Can EGCG Alleviate Symptoms of Down Syndrome by Altering Proteolytic Activity?

Abstract
:
{kind=link}
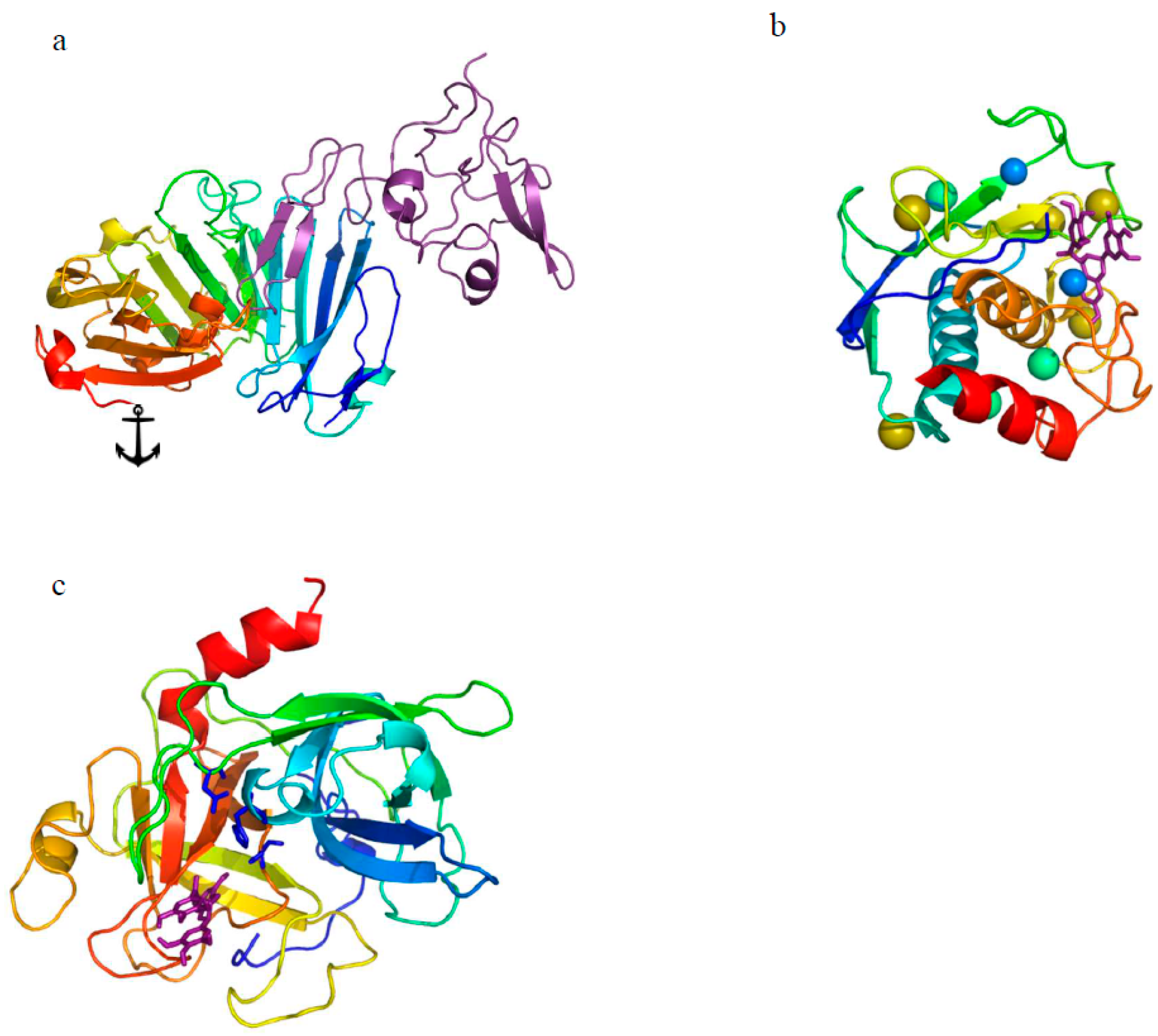
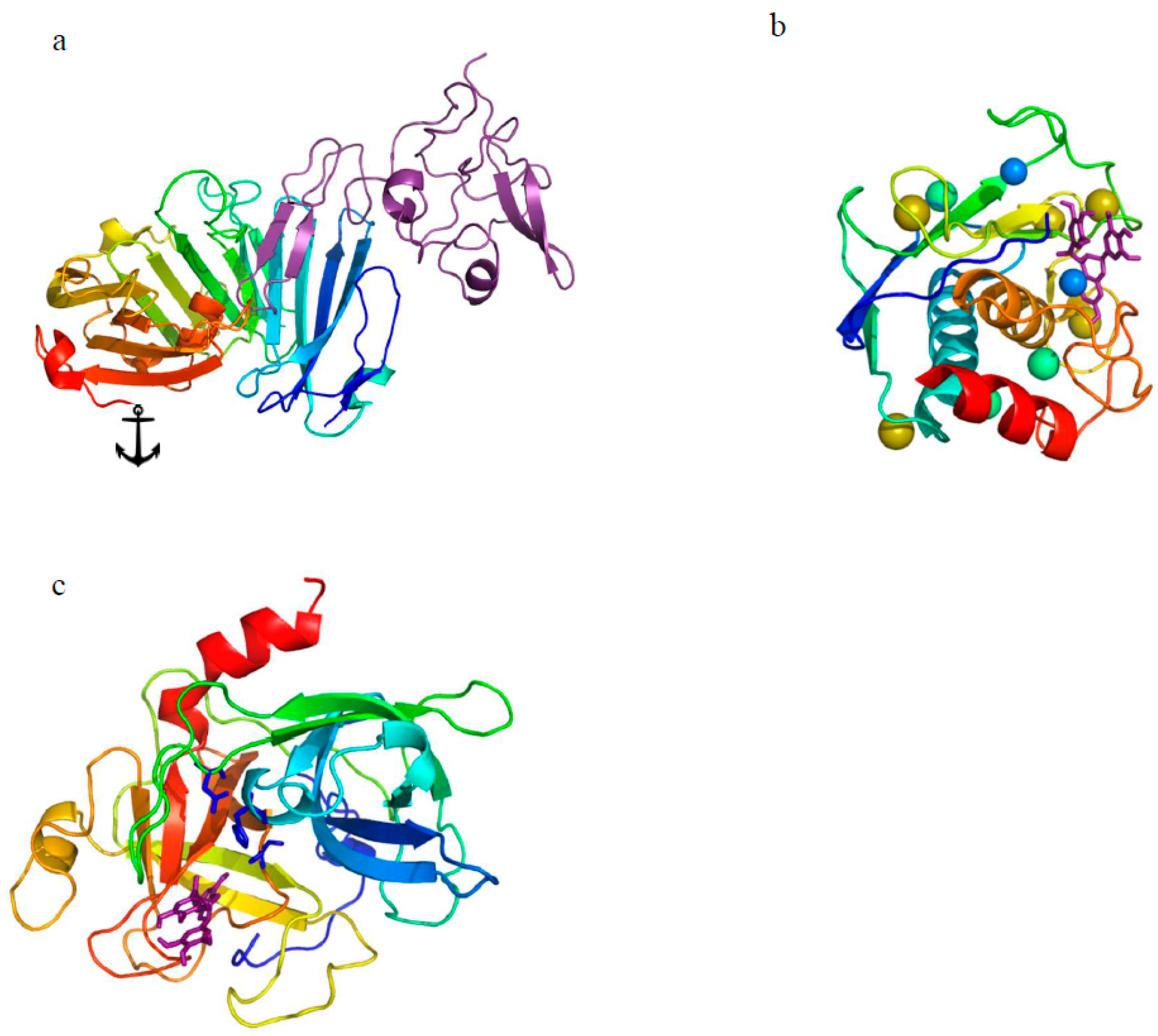{kind=link}
{kind=link}
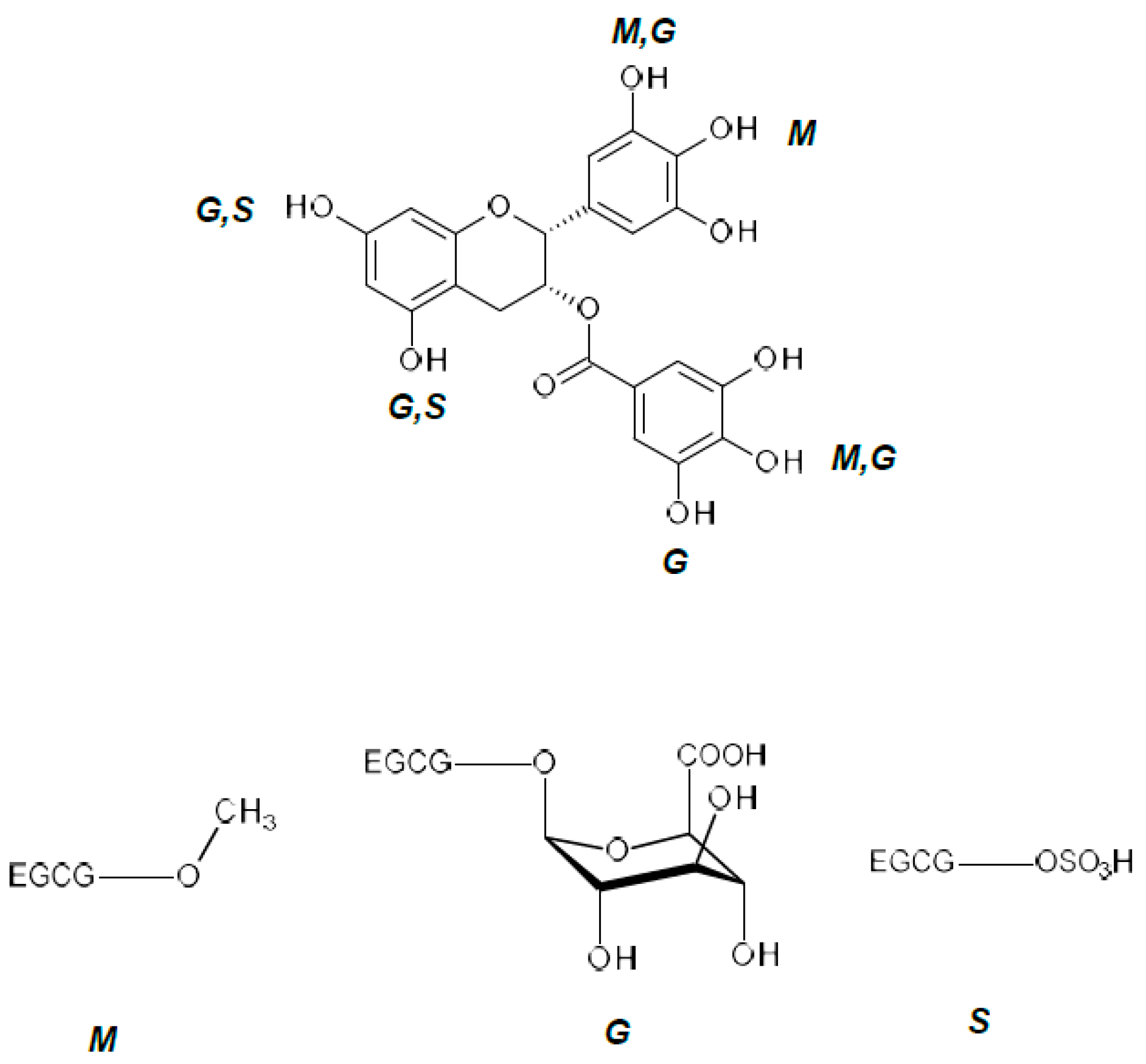{kind=link}
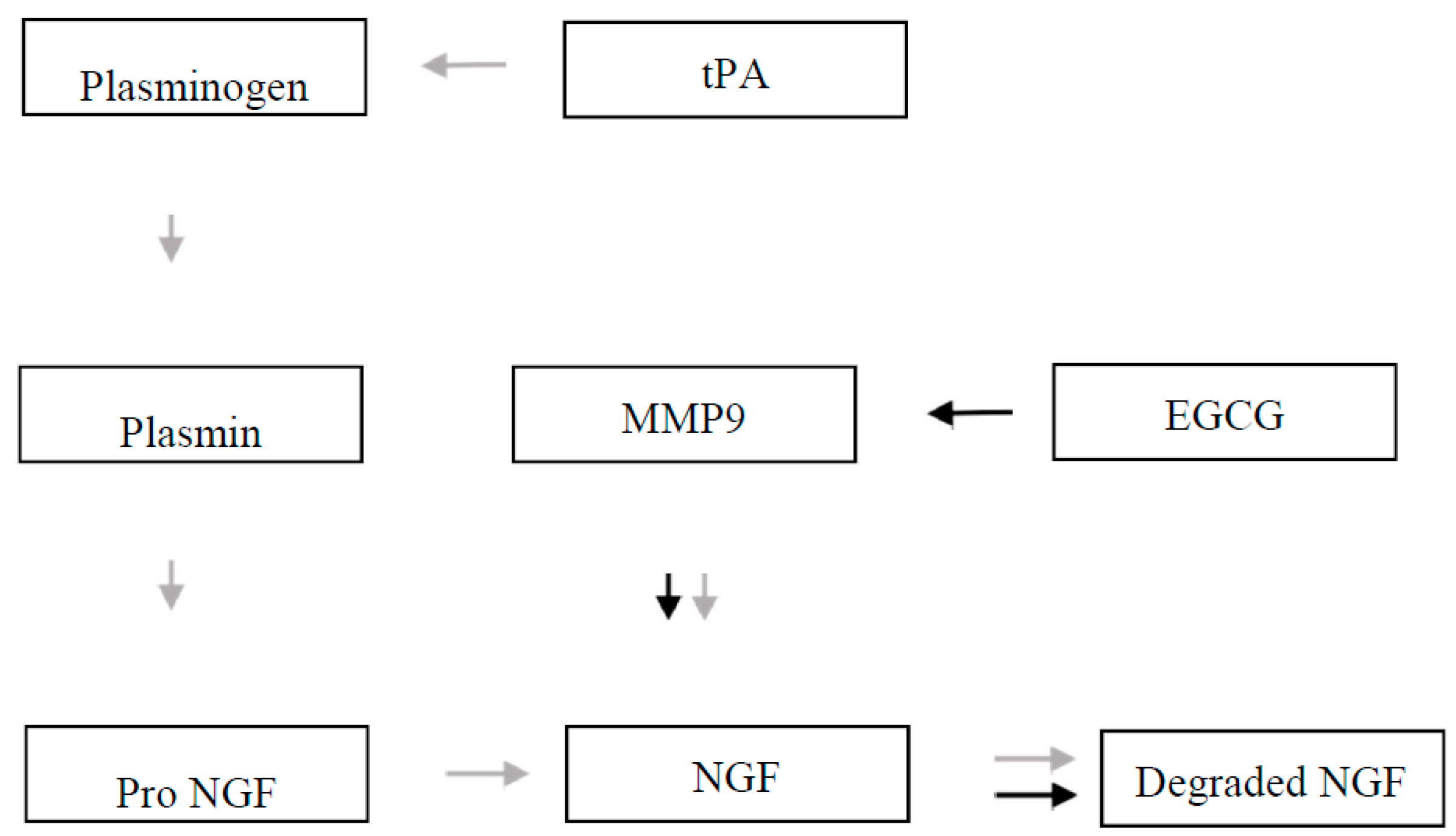{kind=link}
1. Introduction
2. The Plasminogen Activator System (PAS)
3. Matrix Metalloproteinases
4. Tissue Inhibitors of Metalloproteinases
5. Plasminogen Activator System in Down Syndrome
6. Tissue Plasminogen Activator in Down Syndrome
7. Plasminogen Activator Inhibitor Type 1 in Down Syndrome
8. Urokinase in Down Syndrome
9. Metrics Proteins
10. Matrix Metalloproteinases and Down Syndrome
11. Epigallocatechin Gallate
12. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Antonarakis, S.E. Down syndrome and the complexity of genome dosage imbalance. Nat. Rev. Genet. 2017, 18, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Kazemi, M.; Salehi, M.; Kheirollahi, M. Down Syndrome: Current Status, Challenges and Future Perspectives. Int. J. Mol. Cell. Med. 2016, 5, 125–133. [Google Scholar] [PubMed]
- Sturgeon, X.; Le, T.; Ahmed, M.M.; Gardiner, K.J. Pathways to cognitive deficits in Down syndrome. Prog. Brain Res. 2012, 197, 73–100. [Google Scholar] [PubMed]
- Hyman, S.L.; Levy, S.E. Introduction: Novel therapies in developmental disabilities—Hope, reason, and evidence. Ment. Retard. Dev. Disabil. Res. Rev. 2005, 11, 107–109. [Google Scholar] [CrossRef] [PubMed]
- Roizen, N.J. Complementary and alternative therapies for Down syndrome. Ment. Retard. Dev. Disabil. Res. Rev. 2005, 11, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Prussing, E.; Sobo, E.J.; Walker, E.; Dennis, K.; Kurtin, P.S. Communicating with pediatricians about complementary/alternative medicine: Perspectives from parents of children with down syndrome. Ambul. Pediatr. 2004, 4, 488–494. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, R.; de Sola, S.; Hernandez, G.; Farre, M.; Pujol, J.; Rodriguez, J.; Espadaler, J.M.; Langohr, K.; Cuenca-Royo, A.; Principe, A.; et al. Safety and efficacy of cognitive training plus epigallocatechin-3-gallate in young adults with Down’s syndrome (TESDAD): A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016, 15, 801–810. [Google Scholar] [CrossRef]
- Bruno, M.A.; Mufson, E.J.; Wuu, J.; Cuello, A.C. Increased matrix metalloproteinase 9 activity in mild cognitive impairment. J. Neuropathol. Exp. Neurol. 2009, 68, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Iulita, M.F.; Do Carmo, S.; Ower, A.K.; Fortress, A.M.; Flores Aguilar, L.; Hanna, M.; Wisniewski, T.; Granholm, A.C.; Buhusi, M.; Busciglio, J.; et al. Nerve growth factor metabolic dysfunction in Down’s syndrome brains. Brain 2014, 137, 860–872. [Google Scholar] [CrossRef] [PubMed]
- Jankun, J.; Keck, R.W.; Selman, S.H. Epigallocatechin-3-gallate prevents tumor cell implantation/growth in an experimental rat bladder tumor model. Int. J. Oncol. 2014, 44, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Kobalka, A.J.; Keck, R.W.; Jankun, J. Synergistic anticancer activity of biologicals from green and black tea on DU 145 human prostate cancer cells. Cent. Eur. J. Immunol. 2015, 40, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J. The urokinase plasminogen activator system: Role in malignancy. Curr. Pharm. Des. 2004, 10, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Choong, P.F.; Nadesapillai, A.P. Urokinase plasminogen activator system: A multifunctional role in tumor progression and metastasis. Clin. Orthop. Relat. Res. 2003, 415, S46–S58. [Google Scholar] [CrossRef]
