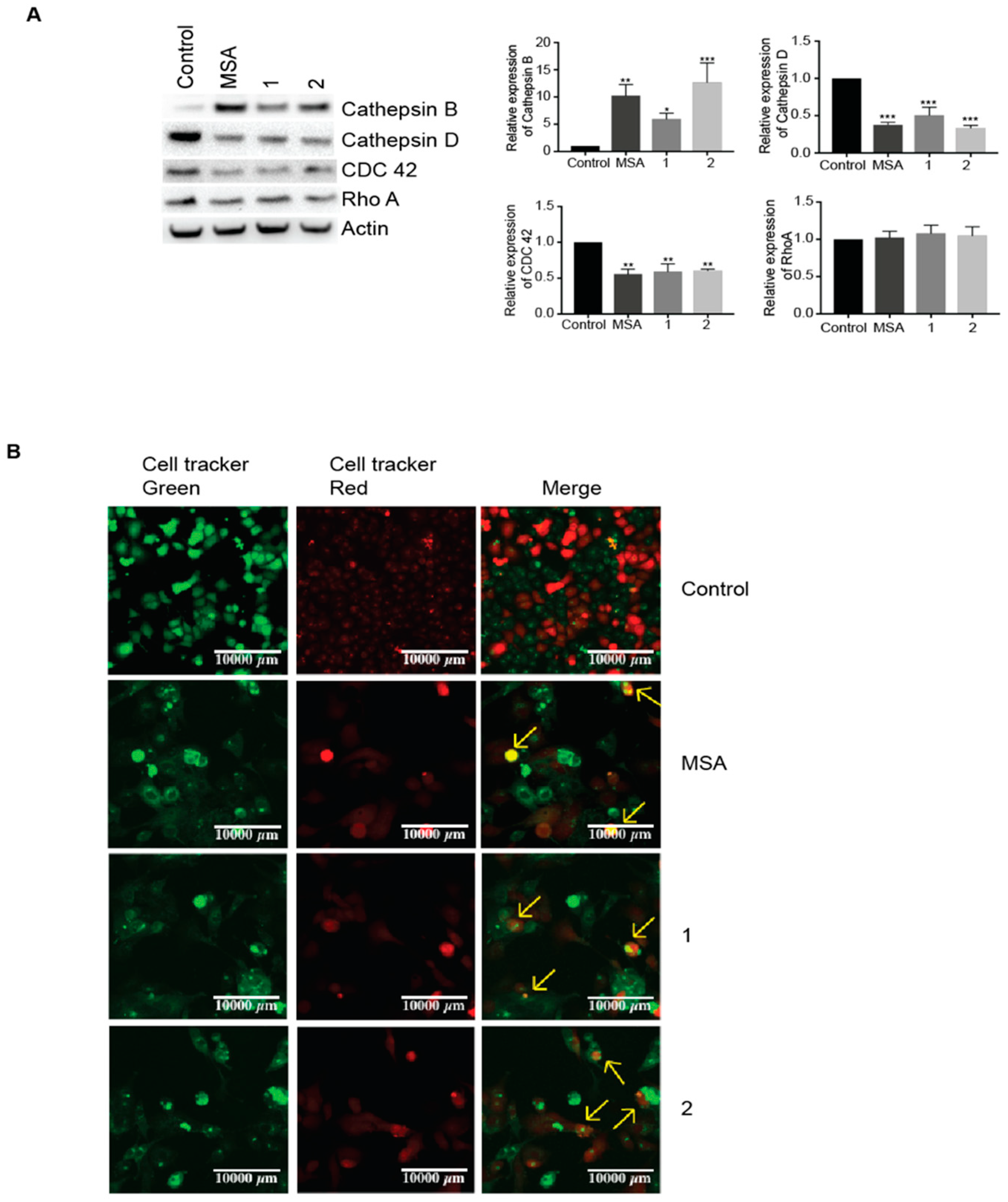
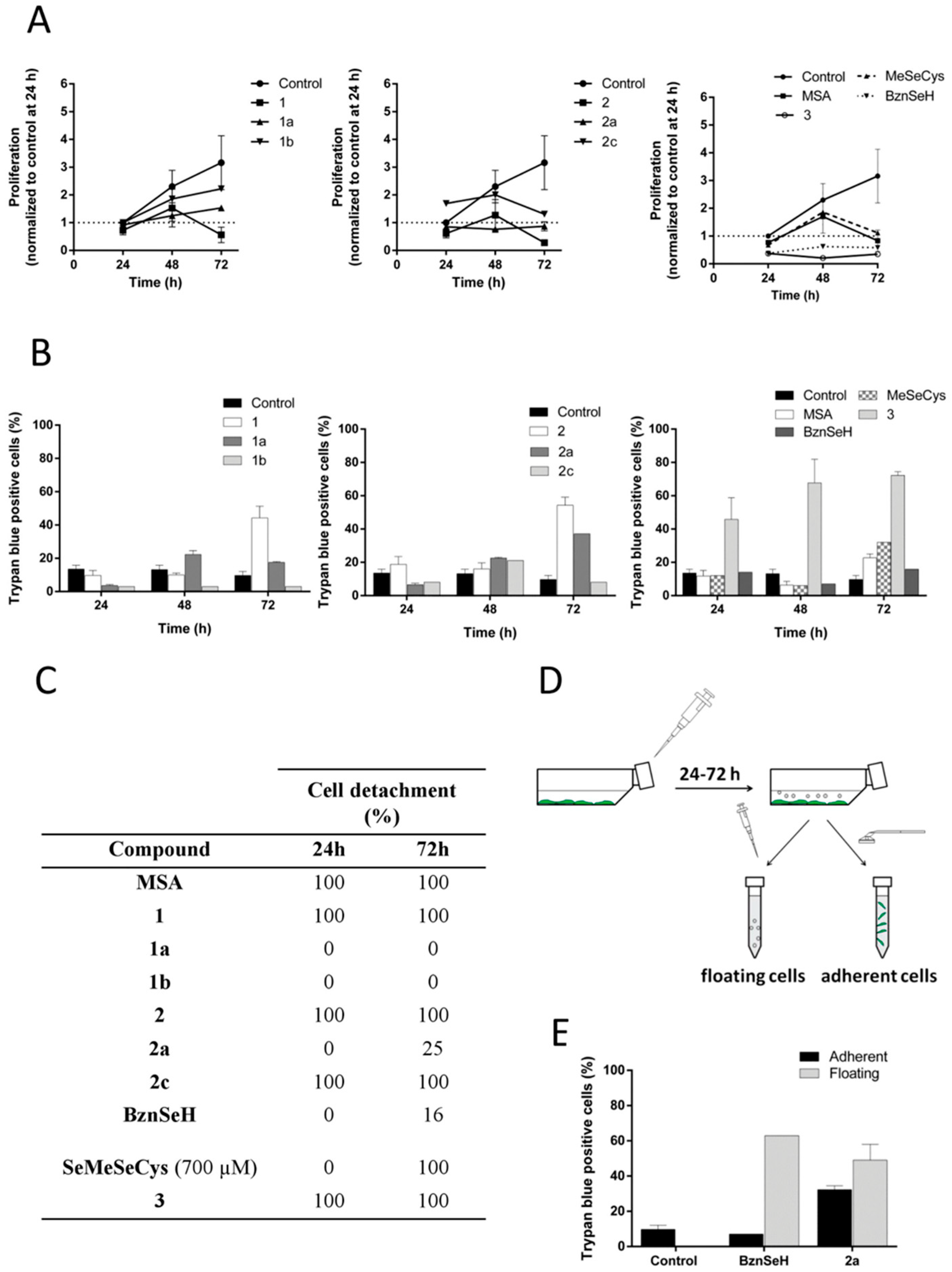
2.2. MSA, and Compounds 1 and 2 Induce Cell Detachment and Compromise Reattachment Abilities by Promoting an Aberrant Adhesive Repertoire
