Thioredoxin 2 Offers Protection against Mitochondrial Oxidative Stress in H9c2 Cells and against Myocardial Hypertrophy Induced by Hyperglycemia


Abstract
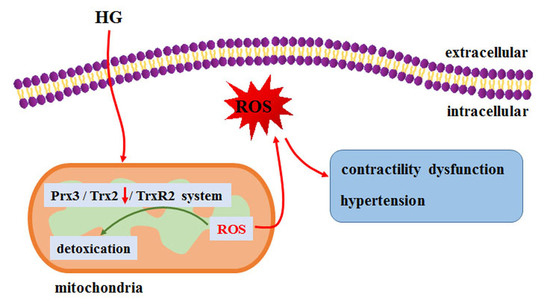
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
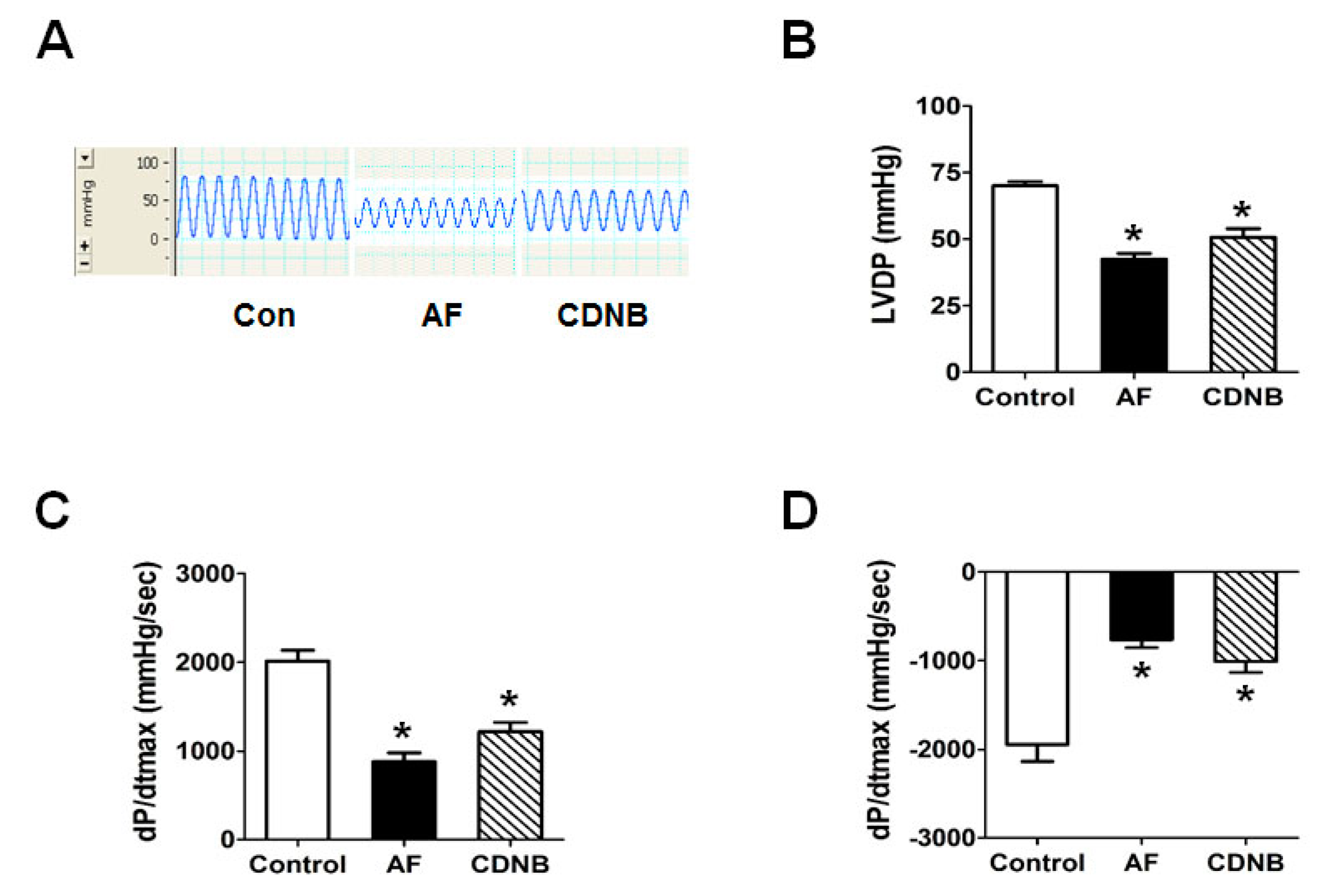
2.1. Inhibition of TrxR Impaired the Left Ventricle Contractile Functions of Isolated Heart
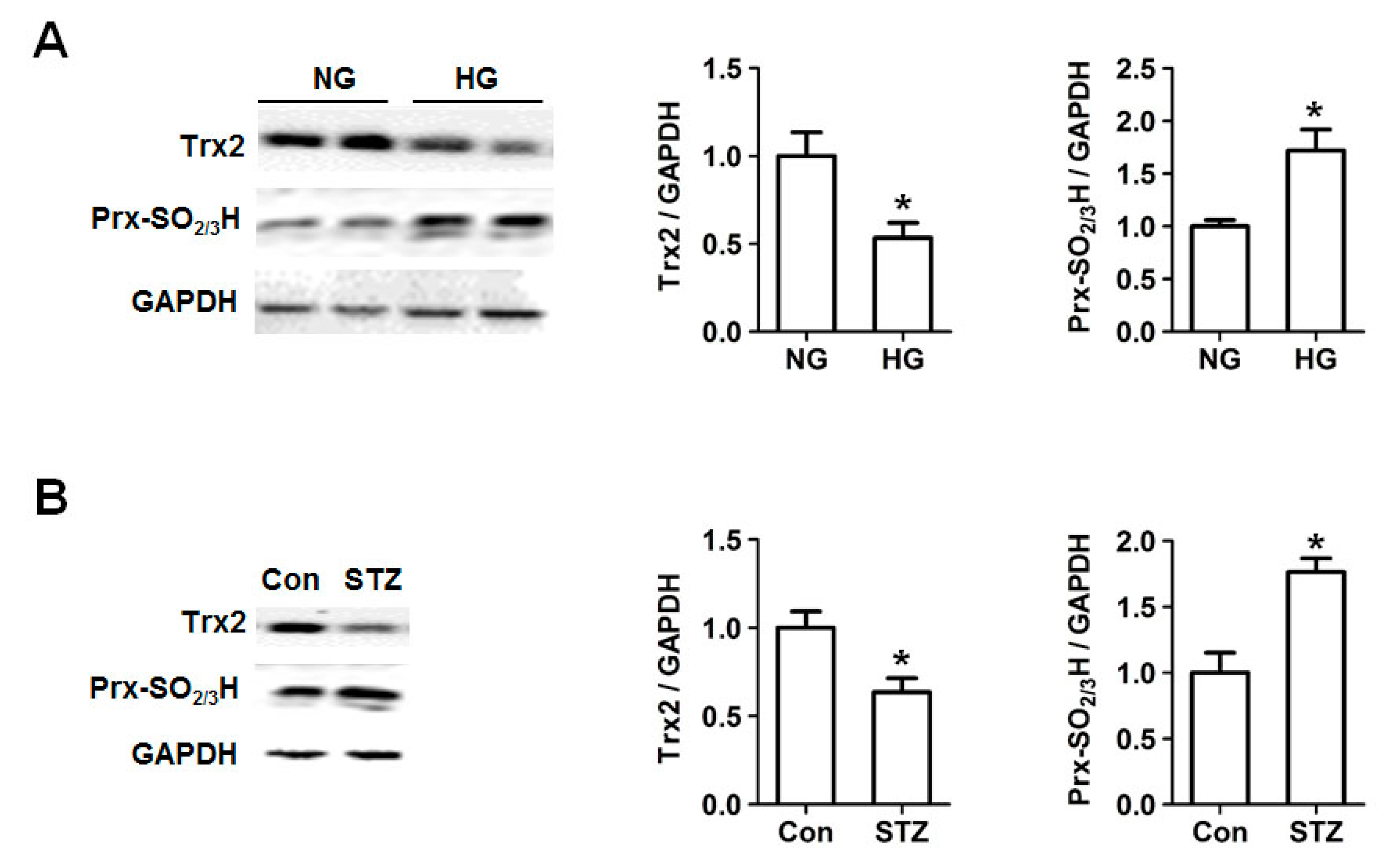
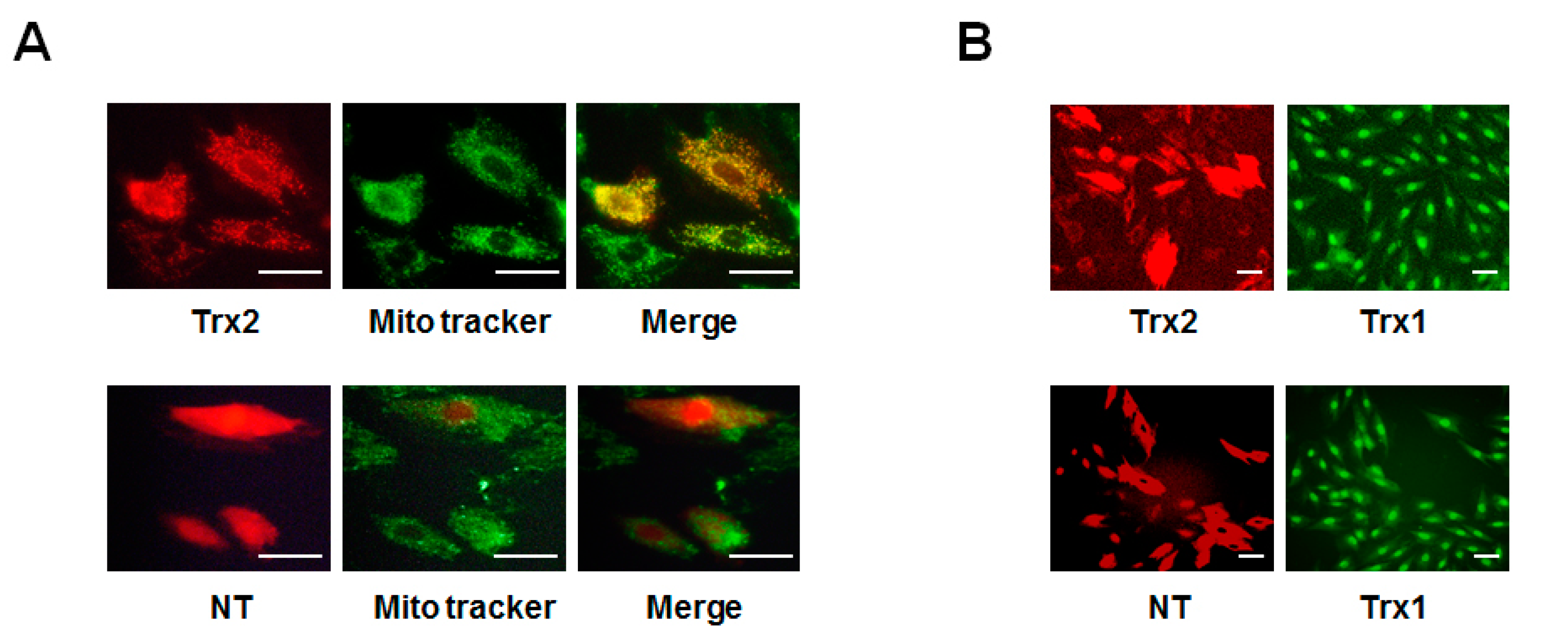
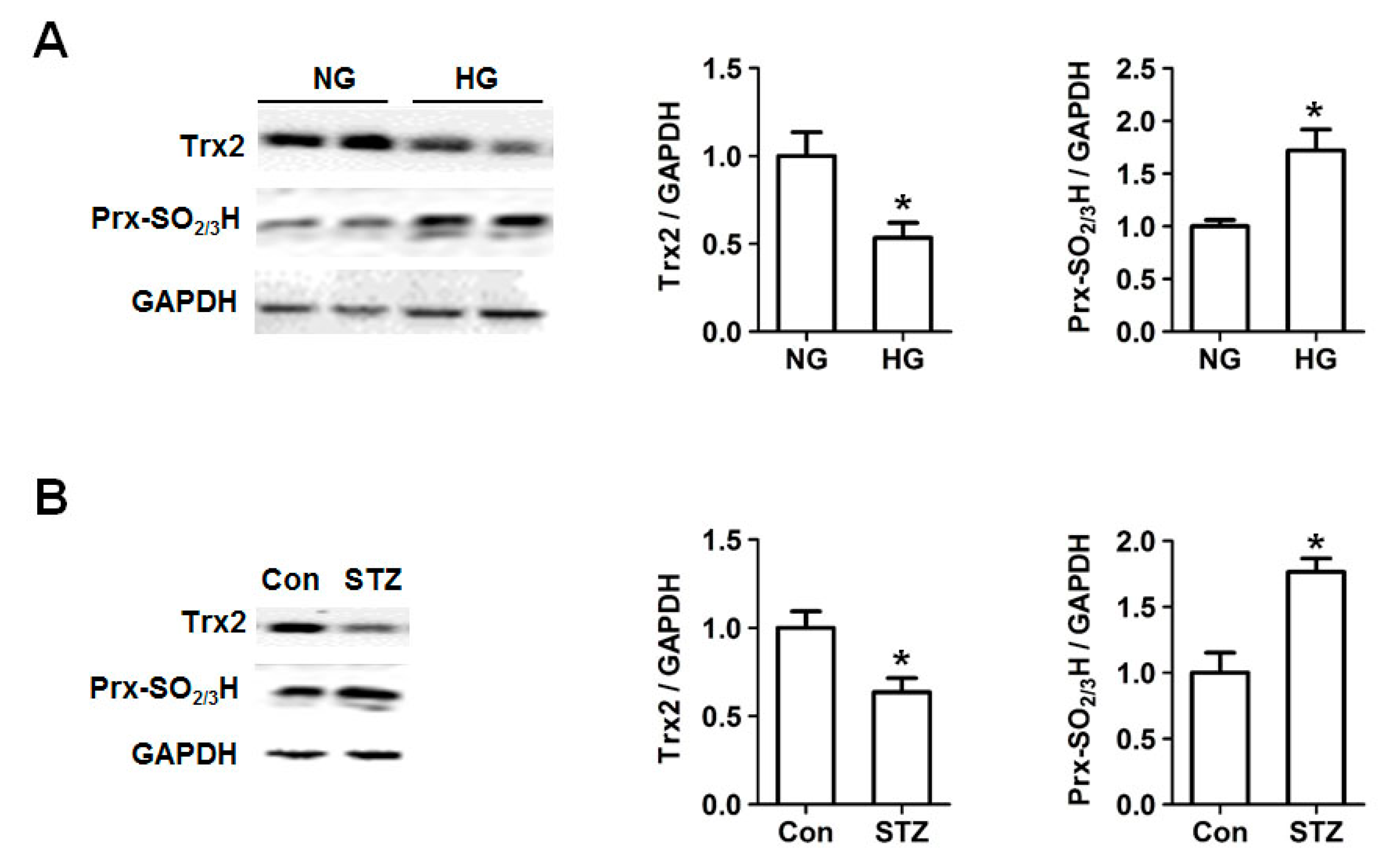
2.2. Decreased Trx2 Expression and Increased Prx Hyperoxidation in Cardiac Cells under Hyperglycemic Conditions
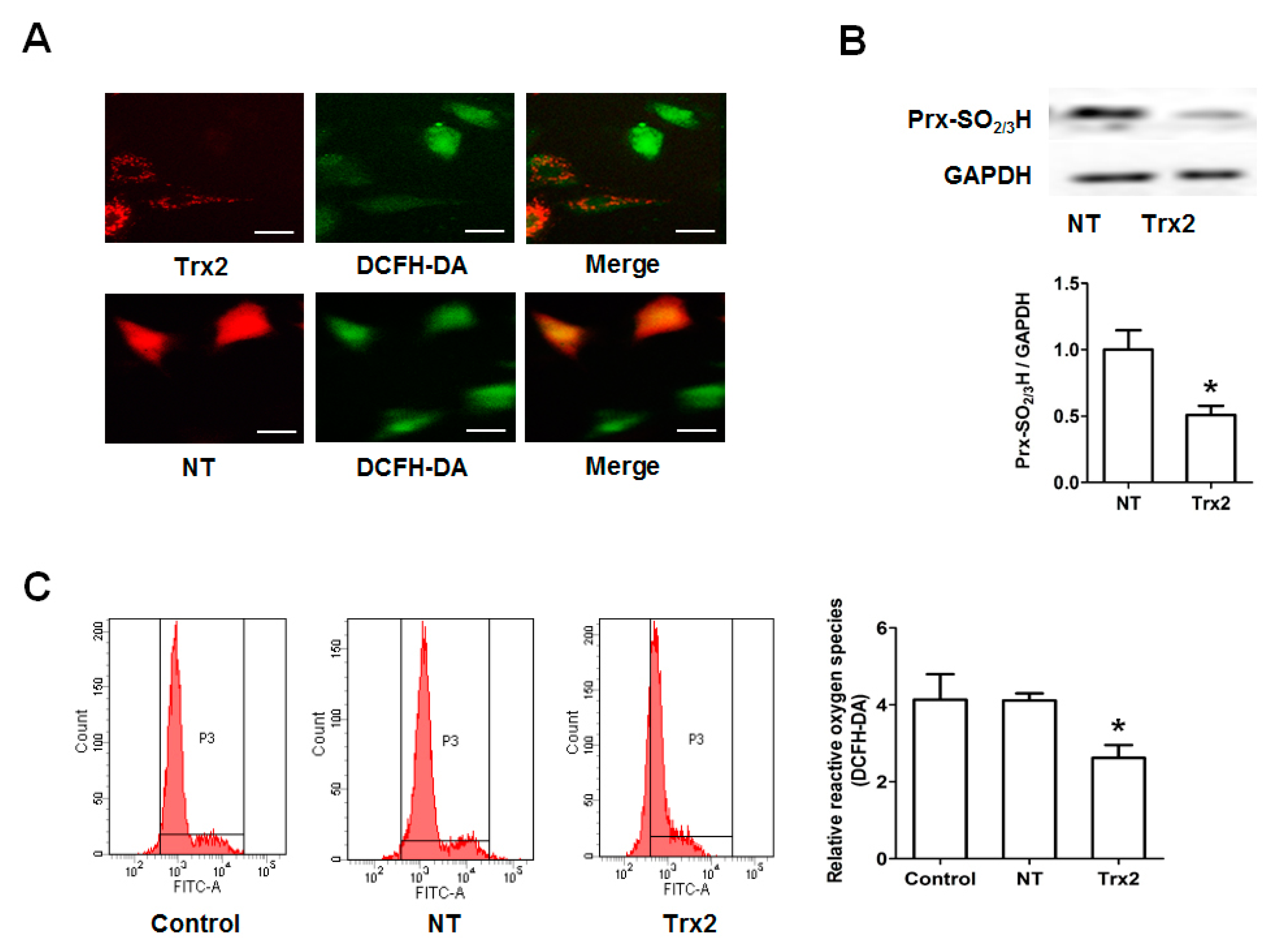
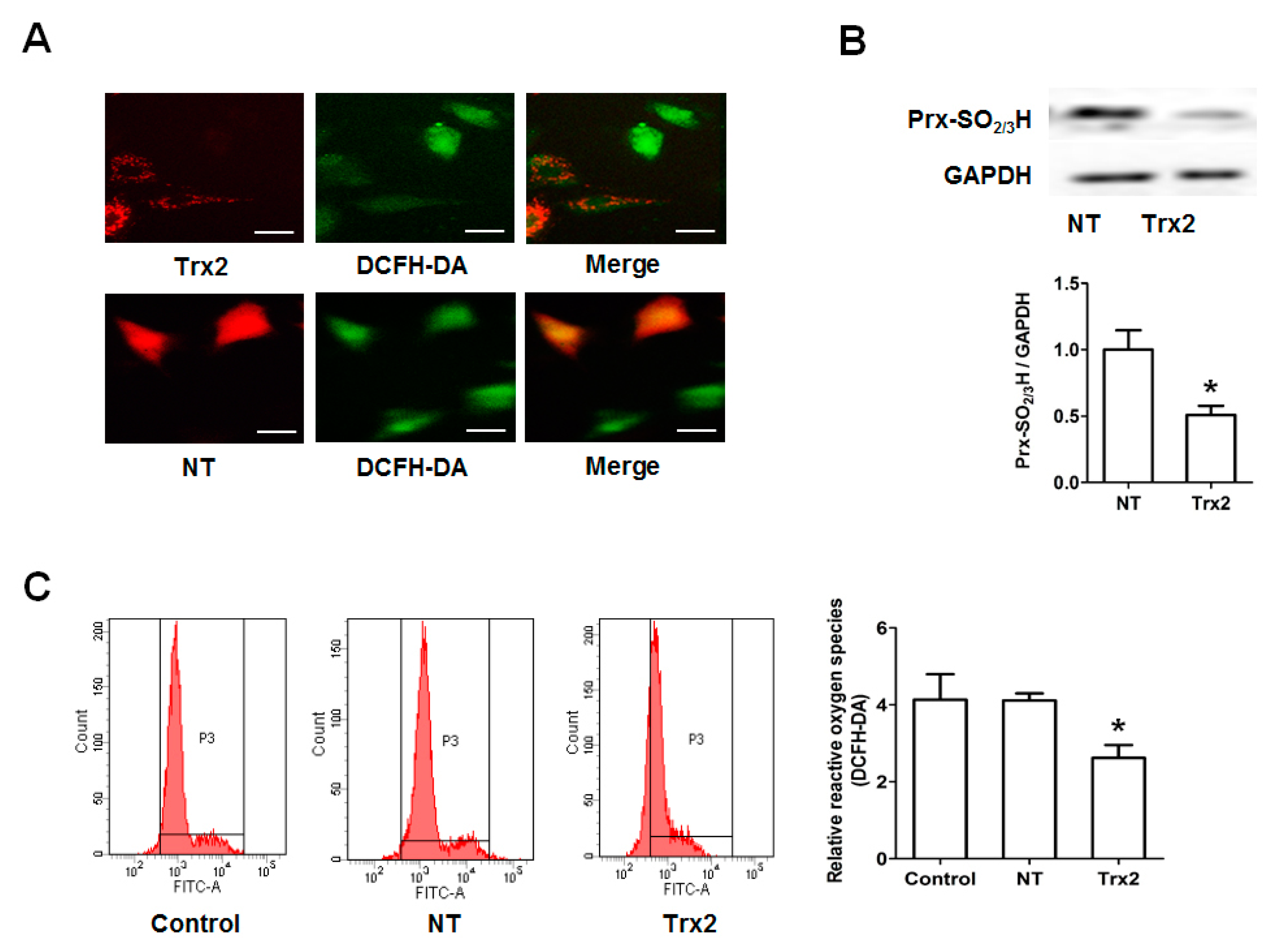
2.3. Protective Effect of Trx2 on ROS Generation and Prx Hyperoxidation
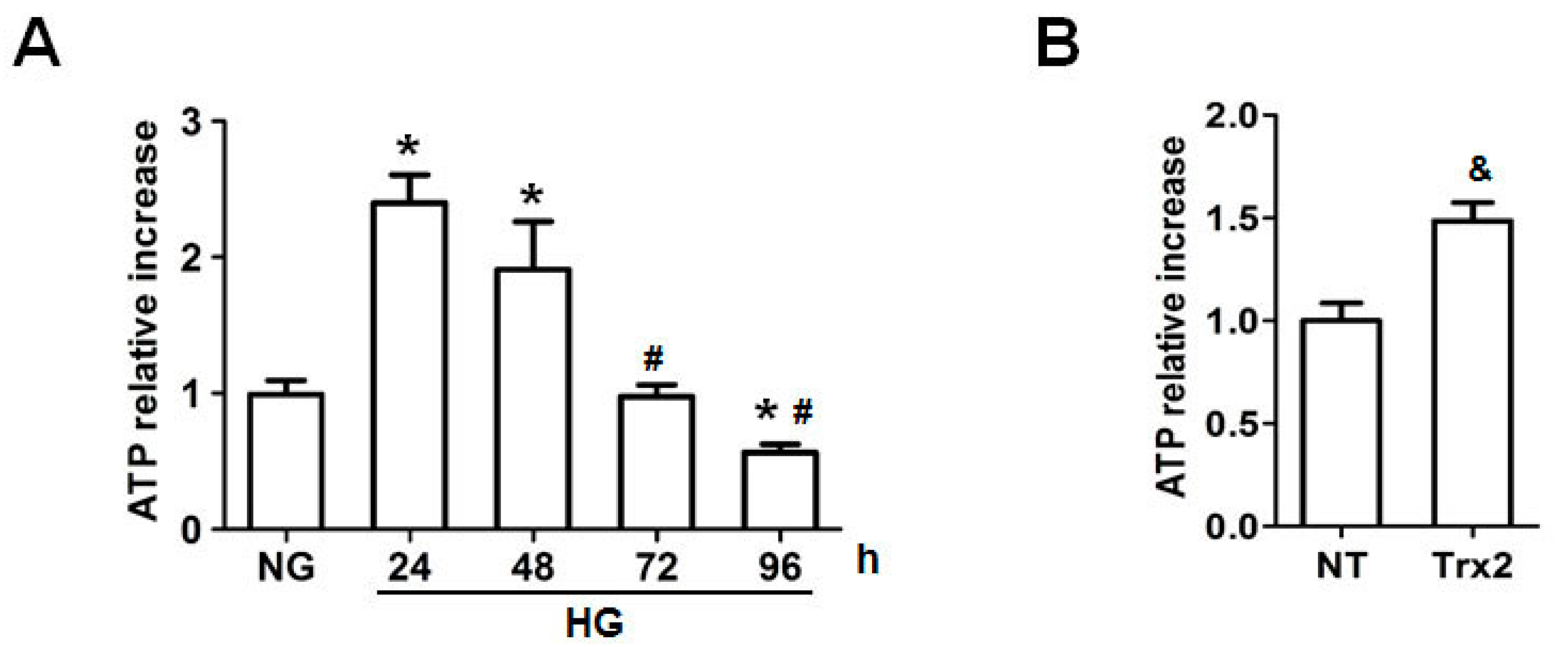
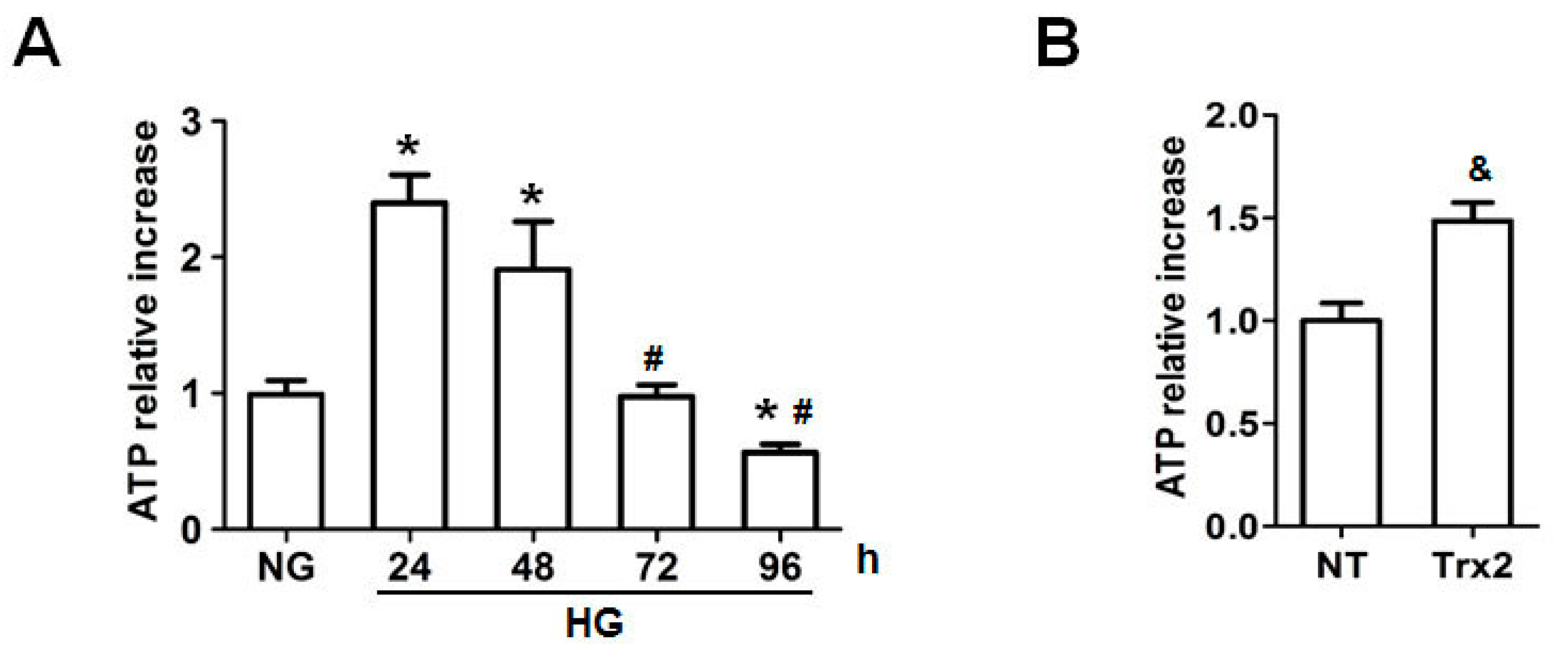
2.4. Trx2 Overexpression Reversed the Decreased ATP Generation
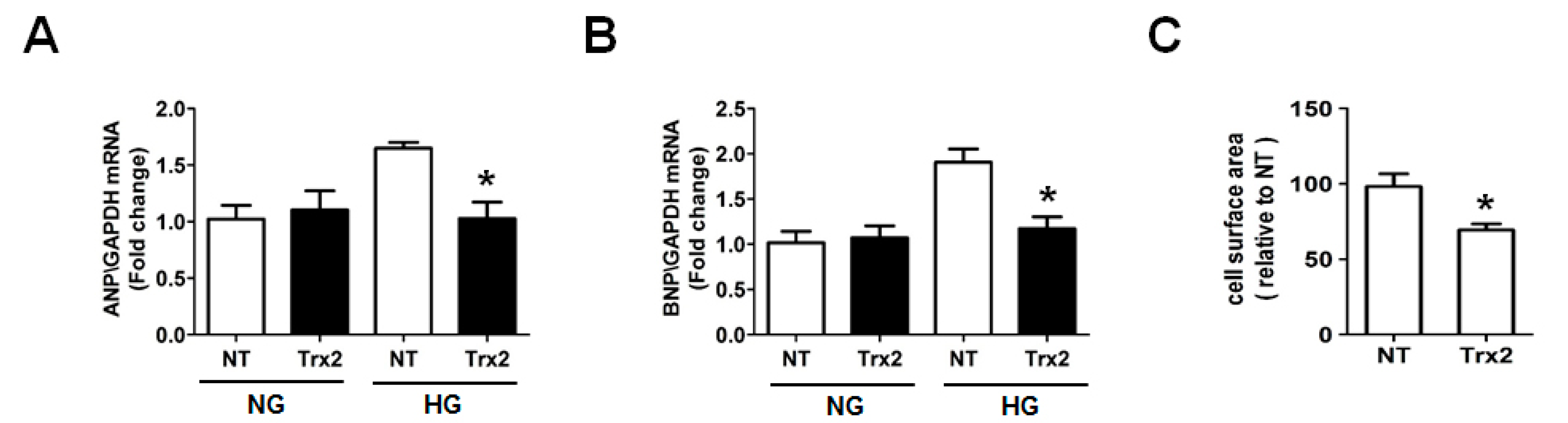
2.5. Overexpression Trx2 Attenuated the Hypertrophy Induced by HG in H9c2 Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animals
4.3. Animal Model
4.4. Assessment of Left Ventricular Function in Langendorff-Perfused Hearts
4.5. Cell Culture
4.6. Western Blot Analysis
4.7. Transfection of Trx2-pmCherry Plasmid
