Biophysical and Computational Studies of the vCCI:vMIP-II Complex

Abstract
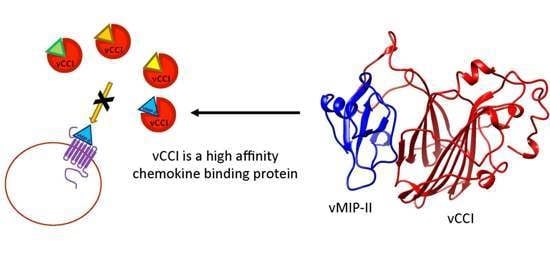
:
1. Introduction
2. Results
2.1. Folded Viral CC Chemokine Inhibitor(vCCI)/35K Can Be Produced from E. coli
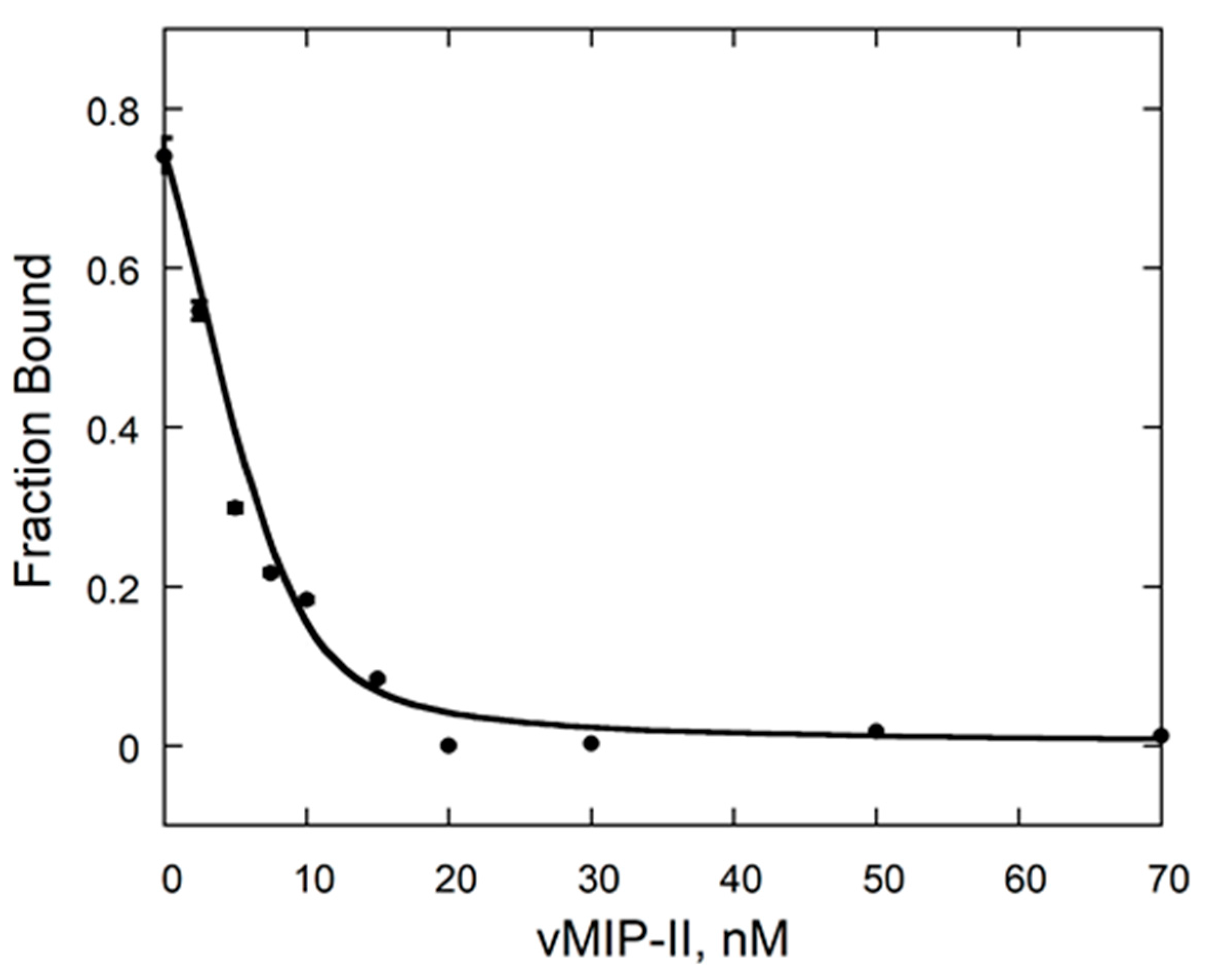
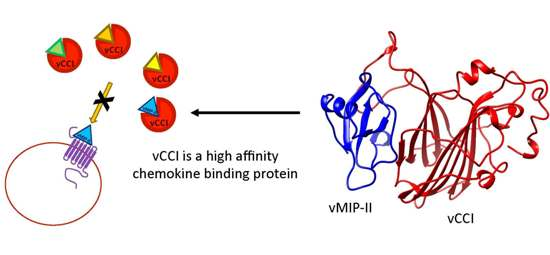
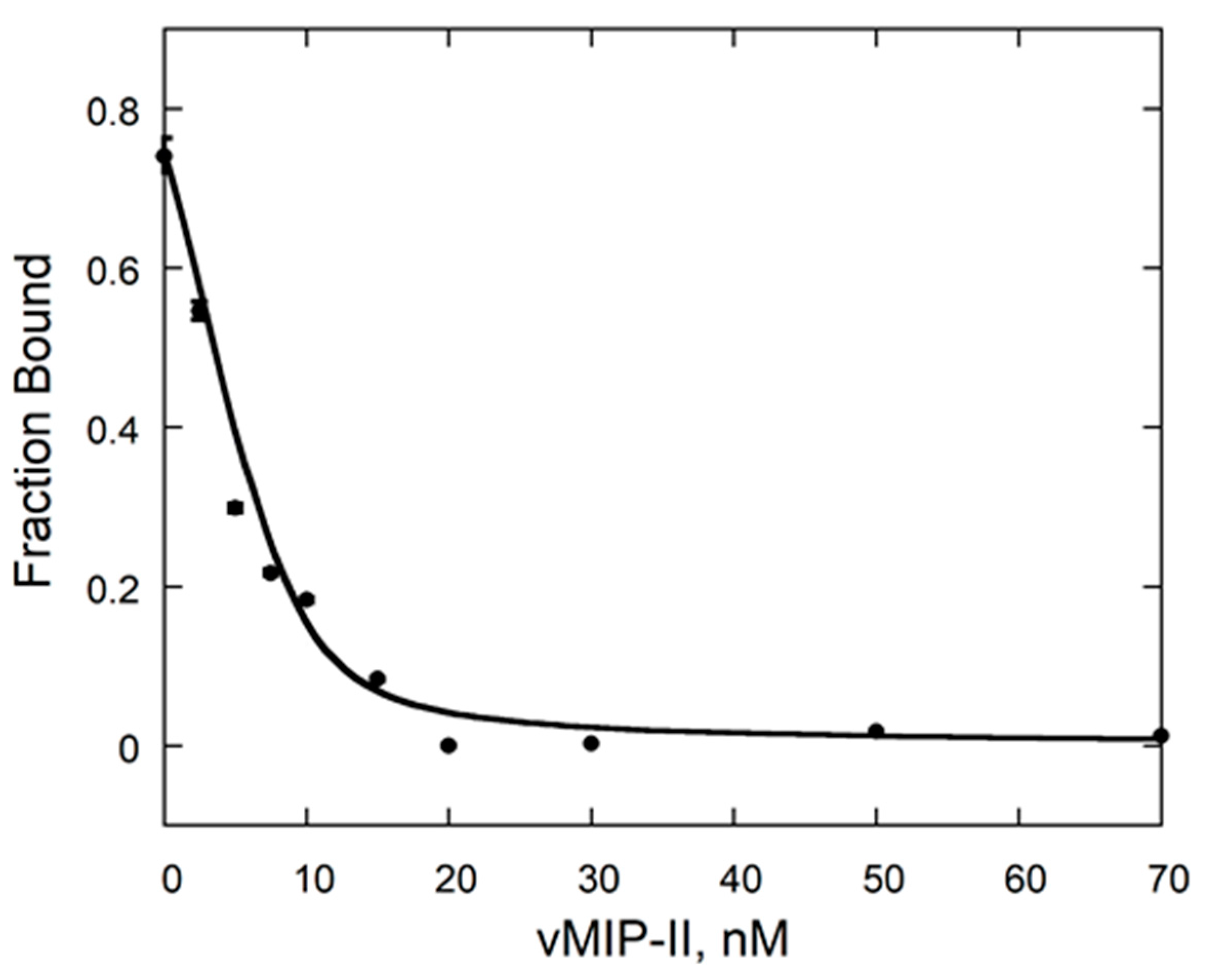
2.2. vCCI:vMIP-II Produce a High Affinity Complex
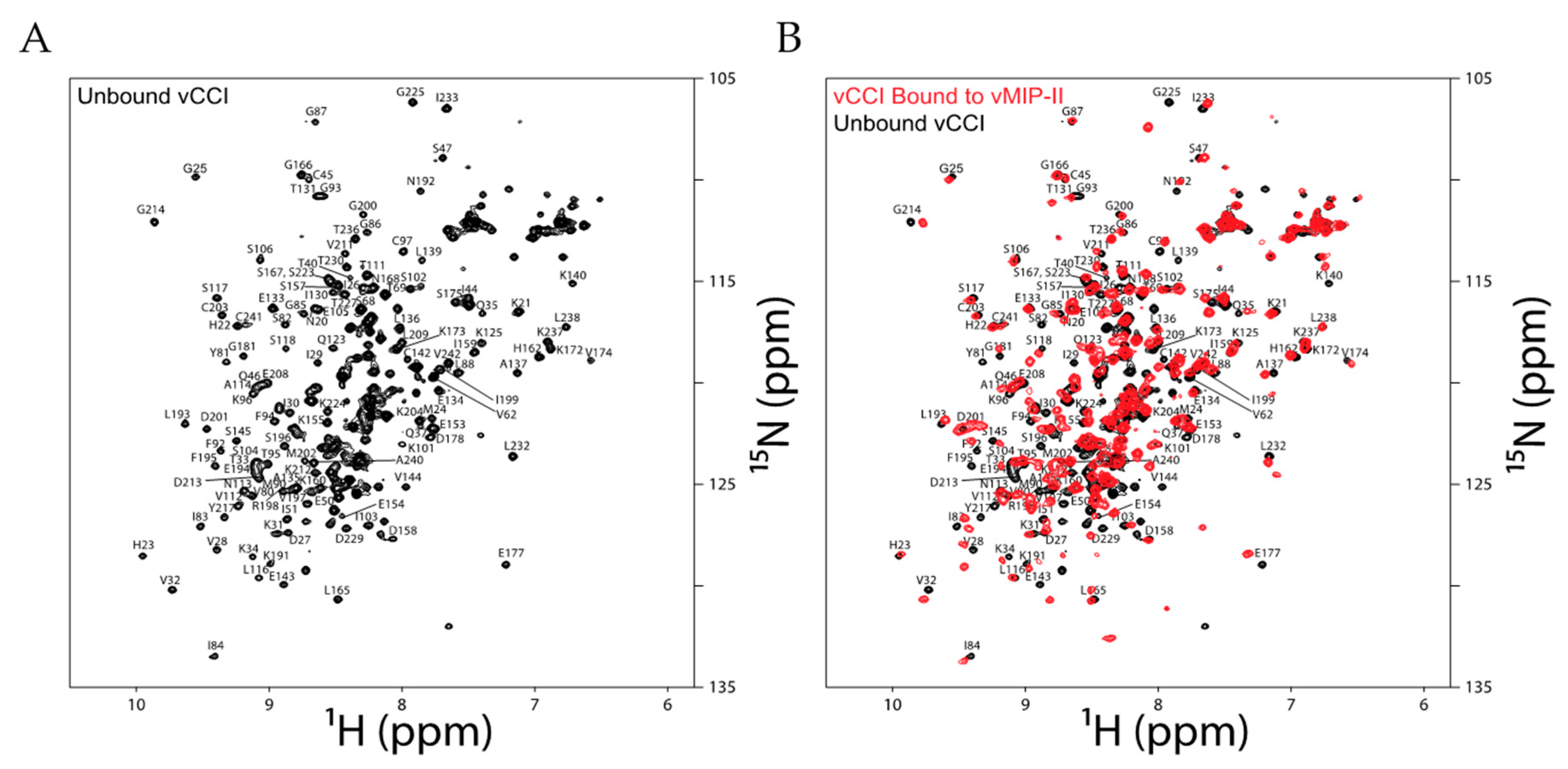
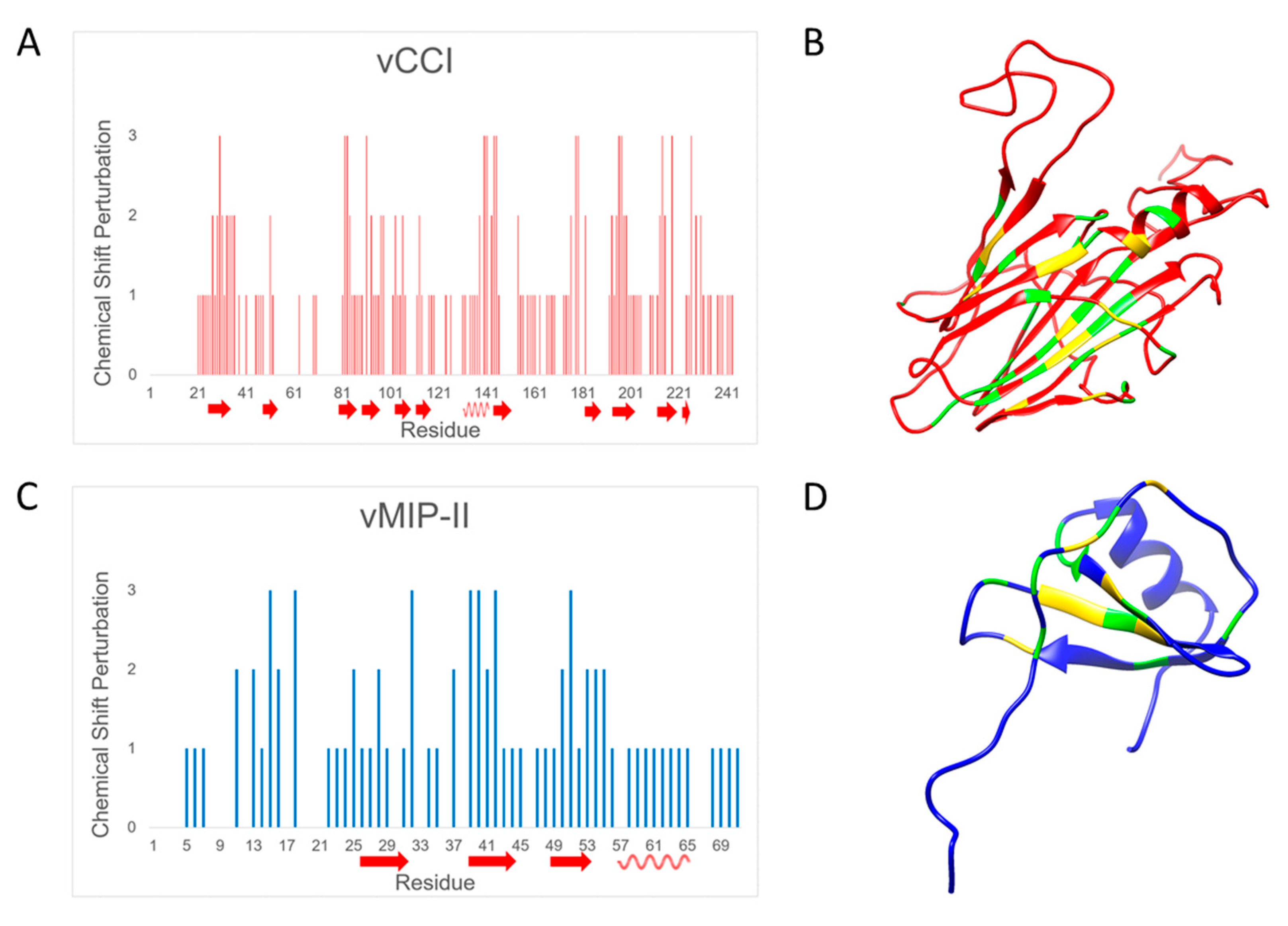
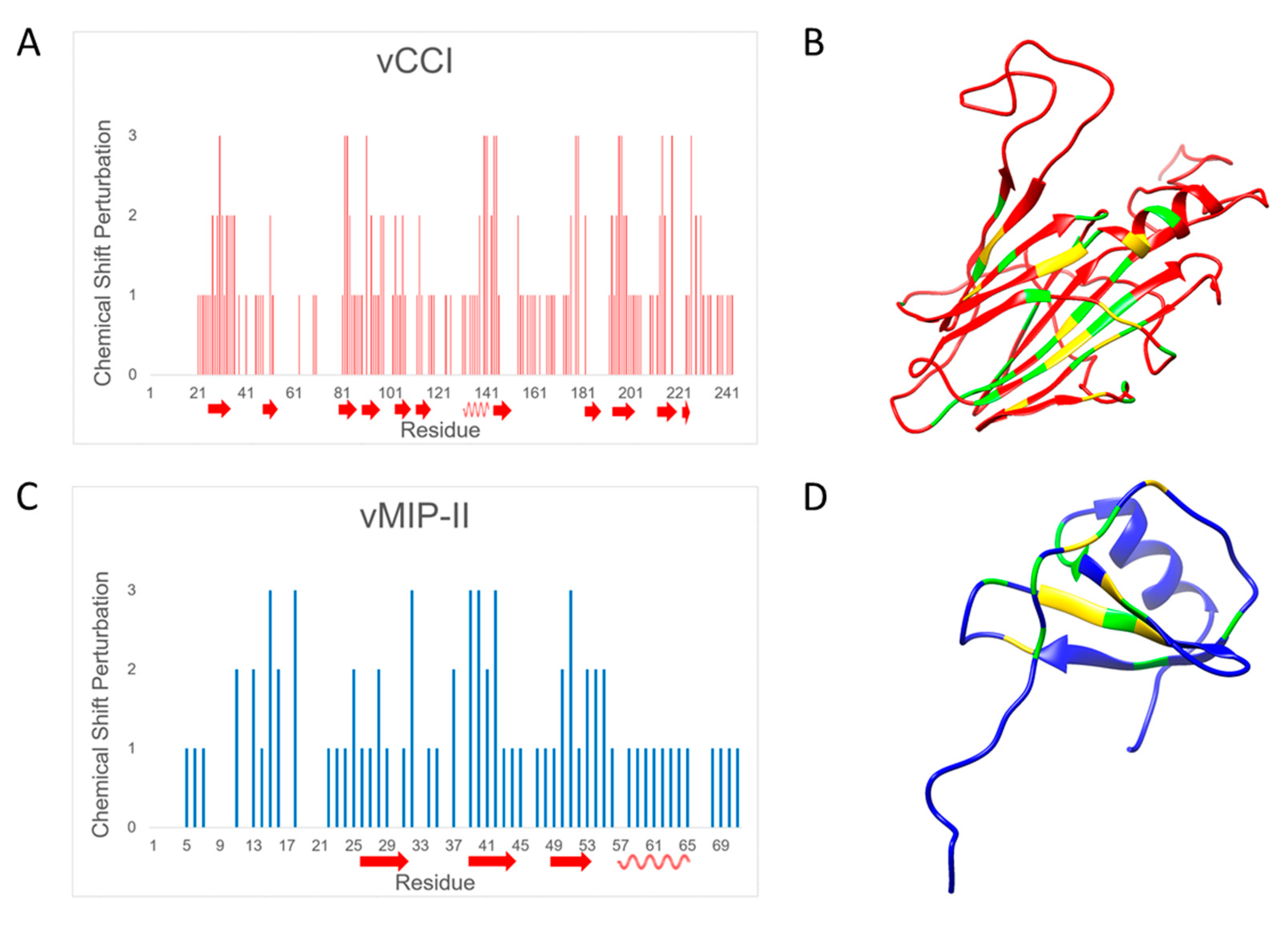
2.3. Changes in Chemical Shift Suggest vCCI:vMIP-II Interaction Is Similar to Other vCCI:Chemokine Complexes
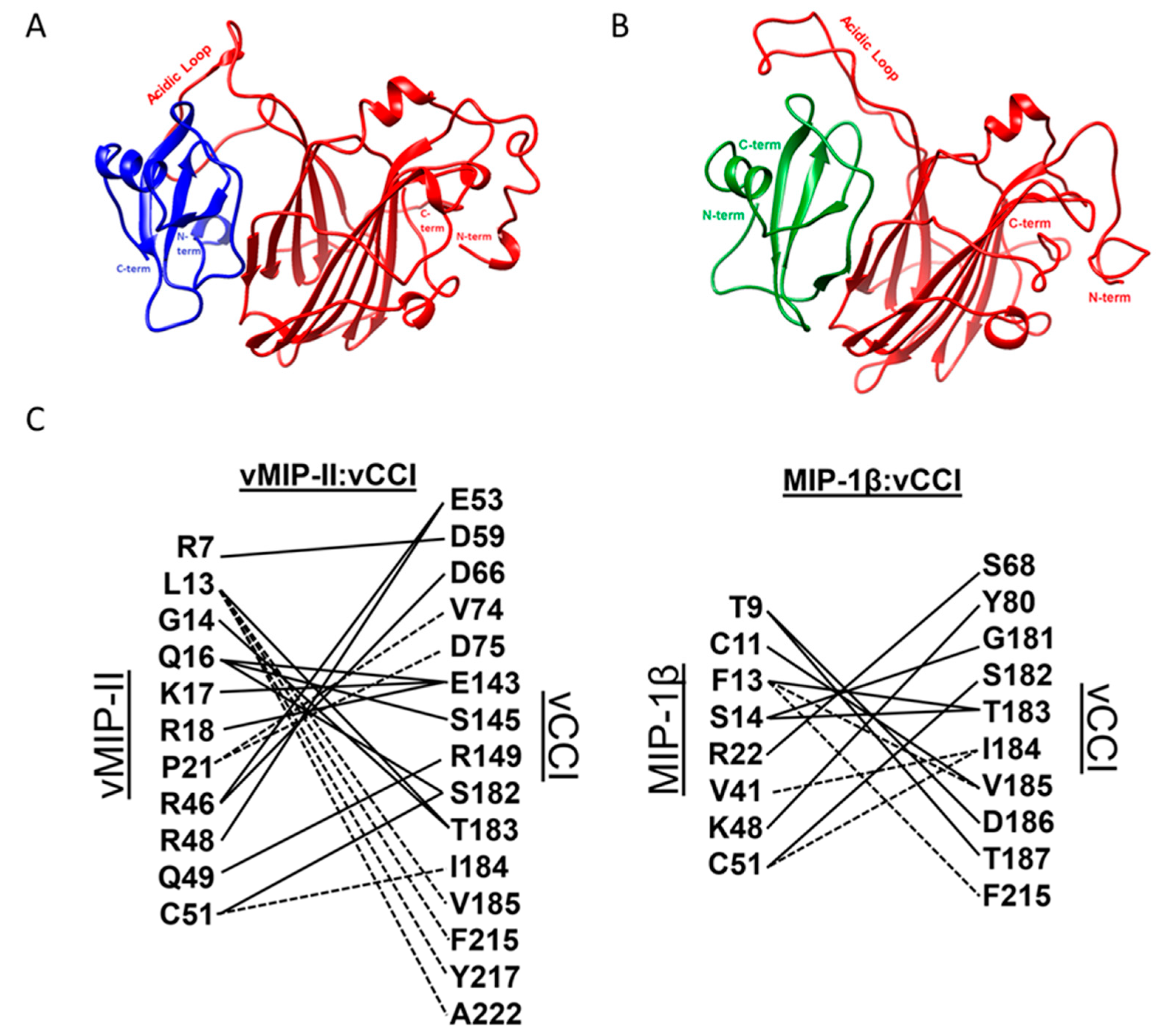
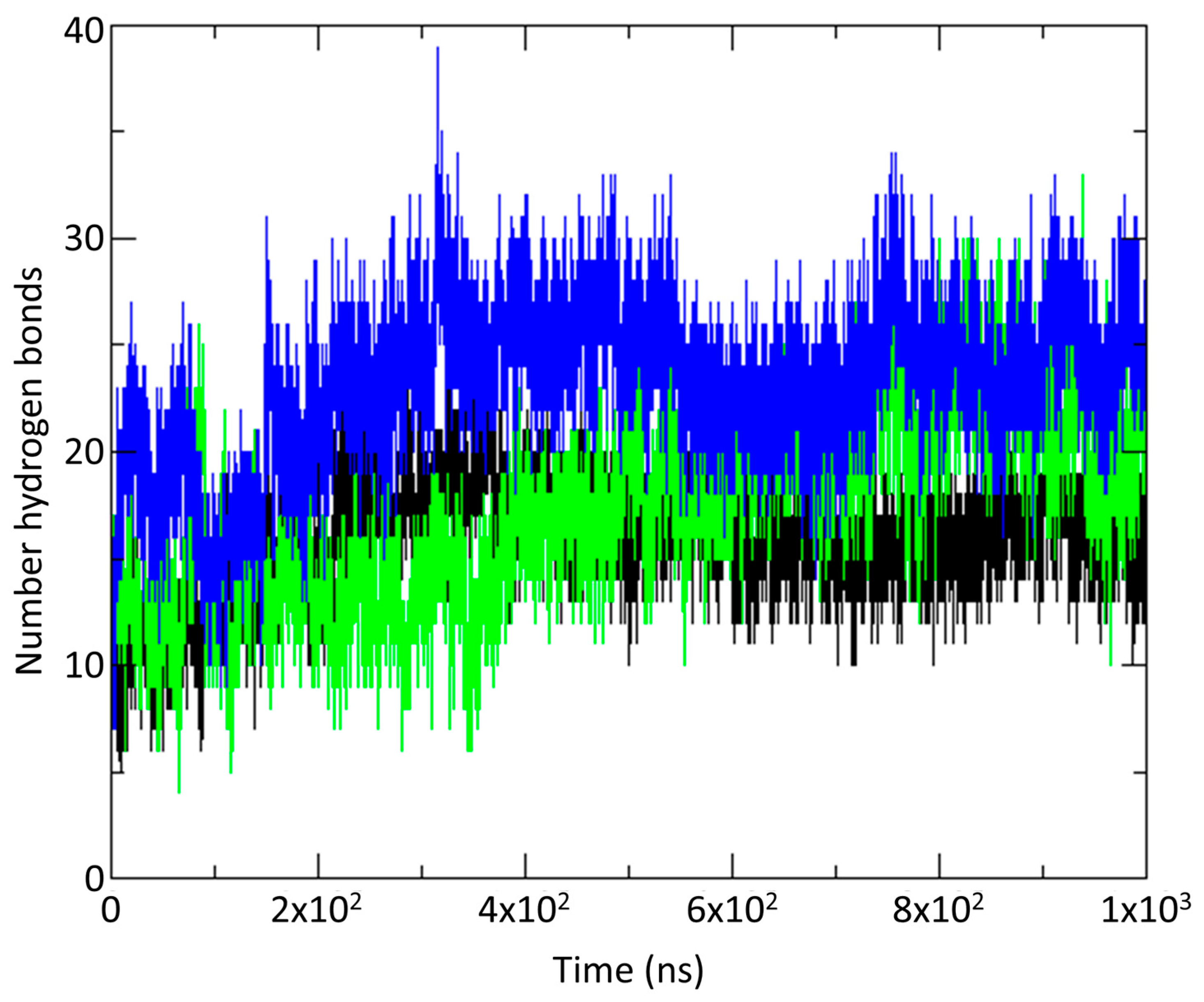
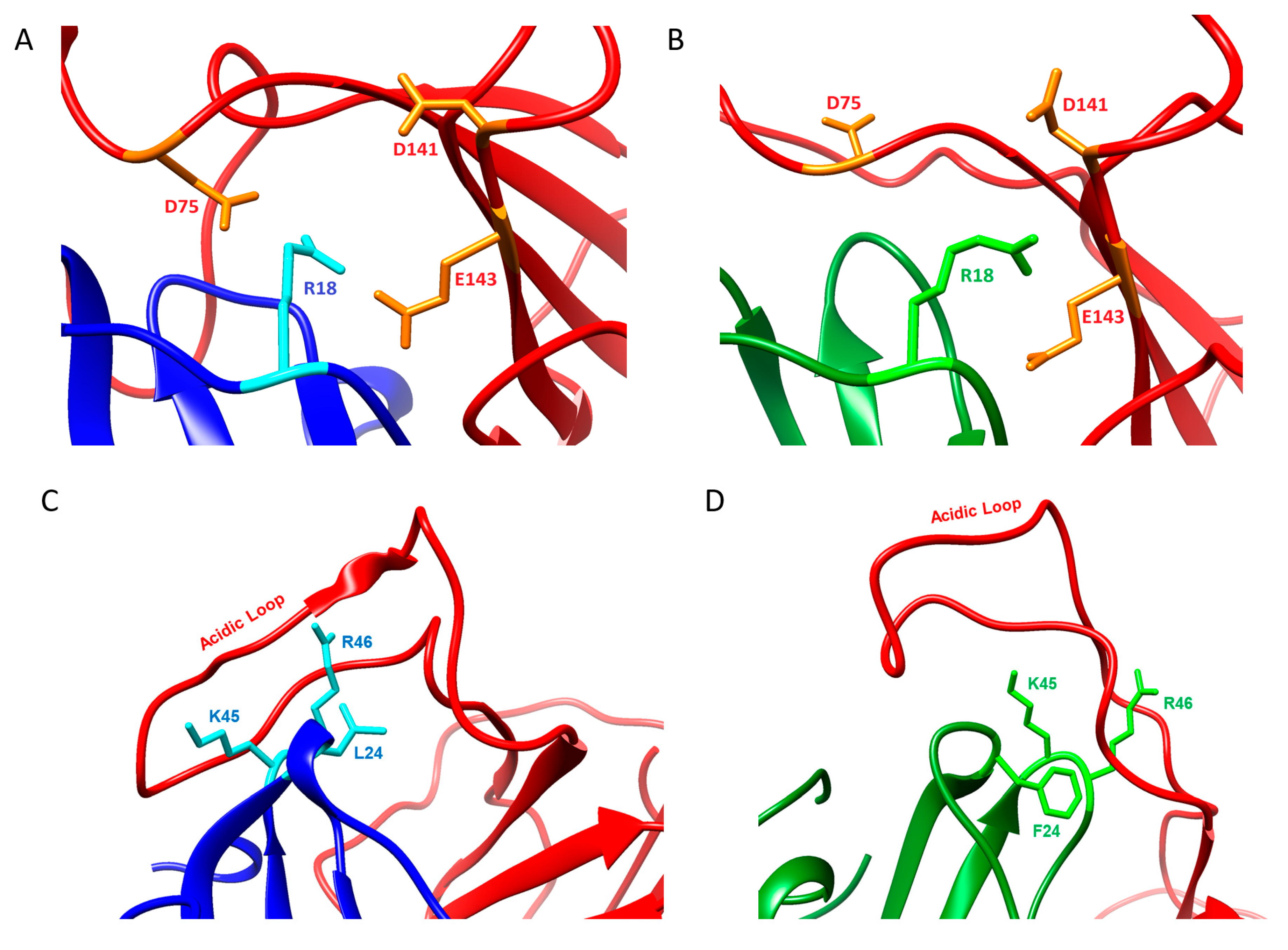
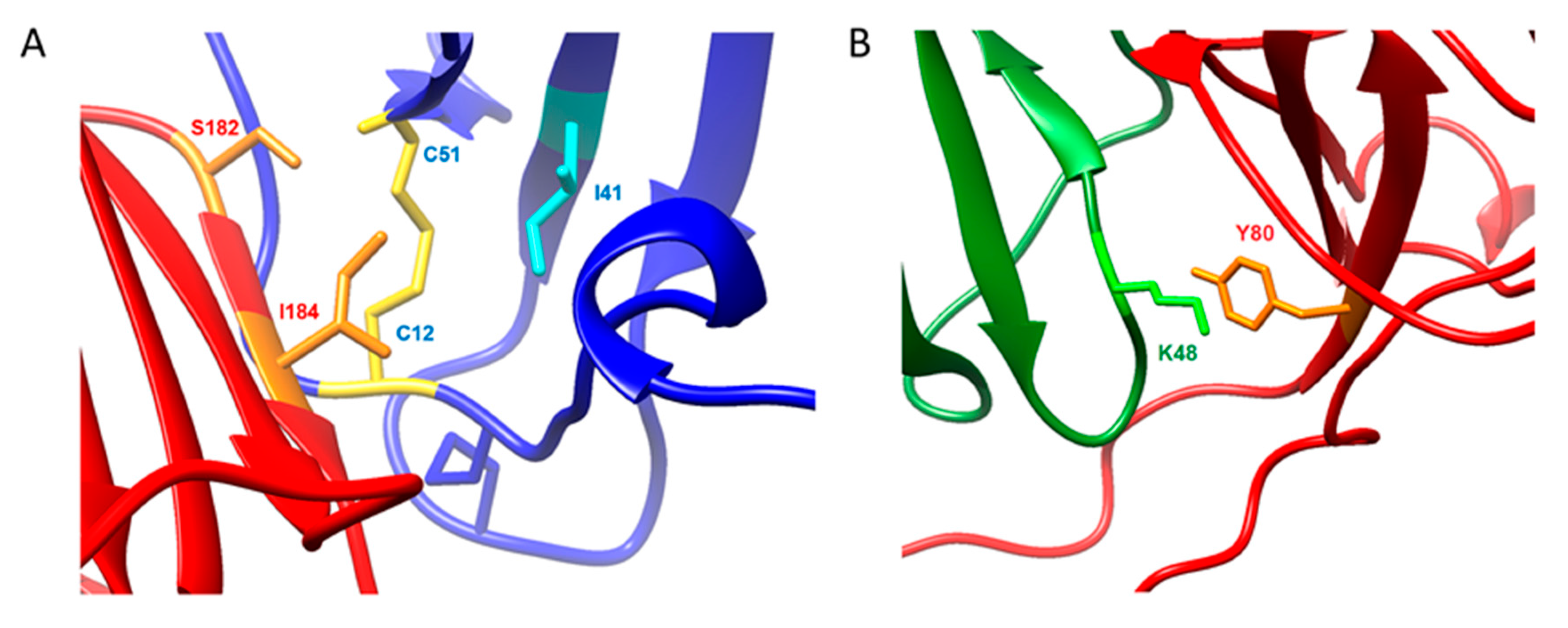
2.4. Molecular Dynamics Simulations on vCCI:vMIP-II
3. Discussion
4. Materials and Methods
4.1. Protein Purification
4.1.1. Purifying vCCI from E. coli