- VanMeter, T.E.; Rooprai, H.K.; Kibble, M.M.; Fillmore, H.L.; Broaddus, W.C.; Pilkington, G.J. The role of matrix metalloproteinase genes in glioma invasion: Co-dependent and interactive proteolysis. J. Neurooncol. 2001, 53, 213–235. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Uno, S.; Watanabe, Y.; Arakawa, K.; Nakagawa, S. Expression of nerve growth factor-induced type 1 plasminogen activator inhibitor (PAI-1) mRNA is inhibited by genistein and wortmannin. Neuroreport 2000, 11, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Blasi, F.; Verde, P. Urokinase-dependent cell surface proteolysis and cancer. Semin. Cancer Biol. 1990, 1, 117–126. [Google Scholar] [PubMed]
- Jankun, J.; Skrzypczak-Jankun, E. Yin and yang of the plasminogen activator inhibitor. Polskie Arch. Med. Wewn. 2009, 119, 410–417. [Google Scholar]
- Matteucci, E.; Modora, S.; Simone, M.; Desiderio, M.A. Hepatocyte growth factor induces apoptosis through the extrinsic pathway in hepatoma cells: Favouring role of hypoxia-inducible factor-1 deficiency. Oncogene 2003, 22, 4062–4073. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, D.; Camardella, L.; Ulivi, V.; Guasco, G.; Manduca, P. Trimer carboxyl propeptide of collagen I produced by mature osteoblasts is chemotactic for endothelial cells. J. Biol. Chem. 2000, 275, 32658–32663. [Google Scholar] [CrossRef] [PubMed]
- Inui, M.; Nishi, N.; Yasumoto, A.; Takenaka, I.; Miyanaka, H.; Matsumoto, K.; Nakamura, T.; Wada, F. Enhanced gene expression of transforming growth factor-alpha and c-met in rat urinary bladder cancer. Urol. Res. 1996, 24, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Takeichi, M.; Watabe, M.; Shibamoto, S.; Ito, F. Cadherin-dependent organization and disorganization of epithelial architecture. Princess Takamatsu Symp. 1994, 24, 28–37. [Google Scholar] [PubMed]
- Naldini, L.; Tamagnone, L.; Vigna, E.; Sachs, M.; Hartmann, G.; Birchmeier, W.; Daikuhara, Y.; Tsubouchi, H.; Blasi, F.; Comoglio, P.M. Extracellular proteolytic cleavage by urokinase is required for activation of hepatocyte growth factor/scatter factor. EMBO J. 1992, 11, 4825–4833. [Google Scholar] [PubMed]
- Barinka, C.; Parry, G.; Callahan, J.; Shaw, D.E.; Kuo, A.; Bdeir, K.; Cines, D.B.; Mazar, A.; Lubkowski, J. Structural basis of interaction between urokinase-type plasminogen activator and its receptor. J. Mol. Biol. 2006, 363, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Tochowicz, A.; Maskos, K.; Huber, R.; Oltenfreiter, R.; Dive, V.; Yiotakis, A.; Zanda, M.; Pourmotabbed, T.; Bode, W.; Goettig, P. Crystal structures of MMP-9 complexes with five inhibitors: Contribution of the flexible Arg424 side-chain to selectivity. J. Mol. Biol. 2007, 371, 989–1006. [Google Scholar] [CrossRef] [PubMed]
- Fish, P.V.; Barber, C.G.; Brown, D.G.; Butt, R.; Collis, M.G.; Dickinson, R.P.; Henry, B.T.; Horne, V.A.; Huggins, J.P.; King, E.; et al. Selective urokinase-type plasminogen activator inhibitors. 4. 1-(7-sulfonamidoisoquinolinyl)guanidines. J. Med. Chem. 2007, 50, 2341–2351. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.P.; Hansch, C. Matrix metalloproteinases (MMPs): Chemical-biological functions and (Q)SARs. Bioorg. Med. Chem. 2007, 15, 2223–2268. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, P.; Libert, C. Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration and inflammation. J. Leukoc. Biol. 2007, 82, 1375–1381. [Google Scholar] [CrossRef] [PubMed]
- Hosono, T.; Ito, A.; Sato, T.; Nagase, H.; Mori, Y. Translational augmentation of pro-matrix metalloproteinase 3 (prostromelysin 1) and tissue inhibitor of metalloproteinases (TIMP)-1 mRNAs induced by epidermal growth factor in human uterine cervical fibroblasts. FEBS Lett. 1996, 381, 115–118. [Google Scholar] [CrossRef]
- Nagase, H.; Suzuki, K.; Itoh, Y.; Kan, C.C.; Gehring, M.R.; Huang, W.; Brew, K. Involvement of tissue inhibitors of metalloproteinases (TIMPS) during matrix metalloproteinase activation. Adv. Exp. Med. Biol. 1996, 389, 23–31. [Google Scholar] [PubMed]
- Sarkar, J.; Nandy, S.K.; Chowdhury, A.; Chakraborti, T.; Chakraborti, S. Inhibition of MMP-9 by green tea catechins and prediction of their interaction by molecular docking analysis. Biomed. Pharmacother. 2016, 84, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, M.W.; Michel, T.M. Matrix metalloproteinases in autism spectrum disorders. J. Mol. Psychiatry 2013, 1, 16. [Google Scholar] [CrossRef] [PubMed]
- Galis, Z.S.; Khatri, J.J. Matrix metalloproteinases in vascular remodeling and atherogenesis: The good, the bad, and the ugly. Circ. Res. 2002, 90, 251–262. [Google Scholar] [PubMed]
- Aharinejad, S.; Krenn, K.; Zuckermann, A.; Schafer, R.; Paulus, P.; Seebacher, G.; Wolner, E.; Grimm, M. Matrix metalloproteases and their tissue inhibitor in cardiac transplantation. Eur. J. Cardiothorac. Surg. 2007, 32, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Amalinei, C.; Caruntu, I.D.; Giusca, S.E.; Balan, R.A. Matrix metalloproteinases involvement in pathologic conditions. Rom. J. Morphol. Embryol. 2010, 51, 215–228. [Google Scholar] [PubMed]
- Visse, R.; Nagase, H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: Structure, function, and biochemistry. Circ. Res. 2003, 92, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Brew, K.; Dinakarpandian, D.; Nagase, H. Tissue inhibitors of metalloproteinases: Evolution, structure and function. Biochim. Biophys. Acta 2000, 1477, 267–283. [Google Scholar] [CrossRef]
- Bigg, H.F.; Shi, Y.E.; Liu, Y.E.; Steffensen, B.; Overall, C.M. Specific, high affinity binding of tissue inhibitor of metalloproteinases-4 (TIMP-4) to the COOH-terminal hemopexin-like domain of human gelatinase A. TIMP-4 binds progelatinase A and the COOH-terminal domain in a similar manner to TIMP-2. J. Biol. Chem. 1997, 272, 15496–15500. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.R.; Handsley, M.M.; Pennington, C.J. The ADAM metalloproteinases. Mol. Asp. Med. 2008, 29, 258–289. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, G.I.; Strongin, A.; Collier, I.E.; Genrich, L.T.; Marmer, B.L. Interaction of 92-kDa type IV collagenase with the tissue inhibitor of metalloproteinases prevents dimerization, complex formation with interstitial collagenase, and activation of the proenzyme with stromelysin. J. Biol. Chem. 1992, 267, 4583–4591. [Google Scholar] [PubMed]
- Candelario-Jalil, E.; Yang, Y.; Rosenberg, G.A. Diverse roles of matrix metalloproteinases and tissue inhibitors of metalloproteinases in neuroinflammation and cerebral ischemia. Neuroscience 2009, 158, 983–994. [Google Scholar] [CrossRef] [PubMed]
- Mori, C.; Spooner, E.T.; Wisniewsk, K.E.; Wisniewski, T.M.; Yamaguch, H.; Saido, T.C.; Tolan, D.R.; Selkoe, D.J.; Lemere, C.A. Intraneuronal Abeta42 accumulation in Down syndrome brain. Amyloid 2002, 9, 88–102. [Google Scholar] [PubMed]
- Stefos, G.C.; Soppa, U.; Dierssen, M.; Becker, W. NGF upregulates the plasminogen activation inhibitor-1 in neurons via the calcineurin/NFAT pathway and the Down syndrome-related proteins DYRK1A and RCAN1 attenuate this effect. PLoS ONE 2013, 8, e67470. [Google Scholar] [CrossRef] [PubMed]
- Zamolodchikov, D.; Berk-Rauch, H.E.; Oren, D.A.; Stor, D.S.; Singh, P.K.; Kawasaki, M.; Aso, K.; Strickland, S.; Ahn, H.J. Biochemical and structural analysis of the interaction between beta-amyloid and fibrinogen. Blood 2016, 128, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- ElAli, A.; Bordeleau, M.; Theriault, P.; Filali, M.; Lampron, A.; Rivest, S. Tissue-Plasminogen Activator Attenuates Alzheimer’s Disease-Related Pathology Development in APPswe/PS1 Mice. Neuropsychopharmacology 2016, 41, 1297–1307. [Google Scholar] [CrossRef] [PubMed]
- Bi Oh, S.; Suh, N.; Kim, I.; Lee, J.Y. Impacts of aging and amyloid-beta deposition on plasminogen activators and plasminogen activator inhibitor-1 in the Tg2576 mouse model of Alzheimer’s disease. Brain Res. 2015, 1597, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Martorana, A.; Sancesario, G.M.; Esposito, Z.; Nuccetelli, M.; Sorge, R.; Formosa, A.; Dinallo, V.; Bernardi, G.; Bernardini, S.; Sancesario, G. Plasmin system of Alzheimer’s disease patients: CSF analysis. J. Neural Transm. 2012, 119, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Iulita, M.F.; Cuello, A.C. The NGF Metabolic Pathway in the CNS and its Dysregulation in Down Syndrome and Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 53–67. [Google Scholar] [CrossRef] [PubMed]
- El Zein, N.; D’Hondt, S.; Sariban, E. Crosstalks between the receptors tyrosine kinase EGFR and TrkA and the GPCR, FPR, in human monocytes are essential for receptors-mediated cell activation. Cell. Signal. 2010, 22, 1437–1447. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Takabe, A.; Shimizu, F.; Takahashi, M.; Matsuo, O.; Mitsui, S. Tissue plasminogen activator modulates emotion in a social context. Behav. Brain Res. 2015, 281, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Soeda, S.; Koyanagi, S.; Kuramoto, Y.; Kimura, M.; Oda, M.; Kozako, T.; Hayashida, S.; Shimeno, H. Anti-apoptotic roles of plasminogen activator inhibitor-1 as a neurotrophic factor in the central nervous system. Thromb. Haemost. 2008, 100, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Salehi, A.; Faizi, M.; Belichenko, P.V.; Mobley, W.C. Using mouse models to explore genotype-phenotype relationship in Down syndrome. Ment. Retard. Dev. Disabil. Res. Rev. 2007, 13, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Sacks, H.S.; Fain, J.N.; Cheema, P.; Bahouth, S.W.; Garrett, E.; Wolf, R.Y.; Wolford, D.; Samaha, J. Depot-specific overexpression of proinflammatory, redox, endothelial cell, and angiogenic genes in epicardial fat adjacent to severe stable coronary atherosclerosis. Metab. Syndr. Relat. Disord. 2011, 9, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Merino, P.; Diaz, A.; Yepes, M. Urokinase-type plasminogen activator (uPA) and its receptor (uPAR) promote neurorepair in the ischemic brain. Recept. Clin. Investig. 2017, 4, e1552. [Google Scholar]
- Riemenschneider, M.; Konta, L.; Friedrich, P.; Schwarz, S.; Taddei, K.; Neff, F.; Padovani, A.; Kolsch, H.; Laws, S.M.; Klopp, N.; et al. A functional polymorphism within plasminogen activator urokinase (PLAU) is associated with Alzheimer’s disease. Hum. Mol. Genet. 2006, 15, 2446–2456. [Google Scholar] [CrossRef] [PubMed]
- Farias-Eisner, R.; Vician, L.; Reddy, S.; Basconcillo, R.; Rabbani, S.A.; Wu, Y.Y.; Bradshaw, R.A.; Herschman, H.R. Expression of the urokinase plasminogen activator receptor is transiently required during “priming” of PC12 cells in nerve growth factor-directed cellular differentiation. J. Neurosci. Res. 2001, 63, 341–346. [Google Scholar] [CrossRef]
- Jayakumar, A.R.; Apeksha, A.; Norenberg, M.D. Role of Matricellular Proteins in Disorders of the Central Nervous System. Neurochem. Res. 2017, 42, 858–875. [Google Scholar] [CrossRef] [PubMed]
- Docagne, F.; Nicole, O.; Gabriel, C.; Fernandez-Monreal, M.; Lesne, S.; Ali, C.; Plawinski, L.; Carmeliet, P.; MacKenzie, E.T.; Buisson, A.; et al. Smad3-dependent induction of plasminogen activator inhibitor-1 in astrocytes mediates neuroprotective activity of transforming growth factor-beta 1 against NMDA-induced necrosis. Mol. Cell. Neurosci. 2002, 21, 634–644. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Cui, J.; Lee, J.C.; Ding, S.; Chalimoniuk, M.; Simonyi, A.; Sun, A.Y.; Gu, Z.; Weisman, G.A.; Wood, W.G.; et al. Prolonged exposure of cortical neurons to oligomeric amyloid-beta impairs NMDA receptor function via NADPH oxidase-mediated ROS production: Protective effect of green tea (−)-epigallocatechin-3-gallate. ASN Neuro 2011, 3, e00050. [Google Scholar] [PubMed]
- Abdallah, M.W.; Pearce, B.D.; Larsen, N.; Greaves-Lord, K.; Norgaard-Pedersen, B.; Hougaard, D.M.; Mortensen, E.L.; Grove, J. Amniotic fluid MMP-9 and neurotrophins in autism spectrum disorders: An exploratory study. Autism Res. 2012, 5, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Dziembowska, M.; Pretto, D.I.; Janusz, A.; Kaczmarek, L.; Leigh, M.J.; Gabriel, N.; Durbin-Johnson, B.; Hagerman, R.J.; Tassone, F. High MMP-9 activity levels in fragile X syndrome are lowered by minocycline. Am. J. Med. Genet. Part A 2013, 161, 1897–1903. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, M.; Sapone, A.; Senger, S.; Camhi, S.S.; Kadzielski, S.M.; Buie, T.M.; Kelly, D.L.; Cascella, N.; Fasano, A. Blood-brain barrier and intestinal epithelial barrier alterations in autism spectrum disorders. Mol. Autism 2016, 7, 49. [Google Scholar] [CrossRef] [PubMed]
- Lepeta, K.; Kaczmarek, L. Matrix Metalloproteinase-9 as a Novel Player in Synaptic Plasticity and Schizophrenia. Schizophr. Bull. 2015, 41, 1003–1009. [Google Scholar] [CrossRef] [PubMed]
- Stamenkovic, V.; Stamenkovic, S.; Jaworski, T.; Gawlak, M.; Jovanovic, M.; Jakovcevski, I.; Wilczynski, G.M.; Kaczmarek, L.; Schachner, M.; Radenovic, L.; et al. The extracellular matrix glycoprotein tenascin-C and matrix metalloproteinases modify cerebellar structural plasticity by exposure to an enriched environment. Brain Struct. Funct. 2017, 222, 393–415. [Google Scholar] [CrossRef] [PubMed]
- Vafadari, B.; Salamian, A.; Kaczmarek, L. MMP-9 in translation: From molecule to brain physiology, pathology, and therapy. J. Neurochem. 2016, 139 (Suppl. 2), 91–114. [Google Scholar] [CrossRef] [PubMed]
- Gundimeda, U.; McNeill, T.H.; Fan, T.K.; Deng, R.; Rayudu, D.; Chen, Z.; Cadenas, E.; Gopalakrishna, R. Green tea catechins potentiate the neuritogenic action of brain-derived neurotrophic factor: Role of 67-kDa laminin receptor and hydrogen peroxide. Biochem. Biophys. Res. Commun. 2014, 445, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Chow, H.H.; Cai, Y.; Hakim, I.A.; Crowell, J.A.; Shahi, F.; Brooks, C.A.; Dorr, R.T.; Hara, Y.; Alberts, D.S. Pharmacokinetics and safety of green tea polyphenols after multiple-dose administration of epigallocatechin gallate and polyphenon E in healthy individuals. Clin. Cancer Res. 2003, 9, 3312–3319. [Google Scholar] [PubMed]
- Furst, R.; Zundorf, I. Plant-derived anti-inflammatory compounds: Hopes and disappointments regarding the translation of preclinical knowledge into clinical progress. Mediat. Inflamm. 2014, 2014, 146832. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Shankar, S.; Srivastava, R.K. Green tea catechin, epigallocatechin-3-gallate (EGCG): Mechanisms, perspectives and clinical applications. Biochem. Pharmacol. 2011, 82, 1807–1821. [Google Scholar] [CrossRef] [PubMed]
- Garbisa, S.; Sartor, L.; Biggin, S.; Salvato, B.; Benelli, R.; Albini, A. Tumor gelatinases and invasion inhibited by the green tea flavanol epigallocatechin-3-gallate. Cancer 2001, 91, 822–832. [Google Scholar] [CrossRef]
- Roomi, M.W.; Ivanov, V.; Kalinovsky, T.; Niedzwiecki, A.; Rath, M. Inhibition of cell invasion and MMP production by a nutrient mixture in malignant liposarcoma cell line SW-872. Med. Oncol. 2007, 24, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Swiercz, R.; Skrzypczak-Jankun, E.; Merrell, M.M.; Selman, S.H.; Jankun, J. Angiostatic activity of synthetic inhibitors of urokinase type plasminogen activator. Oncol. Rep. 1999, 6, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Sang, S.; You, H.; Lee, M.J.; Hong, J.; Chin, K.V.; Yang, C.S. Mechanism of action of (−)-epigallocatechin-3-gallate: Auto-oxidation-dependent inactivation of epidermal growth factor receptor and direct effects on growth inhibition in human esophageal cancer KYSE 150 cells. Cancer Res. 2005, 65, 8049–8056. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhu, Q.Y.; Tsang, D.; Huang, Y. Degradation of green tea catechins in tea drinks. J. Agric. Food Chem. 2001, 49, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Kim, C.W.; Kim, J.K.; Shin, H.J.; Baik, J.H. GCG-rich tea catechins are effective in lowering cholesterol and triglyceride concentrations in hyperlipidemic rats. Lipids 2008, 43, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhou, W.; Jiang, X. Reaction kinetics of degradation and epimerization of epigallocatechin gallate (EGCG) in aqueous system over a wide temperature range. J. Agric. Food Chem. 2008, 56, 2694–2701. [Google Scholar] [CrossRef] [PubMed]