4.8. Intracellular ROS Level and Analysis
4.9. ATP-Luminescent Measurements
4.10. Real-Time RT-PCR
4.11. Measurement of Cell Surface Area
4.12. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fonarow, G.C.; Srikanthan, P. Diabetic cardiomyopathy. Endocrinol. Metab. Clin. N. Am. 2006, 35, 575–599. [Google Scholar] [CrossRef] [PubMed]
- Ruddy, T.D.; Shumak, S.L.; Liu, P.P.; Barnie, A.; Seawright, S.J.; McLaughlin, P.R.; Zinman, B. The relationship of cardiac diastolic dysfunction to concurrent hormonal and metabolic status in type I diabetes mellitus. J. Clin. Endocrinol. Metab. 1988, 66, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Severson, D.L. Diabetic cardiomyopathy: Recent evidence from mouse models of type 1 and type 2 diabetes. Can. J. Physiol. Pharmacol. 2004, 82, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Karvounis, H.I.; Papadopoulos, C.E.; Zaglavara, T.A.; Nouskas, I.G.; Gemitzis, K.D.; Parharidis, G.E.; Louridas, G.E. Evidence of left ventricular dysfunction in asymptomatic elderly patients with non-insulin-dependent diabetes mellitus. Angiology 2004, 55, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, O.; Ramirez, E.; Picatoste, B.; Egido, J.; Tunon, J. Alteration of energy substrates and ROS production in diabetic cardiomyopathy. Mediat. Inflamm. 2013, 2013, 461967. [Google Scholar] [CrossRef] [PubMed]
- Eklund, H.; Gleason, F.K.; Holmgren, A. Structural and functional relations among Thioredoxins of different species. Proteins 1991, 11, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Laurent, T.C.; Moore, E.C.; Reichard, P. Enzymatic Synthesis of Deoxyribonucleotides. Iv. Isolation and Characterization of Thioredoxin, the Hydrogen Donor from Escherichia coli B. J. Biol. Chem. 1964, 239, 3436–3444. [Google Scholar] [PubMed]
- Irwin, M.E.; Rivera-Del Valle, N.; Chandra, J. Redox control of leukemia: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 1349–1383. [Google Scholar] [CrossRef] [PubMed]
- Krapfenbauer, K.; Engidawork, E.; Cairns, N.; Fountoulakis, M.; Lubec, G. Aberrant expression of peroxiredoxin subtypes in neurodegenerative disorders. Brain Res. 2003, 967, 152–160. [Google Scholar] [CrossRef]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef] [PubMed]
- Schenk, H.; Vogt, M.; Droge, W.; Schulze-Osthoff, K. Thioredoxin as a potent costimulus of cytokine expression. J. Immunol. 1996, 156, 765–771. [Google Scholar] [PubMed]
- Berggren, M.I.; Husbeck, B.; Samulitis, B.; Baker, A.F.; Gallegos, A.; Powis, G. Thioredoxin peroxidase-1 (peroxiredoxin-1) is increased in thioredoxin-1 transfected cells and results in enhanced protection against apoptosis caused by hydrogen peroxide but not by other agents including dexamethasone, etoposide, and doxorubicin. Arch. Biochem. Biophys. 2001, 392, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Kang, S.W.; Netto, L.E.; Seo, M.S.; Stadtman, E.R. A family of novel peroxidases, peroxiredoxins. Biofactors 1999, 10, 207–209. [Google Scholar] [CrossRef] [PubMed]
- Pekkari, K.; Holmgren, A. Truncated thioredoxin: Physiological functions and mechanism. Antioxid. Redox Signal. 2004, 6, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Pekkari, K.; Gurunath, R.; Arner, E.S.; Holmgren, A. Truncated thioredoxin is a mitogenic cytokine for resting human peripheral blood mononuclear cells and is present in human plasma. J. Biol. Chem. 2000, 275, 37474–37480. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, A. Antioxidant function of thioredoxin and glutaredoxin systems. Antioxid. Redox Signal. 2000, 2, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.X.; Anilkumar, N.; Zhang, M.; Brewer, A.C.; Shah, A.M. Redox signaling in cardiac myocytes. Free Radic. Biol. Med. 2011, 50, 777–793. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, S.M.; Lee, R.T. Thioredoxin and thioredoxin target proteins: From molecular mechanisms to functional significance. Antioxid. Redox Signal. 2013, 18, 1165–1207. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Jakupoglu, C.; Moreno, S.G.; Lippl, S.; Banjac, A.; Schneider, M.; Beck, H.; Hatzopoulos, A.K.; Just, U.; Sinowatz, F.; et al. Essential role for mitochondrial thioredoxin reductase in hematopoiesis, heart development, and heart function. Mol. Cell. Biol. 2004, 24, 9414–9423. [Google Scholar] [CrossRef] [PubMed]
- Nonn, L.; Williams, R.R.; Erickson, R.P.; Powis, G. The absence of mitochondrial Thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Mol. Cell. Biol. 2003, 23, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Hosoi, F.; Yamaguchi-Iwai, Y.; Nakamura, H.; Masutani, H.; Ueda, S.; Nishiyama, A.; Takeda, S.; Wada, H.; Spyrou, G.; et al. Thioredoxin-2 (Trx-2) is an essential gene regulating mitochondria-dependent apoptosis. EMBO J. 2002, 21, 1695–1703. [Google Scholar] [CrossRef] [PubMed]
- Jakupoglu, C.; Przemeck, G.K.; Schneider, M.; Moreno, S.G.; Mayr, N.; Hatzopoulos, A.K.; de Angelis, M.H.; Wurst, W.; Bornkamm, G.W.; Brielmeier, M.; et al. Cytoplasmic thioredoxin reductase is essential for embryogenesis but dispensable for cardiac development. Mol. Cell. Biol. 2005, 25, 1980–1988. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Zhou, H.J.; Zhang, H.; Huang, Y.; Hinojosa-Kirschenbaum, F.; Fan, P.; Yao, L.; Belardinelli, L.; Tellides, G.; Giordano, F.J.; et al. Thioredoxin-2 inhibits mitochondrial reactive oxygen species generation and apoptosis stress kinase-1 activity to maintain cardiac function. Circulation 2015, 131, 1082–1097. [Google Scholar] [CrossRef] [PubMed]
- Widder, J.D.; Fraccarollo, D.; Galuppo, P.; Hansen, J.M.; Jones, D.P.; Ertl, G.; Bauersachs, J. Attenuation of angiotensin II—Induced vascular dysfunction and hypertension by overexpression of Thioredoxin 2. Hypertension 2009, 54, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Luo, Y.; Zhang, W.; He, Y.; Dai, S.; Zhang, R.; Huang, Y.; Bernatchez, P.; Giordano, F.J.; Shadel, G.; et al. Endothelial-specific expression of mitochondrial thioredoxin improves endothelial cell function and reduces atherosclerotic lesions. Am. J. Pathol. 2007, 170, 1108–1120. [Google Scholar] [CrossRef] [PubMed]
- Dong, F.; Li, Q.; Sreejayan, N.; Nunn, J.M.; Ren, J. Metallothionein prevents high-fat diet induced cardiac contractile dysfunction: Role of peroxisome proliferator activated receptor γ coactivator 1α and mitochondrial biogenesis. Diabetes 2007, 56, 2201–2212. [Google Scholar] [CrossRef] [PubMed]
- Kayama, Y.; Raaz, U.; Jagger, A.; Adam, M.; Schellinger, I.N.; Sakamoto, M.; Suzuki, H.; Toyama, K.; Spin, J.M.; Tsao, P.S. Diabetic Cardiovascular Disease Induced by Oxidative Stress. Int. J. Mol. Sci. 2015, 16, 25234–25263. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.M.; Jones, D.P. Redox compartmentalization in eukaryotic cells. Biochim. Biophys. Acta 2008, 1780, 1273–1290. [Google Scholar] [CrossRef] [PubMed]
- Arkat, S.; Umbarkar, P.; Singh, S.; Sitasawad, S.L. Mitochondrial Peroxiredoxin-3 protects against hyperglycemia induced myocardial damage in Diabetic cardiomyopathy. Free Radic. Biol. Med. 2016, 97, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Rovira-Llopis, S.; Banuls, C.; Diaz-Morales, N.; Hernandez-Mijares, A.; Rocha, M.; Victor, V.M. Mitochondrial dynamics in type 2 diabetes: Pathophysiological implications. Redox Biol. 2017, 11, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, S.; Kuroda, J.; Ago, T.; Zhai, P.; Park, J.Y.; Xie, L.H.; Tian, B.; Sadoshima, J. Increased oxidative stress in the nucleus caused by Nox4 mediates oxidation of HDAC4 and cardiac hypertrophy. Circ. Res. 2013, 112, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Rosca, M.G.; Tandler, B.; Hoppel, C.L. Mitochondria in cardiac hypertrophy and heart failure. J. Mol. Cell. Cardiol. 2013, 55, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Guo, Y.; Lou, Y.; Li, Q.; Cai, X.; Zhong, X.; Li, H. Sulfiredoxin involved in the protection of peroxiredoxins against hyperoxidation in the early hyperglycaemia. Exp. Cell Res. 2017, 352, 273–280. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Xu, C.; Li, Q.; Gao, X.; Sugano, E.; Tomita, H.; Yang, L.; Shi, S. Thioredoxin 2 Offers Protection against Mitochondrial Oxidative Stress in H9c2 Cells and against Myocardial Hypertrophy Induced by Hyperglycemia. Int. J. Mol. Sci. 2017, 18, 1958. https://doi.org/10.3390/ijms18091958
Li H, Xu C, Li Q, Gao X, Sugano E, Tomita H, Yang L, Shi S. Thioredoxin 2 Offers Protection against Mitochondrial Oxidative Stress in H9c2 Cells and against Myocardial Hypertrophy Induced by Hyperglycemia. International Journal of Molecular Sciences. 2017; 18(9):1958. https://doi.org/10.3390/ijms18091958
Chicago/Turabian StyleLi, Hong, Changqing Xu, Quanfeng Li, Xiuxiang Gao, Erkio Sugano, Hiroshi Tomita, Liming Yang, and Sa Shi. 2017. "Thioredoxin 2 Offers Protection against Mitochondrial Oxidative Stress in H9c2 Cells and against Myocardial Hypertrophy Induced by Hyperglycemia" International Journal of Molecular Sciences 18, no. 9: 1958. https://doi.org/10.3390/ijms18091958