Signaling Cascade Involved in Rapid Stimulation of Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) by Dexamethasone

Abstract
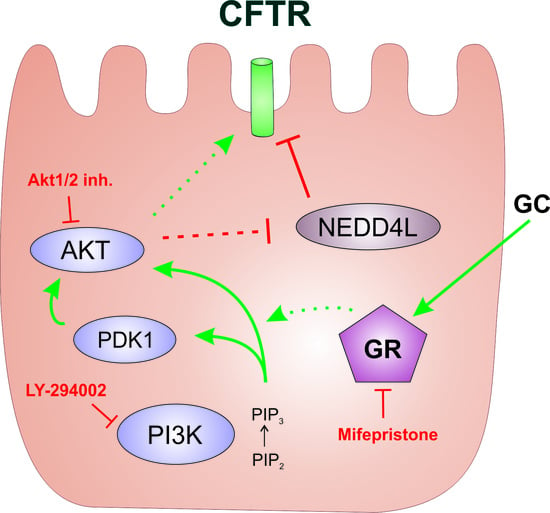
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
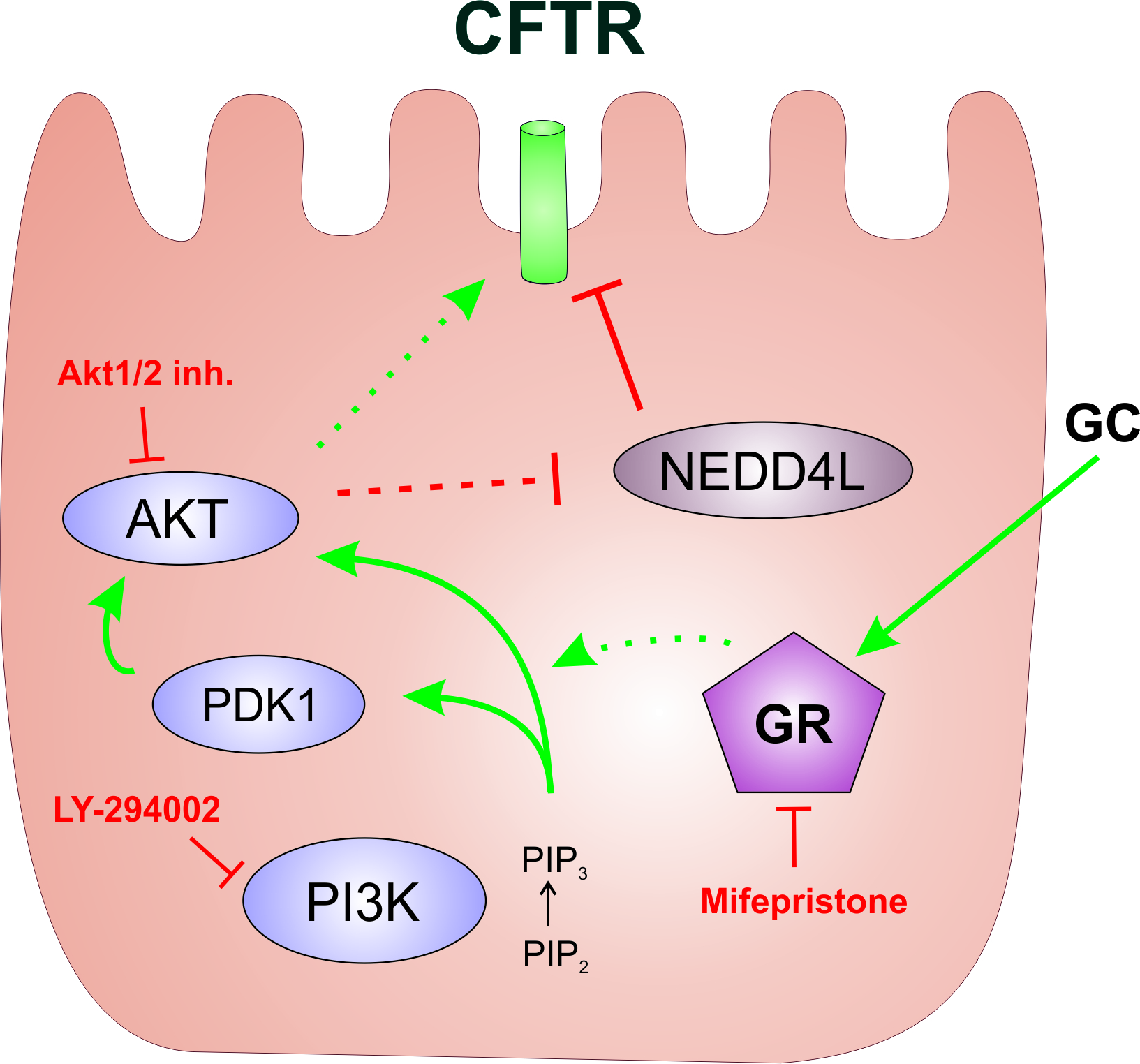
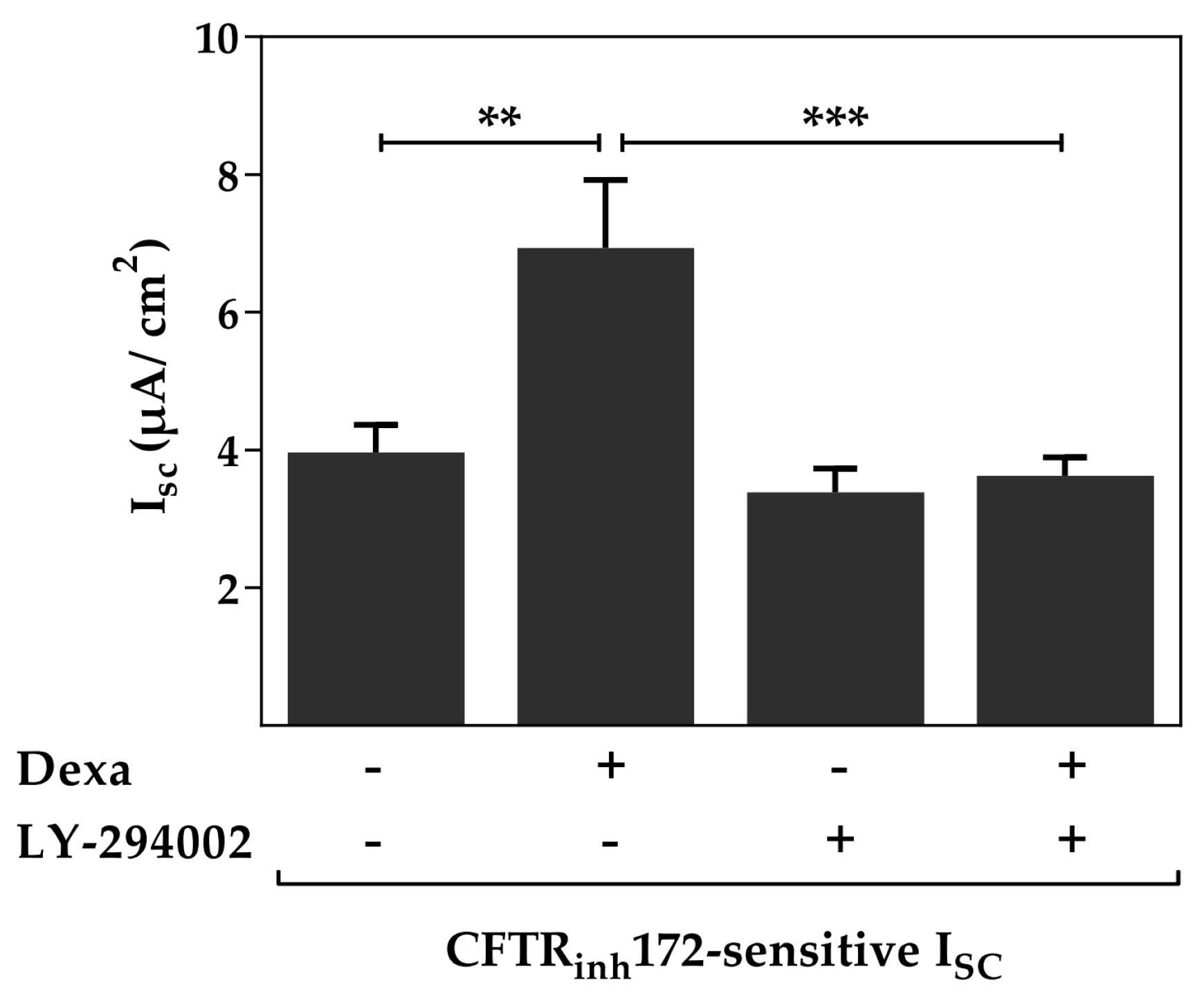
2.1. The Phosphoinositide 3-Kinase (PI3K) Pathway Is Involved in the Dexa-Stimulated Cystic Fibrosis Transmembrane Conductance Regulator Activity
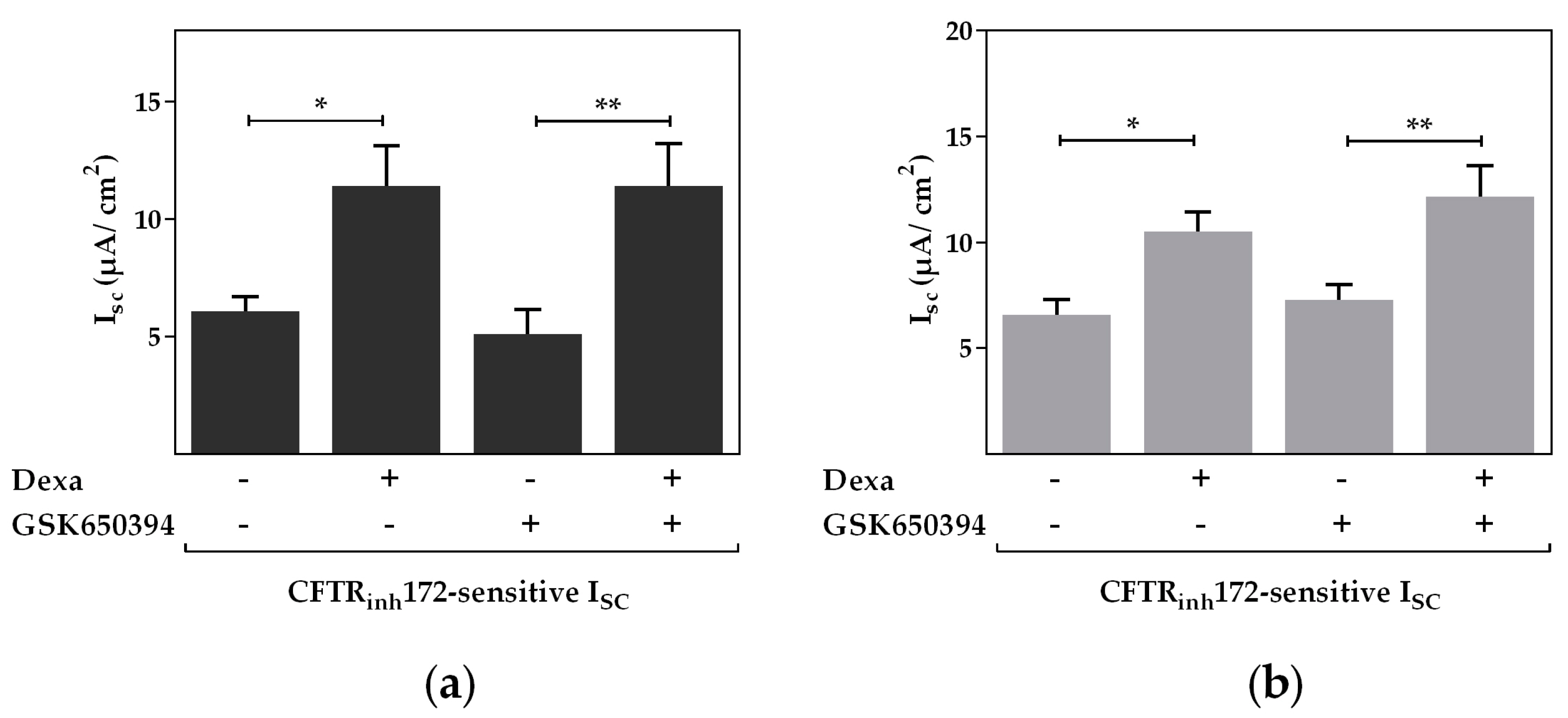
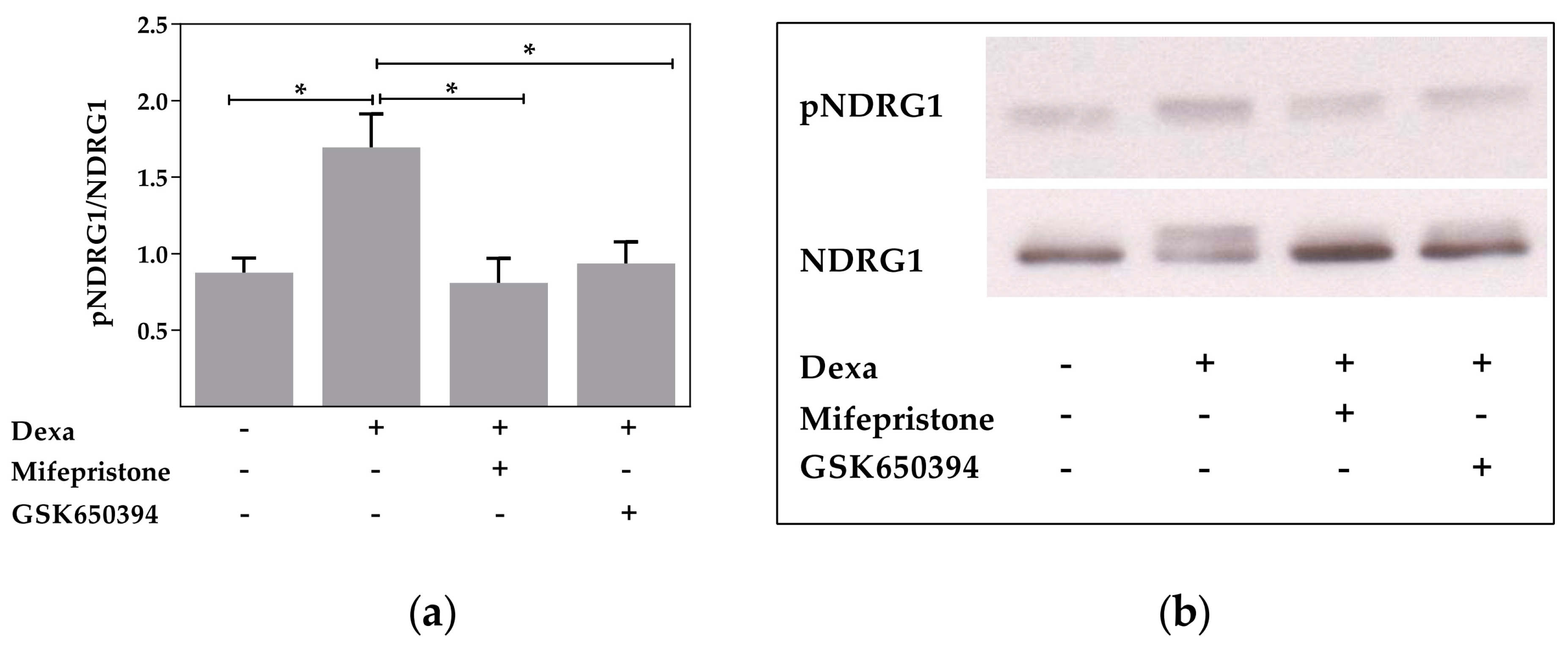
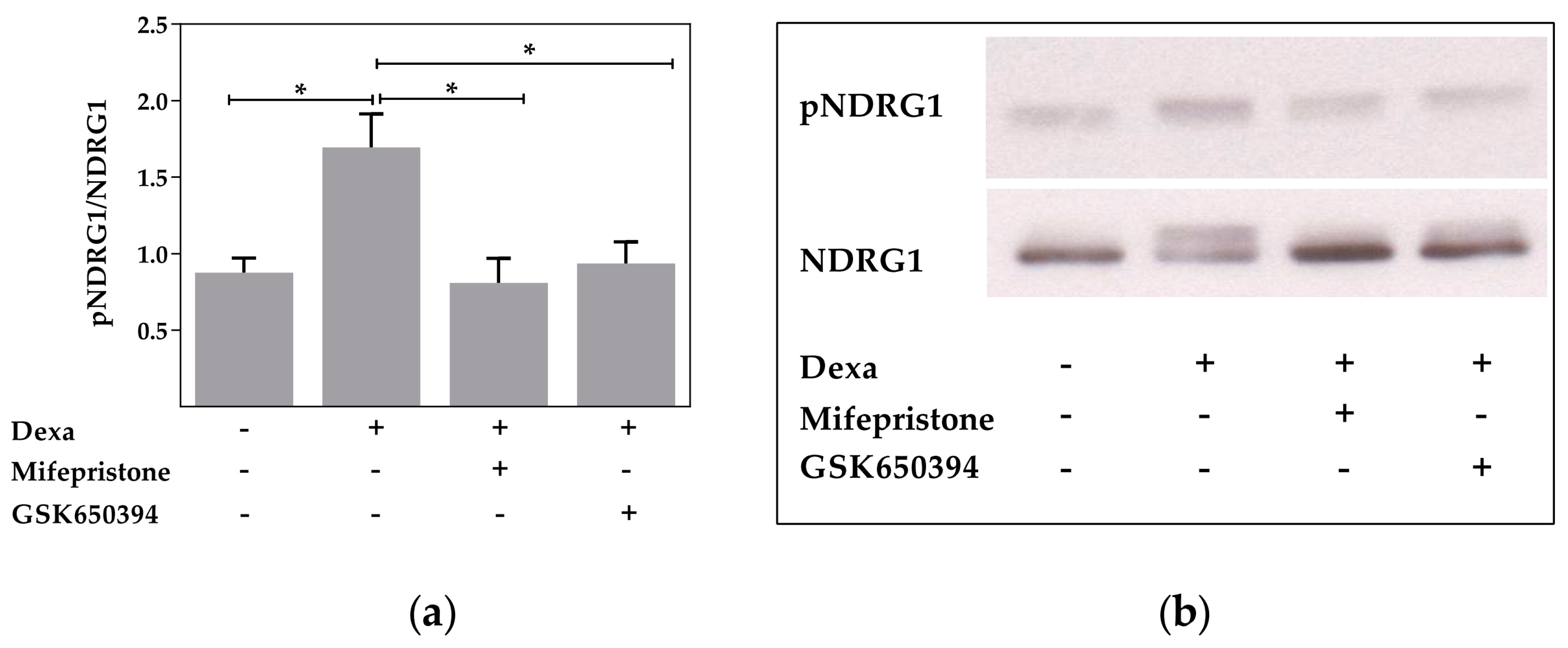
2.2. Serum and Glucocorticoid Dependent Kinase 1 Is Not Involved in the Dexa-Stimulated CFTR Activity
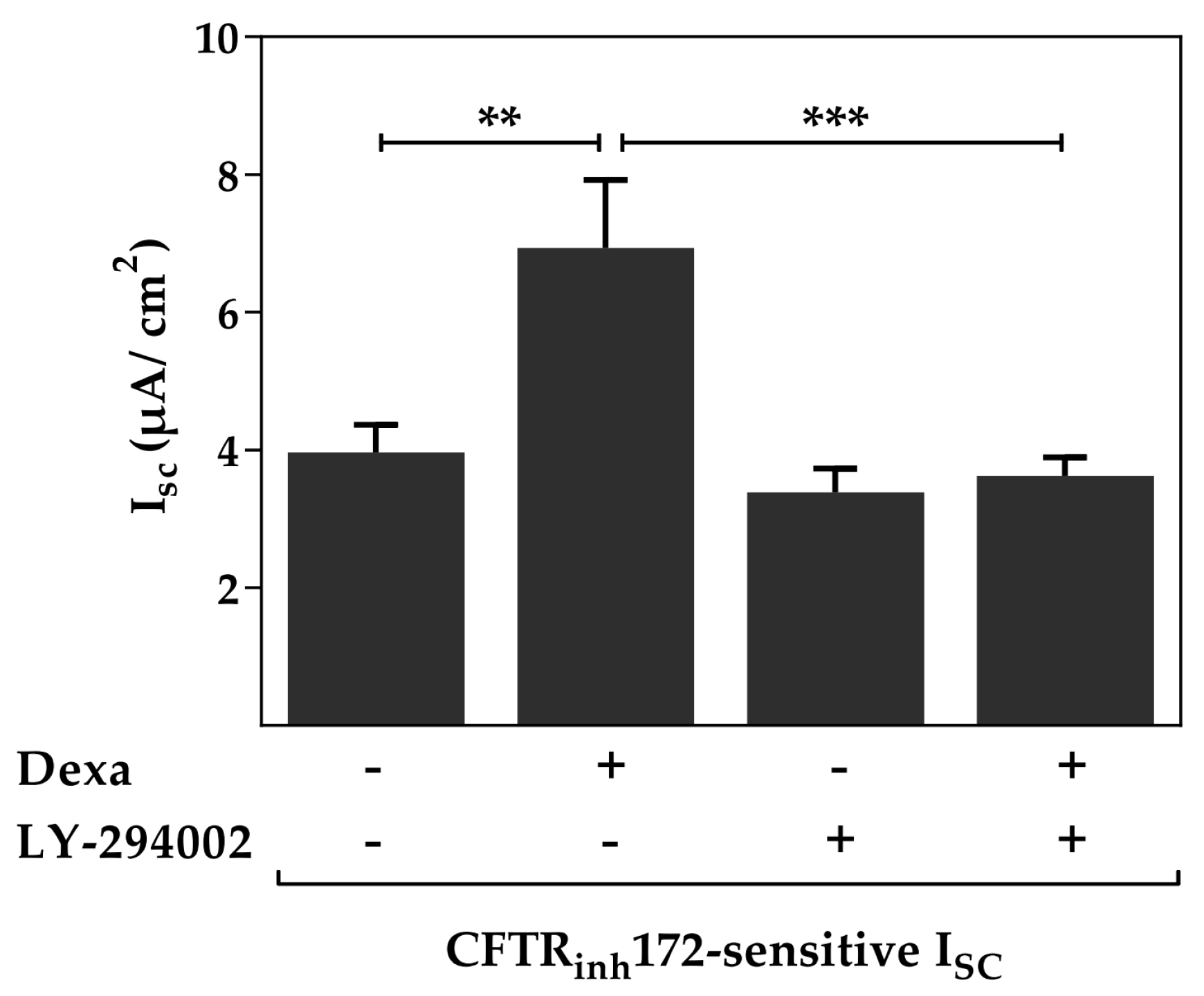
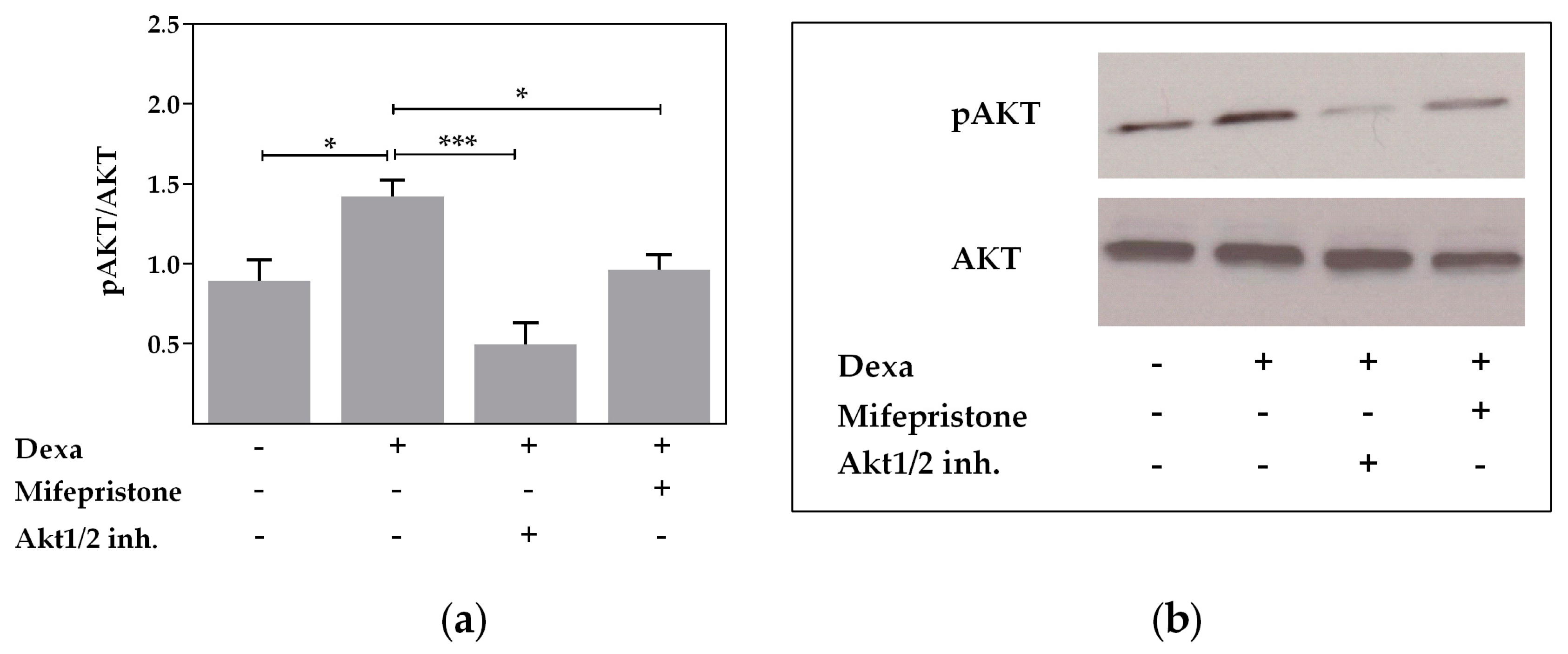
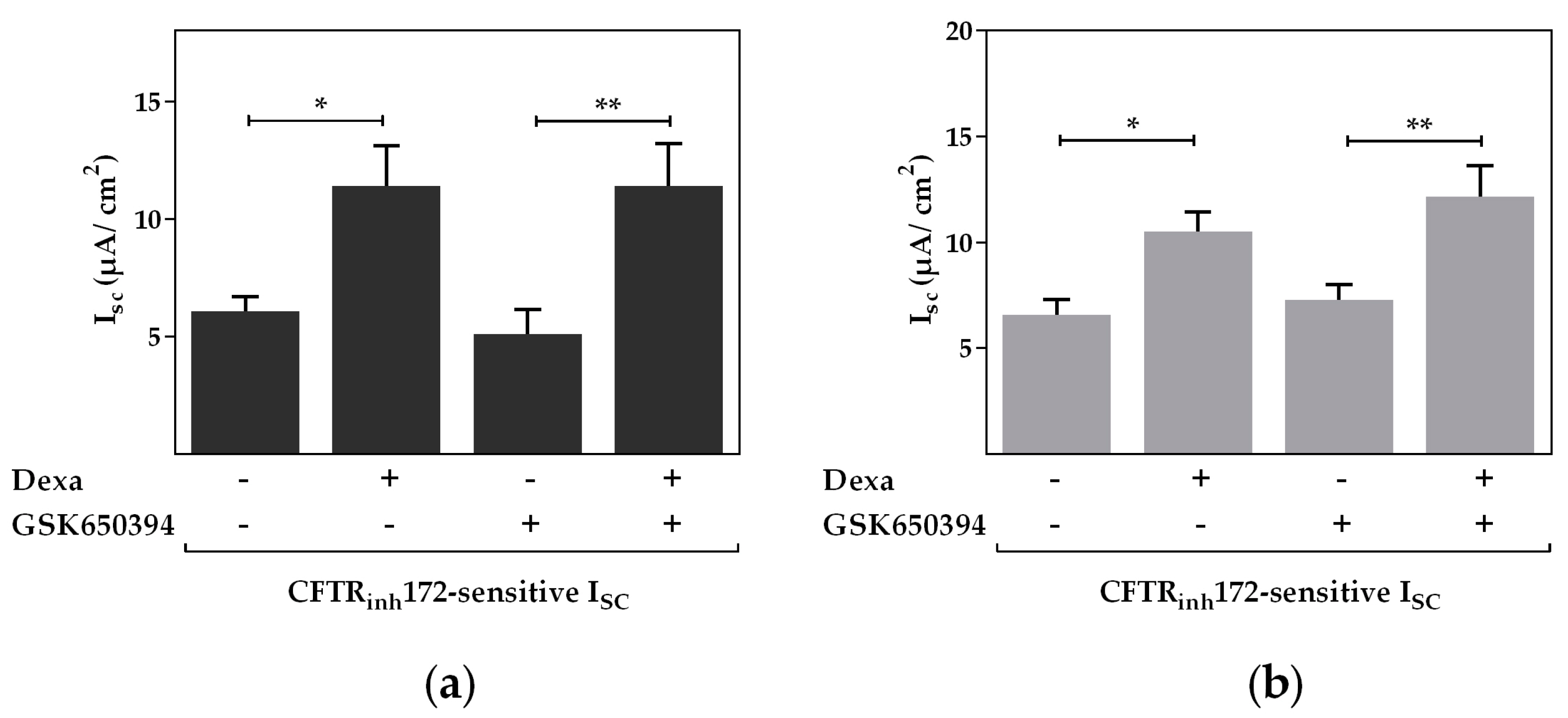
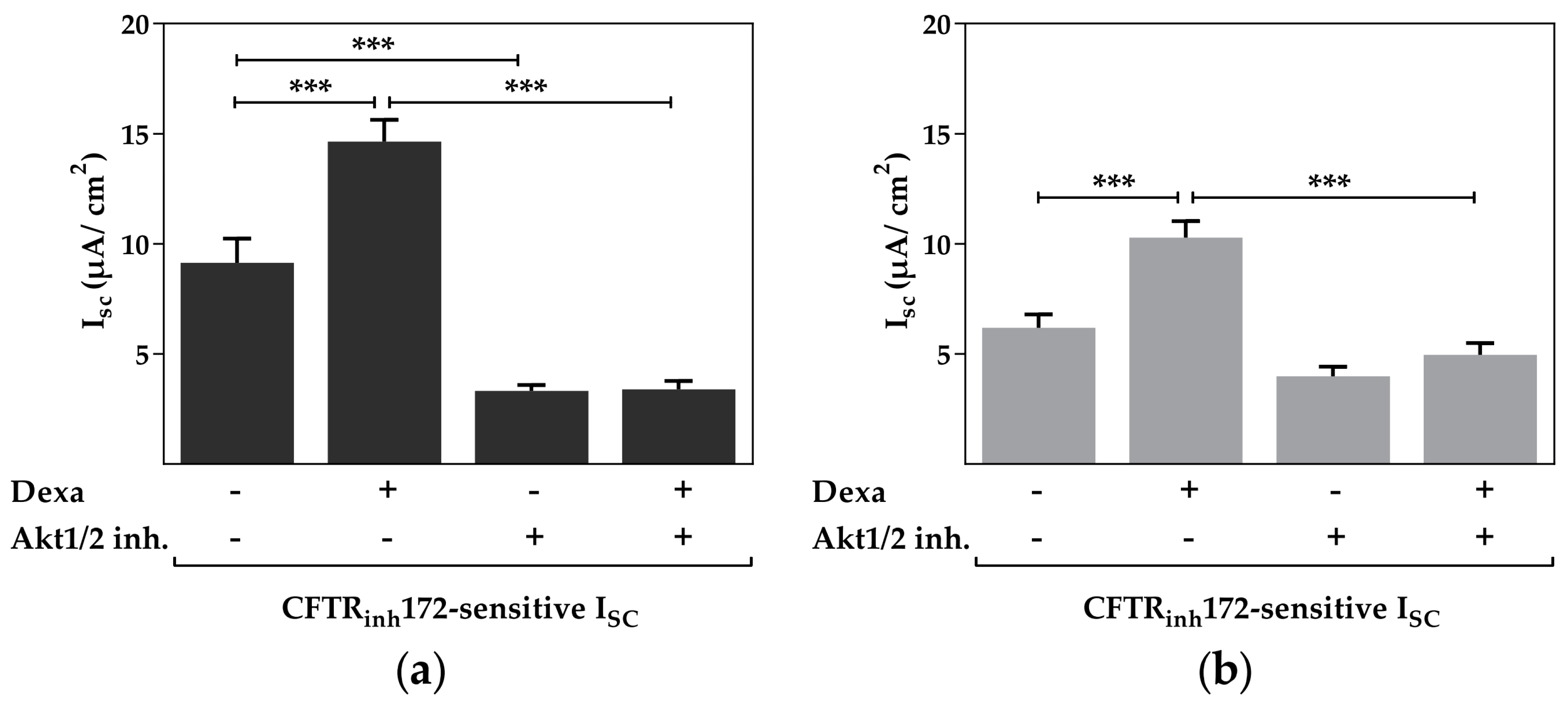
2.3. Protein Kinase B Activity Is Involved in the Dexa-Stimulated CFTR Activity
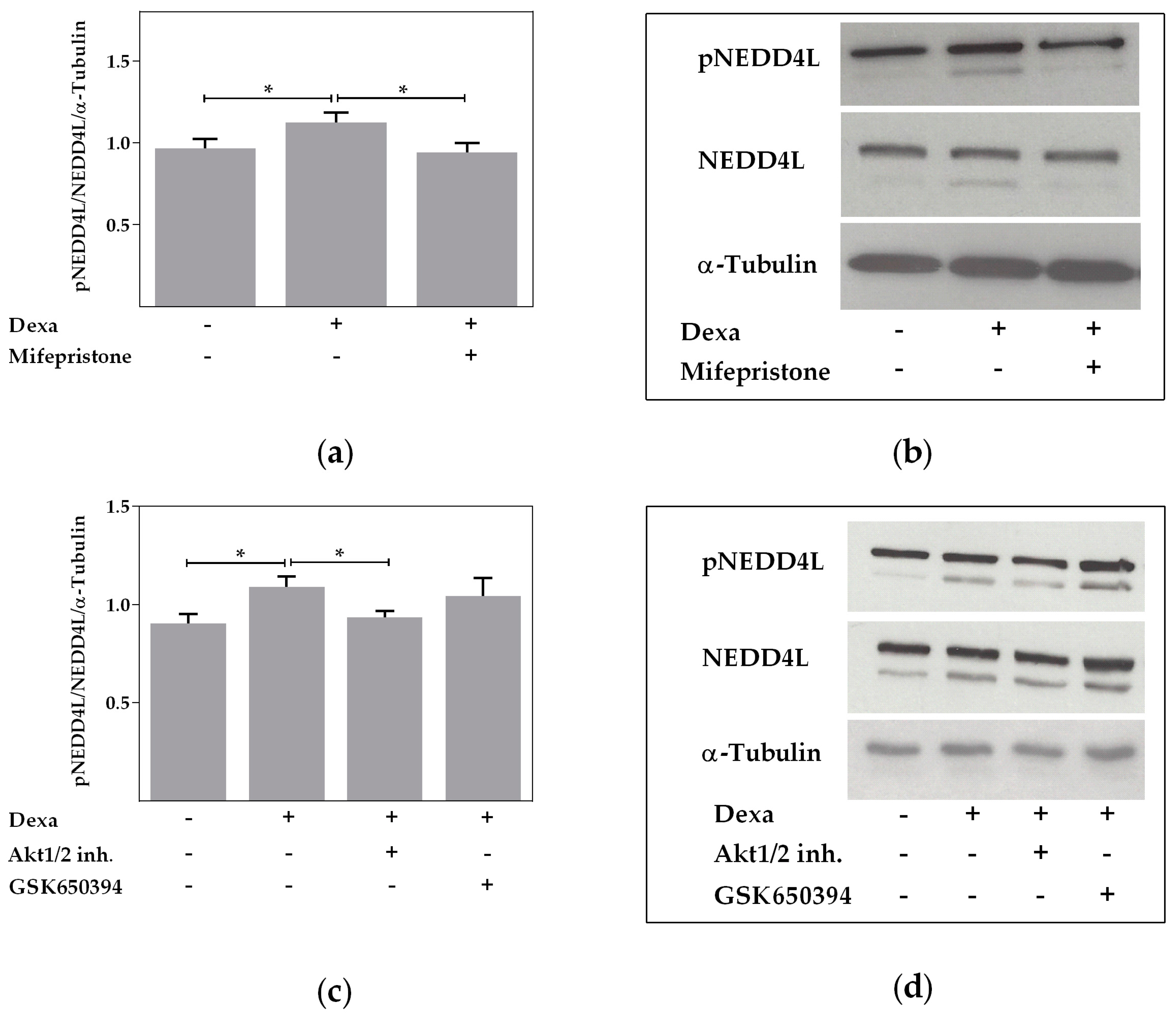
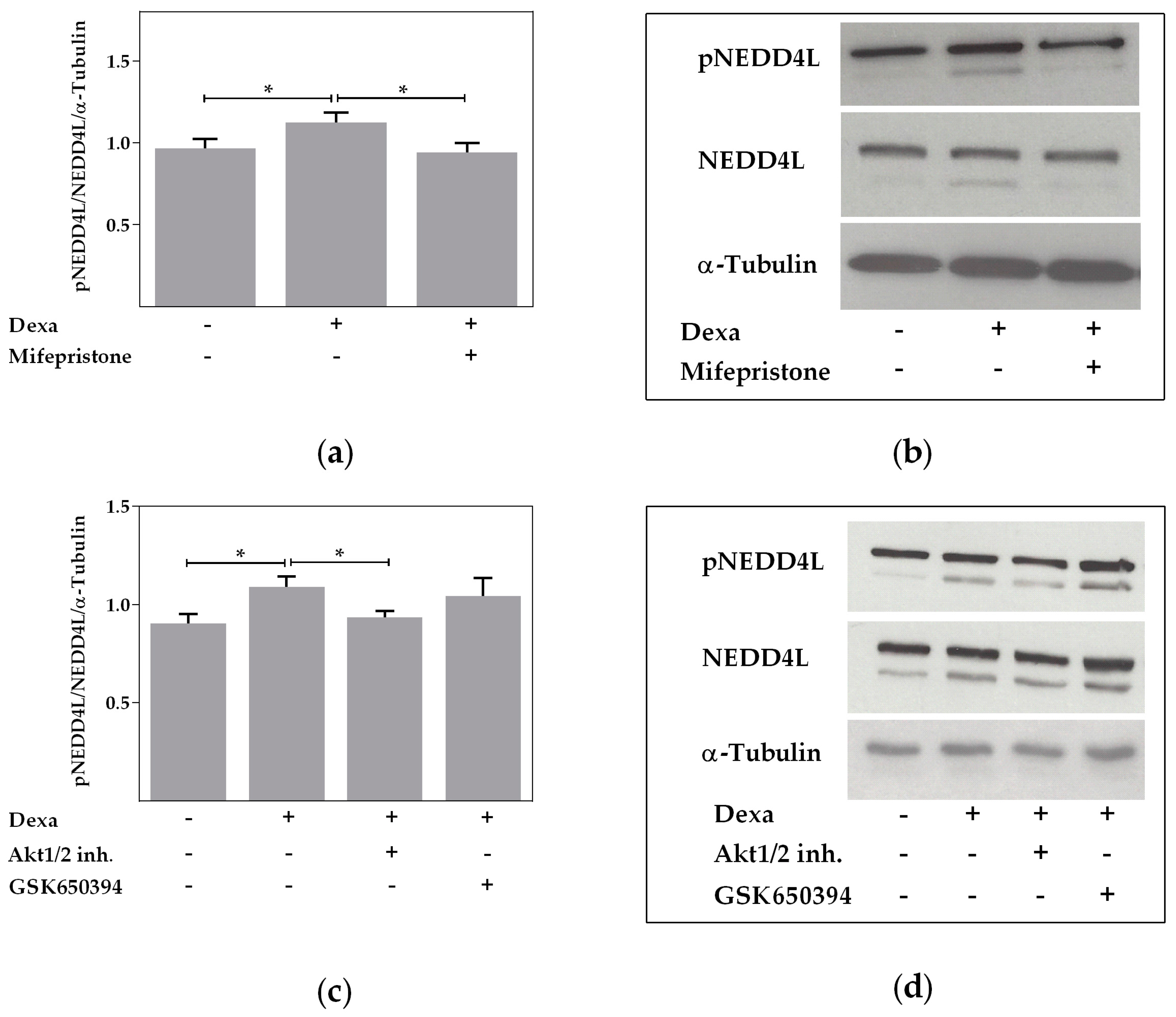
2.4. Neural Precursor Cell Expressed, Developmentally Downregulated 4-Like Activity Is Reduced by Dexa
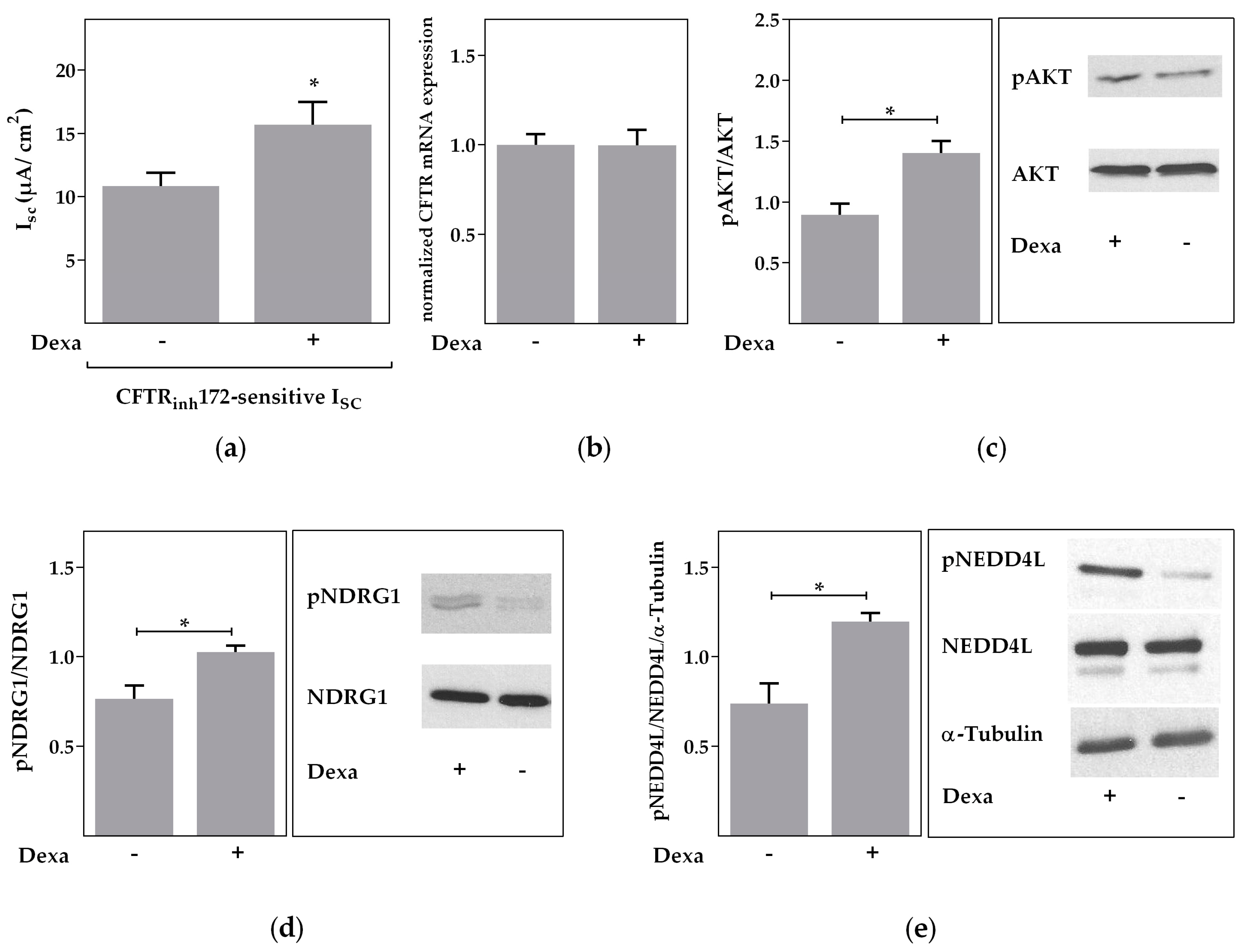
2.5. Rapid Effects of Dexa on CFTR Activity
3. Discussion
4. Materials and Methods
4.1. Tissue Preparation
4.2. Isolation of Primary Airway Epithelial Cells
4.3. Culture of Cell Lines
4.4. Electrophysiological Measurements
4.5. Western Blot Analyses
4.6. Measurement of CFTR mRNA Expression
4.7. Statistical Analyses
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AKT | Protein kinase B |
| cAMP | Cyclic adenosine monophosphate |
| CFTR | Cystic fibrosis transmembrane conductance regulator |
| COPD | Chronic obstructive pulmonary disease |
| Dexa | Dexamethasone |
| GCs | Glucocorticoids |
| GR | Glucocorticoid receptor |
| ISC | Short-circuit current |
| MCC | Mucociliary clearance |
| NDRG1 | N-myc downregulated gene 1 |
| NEDD4L | Neural precursor cell expressed, developmentally downregulated 4-like |
| mTORC2 | Mammalian target of rapamycin complex 2 |
| PDK1 | Phosphoinositide-dependent kinase |
| PI3K | Phosphoinositide 3-kinase |
| Rte | Transepithelial resistance |
| SGK1 | Serum and glucocorticoid dependent kinase 1 |
| Vte | Transepithelial voltage |
References
- Pujols, L.; Mullol, J.; Perez, M.; Roca-Ferrer, J.; Juan, M.; Xaubet, A.; Cidlowski, J.A.; Picado, C. Expression of the human glucocorticoid receptor alpha and beta isoforms in human respiratory epithelial cells and their regulation by dexamethasone. Am. J. Respir. Cell Mol. Biol. 2001, 24, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Jewell, C.M.; Yudt, M.R.; Bofetiado, D.M.; Cidlowski, J.A. The dominant negative activity of the human glucocorticoid receptor beta isoform. Specificity and mechanisms of action. J. Biol. Chem. 1999, 274, 27857–27866. [Google Scholar] [CrossRef] [PubMed]
- Stahn, C.; Buttgereit, F. Genomic and nongenomic effects of glucocorticoids. Nat. Clin. Pract. Rheumatol. 2008, 4, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Pilewski, J.M.; Frizzell, R.A. Role of CFTR in airway disease. Physiol. Rev. 1999, 79, S215–S255. [Google Scholar]
- Tarran, R.; Button, B.; Boucher, R.C. Regulation of normal and cystic fibrosis airway surface liquid volume by phasic shear stress. Annu. Rev. Physiol. 2006, 68, 543–561. [Google Scholar] [CrossRef] [PubMed]
- Livraghi, A.; Randell, S.H. Cystic fibrosis and other respiratory diseases of impaired mucus clearance. Toxicol. Pathol. 2007, 35, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Dransfield, M.T.; Wilhelm, A.M.; Flanagan, B.; Courville, C.; Tidwell, S.L.; Raju, S.V.; Gaggar, A.; Steele, C.; Tang, L.P.; Liu, B.; et al. Acquired cystic fibrosis transmembrane conductance regulator dysfunction in the lower airways in COPD. Chest 2013, 144, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Sloane, P.A.; Shastry, S.; Wilhelm, A.; Courville, C.; Tang, L.P.; Backer, K.; Levin, E.; Raju, S.V.; Li, Y.; Mazur, M.; et al. A pharmacologic approach to acquired cystic fibrosis transmembrane conductance regulator dysfunction in smoking related lung disease. PLoS ONE 2012, 7, e39809. [Google Scholar] [CrossRef] [PubMed]
- Laube, M.; Bossmann, M.; Thome, U.H. Glucocorticoids Distinctively modulate the CFTR channel with possible implications in lung development and transition into extrauterine life. PLoS ONE 2015, 10, e0124833. [Google Scholar] [CrossRef] [PubMed]
- Kayahara, M.; Berry, A.A.; Ray, D.W. Non-genomic effects of the glucocorticoid receptor—The effect of glucocorticoids on activation of c-src and PKB/Akt. In Endocrine Abstracts, Proceedings of the 196th Meeting of the Society for Endocrinology, London, UK, 7–9 November 2005; Bioscientifica: Bristol, UK, 2005; Volume 10, p. 88. [Google Scholar]
- Kino, T.; Charmandari, E.; Chrousos, G.P. Glucocorticoid receptor: Implications for rheumatic diseases. Clin. Exp. Rheumatol. 2011, 29, 32–41. [Google Scholar]
- Makara, G.B.; Haller, J. Non-genomic effects of glucocorticoids in the neural system: Evidence, mechanisms and implications. Prog. Neurobiol. 2001, 65, 367–390. [Google Scholar] [CrossRef]
- Prota, L.F.M.; Cebotaru, L.; Cheng, J.; Wright, J.; Vij, N.; Morales, M.M.; Guggino, W.B. Dexamethasone regulates CFTR expression in Calu-3 cells with the involvement of chaperones HSP70 and HSP90. PLoS ONE 2012, 7, e47405. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, R.C.; Lockwood, S.R.; Lide, E.; Bauer, R.; Suaud, L.; Grumbach, Y. Regulation of endogenous ENaC functional expression by CFTR and ΔF508-CFTR in airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L88–L101. [Google Scholar] [CrossRef] [PubMed]
- Bomberger, J.M.; Coutermarsh, B.A.; Barnaby, R.L.; Sato, J.D.; Chapline, M.C.; Stanton, B.A. Serum and glucocorticoid-inducible kinase1 increases plasma membrane wt-CFTR in human airway epithelial cells by inhibiting its endocytic retrieval. PLoS ONE 2014, 9, e89599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, C.; Klammt, J.; Thome, U.H.; Laube, M. The interaction of glucocorticoids and progesterone distinctively affects epithelial sodium transport. Lung 2014, 192, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Tuo, B.; Wen, G.; Zhang, Y.; Liu, X.; Wang, X.; Liu, X.; Dong, H. Involvement of phosphatidylinositol 3-kinase in cAMP- and cGMP-induced duodenal epithelial CFTR activation in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, 15. [Google Scholar] [CrossRef] [PubMed]
- Gehring, E.-M.; Lam, R.S.; Siraskar, G.; Koutsouki, E.; Seebohm, G.; Ureche, O.N.; Ureche, L.; Baltaev, R.; Tavare, J.M.; Lang, F. PIKfyve upregulates CFTR activity. Biochem. Biophys. Res. Commun. 2009, 390, 952–957. [Google Scholar] [CrossRef] [PubMed]
- Luz, S.; Kongsuphol, P.; Mendes, A.I.; Romeiras, F.; Sousa, M.; Schreiber, R.; Matos, P.; Jordan, P.; Mehta, A.; Amaral, M.D.; et al. Contribution of casein kinase 2 and spleen tyrosine kinase to CFTR trafficking and protein kinase A-induced activity. Mol. Cell. Biol. 2011, 31, 4392–4404. [Google Scholar] [CrossRef] [PubMed]
- Faria, D.; Schreiber, R.; Kunzelmann, K. CFTR is activated through stimulation of purinergic P2Y2 receptors. Pflugers Arch. 2009, 457, 1373–1380. [Google Scholar] [CrossRef] [PubMed]
- Mendes, A.I.; Matos, P.; Moniz, S.; Luz, S.; Amaral, M.D.; Farinha, C.M.; Jordan, P. Antagonistic regulation of cystic fibrosis transmembrane conductance regulator cell surface expression by protein kinases WNK4 and spleen tyrosine kinase. Mol. Cell. Biol. 2011, 31, 4076–4086. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Artunc, F.; Vallon, V. The physiological impact of the serum and glucocorticoid-inducible kinase SGK1. Curr. Opin. Nephrol. Hypertens. 2009, 18, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Viard, P.; Butcher, A.J.; Halet, G.; Davies, A.; Nurnberg, B.; Heblich, F.; Dolphin, A.C. PI3K promotes voltage-dependent calcium channel trafficking to the plasma membrane. Nat. Neurosci. 2004, 7, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.A.; Ott, M.; Klingel, K.; Beck, S.; Melzig, J.; Friedrich, B.; Wild, K.N.; Broer, S.; Moschen, I.; Albers, A.; et al. Effects of the serine/threonine kinase SGK1 on the epithelial Na+ channel (ENaC) and CFTR: Implications for cystic fibrosis. Cell. Physiol. Biochem. 2001, 11, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Sato, J.D.; Chapline, M.C.; Thibodeau, R.; Frizzell, R.A.; Stanton, B.A. Regulation of human cystic fibrosis transmembrane conductance regulator (CFTR) by serum- and glucocorticoid-inducible kinase (SGK1). Cell. Physiol. Biochem. 2007, 20, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Caohuy, H.; Jozwik, C.; Pollard, H.B. Rescue of F508-CFTR by the SGK1/NEDD4–2 signaling pathway. J. Biol. Chem. 2009, 284, 25241–25253. [Google Scholar] [CrossRef] [PubMed]
- Mattes, C.; Laube, M.; Thome, U.H. Rapid elevation of sodium transport through insulin is mediated by AKT in alveolar cells. Physiol. Rep. 2014, 2, e00269. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.M.; Mansley, M.K.; Getty, J.; Husband, E.M.; Inglis, S.K.; Hansen, M.K. Effects of peroxisome proliferator-activated receptor gamma agonists on Na+ transport and activity of the kinase SGK1 in epithelial cells from lung and kidney. Br. J. Pharmacol. 2010, 159, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Snyder, P.M. Serum and Glucocorticoid-regulated kinase modulates NEDD4-2-mediated inhibition of the epithelial Na+ channel. J. Biol. Chem. 2002, 277, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Barnett, S.F.; Defeo-Jones, D.; Fu, S.; Hancock, P.J.; Haskell, K.M.; Jones, R.E.; Kahana, J.A.; Kral, A.M.; Leander, K.; Lee, L.L.; et al. Identification and characterization of pleckstrin-homology-domain-dependent and isoenzyme-specific AKT inhibitors. Biochem. J. 2005, 385, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; Mclauchlan, H.; Klevernic, I.; Arthur, J.S.C.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297. [Google Scholar] [CrossRef] [PubMed]
- Mansley, M.K.; Wilson, S.M. Effects of nominally selective inhibitors of the kinases PI3K, SGK1 and PKB on the insulin-dependent control of epithelial Na+ absorption. Br. J. Pharmacol. 2010, 161, 571–588. [Google Scholar] [CrossRef] [PubMed]
- Staub, O.; Gautschi, I.; Ishikawa, T.; Breitschopf, K.; Ciechanover, A.; Schild, L.; Rotin, D. Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J. 1997, 16, 6325–6336. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Koshy, S.; Folkesson, H.G. Involvement of αENaC and NEDD4-2 in the conversion from lung fluid secretion to fluid absorption at birth in the rat as assayed by RNA interference analysis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L1069–L1078. [Google Scholar] [CrossRef] [PubMed]
- Goulet, C.C.; Volk, K.A.; Adams, C.M.; Prince, L.S.; Stokes, J.B.; Snyder, P.M. Inhibition of the epithelial Na+ channel by interaction of NEDD4 with a PY motif deleted in Liddle’s syndrome. J. Biol. Chem. 1998, 273, 30012–30017. [Google Scholar] [CrossRef] [PubMed]
- Nagaki, K.; Yamamura, H.; Shimada, S.; Saito, T.; Hisanaga, S.; Taoka, M.; Isobe, T.; Ichimura, T. 14-3-3 Mediates phosphorylation-dependent inhibition of the interaction between the ubiquitin E3 ligase NEDD4-2 and epithelial Na+ channels. Biochemistry 2006, 45, 6733–6740. [Google Scholar] [CrossRef] [PubMed]
- Debonneville, C.; Flores, S.Y.; Kamynina, E.; Plant, P.J.; Tauxe, C.; Thomas, M.A.; Munster, C.; Chraibi, A.; Pratt, J.H.; Horisberger, J.D.; et al. Phosphorylation of NEDD4-2 by SGK1 regulates epithelial Na+ channel cell surface expression. EMBO J. 2001, 20, 7052–7059. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, K.; Chapline, C.; Sato, J.D.; Stanton, B.A. NEDD4-2 does not regulate wt-CFTR in human airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, 7. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.-H.; Dinudom, A.; Sanchez-Perez, A.; Kumar, S.; Cook, D.I. AKT mediates the effect of insulin on epithelial sodium channels by inhibiting NEDD4-2. J. Biol. Chem. 2007, 282, 29866–29873. [Google Scholar] [CrossRef] [PubMed]
- Falkenstein, E.; Norman, A.W.; Wehling, M. Mannheim classification of nongenomically initiated (rapid) steroid action(s). J. Clin. Endocrinol. Metab. 2000, 85, 2072–2075. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.Z.; Cidlowski, J.A. The origin and functions of multiple human glucocorticoid receptor isoforms. Ann. N. Y. Acad. Sci. 2004, 1024, 102–123. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.L.; Burns, K.A.; Bayle, J.Y.; Boucher, R.C.; van Scott, M.R. Sodium- and chloride-conductive pathways in cultured mouse tracheal epithelium. Am. J. Physiol. 1992, 263, 25. [Google Scholar]
- Thome, U.H.; Davis, I.C.; Nguyen, S.V.; Shelton, B.J.; Matalon, S. Modulation of sodium transport in fetal alveolar epithelial cells by oxygen and corticosterone. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, 85. [Google Scholar] [CrossRef] [PubMed]
- Inglis, S.K.; Gallacher, M.; Brown, S.G.; McTavish, N.; Getty, J.; Husband, E.M.; Murray, J.T.; Wilson, S.M. SGK1 activity in Na+ absorbing airway epithelial cells monitored by assaying NDRG1-Thr346/356/366 phosphorylation. Pflugers Arch. 2009, 457, 1287–1301. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.T.; Cummings, L.A.; Bloomberg, G.B.; Cohen, P. Identification of different specificity requirements between SGK1 and PKBα. FEBS Lett. 2005, 579, 991–994. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bossmann, M.; Ackermann, B.W.; Thome, U.H.; Laube, M. Signaling Cascade Involved in Rapid Stimulation of Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) by Dexamethasone. Int. J. Mol. Sci. 2017, 18, 1807. https://doi.org/10.3390/ijms18081807
Bossmann M, Ackermann BW, Thome UH, Laube M. Signaling Cascade Involved in Rapid Stimulation of Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) by Dexamethasone. International Journal of Molecular Sciences. 2017; 18(8):1807. https://doi.org/10.3390/ijms18081807
Chicago/Turabian StyleBossmann, Miriam, Benjamin W. Ackermann, Ulrich H. Thome, and Mandy Laube. 2017. "Signaling Cascade Involved in Rapid Stimulation of Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) by Dexamethasone" International Journal of Molecular Sciences 18, no. 8: 1807. https://doi.org/10.3390/ijms18081807