4.1.2. Expression and Purification of vMIP-II
4.2. Nuclear Magnetic Resonance (NMR) Spectroscopy
4.3. Isothermal Titration Calorimetry (ITC)
4.4. Fluorescence Anisotropy
4.5. Molecular Dynamics
4.6. Figure Preparation
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| vCCI | Viral CC Chemokine Inhibitor; also called 35K or vCCI/35K |
| vMIP-II | Viral macrophage inflammatory protein II |
| E. coli | Escherichia coli |
| NMR | Nuclear magnetic resonance |
| HSQC | Heteronuclear single quantum coherence |
| NaOP | Sodium phosphate |
| HHV-8 | Human herpesvirus 8, Kaposi’s sarcoma-associated herpesvirus |
| MIP-1β | Macrophage inflammatory protein β, also known as CCL4 |
| CCL4 | CC chemokine ligand 4, also known as MIP-1β |
| IPTG | Isopropyl β-D-1-thiogalactopyranoside |
| DSS | 4,4-dimethyl-4-silapentane-1-sulfonic acid |
| β-ME | β-mercaptoethanol |
| RMSD | Root mean squared deviation |
| RMSF | Root mean squared fluctuation (for specific residues) |
| MD | Molecular dynamics |
References
- Graham, K.A.; Lalani, A.S.; Macen, J.L.; Ness, T.L.; Barry, M.; Liu, L.-Y.; Lucas, A.; Clark-Lewis, I.; Moyer, R.W.; Mcfadden, G. The T1/35kDa family of poxvirus-secreted proteins bind chemokines and modulate leukocyte influx into virus-infected tissues. Virology 1997, 229, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef] [PubMed]
- Heidarieh, H.; Hernáez, B.; Alcamí, A. Immune modulation by virus-encoded secreted chemokine binding proteins. Virus Res. 2015, 209, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Lalani, A.S.; Graham, K.; Mossman, K.; Rajarathnam, K.; Clark-Lewis, I.; Kelvin, D.; McFadden, G. The purified myxoma virus γ interferon receptor homolog M-T7 interacts with the heparin-binding domains of chemokines. J. Virol. 1997, 71, 4356–4363. [Google Scholar] [PubMed]
- Hughes, D.J.; Kipar, A.; Leeming, G.H.; Bennet, E.; Howarth, D.; Commerson, J.A.; Papoula-Pereira, R.; Flanagan, B.F.; Sample, J.T.; Stewart, J.P. Chemokine Binding Protein M3 of Murine Gammaherpesvirus 68 Modulates the Host Response to Infection in a Natural Host. PLoS Pathog. 2011, 7, e1001321. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.M.; Nelson, C.A.; Van Berkel, V.; Lau, E.K.; Studts, J.M.; Brett, T.J.; Speck, S.H.; Handel, T.M.; Virgin, H.W. Structural basis of chemokine sequestration by a herpesvirus decoy receptor. Cell 2002, 111, 343–356. [Google Scholar] [CrossRef]
- Webb, L.M.C.; Smith, V.P.; Alcami, A. The gammaherpesvirus chemokine binding protein can inhibit the interaction of chemokines with glycosaminoglycans. FASEB J. 2004, 18, 571–573. [Google Scholar] [CrossRef] [PubMed]
- Alexander-Brett, J.M.; Fremont, D.H. Dual GPCR and GAG mimicry by the M3 chemokine decoy receptor. J. Exp. Med. 2007, 204, 3157–3172. [Google Scholar] [CrossRef] [PubMed]
- Alejo, A.; Ruiz-Argüello, M.B.; Ho, Y.; Smith, V.P.; Saraiva, M.; Alcami, A. A chemokine-binding domain in the tumor necrosis factor receptor from variola (smallpox) virus. Proc. Natl. Acad. Sci. USA 2006, 103, 5995–6000. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.M.; Dairaghi, D.J.; Deitz, M.; Tsang, M.; Schall, T.J. Comprehensive mapping of poxvirus vCCI chemokine-binding protein. Expanded range of ligand interactions and unusual dissociation kinetics. J. Biol. Chem. 2002, 277, 2785–2789. [Google Scholar] [CrossRef] [PubMed]
- Kuo, N.-W.; Gao, Y.-G.; Schill, M.S.; Isern, N.; Dupureur, C.M.; LiWang, P.J. Structural insights into the interaction between a potent anti-inflammatory protein, viral CC chemokine inhibitor (vCCI), and the human CC chemokine, eotaxin-1. J. Biol. Chem. 2014, 289, 6592–6603. [Google Scholar] [CrossRef] [PubMed]
- Carfí, A.; Smith, C.A.; Smolak, P.J.; McGrew, J.; Wiley, D.C. Structure of a soluble secreted chemokine inhibitor vCCI (p35) from cowpox virus. Proc. Natl. Acad. Sci. USA 1999, 96, 12379–12383. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; DeRider, M.; McCornack, M.A.; Jao, S.C.; Isern, N.; Ness, T.; Moyer, R.; LiWang, P.J. Solution structure of the complex between poxvirus-encoded CC chemokine inhibitor vCCI and human MIP-1β. Proc. Natl. Acad. Sci. USA 2006, 103, 13985–13990. [Google Scholar] [CrossRef] [PubMed]
- Arnold, P.L.; Fremont, D.H. Structural Determinants of chemokine binding by an ectromelia virus-encoded decoy receptor. J. Virol. 2006, 80, 7439–7449. [Google Scholar] [CrossRef] [PubMed]
- White, G.E.; Mcneill, E.; Christou, I.; Channon, K.M.; Greaves, D.R. Site-directed mutagenesis of the CC chemokine binding protein 35K-Fc reveals residues essential for activity and mutations that increase the potency of CC chemokine blockade. Mol. Pharmacol. 2011, 80, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Beck, C.G.; Studer, C.; Zuber, J.F.; Demange, B.J.; Manning, U.; Urfer, R. The viral CC chemokine-binding protein vCCI inhibits monocyte chemoattractant protein-1 activity by masking its CCR2B-binding site. J. Biol. Chem. 2001, 276, 43270–43276. [Google Scholar] [CrossRef] [PubMed]
- Seet, B.T.; Singh, R.; Paavola, C.; Lau, E.K.; Handel, T.M.; McFadden, G. Molecular determinants for CC-chemokine recognition by a poxvirus CC-chemokine inhibitor. Proc. Natl. Acad. Sci. USA 2001, 98, 9008–9013. [Google Scholar] [CrossRef] [PubMed]
- Kledal, T.N.; Rosenkilde, M.M.; Coulin, F.; Simmons, G.; Johnsen, A.H.; Alouani, S.; Power, C.A.; Luttichau, H.R.; Gerstoft, J.; Clapham, P.R.; et al. A broad-spectrum chemokine antagonist encoded by Kaposi’s sarcoma-associated herpesvirus. Science 1997, 277, 1656–1659. [Google Scholar] [CrossRef] [PubMed]
- Sozzani, S.; Luini, W.; Bianchi, G.; Allavena, P.; Wells, T.N.C.; Napolitano, M.; Bernardini, G.; Vecchi, A.; D’Ambrosio, D.; Mazzeo, D.; et al. The viral chemokine macrophage inflammatory protein-II is a selective Th2 chemoattractant. Blood 1998, 92, 4036–4039. [Google Scholar] [PubMed]
- Weber, K.S.; Gröne, H.J.; Röcken, M.; Klier, C.; Gu, S.; Wank, R.; Proudfoot, A.E.; Nelson, P.J.; Weber, C. Selective recruitment of Th2-type cells and evasion from a cytotoxic immune response mediated by viral macrophage inhibitory protein-II. Eur. J. Immunol. 2001, 31, 2458–2466. [Google Scholar] [CrossRef]
- Liwang, A.C.; Wang, Z.X.; Sun, Y.; Peiper, S.C.; Liwang, P.J. The solution structure of the anti-HIV chemokine vMIP-II. Protein Sci. 1999, 8, 2270–2280. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Liwang, P.J. Characterization of the interactions of vMIP-II, and a dimeric variant of vMIP-II, with glycosaminoglycans. Biochemistry 2010, 49, 7012–7022. [Google Scholar] [CrossRef] [PubMed]
- