Transcriptome Analysis in Haematococcus pluvialis: Astaxanthin Induction by High Light with Acetate and Fe2+

Abstract
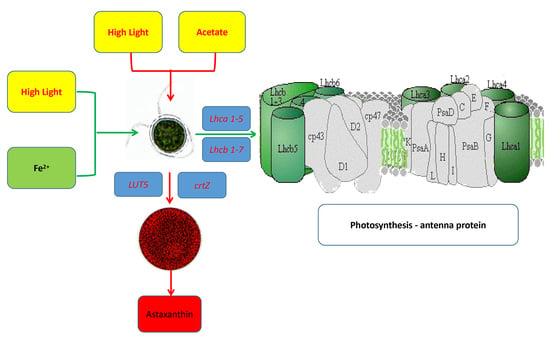
:
1. Introduction
2. Result
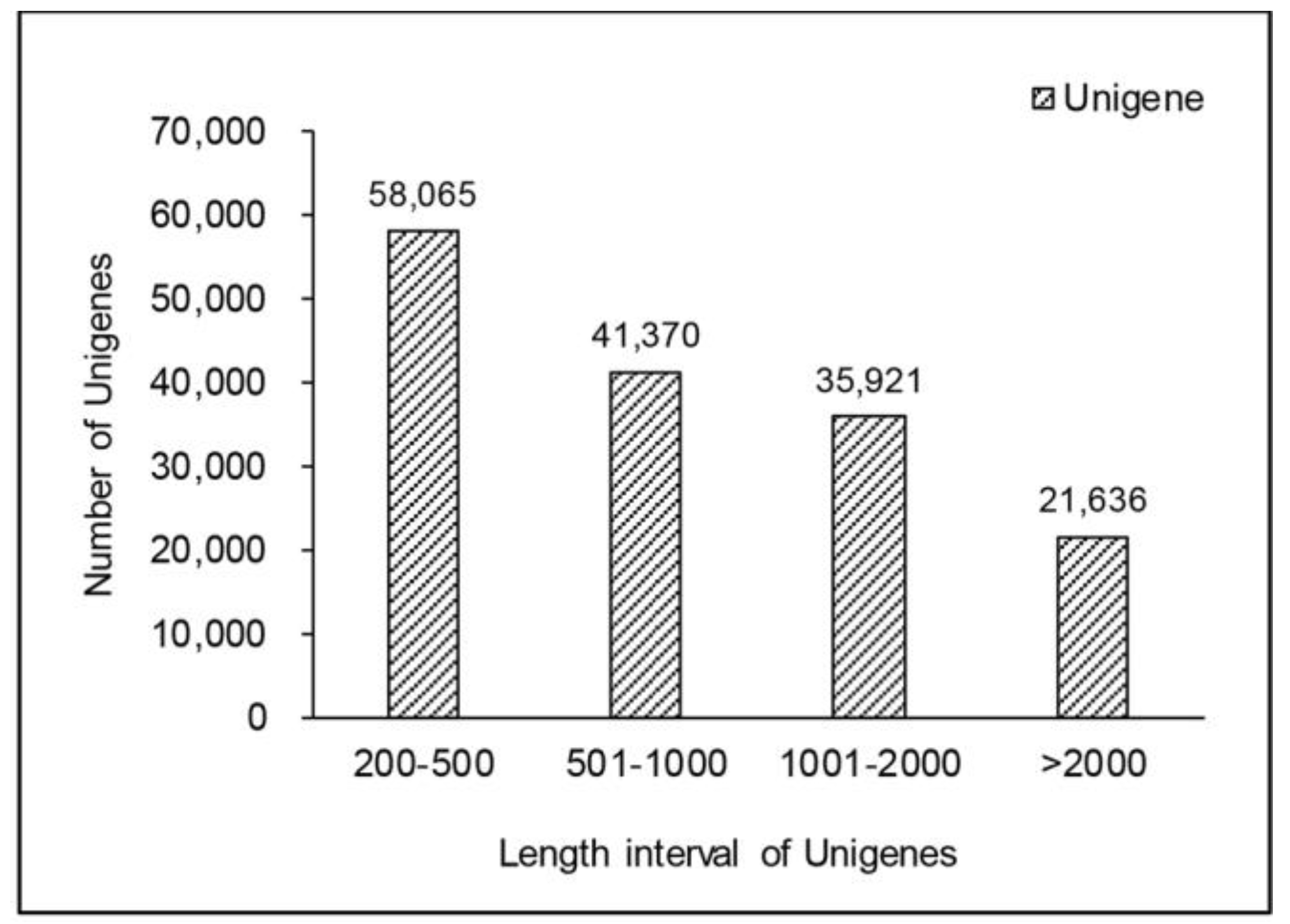
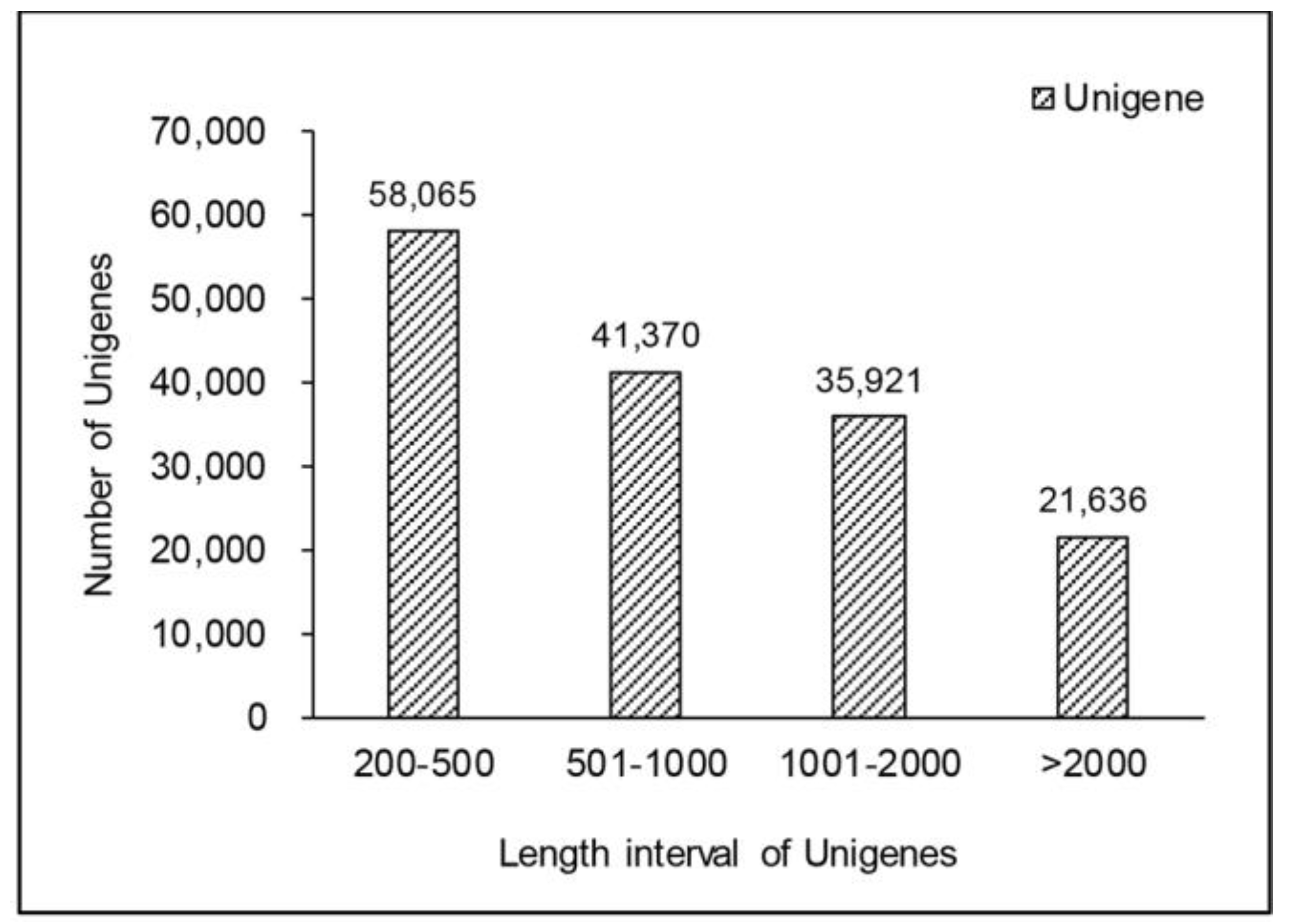
2.1. Short-Read De Novo Sequencing and Assembly
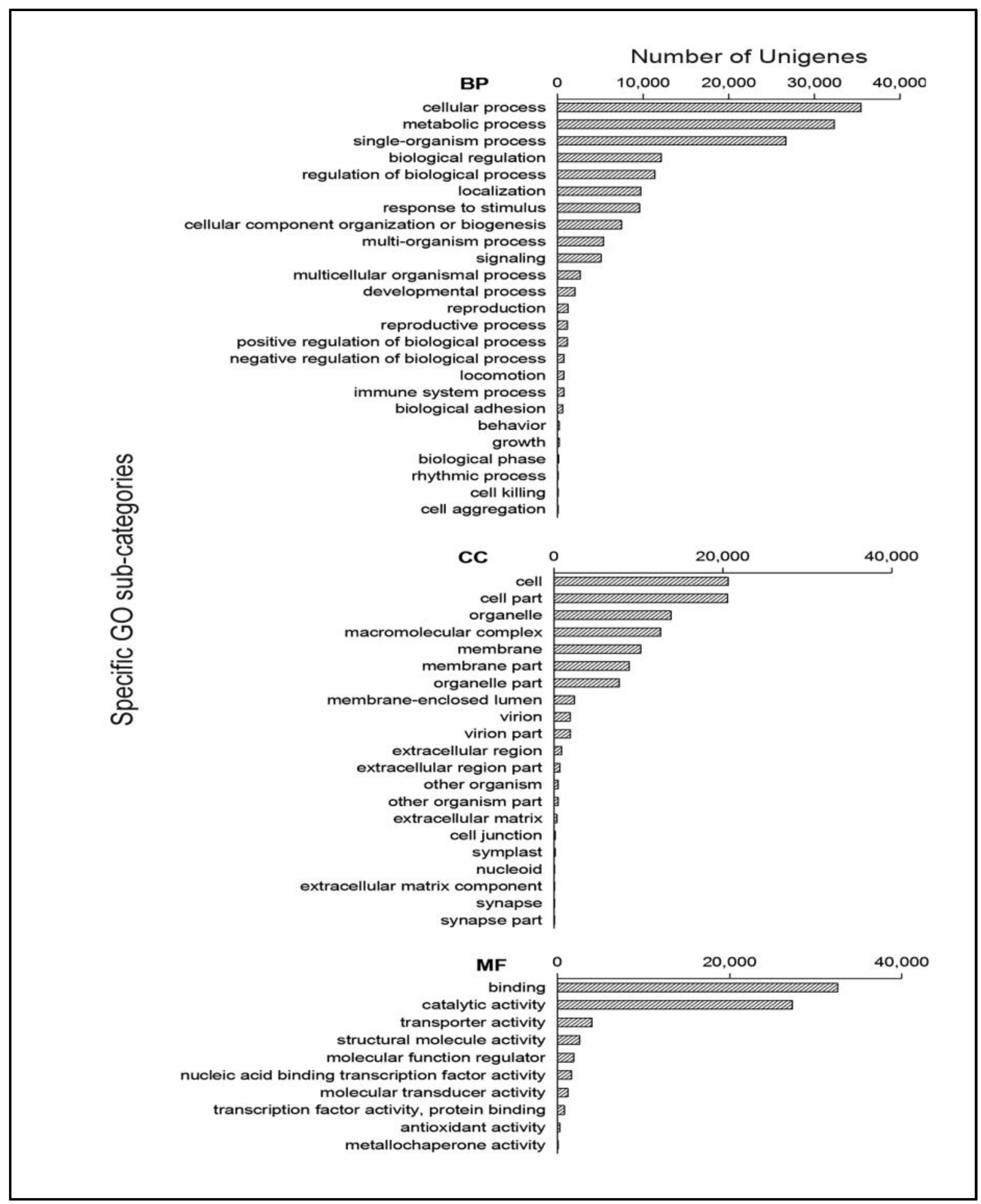
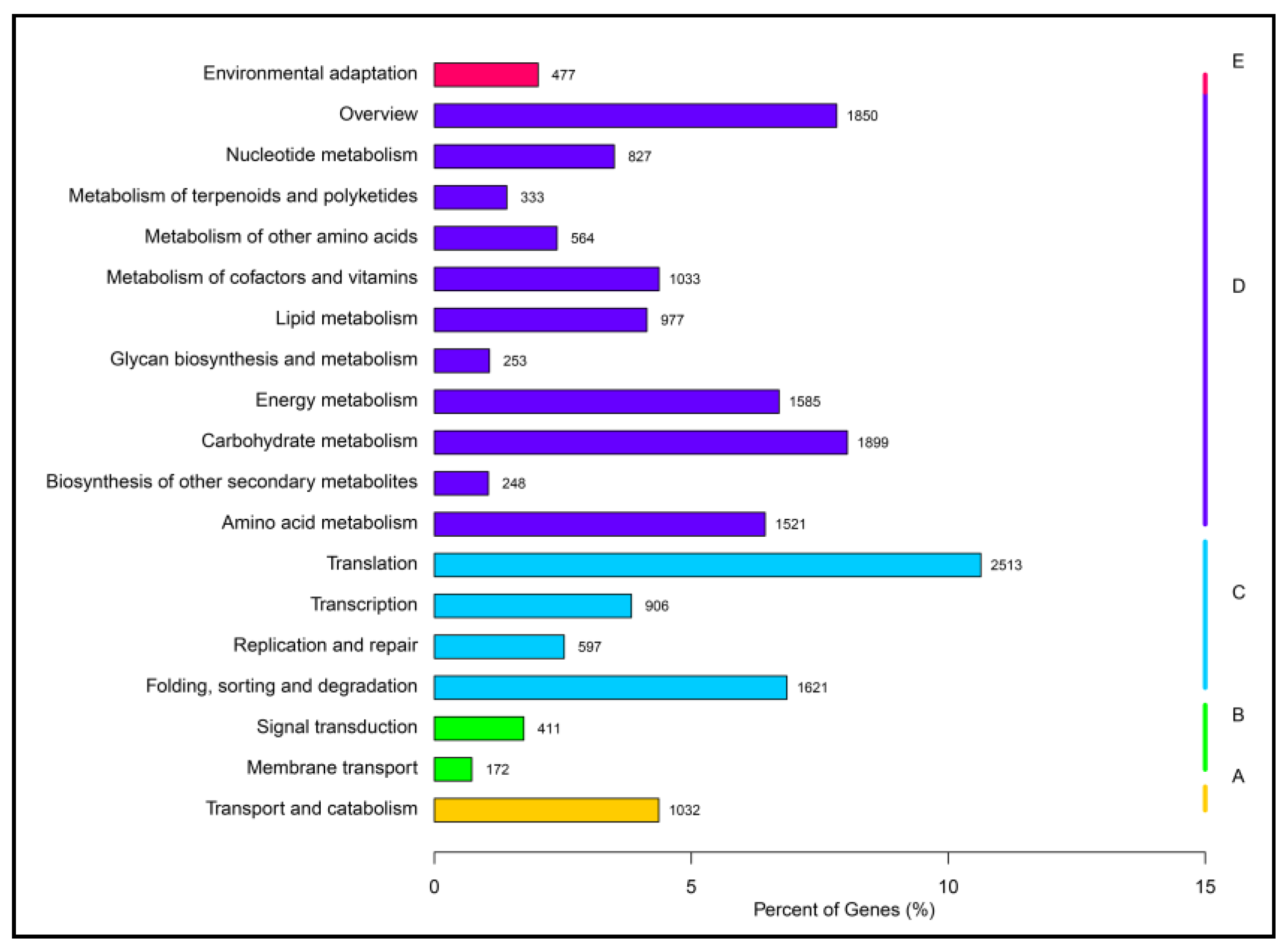
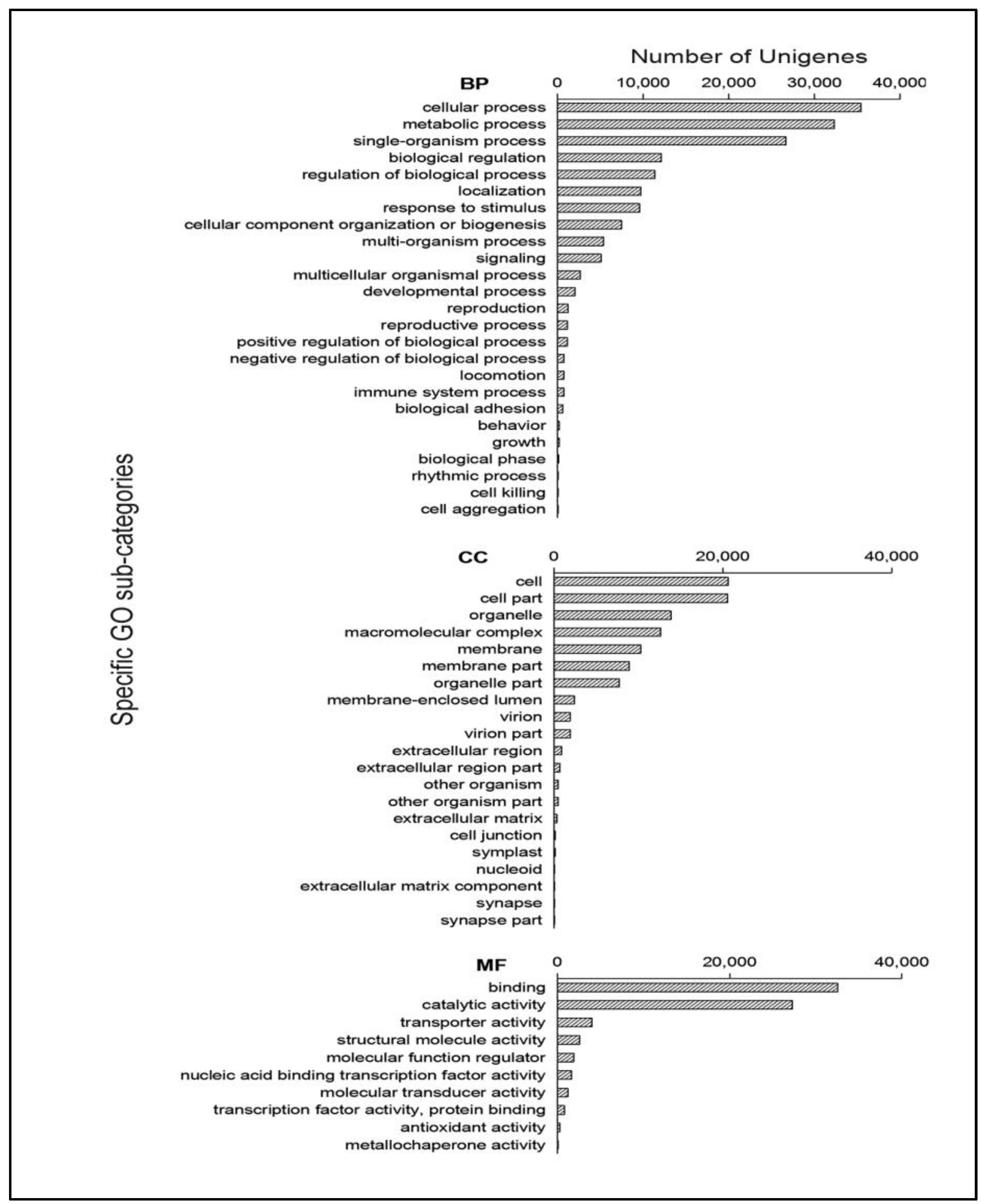
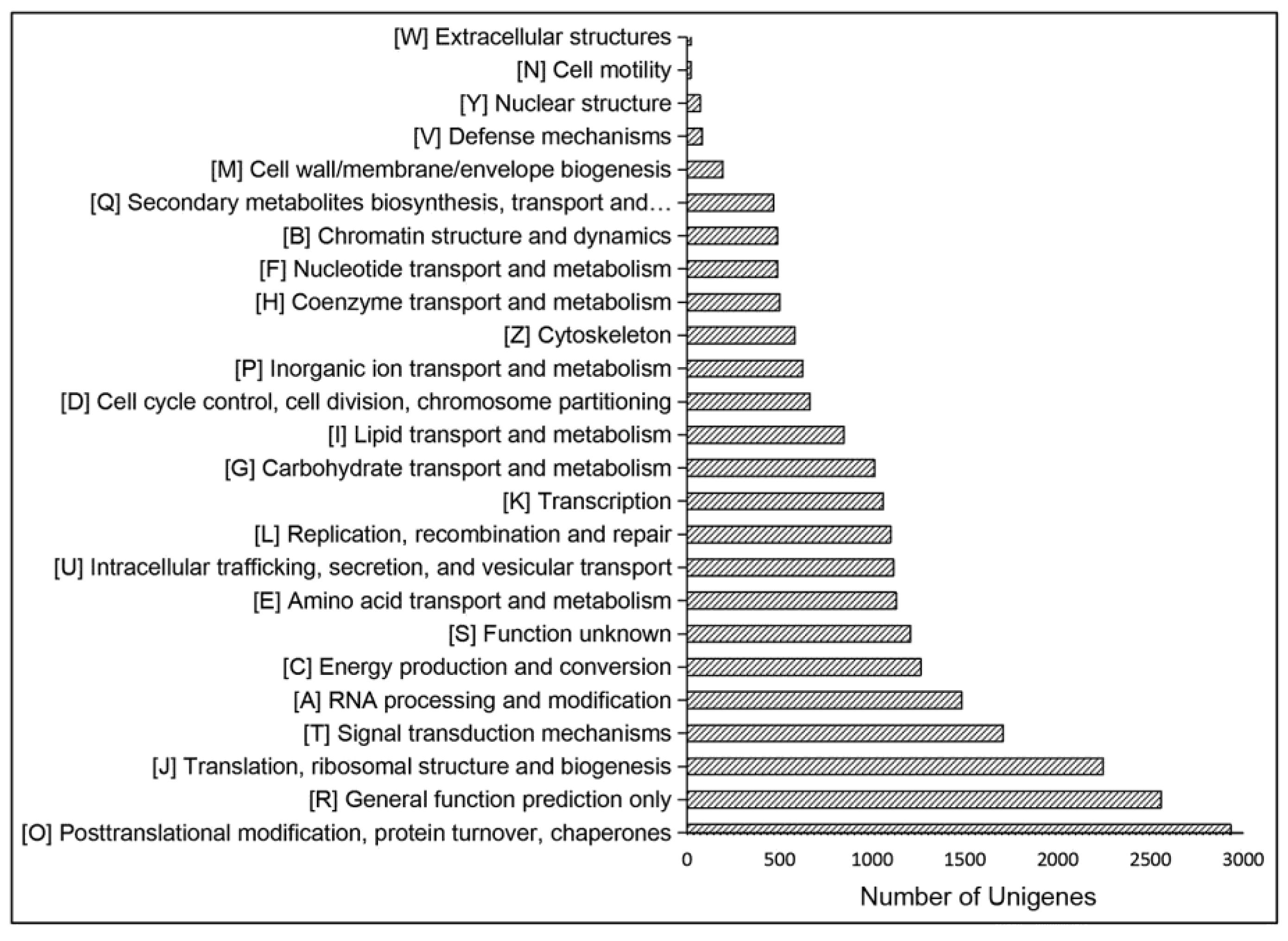
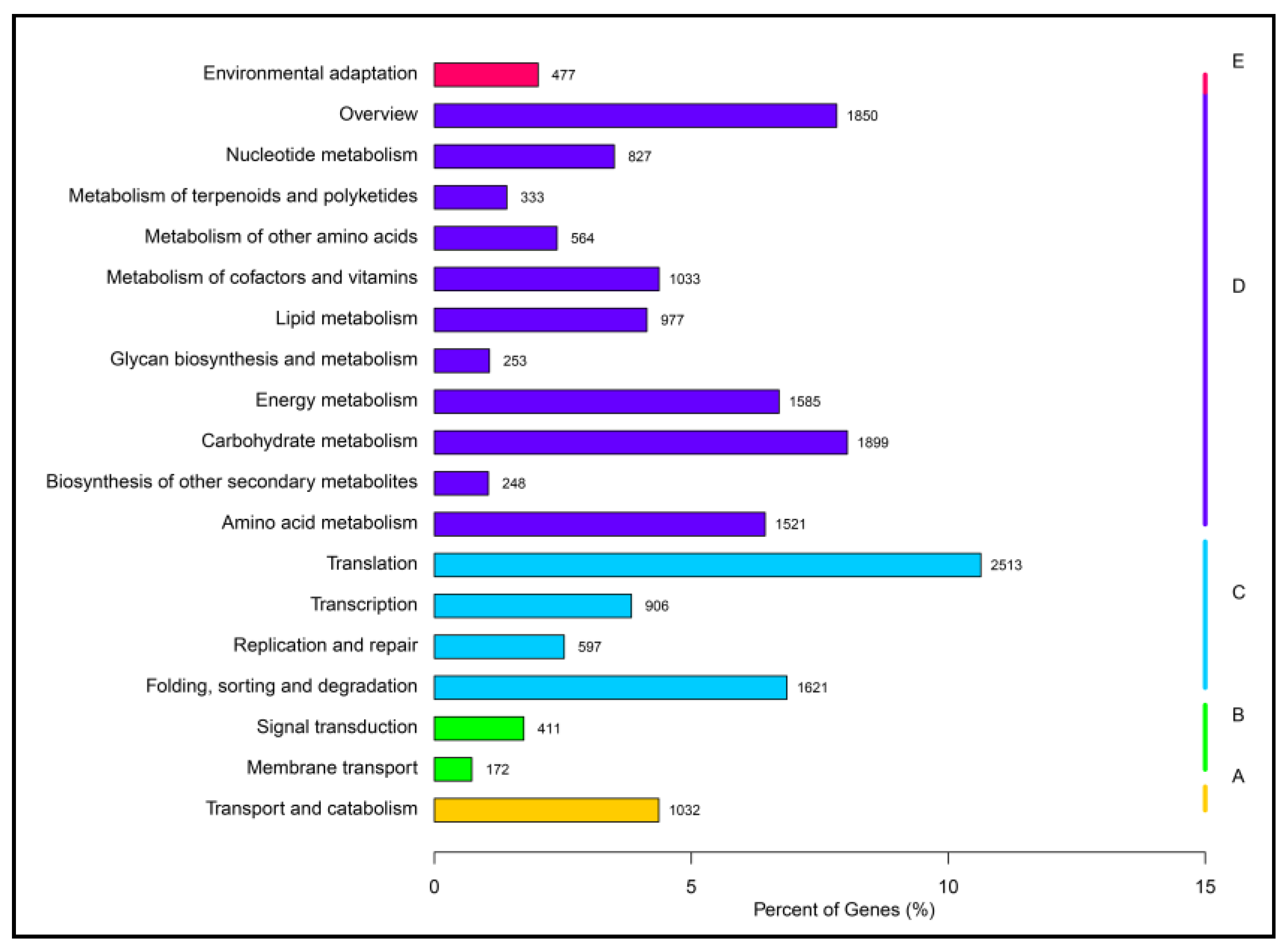
2.2. Function Annotation
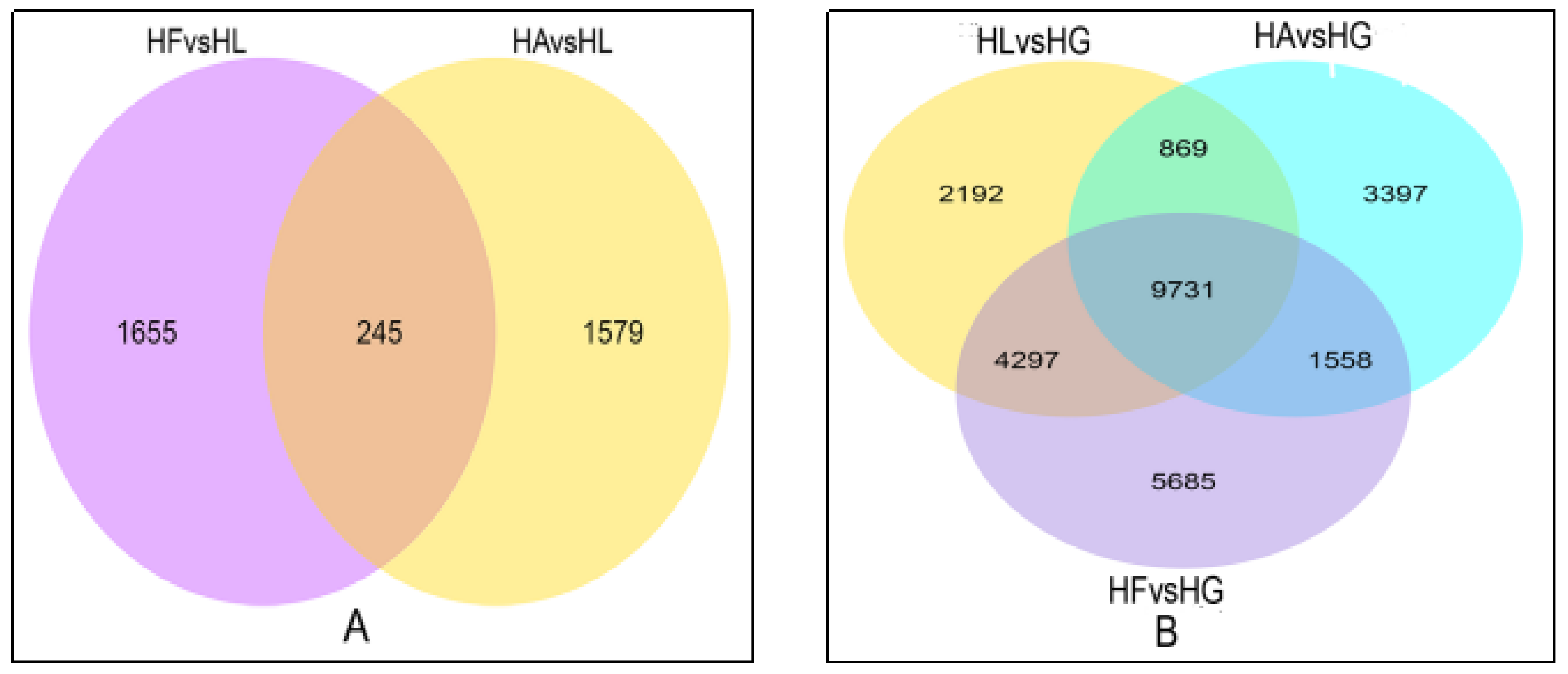
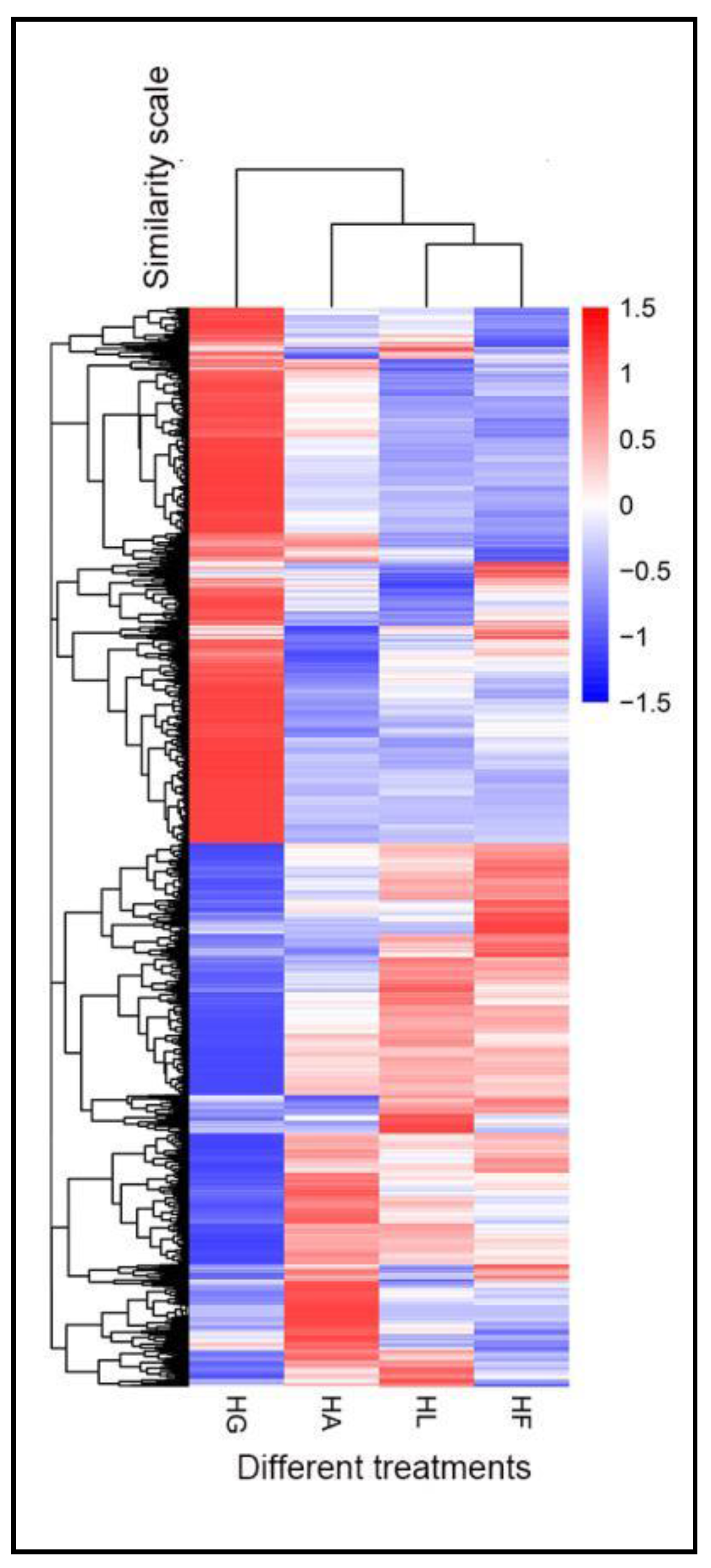
2.3. Analysis of DEGs
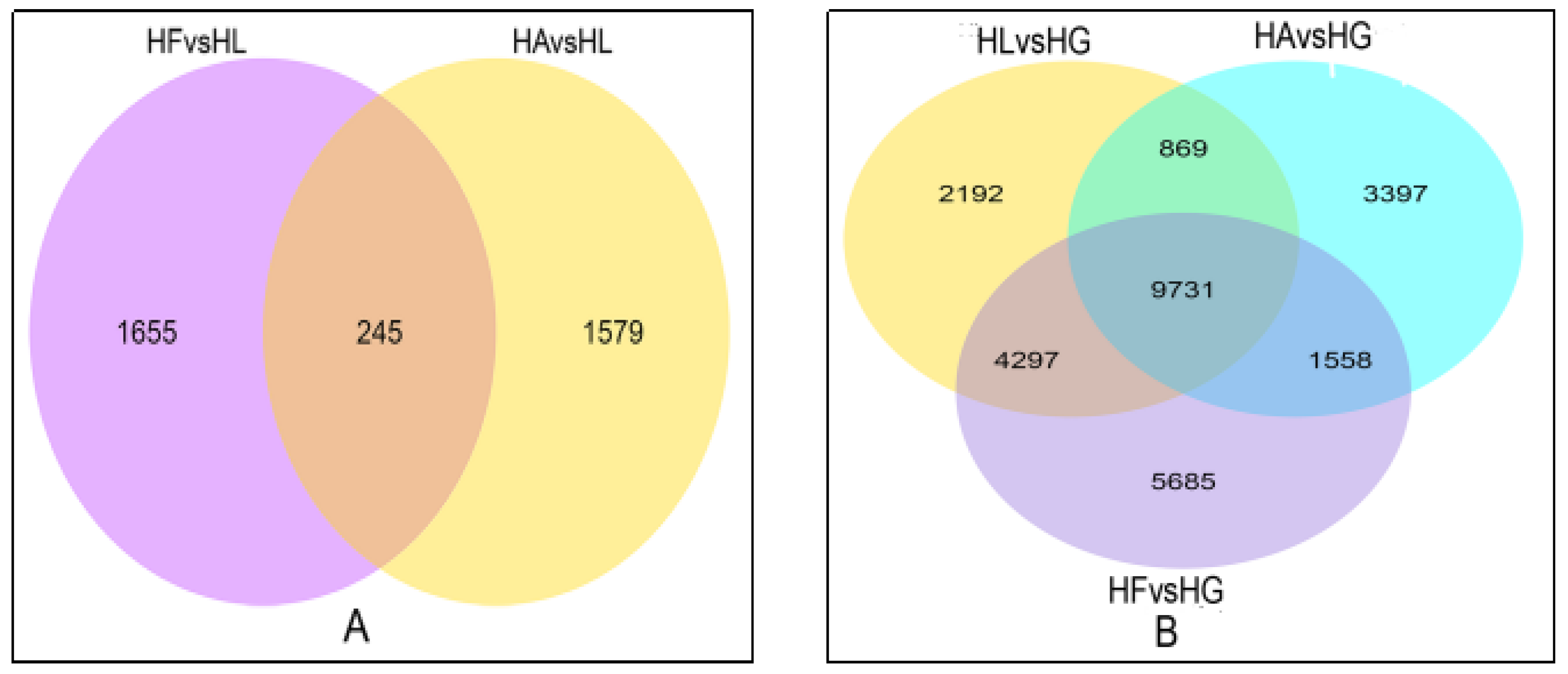
2.3.1. DEG Profiles in Response to the Treatments
2.3.2. DEG Analysis in the High Light Treatment
2.3.3. DEG Analysis for Addition of Acetate under High Light Condition
2.3.4. DEG Analysis for Addition of Fe2+ under High Light Condition
2.3.5. KEGG Analysis of DEGs between Two Groups: Addition of Acetate or Fe2+
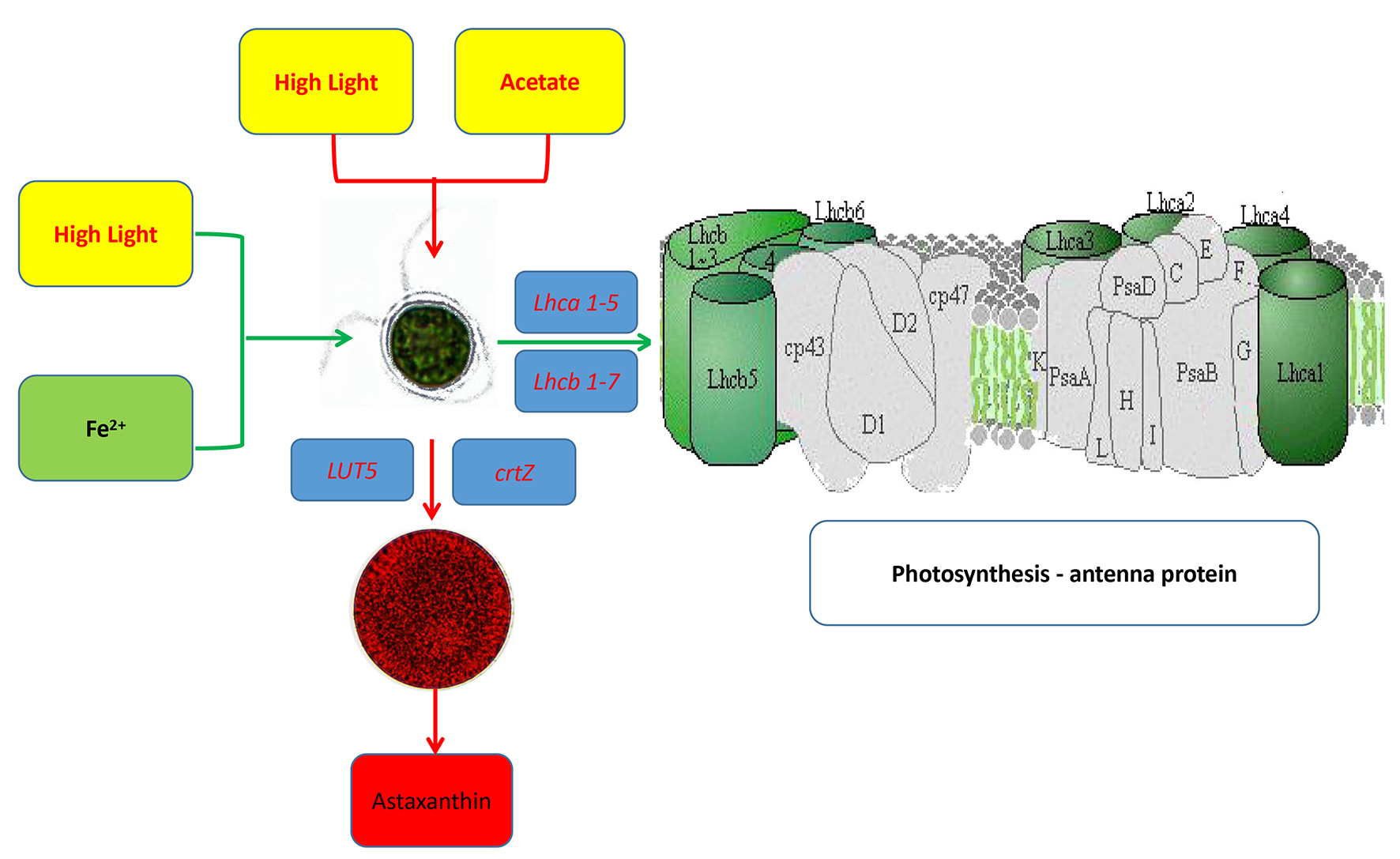
2.4. The Effects of Induction Treatment on Specific Biological Pathways
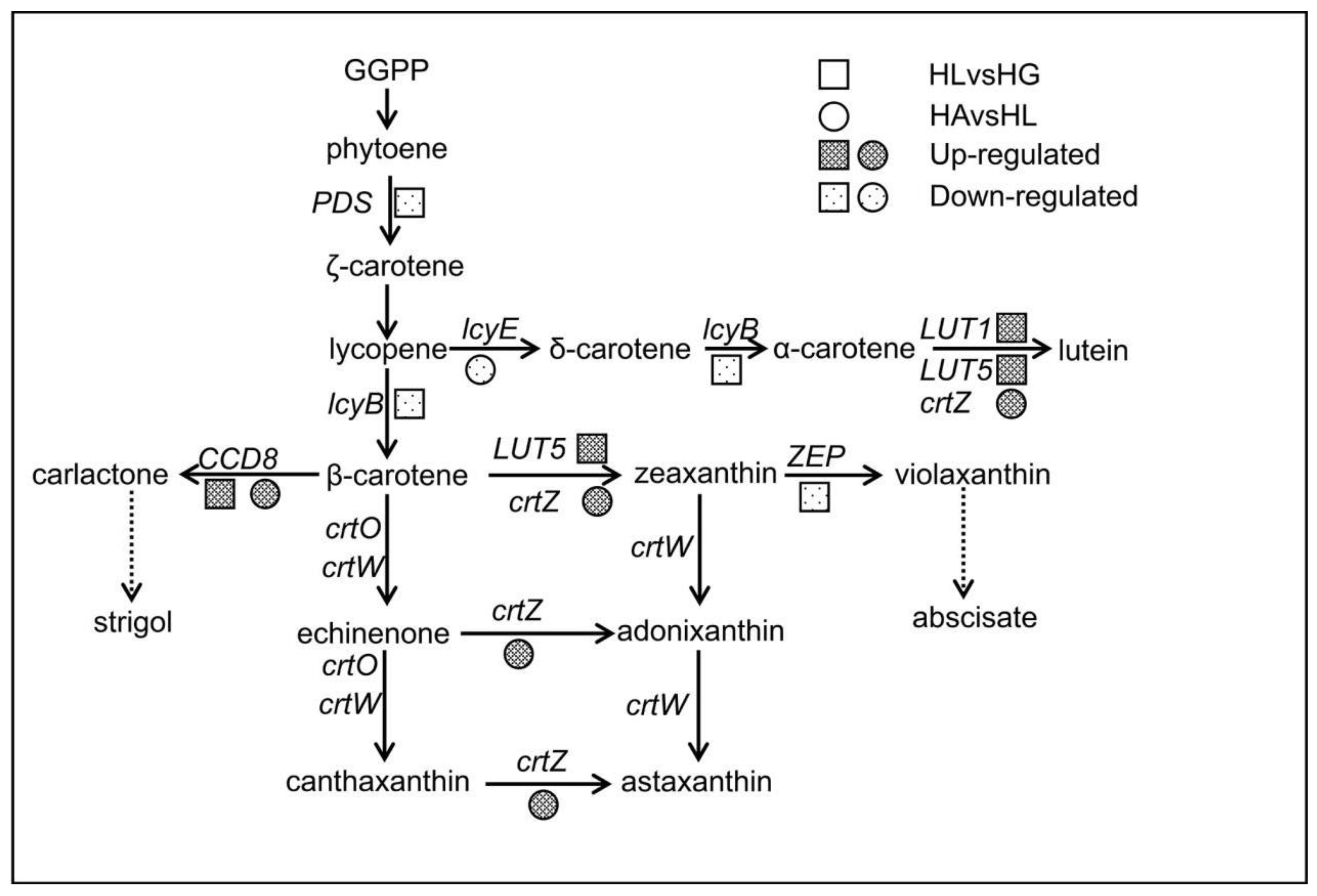
2.4.1. The Effects on the Carotenoid Biosynthesis Pathway
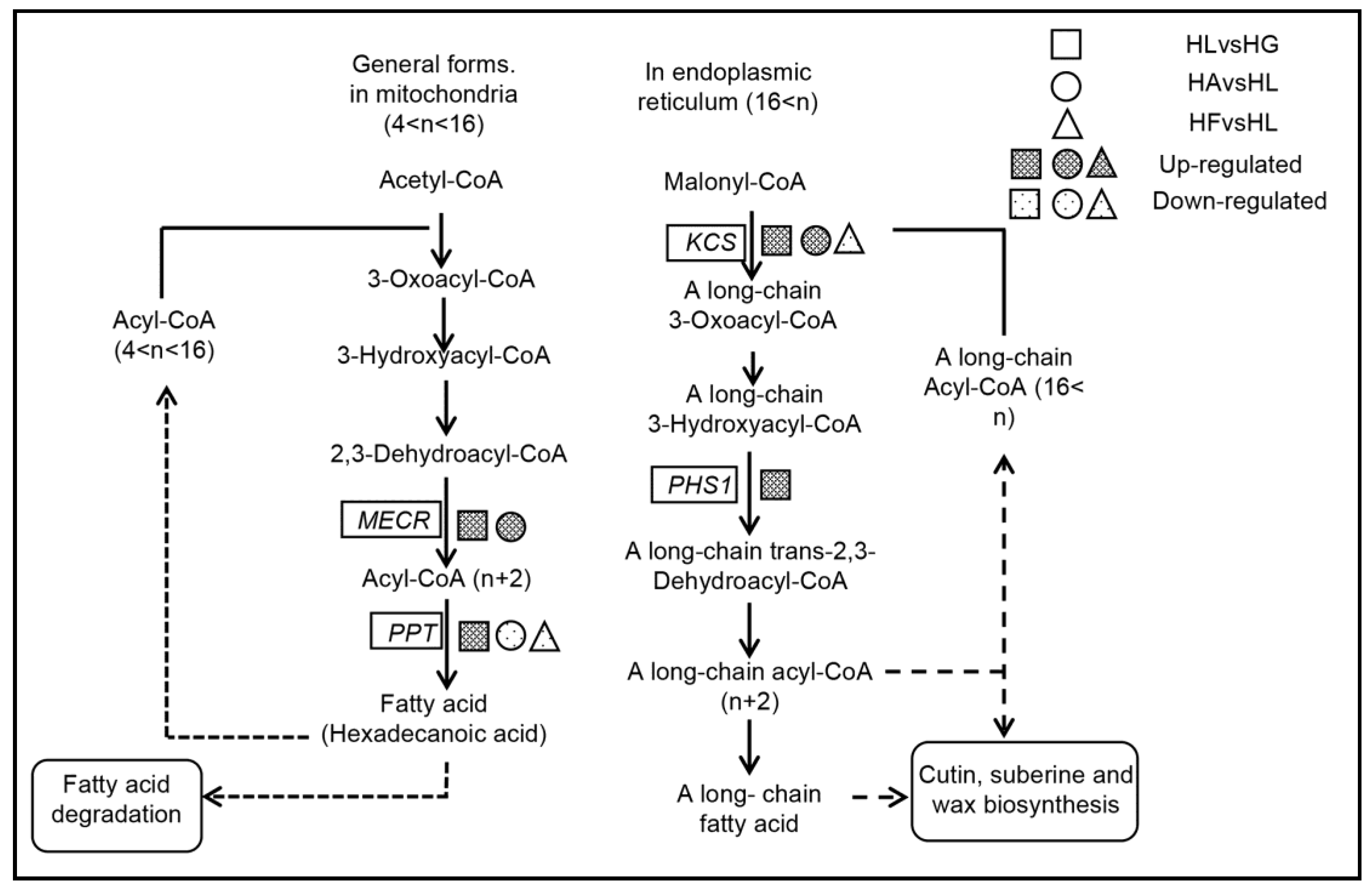
2.4.2. The Effects on the Fatty Acid Elongation Pathway
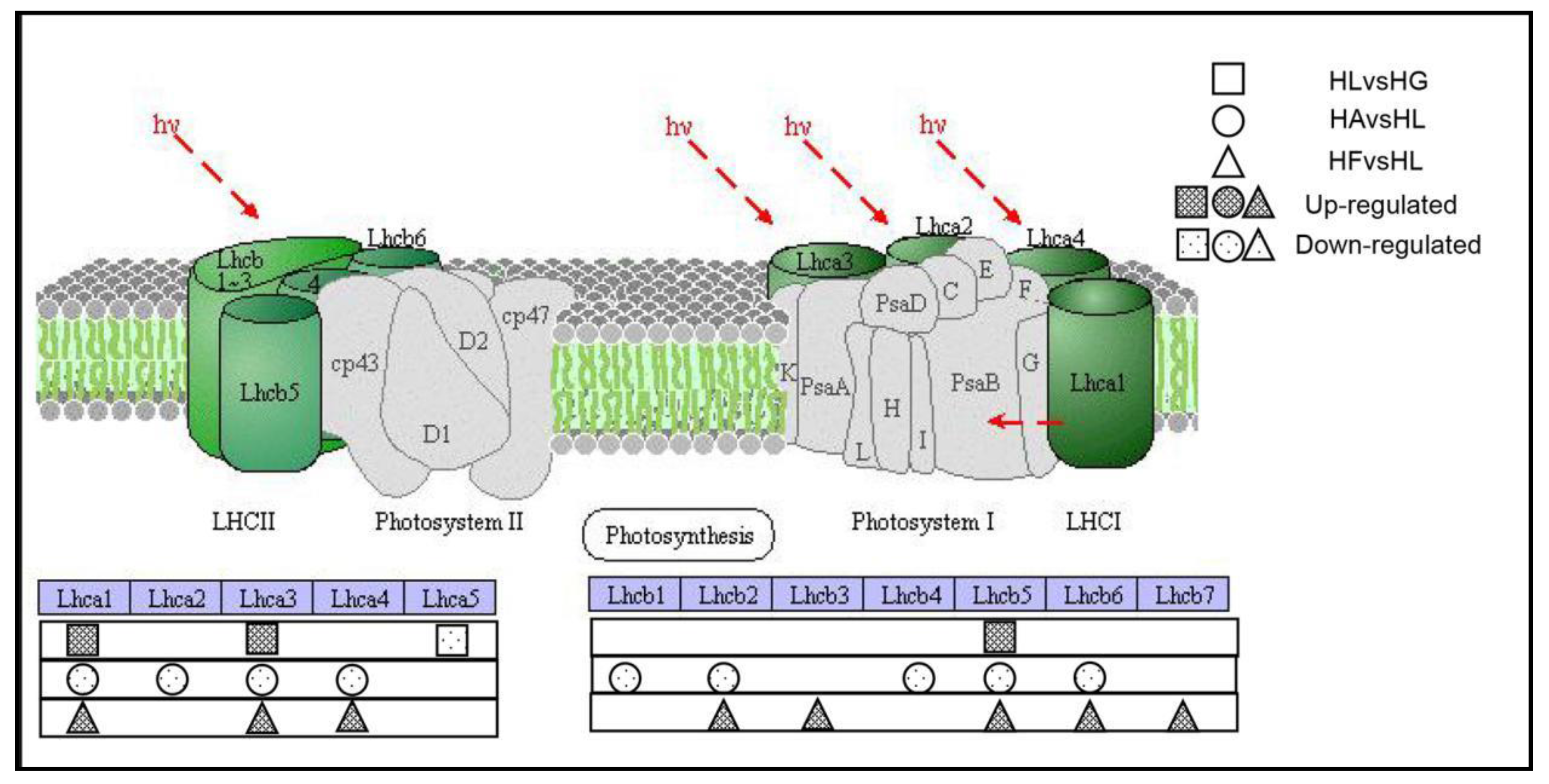
2.4.3. The Effects on Photosynthesis-Antenna Proteins Pathway
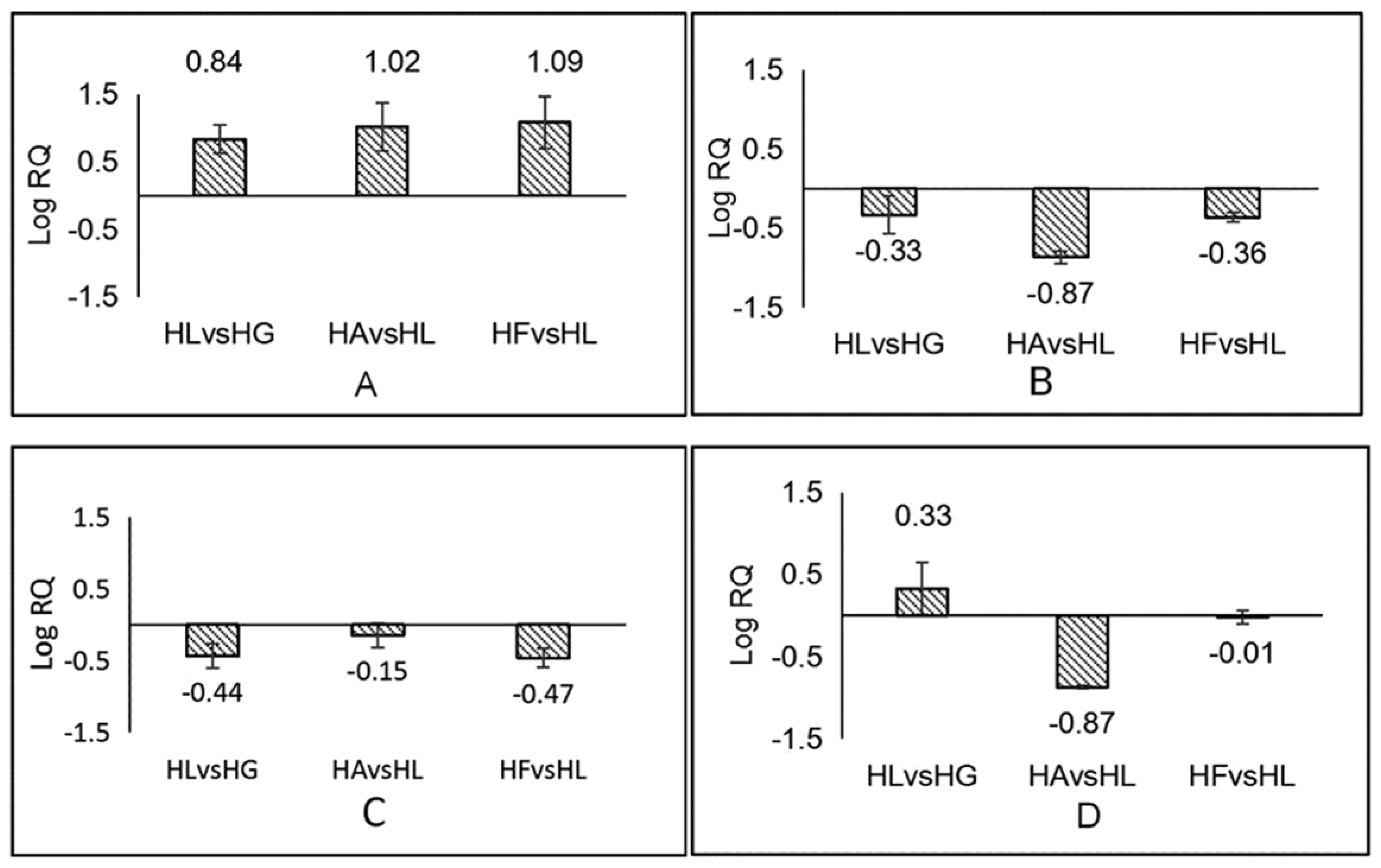
2.5. Analysis of the Results of Real-Time Quantitative PCR
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. RNA Extraction and cDNA Library Construction
4.3. Quality Controls, De Novo Assembly and Clustering
4.4. Gene Function Annotation and Classification
4.5. Analysis of Differentially Expressed Genes (DEGs)
4.6. DEG Validation and Expression Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Guerin, M.; Huntley, M.E.; Olaizola, M. Haematococcus astaxanthin: Applications for human health and nutrition. Trends Biotechnol. 2003, 21, 210–216. [Google Scholar] [CrossRef]
- Yuan, J.P.; Peng, J.; Yin, K.; Wang, J.H. Potential health-promoting effects of astaxanthin: A high-value carotenoid mostly from microalgae. Mol. Nutr. Food Res. 2011, 55, 150–165. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhu, K.; Xu, Q.; Wang, G.; Zhang, J.; Cao, R.; Ye, J.; Yu, X. The antidepressant-like effect of trans-astaxanthin involves the serotonergic system. Oncotarget 2017, 8, 25552–25563. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.L.; Han, X.D.; Li, Y.; Chu, X.F.; Miao, W.M.; Zhang, J.L.; Fan, S.J. Astaxanthin attenuates total body irradiation-induced hematopoietic system injury in mice via inhibition of oxidative stress and apoptosis. Stem Cell Res. Ther. 2017, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Steinbrenner, J.; Linden, H. Light induction of carotenoid biosynthesis genes in the green alga Haematococcus pluvialis: Regulation by photosynthetic redox control. Plant Mol. Biol. 2003, 52, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Steinbrenner, J.; Sandmann, G. Transformation of the green alga Haematococcus pluvialis with a phytoene desaturase for accelerated astaxanthin biosynthesis. Appl. Environ. Microbiol. 2006, 72, 7477–7484. [Google Scholar] [CrossRef] [PubMed]
- Gwak, Y.; Hwang, Y.S.; Wang, B.; Kim, M.; Jeong, J.; Lee, C.G.; Hu, Q.; Han, D.; Jin, E.S. Comparative analyses of lipidomes and transcriptomes reveal a concerted action of multiple defensive systems against photooxidative stress in Haematococcus pluvialis. J. Exp. Bot. 2014, 65, 4317–4334. [Google Scholar]
- Boussiba, S. Carotenogenesis in the green alga Haematococcus pluvialis: Cellular physiology and stress response. Physiol. Plant. 2000, 108, 111–117. [Google Scholar] [CrossRef]
- Gao, Z.; Li, Y.; Wu, G.; Li, G.; Sun, H.; Deng, S.; Shen, Y.; Chen, G.; Zhang, R.; Meng, C. Transcriptome analysis in Haematococcus pluvialis: Astaxanthin induction by salicylic acid (SA) and jasmonic acid (JA). PLoS ONE 2015, 10, e0140609. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wang, B.; Han, D.; Sommerfeld, M.; Lu, Y.; Chen, F.; Hu, Q. Molecular mechanisms of the coordination between astaxanthin and fatty acid biosynthesis in Haematococcus pluvialis (chlorophyceae). Plant J. Cell Mol. Biol. 2015, 81, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Okada, T. Protective role of astaxanthin against u.v.-B irradiation in the green alga Haematococcus pluvialis. Biotechnol. Lett. 2000, 22, 177–181. [Google Scholar]
- Jeon, Y.C.; Cho, C.W.; Yun, Y.S. Combined effects of light intensity and acetate concentration on the growth of unicellular microalga Haematococcus pluvialis. Enzyme Microb. Technol. 2006, 39, 490–495. [Google Scholar] [CrossRef]
- Kobayashi, M.; Kakizono, T.; Nagai, S. Enhanced carotenoid biosynthesis by oxidative stress in acetate-induced cyst cells of a green unicellular alga, Haematococcus pluvialis. Appl. Environ. Microbiol. 1993, 59, 867. [Google Scholar] [PubMed]
- Hong, M.E.; Choi, Y.Y.; Sang, J.S. Effect of red cyst cell inoculation and iron (ii) supplementation on autotrophic astaxanthin production by Haematococcus pluvialis, under outdoor summer conditions. J. Biotechnol. 2015, 218, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Hess, S.K.; Lepetit, B.; Kroth, P.G.; Mecking, S. Production of chemicals from microalgae lipids–status and perspectives. Eur. J. Lipid Sci. Technol. 2017, 120, 1700062. [Google Scholar] [CrossRef]
- Goss, R.; Wilhelm, C.; Wada, H.; Murata, N. (Eds.) Lipids in Photosynthesis: Essential and Regulatory Functions; Springer: Dordrecht, The Netherlands, 2010; pp. 117–137. [Google Scholar]
- Zhekisheva, M.; Boussiba, S.; Khozin-Goldberg, I.; Zarka, A.; Cohen, Z. Accumulation of oleic acid in Haematococcus pluvialis (chlorophyceae) under nitrogen starvation or high light is correlated with that of astaxanthin esters1. J. Phycol. 2010, 38, 325–331. [Google Scholar] [CrossRef]
- Damiani, M.C.; Popovich, C.A.; Diana, C.; Leonardi, P.I. Lipid analysis in Haematococcus pluvialis to assess its potential use as a biodiesel feedstock. Bioresour. Technol. 2010, 101, 3801–3807. [Google Scholar]
- Morozova, O.; Hirst, M.; Marra, M.A. Applications of new sequencing technologies for transcriptome analysis. Ann. Rev. Genom. Hum. Genet. 2009, 10, 135–151. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zang, X.; Wu, F.; Jin, Y.; Wang, H.; Liu, C.; Ding, Y.; He, B.; Xiao, D.; Song, X. Transcriptome sequencing of Gracilariopsis lemaneiformis to analyze the genes related to optically active phycoerythrin synthesis. PLoS ONE 2017, 12, e0170855. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Xian, A.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Davidson, N.M.; Oshlack, A. Corset: Enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biol. 2014, 15, 410. [Google Scholar] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Nat. Preced. 2010. [Google Scholar] [CrossRef] [Green Version]
- Oshlack, A.; Robinson, M.D.; Young, M.D. From RNA-seq reads to differential expression results. Genome Biol. 2010, 11, 220. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T. KEGG for linking genomes to life and the environment. Nucl. Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Clean Reads | Clean Bases | Error (%) | Q20 (%) | GC (%) |
|---|---|---|---|---|---|---|
| HG_1 | 59,081,916 | 57,542,600 | 8.63G | 0.02 | 95.53 | 60.11 |
| HG_2 | 53,033,442 | 51,513,440 | 7.73G | 0.02 | 95.38 | 60.46 |
| HG_3 | 63,249,144 | 61,459,678 | 9.22G | 0.02 | 95.38 | 60.15 |
| HL_1 | 55,314,470 | 53,253,670 | 7.99G | 0.02 | 95.32 | 59.93 |
| HL_2 | 56,631,072 | 55,112,470 | 8.27G | 0.02 | 96.78 | 60.42 |
| HL_3 | 50,409,762 | 49,054,352 | 7.36G | 0.02 | 96.97 | 60.35 |
| HA_1 | 60,762,278 | 58,220,746 | 8.73G | 0.02 | 95.09 | 60.99 |
| HA_2 | 54,539,640 | 51,357,156 | 7.7G | 0.02 | 95.89 | 60.78 |
| HA_3 | 65,324,354 | 62,001,576 | 9.3G | 0.02 | 96.1 | 60.86 |
| HF_1 | 58,086,534 | 55,405,818 | 8.31G | 0.02 | 95.7 | 60.08 |
| HF_2 | 68,968,618 | 65,327,172 | 9.8G | 0.02 | 95.81 | 59.96 |
