A Molecular Modeling Study of the Hydroxyflutamide Resistance Mechanism Induced by Androgen Receptor Mutations

Abstract
:
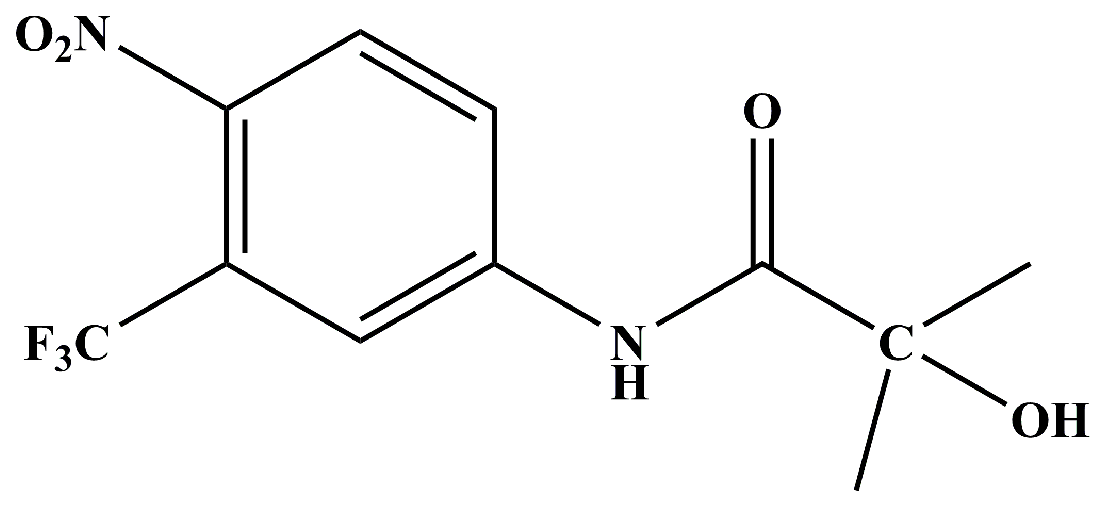
1. Introduction
2. Results and Discussion
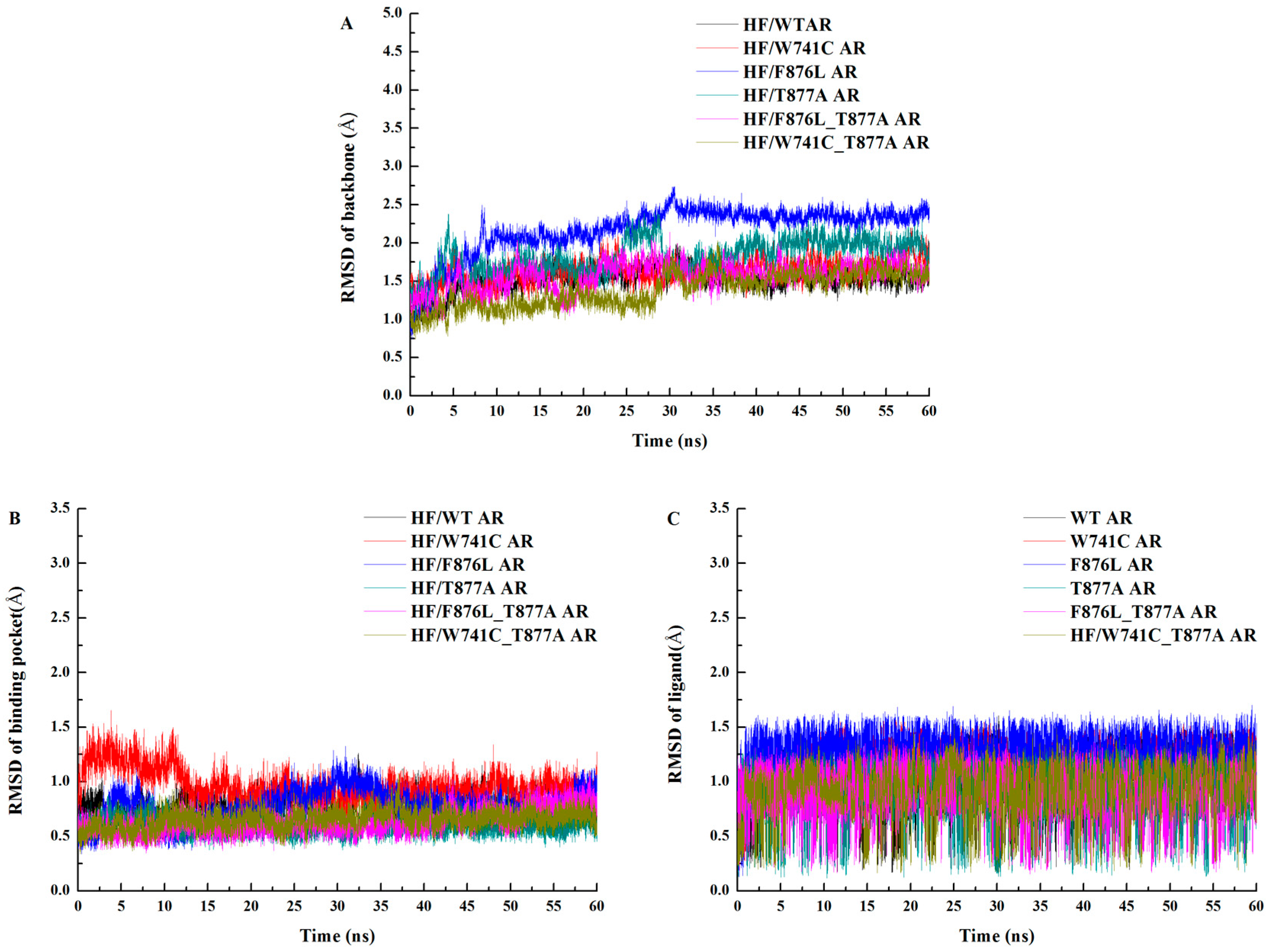
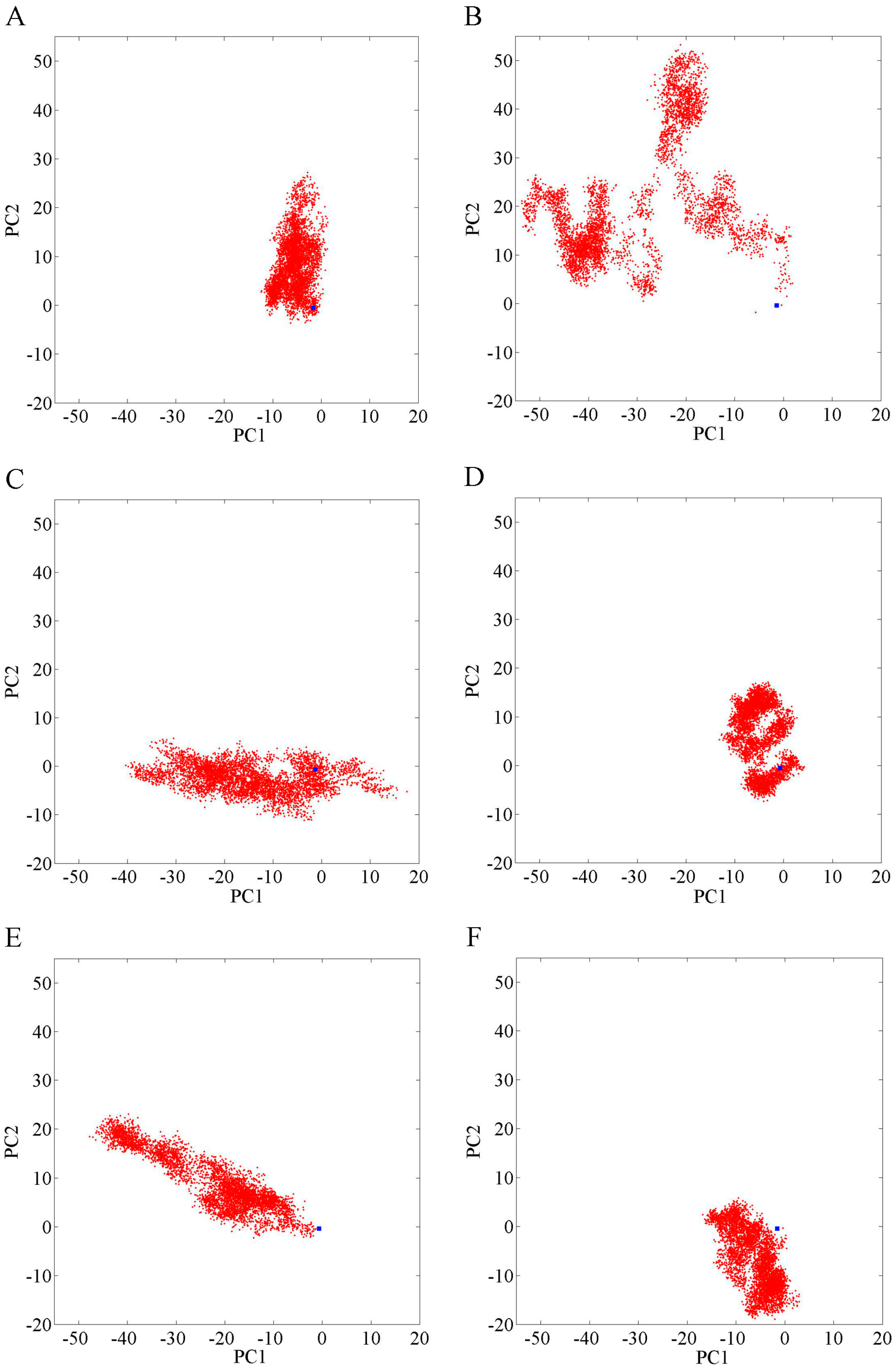
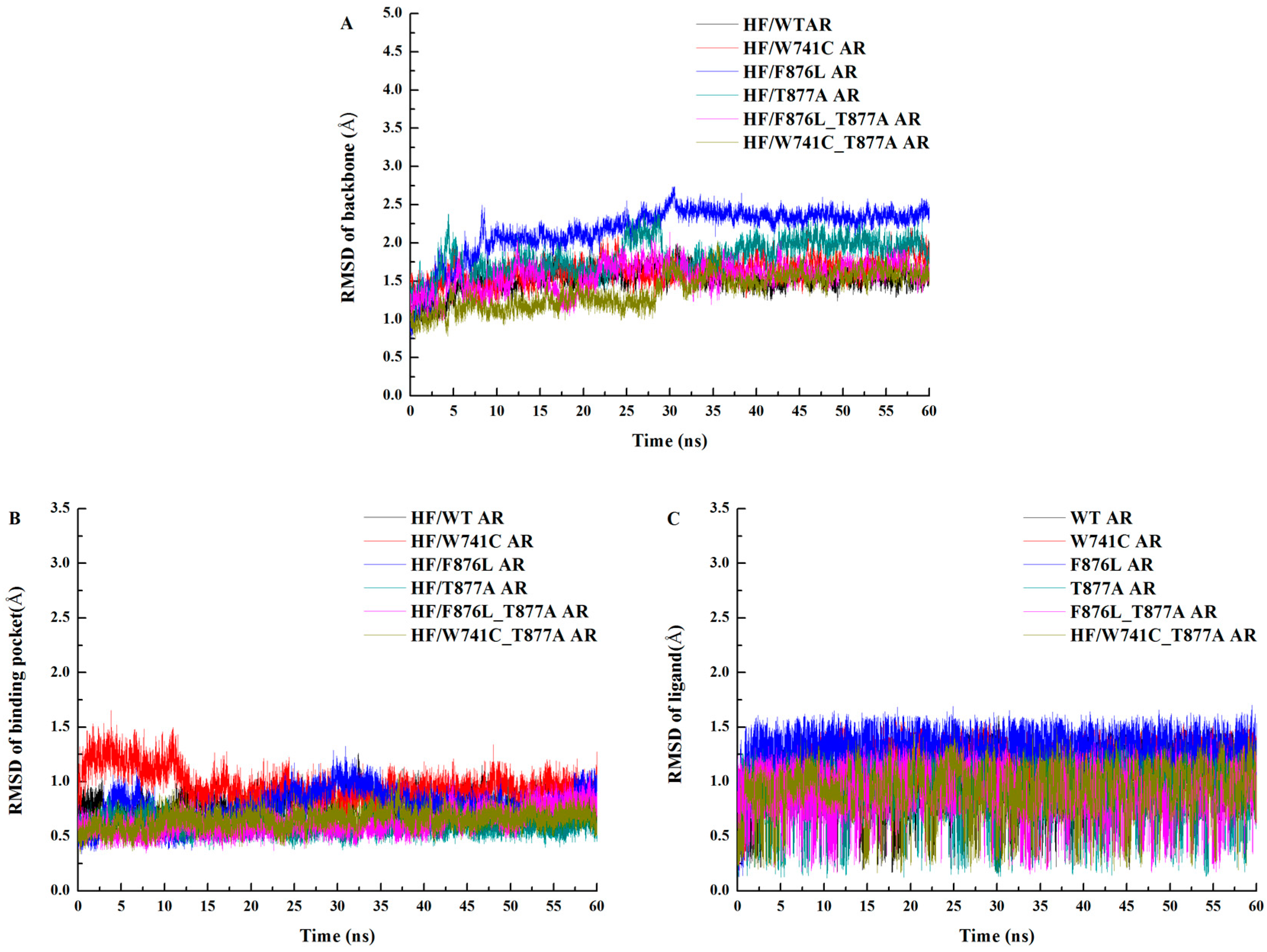
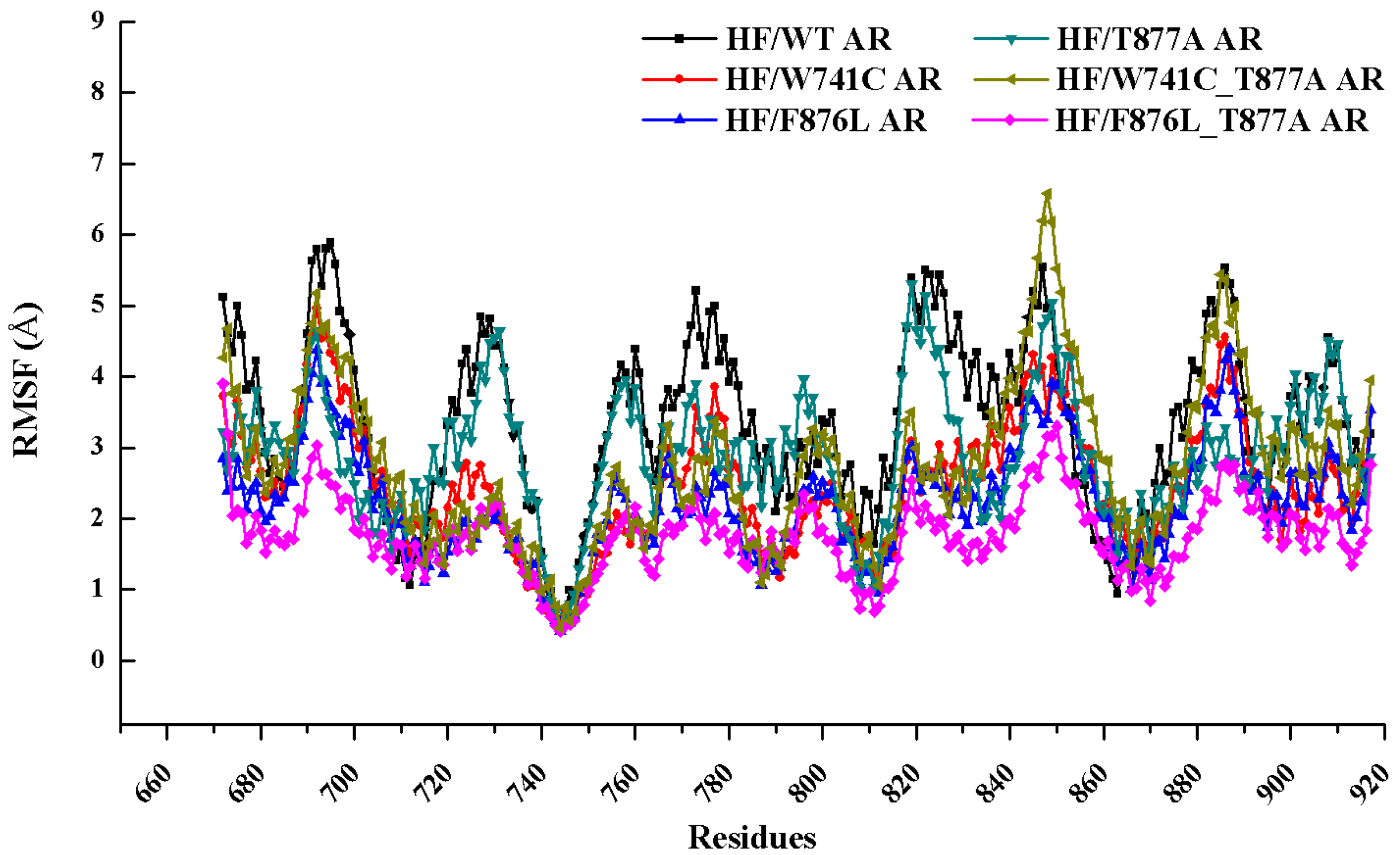
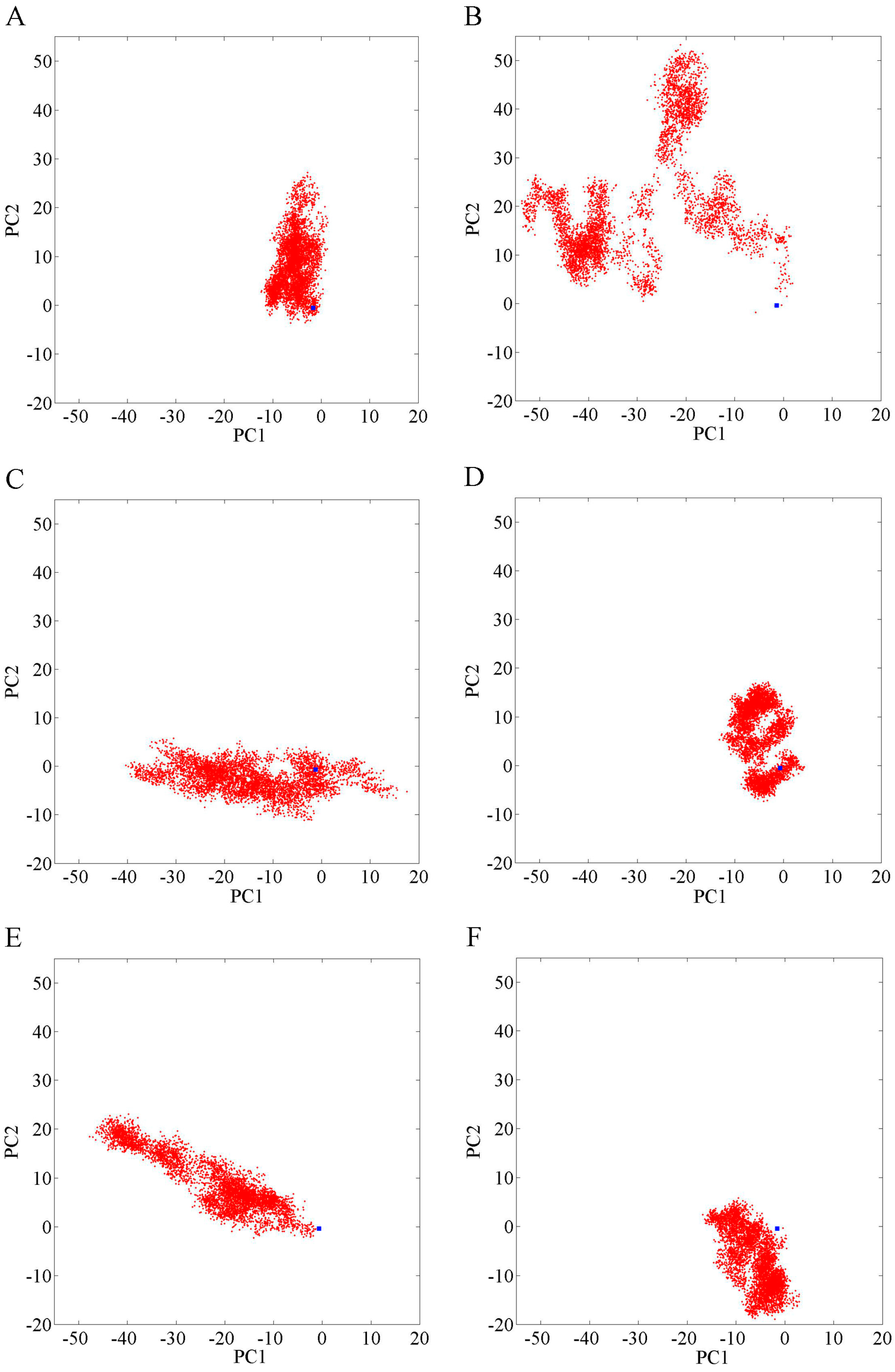
2.1. Overall Structural Properties
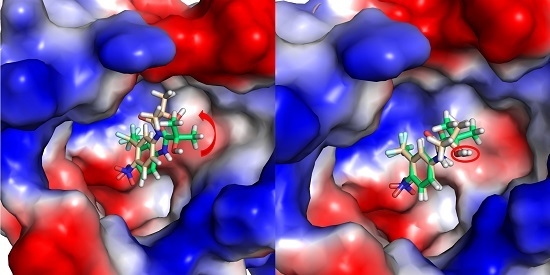
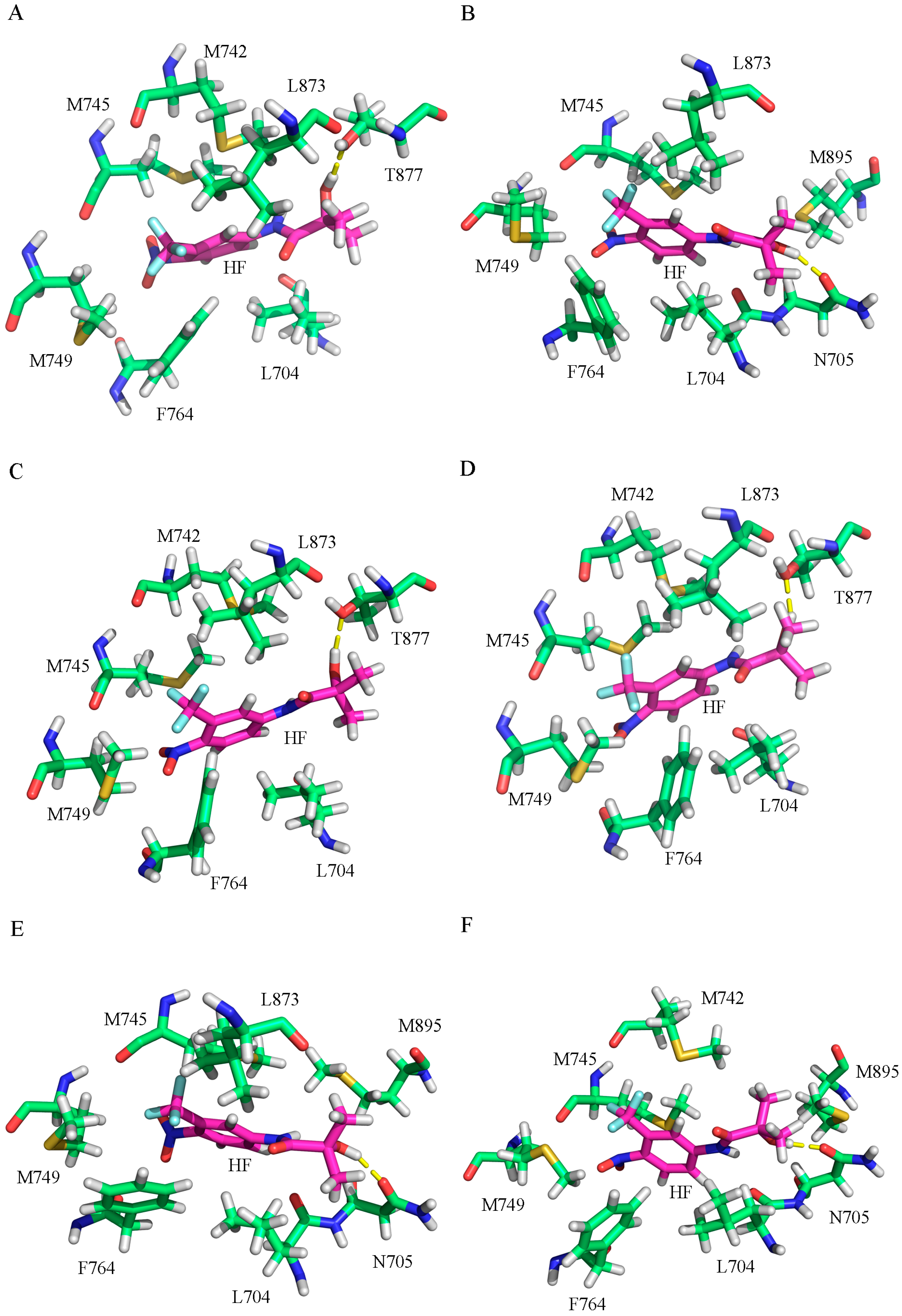
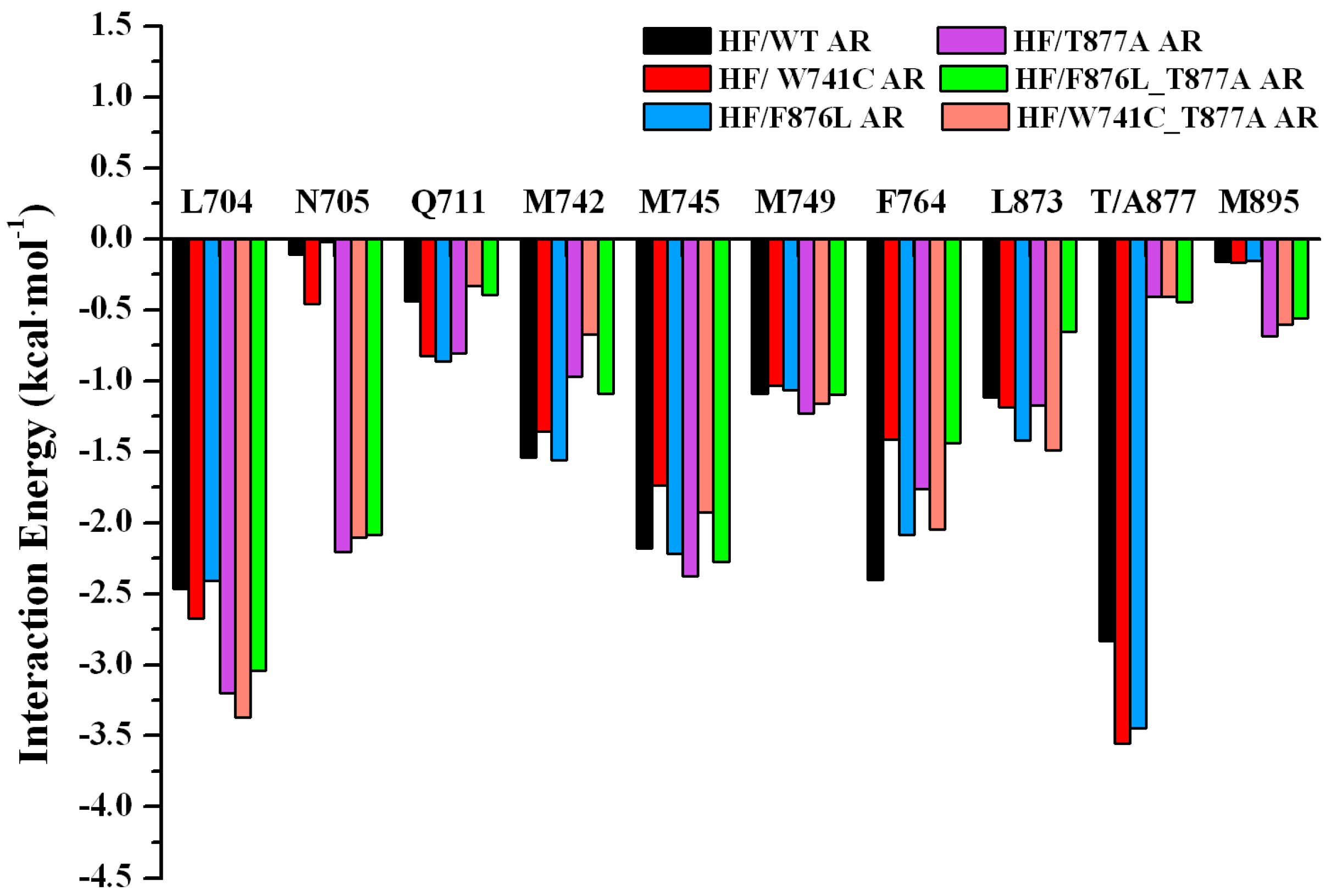
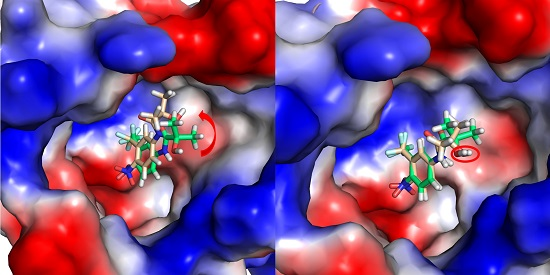
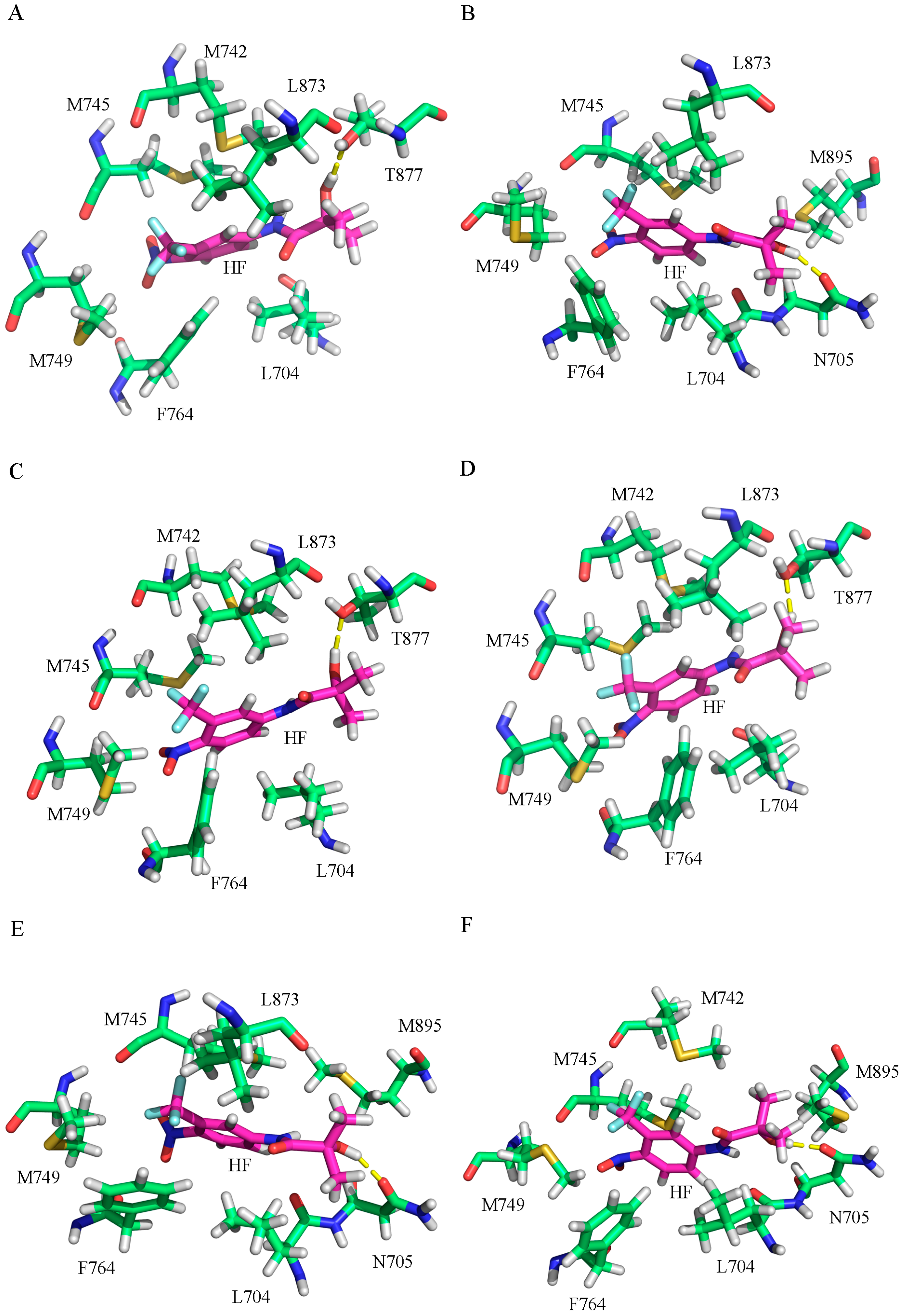
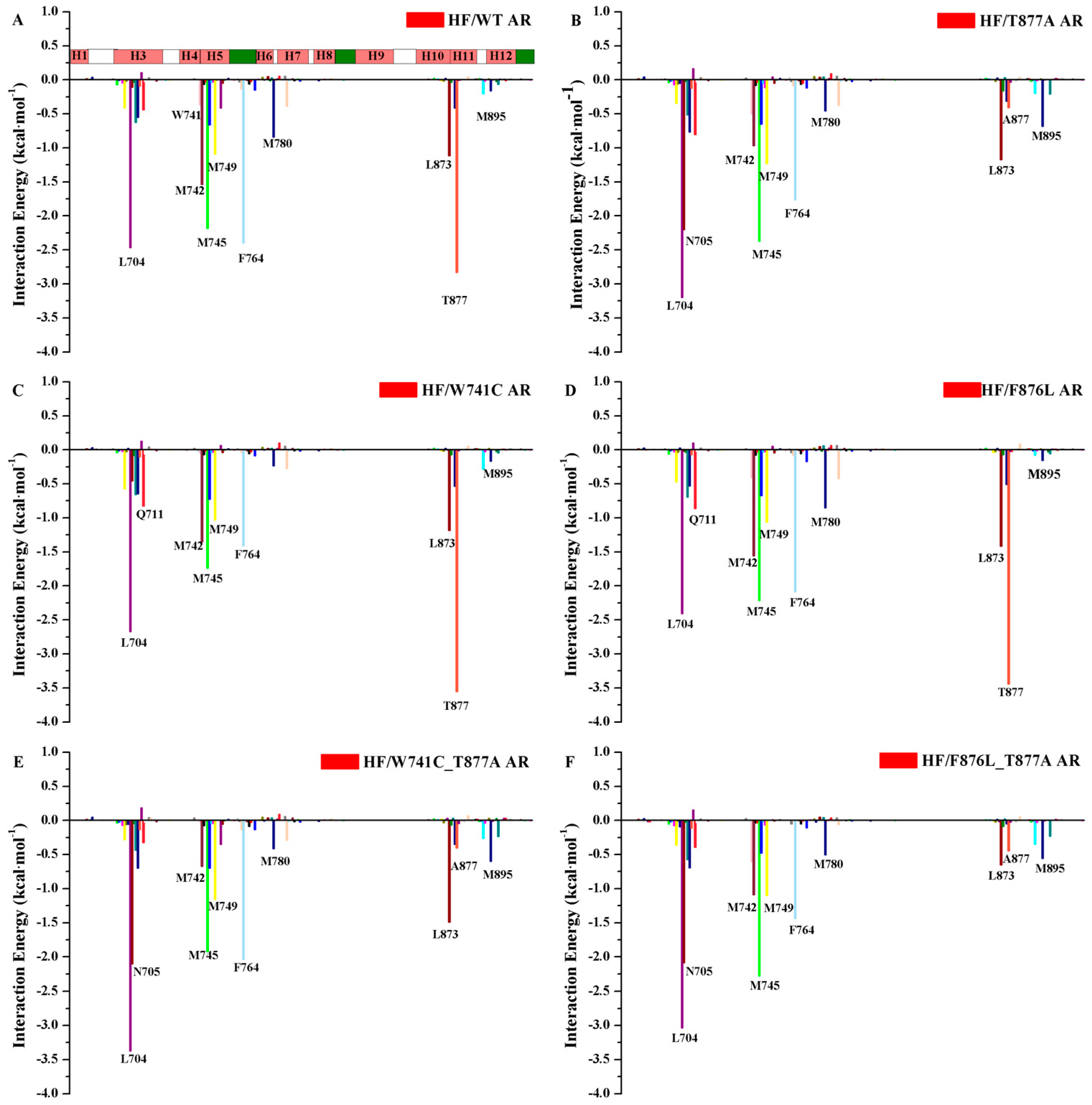
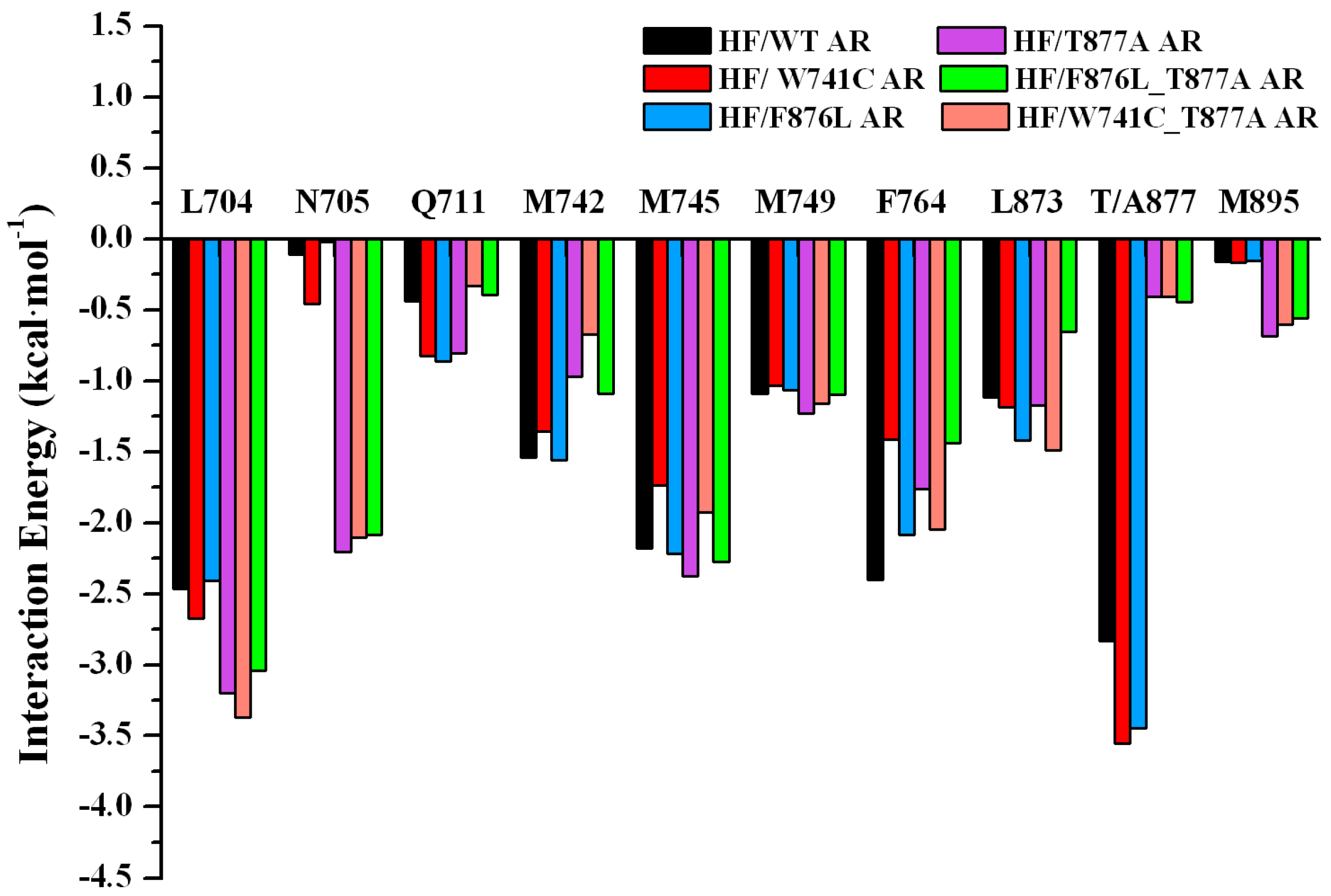
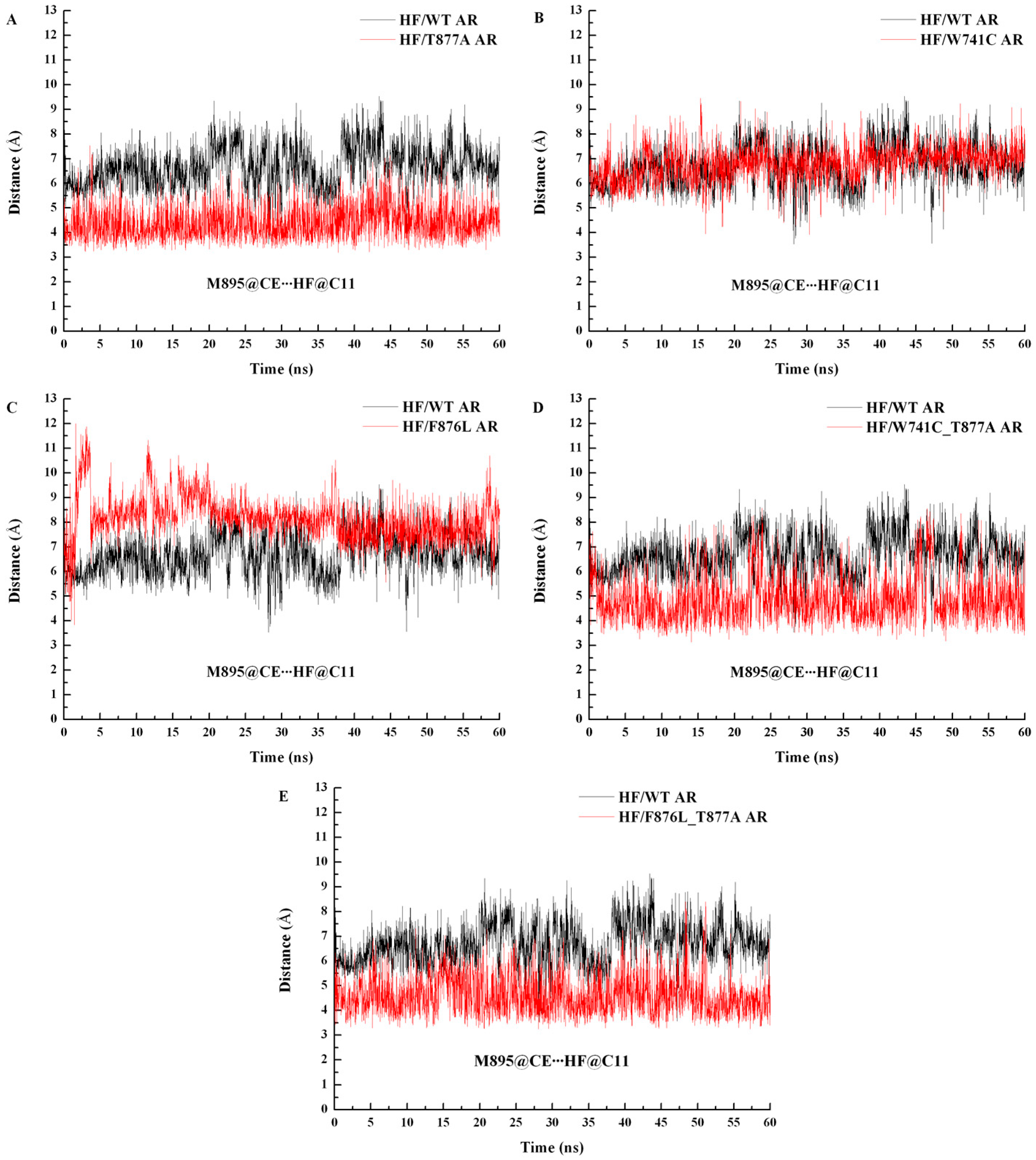
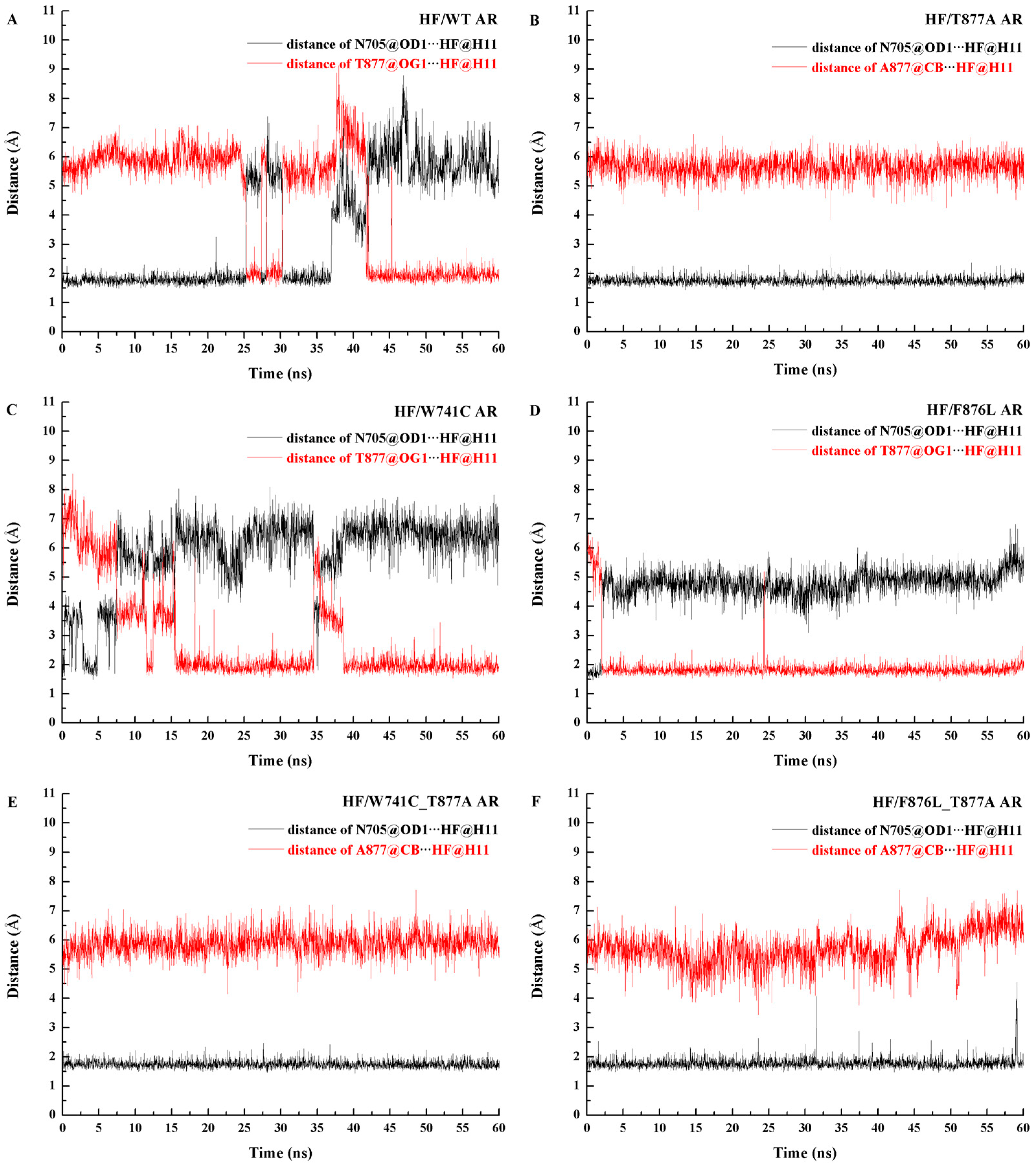
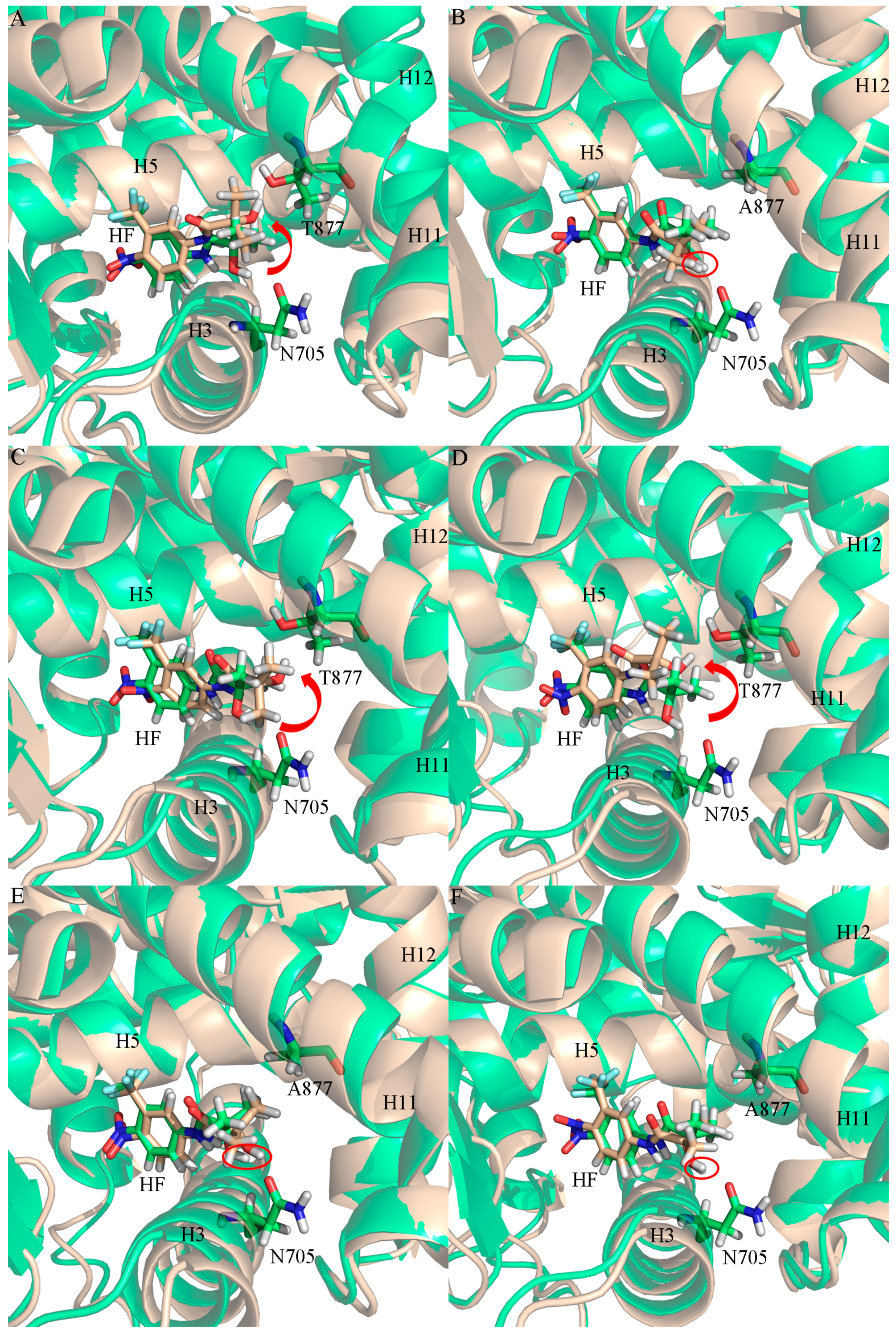
2.2. The Binding Modes of HF with T877A AR
2.3. The Binding Modes of HF with W741C/F876L
2.4. The Binding Modes of HF with W741C_T877A/F876L_T877A
3. Methods
3.1. Preparation of the Starting Structures
3.2. Molecular Dynamics Simulations
3.3. Binding Free Energy Calculations
3.4. Energy Decomposition
3.5. Principal Component Analysis
3.6. MD Trajectory Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2015. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef] [PubMed]
- Center, M.M.; Jemal, A.; Lortet-Tieulent, J.; Ward, E.; Ferlay, J.; Brawley, O.; Bray, F. International variation in prostate cancer incidence and mortality rates. Eur. Urol. 2012, 61, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, A.O.; Blok, L.J.; de Ruiter, P.E.; Doesburg, P.; Steketee, K.; Berrevoets, C.A.; Trapman, J. Mechanism of androgen receptor activation and fuction. J. Steroid Biochem. Mol. Biol. 1999, 69, 307–313. [Google Scholar] [CrossRef]
- Balk, S.P. Androgen receptor as a target in androgen independent prostate cancer. Urology 2002, 60, 132–138. [Google Scholar] [CrossRef]
- Gao, W.; Bohl, C.E.; Dalton, J.T. Chemistry and structural biology of androgen receptor. Chem. Rev. 2005, 105, 3352–3370. [Google Scholar] [CrossRef] [PubMed]
- Mohler, M.L.; Bohl, C.E.; Jones, A.; Coss, C.C.; Narayanan, R.; He, Y.; Hwang, D.J.; Dalton, J.T.; Miller, D.D. Nonsteroidal selective androgen receptor modulators (SARMs): Dissociating the anabolic and anddrogenic activities of the androgen receptor for therapeutic benefit. J. Med. Chem. 2009, 52, 3597–3617. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, R.; Mohler, M.L.; Bohl, C.E.; Miller, D.D.; Dalton, J.T. Selective androgen receptor molulators in prelinical and clinical development. Nucl. Recept. Siqnal. 2008, 6, e010. [Google Scholar]
- He, B.; Kemppainen, J.A.; Voegel, J.J.; Gronemeyer, H.; Wilson, E.M. Activationfunction 2 in the human androgen receptor ligand binding domain mediates interdomain communication with the NH2-terminal domain. J. Biol. Chem. 1999, 274, 37219–37225. [Google Scholar] [CrossRef] [PubMed]
- Georget, V.; Térouanne, B.; Nicolas, J.C.; Sultan, C. Mechanism of antiandrogen action: Key role of hsp90 in conformational change and transcriptional activity of the androgen receptor. Biochemistry 2002, 41, 11824–11831. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, C.; Watanabe, H.; Tanaka, S. An interpretation of positional displacement of the helix12 in nuclear receptors: Preexistent swing-up motion triggered by ligand binding. Biochem. Biophys. Acta 2010, 1804, 1832–1840. [Google Scholar] [CrossRef] [PubMed]
- Crawford, E.D.; DeAntoni, E.P. Current status of combined androgen blockade: Optimal therapy for advanced prostate cancer. J. Clin. Endocrinol. Metab. 1995, 80, 1062–1066. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Steineck, G.; Kelly, W.K. Hormone-refractory (D3) prostate cancer: Refining the concept. Urology 1995, 46, 142–148. [Google Scholar] [CrossRef]
- Feldman, B.J.; Feldman, D. The development of androgen-independent prostate cancer. Nat. Rev. 2001, 1, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Akakura, K.; Komiya, A.; Aida, S.; Shimazaki, J. Codon 877 mutation in the androgen receptor gene in advanced prostate cancer: Relation to antiandrogen withdrawal syndrome. Prostate 1996, 29, 153–158. [Google Scholar] [CrossRef]
- Barrack, E.R. Androgen receptor mutations in prostate cancer. Mount Sinai J. Med. 1996, 63, 403–412. [Google Scholar]
- Labrie, F.; Dupont, A.; Cusan, L.; Manhès, G.; Bergeron, N.; Lacourcière, Y.; Pineault, S.; Bélanger, A.; Monfette, G.; Emond, J. Combination therapy with castration and flutamide: Today’s treatment of choice for prostate cancer. J. Steroid Biochem. 1989, 33, 817–821. [Google Scholar] [CrossRef]
- Veldscholte, J.; Berrevoets, C.A.; Brinkmann, A.O.; Grootegoed, J.A.; Mulder, E. Anti-androgens and the mutated androgen receptor of LNCaP cells: Differential effects on binding affinity, heat-shock protein interaction, and transcription activation. Biochemistry 1992, 31, 2393–2399. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Miyazaki, J.; Yamaoka, H.; Kanzaki, N.; Kusaka, M.; Miyamoto, M. Novel mutations of androgen receptor: A possible mechanism of bicalutamide withdrawal syndrome. Cancer Res. 2003, 63, 149–153. [Google Scholar] [PubMed]
- Balbas, M.D.; Evans, M.J.; Hosfield, D.J.; Wongvipat, J.; Arora, V.K.; Wastson, P.A.; Chen, Y.; Greene, G.L.; Shen, Y.; Sawyers, C.L. Overcoming mutation-based resistance to antiandrogens with rational drug design. Elife 2013, 2, e00499. [Google Scholar] [CrossRef] [PubMed]
- Boqner, J.; Zolqhadr, K.; Hickson, L.; Romer, T.; Yurlova, L. The fluorescent two-hybrid assay for live-cell profiling of androgen receptor modulators. J. Steroid Biochem. Mol. Biol. 2017, 166, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Ozers, M.S.; Marks, B.D.; Gowda, K.; Kupcho, K.R.; Ervin, K.M.; De Rosier, T.; Qadir, N.; Eliason, H.C.; Riddle, S.M.; Shekhani, M.S. The androgen receptor T877A mutant recruits LXXLL and FXXLF peptides differently than wild-type androgen receptor in a time-resolved fluorescence resonance energy transfer assay. Biochemistry 2007, 46, 683–695. [Google Scholar] [CrossRef] [PubMed]
- RCSB Protein Data Bank. Available online: http://www.rcsb.org/pdb (accessed on 29 October 2010).
- Guex, N.; Peitsch, M.C. SWISS-MOIDEL and the Swiss-Pdb viewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Tripos. Sybyl 6.9, Tripos Associates, Inc.: St. Louis, MO, USA, 1999.
- Accelrys Inc. Discovery Studio Version 2.5, Accelrys Inc.: San Diego, CA, USA, 2009.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Cieplak, P.; Cornell, W.D.; Bayly, C.; Kollman, P.A. Application of the multimolecule and multiconformational RESP methodology to bipolymwes: Charge derivation for DNA, RNA, and proteins. J. Comput. Chem. 1995, 16, 1357–1377. [Google Scholar] [CrossRef]
- Fox, T.; Kollman, P.A. Application of the RESP methodology in the parametrization of organic solvents. J. Phys. Chem. B 1998, 102, 8070–8079. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Case, D.A.; Darden, T.A.; Cheatham, T.E.; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M.; et al. AMBER 12, University of California: San Francisco, CA, USA, 2012.
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Eaamann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Partical mesh ewald: An N∙log(N) method for Ewaldsums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods: 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. The accuracy of ranking poses generated from docking. J. Comput. Chem. 2011, 32, 866–877. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Sun, H.; Li, Y.; Wang, J.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 3. The impact of force fields and ligand charge models. J. Phys. Chem. B 2013, 117, 8408–8421. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Shen, Y.; Liu, H.; Yao, X. Molecular dynamics simulation and free energy calculation studies of the binding mechanism of allosteric inhibitors with p38α MAP kinase. J. Chem. Inf. Model. 2011, 51, 3235–3246. [Google Scholar] [CrossRef] [PubMed]
- Sitkoff, D.; Sharp, K.A.; Honig, B. Accurate calculation of hydration free energies using macroscopic solvent models. J. Phys. Chem. 1994, 98, 1978–1988. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Ichiye, T.; Karplus, M. Collective motions in proteins: A covariance analysis of atomic fluctuations in molicular dynamics and normal mode simulations. Proteins 1991, 11, 205–217. [Google Scholar] [CrossRef] [PubMed]
- MMTSB: Multiscale Modeling Tools for Structural Biology. Available online: http://mmtsb.org/ (accessed on 4 May 2016).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complexes | Contribution | ||||||
|---|---|---|---|---|---|---|---|
| ΔEele | ΔEvdw | ΔGp | ΔGnp | ΔEbind | −TΔS | ΔGbind | |
| HF/WT AR | −28.21 | −40.24 | 44.92 | −5.66 | −29.19 | 23.12 | −6.07 |
| HF/T877A AR | −32.34 | −40.16 | 47.45 | −5.63 | −30.67 | 22.25 | −8.42 |
| HF/W741C AR | −23.22 | −38.36 | 41.52 | −5.53 | −25.60 | 23.10 | −2.50 |
| HF/F876L AR | −23.38 | −39.45 | 39.42 | −5.63 | −29.04 | 20.82 | −8.22 |
| HF/W741C_T877A AR | −36.67 | −39.89 | 53.16 | −5.67 | −29.08 | 23.12 | −5.77 |
| HF/F876L_T877A AR | −24.34 | −35.36 | 39.47 | −5.19 | −25.31 | 20.79 | −4.52 |
| Complex | Interaction Pair | Distance (Å) * | Angle (°) * | Occupancy (%) | |||
|---|---|---|---|---|---|---|---|
| 60 ns | 10 ns | 60 ns | 10 ns | 60 ns | 10 ns | ||
| HF/WT AR | HF@O4-H11∙∙∙N705 (OD1) | 2.71 | - | 159.15 | - | 54.41 | - |
| HF@O4-H11∙∙∙T877 (OG1) | 2.87 | 2.87 | 161.89 | 162.05 | 36.78 | 99.38 | |
| HF/T877A AR | HF@O4-H11∙∙∙N705 (OD1) | 2.68 | 2.68 | 160.73 | 160.34 | 99.98 | 99.98 |
| HF/W741C AR | HF@O4-H11∙∙∙T877 (OG1) | 2.89 | 2.86 | 163.49 | 163.88 | 68.22 | 99.52 |
| HF/F876L AR | HF@O4-H11∙∙∙T877 (OG1) | 2.75 | 2.77 | 162.15 | 162.13 | 96.65 | 99.82 |
| HF/W741C_T877A AR | HF@O4-H11∙∙∙N705 (OD1) | 2.67 | 2.67 | 161.27 | 161.54 | 99.98 | 99.96 |
| HF/F876L_T877A AR | HF@O4-H11∙∙∙N705 (OD1) | 2.70 | 2.71 | 160.91 | 160.44 | 99.67 | 98.56 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.-L.; Zhong, H.-Y.; Song, T.-Q.; Li, J.-Z. A Molecular Modeling Study of the Hydroxyflutamide Resistance Mechanism Induced by Androgen Receptor Mutations. Int. J. Mol. Sci. 2017, 18, 1823. https://doi.org/10.3390/ijms18091823
Liu H-L, Zhong H-Y, Song T-Q, Li J-Z. A Molecular Modeling Study of the Hydroxyflutamide Resistance Mechanism Induced by Androgen Receptor Mutations. International Journal of Molecular Sciences. 2017; 18(9):1823. https://doi.org/10.3390/ijms18091823
Chicago/Turabian StyleLiu, Hong-Li, Hai-Yang Zhong, Tian-Qing Song, and Jia-Zhong Li. 2017. "A Molecular Modeling Study of the Hydroxyflutamide Resistance Mechanism Induced by Androgen Receptor Mutations" International Journal of Molecular Sciences 18, no. 9: 1823. https://doi.org/10.3390/ijms18091823