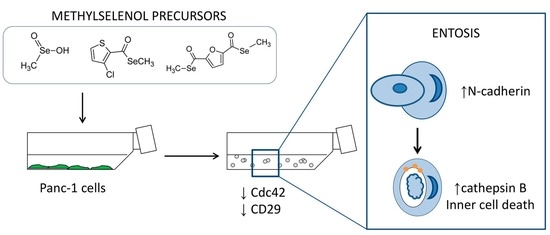
2.3. MSA, Compounds 1 and 2 Induce Entosis.
2.4. Cell Detachment Is Not Restricted to Selenomethylated Compounds and Does Not Correlate with the Cytotoxic Potential of the Compound
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. 2D Viability Assay
4.3. 3D Viability Assay
4.4. Fluorescent Staining
4.5. Western Blotting
4.6. Adhesion Assay
4.7. Flow Cytometry
4.8. Clonogenic Assay
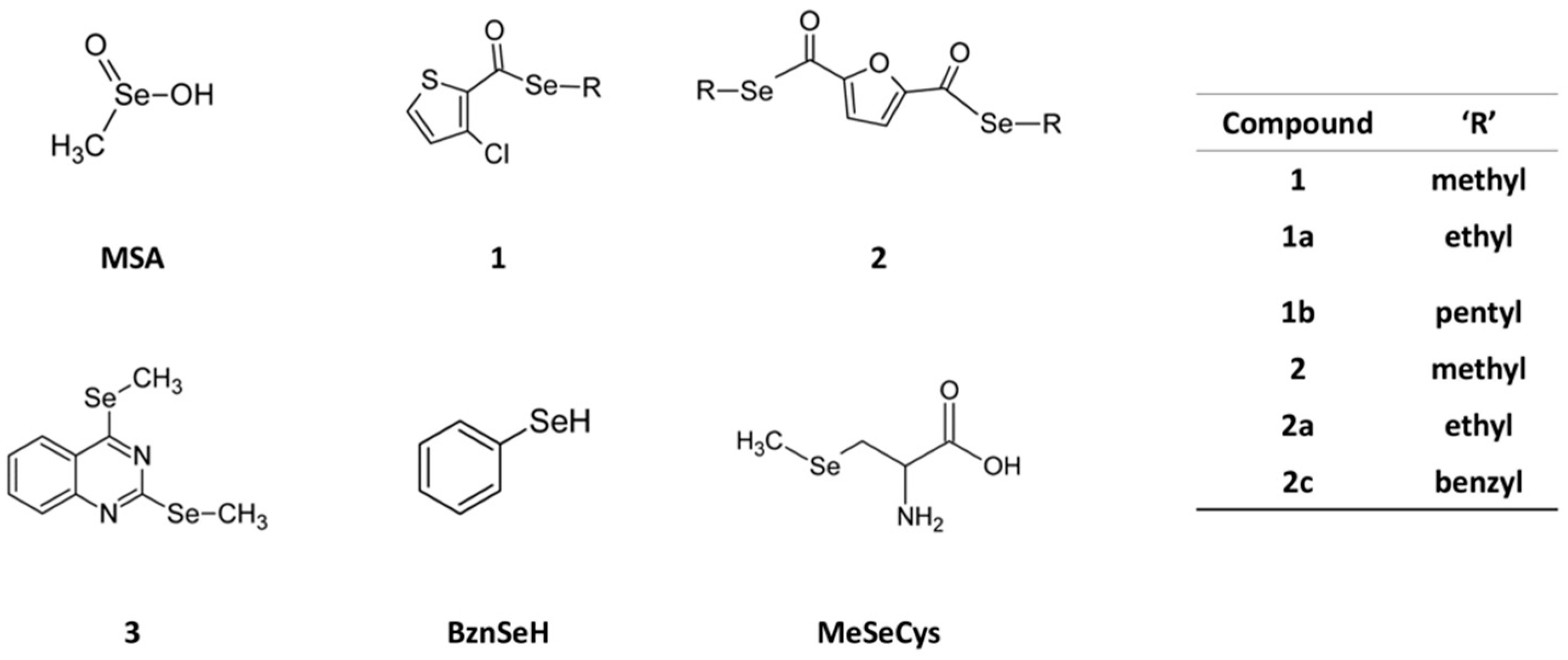
4.9. Chemical Synthesis
4.9.1. Procedure for Compounds 1 and 2
Procedure for Compounds 1a, 2a, 1b and 2c
Ethyl 3-Chlorothiophen-2-Carboselenoate (1a)
Diethyl 2,5-Furandicarboselenoate (2a)
Pentyl 3-Chlorothiophen-2-Carboselenoate (1b)
Dibenzyl 2,5-Furandicarboselenoate (2c)
4.9.2. Procedure for Compound 3
4.10. Timelapse
4.11. Confocal
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BznSeH | Benzeneselenol |
| CatB | Cathepsin B |
| CatD | Cathepsin D |
| CD29 | β1-integrin |
| CDC42 | Cell division control protein 42 homolog |
| PARP | DNA damage-responsive enzymes poly(ADP-ribose) polymerase |
| MSA | Methylseleninic acid |
| MeSeCys | Methylselenocysteine |
| CH3SeH | Methylselenol |
| Nrf2 | nuclear factor erythroid 2 related factor 2 |
| PDAC | Pancreatic ductal adenocarcinoma |
| PI | Propidium iodide |
| ROS | Reactive oxygen species |
| Se | Selenium |
References
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Bijlsma, M.F.; van Laarhoven, H.W.M. The conflicting roles of tumor stroma in pancreatic cancer and their contribution to the failure of clinical trials: A systematic review and critical appraisal. Cancer Metastasis Rev. 2015, 34, 97–114. [Google Scholar] [CrossRef] [PubMed]
- Al Saati, T.; Clerc, P.; Hanoun, N.; Peuget, S.; Lulka, H.; Gigoux, V.; Capilla, F.; Béluchon, B.; Couvelard, A.; Selves, J.; et al. Oxidative Stress Induced by Inactivation of TP53INP1 Cooperates with KrasG12D to Initiate and Promote Pancreatic Carcinogenesis in the Murine Pancreas. Am. J. Pathol. 2013, 182, 1996–2004. [Google Scholar] [CrossRef] [PubMed]
- Kong, B.; Qia, C.; Erkan, M.; Kleeff, J.; Michalski, C.W. Overview on how oncogenic Kras promotes pancreatic carcinogenesis by inducing low intracellular ROS levels. Front. Physiol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.; Mccracken, S.; Mak, T.W. Cancer cell metabolism. Cold Spring Harb. Symp. Quant. Biol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.; Harris, I.; Mak, T. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Sosa, V.; Moline, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; Me, L.L. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef] [PubMed]
- Chaiswing, L.; St Clair, W.H.; St Clair, D.K. Redox Paradox: A novel approach to therapeutics-resistant cancer. Antioxid. Redox Signal. 2018. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.P.; Gandin, V. Selenium compounds as therapeutic agents in cancer. Biochim. Biophys. Acta 2015, 1850, 1642–1660. [Google Scholar] [CrossRef] [PubMed]
- Gandin, V.; Khalkar, P.; Braude, J.; Fernandes, A.P. Organic selenium compounds as potential chemotherapeutic agents for improved cancer treatment. Free Radic. Biol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Argelich, N.; Encío, I.; Plano, D.; Fernandes, A.P.; Palop, J.A.; Sanmartín, C. Novel Methylselenoesters as Antiproliferative Agents. Molecules 2017, 22, 1288. [Google Scholar] [CrossRef] [PubMed]
- Overholtzer, M.; Mailleux, A.A.; Mouneimne, G.; Normand, G.; Schnitt, S.J.; King, R.W.; Cibas, E.S.; Brugge, J.S. A Nonapoptotic Cell Death Process, Entosis, that Occurs by Cell-in-Cell Invasion. Cell 2007, 131, 966–979. [Google Scholar] [CrossRef] [PubMed]
- Durgan, J.; Tseng, Y.Y.; Hamann, J.C.; Domart, M.C.; Collinson, L.; Hall, A.; Overholtzer, M.; Florey, O. Mitosis can drive cell cannibalism through entosis. eLife 2017, 6, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Garanina, A.S.; Kisurina-Evgenieva, O.P.; Erokhina, M.V.; Smirnova, E.A.; Factor, V.M.; Onishchenko, G.E. Consecutive entosis stages in human substrate-dependent cultured cells. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wan, Q.; Liu, J.; Zheng, Z.; Zhu, H.; Chu, X.; Dong, Z.; Huang, S.; Du, Q. Regulation of myosin activation during cell-cell contact formation by Par3-Lgl antagonism: Entosis without matrix detachment. Mol. Biol. Cell 2012, 23, 2076–2091. [Google Scholar] [CrossRef] [PubMed]
- Hamann, J.C.; Surcel, A.; Chen, R.; Teragawa, C.; Albeck, J.G.; Robinson, D.N.; Overholtzer, M. Entosis Is Induced by Glucose Starvation. Cell Rep. 2017, 20, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Florey, O.; Kim, S.E.; Sandoval, C.P.; Haynes, C.M.; Overholtzer, M. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat. Cell Biol. 2011, 13, 1335–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishna, S.; Overholtzer, M. Mechanisms and consequences of entosis. Cell. Mol. Life Sci. 2016, 73, 2379–2386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reymond, N.; Im, J.H.; Garg, R.; Vega, F.M.; Borda d’Agua, B.; Riou, P.; Cox, S.; Valderrama, F.; Muschel, R.J.; Ridley, A.J. Cdc42 promotes transendothelial migration of cancer cells through β1 integrin. J. Cell Biol. 2012, 199, 653–668. [Google Scholar] [CrossRef] [PubMed]
- Longati, P.; Jia, X.; Eimer, J.; Wagman, A.; Witt, M.-R.; Rehnmark, S.; Verbeke, C.; Toftgård, R.; Löhr, M.; Heuchel, R.L. 3D pancreatic carcinoma spheroids induce a matrix-rich, chemoresistant phenotype offering a better model for drug testing. BMC Cancer 2013, 13, 95. [Google Scholar] [CrossRef] [PubMed]
- Seguin, L.; Desgrosellier, J.S.; Weis, S.M.; Cheresh, D.A. Integrins and cancer: Regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol. 2015, 25, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Van Roy, F. Beyond E-cadherin: Roles of other cadherin superfamily members in cancer. Nat. Rev. Cancer 2014, 14, 121–134. [Google Scholar] [CrossRef] [PubMed]
- McGinnis, K.M.; Gnegy, M.E.; Park, Y.H.; Mukerjee, N.; Wang, K.K.W. Procaspase-3 and Poly(ADP)ribose Polymerase (PARP) Are Calpain Substrates. Biochem. Biophys. Res. Commun. 1999, 263, 94–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gobeil, S.; Boucher, C.C.; Nadeau, D.; Poirier, G.G. Characterization of the necrotic cleavage of poly(ADP-ribose) polymerase (PARP-1): Implication of lysosomal proteases. Cell Death Differ. 2001, 8, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Shalini, S.; Dorstyn, L.; Dawar, S.; Kumar, S. Old, new and emerging functions of caspases. Cell Death Differ. 2015, 22, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Chaitanya, G.; Alexander, J.S.; Babu, P. PARP-1 cleavage fragments: Signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal. 2010, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Lugini, L.; Matarrese, P.; Tinari, A.; Lozupone, F.; Federici, C.; Iessi, E.; Gentile, M.; Luciani, F.; Parmiani, G.; Rivoltini, L.; et al. C annibalism of live lymphocytes by human metastatic but not primary melanoma cells. Cancer Res. 2006, 66, 3629–3638. [Google Scholar] [CrossRef] [PubMed]
- Khalkhali-Ellis, Z.; Goossens, W.; Margaryan, N.V.; Hendrix, M.J.C. Cleavage of histone 3 by cathepsin D in the involuting mammary gland. PLoS ONE 2014, 9, e103230. [Google Scholar] [CrossRef] [PubMed]
- Moreno, E.; Plano, D.; Lamberto, I.; Font, M.; Encío, I.; Palop, J.A.; Sanmartín, C. Sulfur and selenium derivatives of quinazoline and pyrido[2,3-d]pyrimidine: Synthesis and study of their potential cytotoxic activity in vitro. Eur. J. Med. Chem. 2012, 47, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Ip, C.; Thompson, H.J.; Zhu, Z.; Ganther, H.E. In vitro and in vivo studies of methylseleninic acid: Evidence that a monomethylated selenium metabolite is critical for cancer chemoprevention. Cancer Res. 2000, 60, 2882–2886. [Google Scholar] [CrossRef] [PubMed]
- Khalkar, P.; Ali, H.A.; Codó, P.; Argelich, N.D.; Martikainen, A.; Arzenani, M.K.; Lehmann, S.; Walfridsson, J.; Ungerstedt, J.; Fernandes, A.P. Selenite and methylseleninic acid epigenetically affects distinct gene sets in myeloid leukemia: A genome wide epigenetic analysis. Free Radic. Biol. Med. 2018, 117, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Shi, J.; Fu, H.; Wei, Z.; Xu, J.; Hu, Z.; Zhang, Y.; Yan, R.; Cai, Q. Integrin B1 modulates tumour resistance to gemcitabine and serves as an independent prognostic factor in pancreatic adenocarcinomas. Tumor Biol. 2016, 37, 12315–12327. [Google Scholar] [CrossRef] [PubMed]