| HF_3 | 66,261,124 | 63,319,584 | 9.5G | 0.02 | 95.45 | 60.15 |
| Annotated Databases | Number of Genes | Percentage (%) |
|---|---|---|
| Annotated in NR | 55,835 | 35.56 |
| Annotated in NT | 13,504 | 8.6 |
| Annotated in KO | 23,632 | 15.05 |
| Annotated in SwissProt | 36,522 | 23.26 |
| Annotated in PFAM | 58,643 | 37.35 |
| Annotated in GO | 60,846 | 38.75 |
| Annotated in KOG | 21,569 | 13.73 |
| Annotated in all Databases | 5918 | 3.76 |
| Annotated in at least one Database | 76,744 | 48.88 |
| Total Unigenes | 156,992 | 100 |
| Comparison Groups | The Number of DEGs | The Number of Up-Regulated DEGs | The Number of Down-Regulated DEGs |
|---|---|---|---|
| HLvsHG | 17,089 | 8243 | 8846 |
| HAvsHG | 15,555 | 7699 | 7856 |
| HAvsHL | 1824 | 1121 | 703 |
| HFvsHG | 21,271 | 10,496 | 10,775 |
| HFvsHL | 1900 | 1182 | 718 |
| HAvsHF | 4864 | 2844 | 2020 |
| KEGG Orthology Category Description | HLvsHG | HAvsHL | HFvsHL |
|---|---|---|---|
| PDS, crtP; 15-cis-phytoene desaturase [EC:1.3.5.5] | down | / | / |
| crtISO, crtH; prolycopene isomerase [EC:5.2.1.13] | down | / | / |
| lcyB, crtL1, crtY; lycopene β-cyclase [EC:5.5.1.19] | down | / | / |
| lcyE, crtL2; lycopene epsilon-cyclase [EC:5.5.1.18] | / | down | / |
| LUT5, CYP97A3; β-ring hydroxylase [EC:1.14.-.-] | up | / | / |
| LUT1, CYP97C1; carotene epsilon-monooxygenase [EC:1.14.99.45] | up | / | / |
| crtZ; β-carotene 3-hydroxylase [EC:1.14.13.129] | ns | up | / |
| CCD8; carlactone synthase/all-trans-10′-apo-β-carotenal 13,14-cleaving dioxygenase [EC:1.13.11.69 1.13.11.70] | up | up | / |
| ZEP, ABA1; zeaxanthin epoxidase [EC:1.14.15.21] | down | / | / |
| KEGG Orthology Category Description | HLvsHG | HAvsHL | HFvsHL |
|---|---|---|---|
| KCS; 3-ketoacyl-CoA synthase [EC:2.3.1.199] | up | up | down |
| PHS1, PAS2; very-long-chain (3R)-3-hydroxyacyl-CoA dehydratase [EC:4.2.1.134] | up | / | / |
| MECR, NRBF1; mitochondrial trans-2-enoyl-CoA reductase [EC:1.3.1.38] | up | up | / |
| PPT; palmitoyl-protein thioesterase [EC:3.1.2.22] | up | down | down |
| KEGG Orthology Category Description | HLvsHG | HFvsHL | HAvsHL |
|---|---|---|---|
| LHCA1; light-harvesting complex I chlorophyll a/b binding protein 1 | up | up | down |
| LHCA2; light-harvesting complex I chlorophyll a/b binding protein 2 | ns | ns | down |
| LHCA3; light-harvesting complex I chlorophyll a/b binding protein 3 | up | up | down |
| LHCA4; light-harvesting complex I chlorophyll a/b binding protein 4 | ns | up | down |
| LHCA5; light-harvesting complex I chlorophyll a/b binding protein 5 | down | / | / |
| LHCB1; light-harvesting complex II chlorophyll a/b binding protein 1 | ns | ns | down |
| LHCB2; light-harvesting complex II chlorophyll a/b binding protein 2 | ns | up | down |
| LHCB3; light-harvesting complex II chlorophyll a/b binding protein 3 | / | up | / |
| LHCB4; light-harvesting complex II chlorophyll a/b binding protein 4 | ns | ns | down |
| LHCB5; light-harvesting complex II chlorophyll a/b binding protein 5 | up | up | down |
| LHCB6; light-harvesting complex II chlorophyll a/b binding protein 6 | / | up | down |
| LHCB7; light-harvesting complex II chlorophyll a/b binding protein 7 | / | up | / |
| Gene Name | Primers |
|---|---|
| Rbcl (ribulose-1,5-bisphosphate carboxylase/oxygenase: Genbank-KR914695.1) | Rbcl1: ACGAATGTTTACGCGGTGGTCT |
| Rbcl2: GGTACACCCAACTCCTTAGCA | |
| CrtZ (carotenoid hydroxylase: Genbank-KP866868.1) | CrtZ1: ATCTTCGCCACCTACCTGAG |
| CrtZ2: CGGGCAGTCCATTGATGATT | |
| LcyB (lycopene beta cyclase: Genbank-AY182008.1) | LcyB1: CTTTGACCCAGCACGACTTG |
| LcyB2: TTAGGAACTTCTCGCCCTCT | |
| CrtISO (carotenoid isomerase: Genbank-KP780062.1) | CrtISO1: GCACCACTAACCTCATCACA |
| CrtISO2: CCTTCCAGCAGTCGTCATAA | |
| ZEP (zeaxanthin epoxidase: Genbank-KP780061.1) | ZEP1: TACCTCAACCCTGCTCCTGA |
| ZEP2: TTCGCCCTTCTCAAGCCTGT |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, B.; Hou, L.; Dong, M.; Shi, J.; Huang, X.; Ding, Y.; Cong, X.; Zhang, F.; Zhang, X.; Zang, X. Transcriptome Analysis in Haematococcus pluvialis: Astaxanthin Induction by High Light with Acetate and Fe2+. Int. J. Mol. Sci. 2018, 19, 175. https://doi.org/10.3390/ijms19010175
He B, Hou L, Dong M, Shi J, Huang X, Ding Y, Cong X, Zhang F, Zhang X, Zang X. Transcriptome Analysis in Haematococcus pluvialis: Astaxanthin Induction by High Light with Acetate and Fe2+. International Journal of Molecular Sciences. 2018; 19(1):175. https://doi.org/10.3390/ijms19010175
Chicago/Turabian StyleHe, Bangxiang, Lulu Hou, Manman Dong, Jiawei Shi, Xiaoyun Huang, Yating Ding, Xiaomei Cong, Feng Zhang, Xuecheng Zhang, and Xiaonan Zang. 2018. "Transcriptome Analysis in Haematococcus pluvialis: Astaxanthin Induction by High Light with Acetate and Fe2+" International Journal of Molecular Sciences 19, no. 1: 175. https://doi.org/10.3390/ijms19010175