- Dryden, G.W.; Lam, A.; Beatty, K.; Qazzaz, H.H.; McClain, C.J. A pilot study to evaluate the safety and efficacy of an oral dose of (−)-epigallocatechin-3-gallate-rich polyphenon E in patients with mild to moderate ulcerative colitis. Inflamm. Bowel Dis. 2013, 19, 1904–1912. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.D.; Lee, M.J.; Lu, H.; Meng, X.; Hong, J.J.; Seril, D.N.; Sturgill, M.G.; Yang, C.S. Epigallocatechin-3-gallate is absorbed but extensively glucuronidated following oral administration to mice. J. Nutr. 2003, 133, 4172–4177. [Google Scholar] [PubMed]
- Mereles, D.; Hunstein, W. Epigallocatechin-3-gallate (EGCG) for clinical trials: More pitfalls than promises? Int. J. Mol. Sci. 2011, 12, 5592–5603. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, I.A.; Bharali, D.J.; Nihal, M.; Adhami, V.M.; Khan, N.; Chamcheu, J.C.; Khan, M.I.; Shabana, S.; Mousa, S.A.; Mukhtar, H. Excellent anti-proliferative and pro-apoptotic effects of (−)-epigallocatechin-3-gallate encapsulated in chitosan nanoparticles on human melanoma cell growth both in vitro and in vivo. Nanomedicine 2014, 10, 1619–1626. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Taylor, E.W.; Wang, Y.; Wan, X.; Zhang, J. Encapsulated nanoepigallocatechin-3-gallate and elemental selenium nanoparticles as paradigms for nanochemoprevention. Int. J. Nanomed. 2012, 7, 1711–1721. [Google Scholar]
- Bourassa, P.; Cote, R.; Hutchandani, S.; Samson, G.; Tajmir-Riahi, H.A. The effect of milk alpha-casein on the antioxidant activity of tea polyphenols. J. Photochem. Photobiol. B 2013, 128, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Haratifar, S.; Meckling, K.A.; Corredig, M. Antiproliferative activity of tea catechins associated with casein micelles, using HT29 colon cancer cells. J. Dairy Sci. 2014, 97, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Jobstl, E.; Howse, J.R.; Fairclough, J.P.; Williamson, M.P. Noncovalent cross-linking of casein by epigallocatechin gallate characterized by single molecule force microscopy. J. Agric. Food Chem. 2006, 54, 4077–4081. [Google Scholar] [CrossRef] [PubMed]
- Jankun, J.; Keck, R.W.; Skrzypczak-Jankun, E.; Swiercz, R. Inhibitors of urokinase reduce size of prostate cancer xenografts in severe combined immunodeficient mice. Cancer Res. 1997, 57, 559–563. [Google Scholar] [PubMed]
- Shankar, S.; Ganapathy, S.; Hingorani, S.R.; Srivastava, R.K. EGCG inhibits growth, invasion, angiogenesis and metastasis of pancreatic cancer. Front. Biosci. 2008, 13, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Stuart, E.C.; Scandlyn, M.J.; Rosengren, R.J. Role of epigallocatechin gallate (EGCG) in the treatment of breast and prostate cancer. Life Sci. 2006, 79, 2329–2336. [Google Scholar] [CrossRef] [PubMed]
- Pervin, M.; Unno, K.; Nakagawa, A.; Takahashi, Y.; Iguchi, K.; Yamamoto, H.; Hoshino, M.; Hara, A.; Takagaki, A.; Nanjo, F.; et al. Blood brain barrier permeability of (−)-epigallocatechin gallate, its proliferation-enhancing activity of human neuroblastoma SH-SY5Y cells, and its preventive effect on age-related cognitive dysfunction in mice. Biochem. Biophys. Rep. 2017, 9, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Unno, K.; Pervin, M.; Nakagawa, A.; Iguchi, K.; Hara, A.; Takagaki, A.; Nanjo, F.; Minami, A.; Nakamura, Y. Blood-Brain Barrier Permeability of Green Tea Catechin Metabolites and their Neuritogenic Activity in Human Neuroblastoma SH-SY5Y Cells. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Meng, X.; Yang, C.S. Enzymology of methylation of tea catechins and inhibition of catechol-O-methyltransferase by (−)-epigallocatechin gallate. Drug Metab. Dispos. 2003, 31, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Wang, X.; Lu, G.; Picinich, S.C. Cancer prevention by tea: Animal studies, molecular mechanisms and human relevance. Nat. Rev. Cancer 2009, 9, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.H.; You, Y.P. Epigallocatechin Gallate Extends Therapeutic Window of Recombinant Tissue Plasminogen Activator Treatment for Brain Ischemic Stroke: A Randomized Double-Blind and Placebo-Controlled Trial. Clin. Neuropharmacol. 2017, 40, 24–28. [Google Scholar] [CrossRef] [PubMed]
- You, Y.P. Epigallocatechin Gallate Extends the Therapeutic Window of Recombinant Tissue Plasminogen Activator Treatment in Ischemic Rats. J. Stroke Cerebrovasc. Dis. 2016, 25, 990–997. [Google Scholar] [CrossRef] [PubMed]
- Stringer, M.; Abeysekera, I.; Thomas, J.; LaCombe, J.; Stancombe, K.; Stewart, R.J.; Dria, K.J.; Wallace, J.M.; Goodlett, C.R.; Roper, R.J. Epigallocatechin-3-gallate (EGCG) consumption in the Ts65Dn model of Down syndrome fails to improve behavioral deficits and is detrimental to skeletal phenotypes. Physiol. Behav. 2017, 177, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Stagni, F.; Giacomini, A.; Emili, M.; Guidi, S.; Ciani, E.; Bartesaghi, R. Epigallocatechin gallate: A useful therapy for cognitive disability in Down syndrome? Neurogenesis 2017, 4, e1270383. [Google Scholar] [CrossRef] [PubMed]
- Catuara-Solarz, S.; Espinosa-Carrasco, J.; Erb, I.; Langohr, K.; Notredame, C.; Gonzalez, J.R.; Dierssen, M. Principal Component Analysis of the Effects of Environmental Enrichment and (−)-epigallocatechin-3-gallate on Age-Associated Learning Deficits in a Mouse Model of Down Syndrome. Front. Behav. Neurosci. 2015, 9, 330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stringer, M.; Abeysekera, I.; Dria, K.J.; Roper, R.J.; Goodlett, C.R. Low dose EGCG treatment beginning in adolescence does not improve cognitive impairment in a Down syndrome mouse model. Pharmacol. Biochem. Behav. 2015, 138, 70–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wyganowska-Świątkowska, M.; Matthews-Kozanecka, M.; Matthews-Brzozowska, T.; Skrzypczak-Jankun, E.; Jankun, J. Can EGCG Alleviate Symptoms of Down Syndrome by Altering Proteolytic Activity? Int. J. Mol. Sci. 2018, 19, 248. https://doi.org/10.3390/ijms19010248
Wyganowska-Świątkowska M, Matthews-Kozanecka M, Matthews-Brzozowska T, Skrzypczak-Jankun E, Jankun J. Can EGCG Alleviate Symptoms of Down Syndrome by Altering Proteolytic Activity? International Journal of Molecular Sciences. 2018; 19(1):248. https://doi.org/10.3390/ijms19010248
Chicago/Turabian StyleWyganowska-Świątkowska, Marzena, Maja Matthews-Kozanecka, Teresa Matthews-Brzozowska, Ewa Skrzypczak-Jankun, and Jerzy Jankun. 2018. "Can EGCG Alleviate Symptoms of Down Syndrome by Altering Proteolytic Activity?" International Journal of Molecular Sciences 19, no. 1: 248. https://doi.org/10.3390/ijms19010248