- Grzesiak, J.J.; Cao, H.S.T.; Burton, D.W.; Kaushal, S.; Vargas, F.; Clopton, P.; Snyder, C.S.; Deftos, L.J.; Hoffman, R.M.; Bouvet, M. Knockdown of the β1 integrin subunit reduces primary tumor growth and inhibits pancreatic cancer metastasis. Int. J. Cancer 2011, 129, 2905–2915. [Google Scholar] [CrossRef] [PubMed]
- Straub, B.K.; Rickelt, S.; Zimbelmann, R.; Grund, C.; Kuhn, C.; Iken, M.; Ott, M.; Schirmacher, P.; Franke, W.W. E-N-cadherin heterodimers define novel adherens junctions connecting endoderm-derived cells. J. Cell Biol. 2011, 195, 873–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cano, C.E.; Sandí, M.J.; Hamidi, T.; Calvo, E.L.; Turrini, O.; Bartholin, L.; Loncle, C.; Secq, V.; Garcia, S.; Lomberk, G.; et al. Homotypic cell cannibalism, a cell-death process regulated by the nuclear protein 1, opposes to metastasis in pancreatic cancer. EMBO Mol. Med. 2012, 4, 964–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.; Li, J.; Shi, C.; Hruban, R.H.; Radice, G.L. N-cadherin functions as a growth suppressor in a model of K-ras-induced PanIN. Oncogene 2016, 35, 3335–3341. [Google Scholar] [CrossRef] [PubMed]
- Camand, E.; Peglion, F.; Osmani, N.; Sanson, M.; Etienne-Manneville, S. N-cadherin expression level modulates integrin-mediated polarity and strongly impacts on the speed and directionality of glial cell migration. J. Cell Sci. 2012, 125, 844–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maldonado, M.D.M.; Dharmawardhane, S. Targeting Rac and Cdc42 GTPases in Cancer. Cancer Res. 2018, 78, 3101–3111. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, F.; Ushida, K.; Mori, K.; Shibanuma, M. Loss of anchorage primarily induces non-apoptotic cell death in a human mammary epithelial cell line under atypical focal adhesion kinase signaling. Cell Death Dis. 2015, 6, e1619. [Google Scholar] [CrossRef] [PubMed]
- Hah, Y.S.; Noh, H.S.; Ha, J.H.; Ahn, J.S.; Hahm, J.R.; Cho, H.Y.; Kim, D.R. Cathepsin D inhibits oxidative stress-induced cell death via activation of autophagy in cancer cells. Cancer Lett. 2012, 323, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Glondu, M.; Liaudet-Coopman, E.; Derocq, D.; Platet, N.; Rochefort, H.; Garcia, M. Down-regulation of cathepsin-D expression by antisense gene transfer inhibits tumor growth and experimental lung metastasis of human breast cancer cells. Oncogene 2002, 21, 5127–5134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.-D.; Kang, E.-S.; Kim, J.-W.; Lee, K.-T.; Lee, K.H.; Park, Y.S.; Park, J.-O.; Lee, J.; Heo, J.S.; Choi, S.H.; et al. Serum CA19-9, cathepsin D, and matrix metalloproteinase-7 as a diagnostic panel for pancreatic ductal adenocarcinoma. Proteomics 2012, 12, 3590–3597. [Google Scholar] [CrossRef] [PubMed]
- Dumartin, L.; Whiteman, H.J.; Weeks, M.E.; Hariharan, D.; Dmitrovic, B.; Iacobuzio-Donahue, C.A.; Brentnall, T.A.; Bronner, M.P.; Feakins, R.M.; Timms, J.F.; et al. AGR2 is a novel surface antigen that promotes the dissemination of pancreatic cancer cells through regulation of cathepsins B and D. Cancer Res. 2011, 71, 7091–7102. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Wang, Z.; Ganther, H.; Lu, J. Caspases as key executors of methyl selenium-induced apoptosis (anoikis) of DU-145 prostate cancer cells. Cancer Res. 2001, 61, 3062–3070. [Google Scholar] [PubMed]
- Kim, A.; Oh, J.H.; Park, J.M.; Chung, A.S. Methylselenol generated from selenomethionine by methioninase downregulates integrin expression and induces caspase-mediated apoptosis of B16F10 melanoma cells. J. Cell. Physiol. 2007, 212, 386–400. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, E.; Plano, D.; Font, M.; Calvo, A.; Prior, C.; Palop, J.A.; Sanmartín, C. Synthesis and antiproliferative activity of novel symmetrical alkylthio- and alkylseleno-imidocarbamates. Eur. J. Med. Chem. 2011, 46, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Franken, N.A.P.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Álvarez, E.; Plano, D.; Font, M.; Calvo, A.; Prior, C.; Jacob, C.; Palop, J.A.; Sanmartín, C. Synthesis and antiproliferative activity of novel selenoester derivatives. Eur. J. Med. Chem. 2014, 73, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Klayman, D.L.; Griffin, T.S. Reaction of Selenium with Sodium Borohydride in Protic Solvents. A Facile Method for the Introduction of Selenium into Organic Molecules. J. Am. Chem. Soc. 1973, 2, 197–199. [Google Scholar] [CrossRef]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalkar, P.; Díaz-Argelich, N.; Antonio Palop, J.; Sanmartín, C.; Fernandes, A.P. Novel Methylselenoesters Induce Programed Cell Death via Entosis in Pancreatic Cancer Cells. Int. J. Mol. Sci. 2018, 19, 2849. https://doi.org/10.3390/ijms19102849
Khalkar P, Díaz-Argelich N, Antonio Palop J, Sanmartín C, Fernandes AP. Novel Methylselenoesters Induce Programed Cell Death via Entosis in Pancreatic Cancer Cells. International Journal of Molecular Sciences. 2018; 19(10):2849. https://doi.org/10.3390/ijms19102849
Chicago/Turabian StyleKhalkar, Prajakta, Nuria Díaz-Argelich, Juan Antonio Palop, Carmen Sanmartín, and Aristi P. Fernandes. 2018. "Novel Methylselenoesters Induce Programed Cell Death via Entosis in Pancreatic Cancer Cells" International Journal of Molecular Sciences 19, no. 10: 2849. https://doi.org/10.3390/ijms19102849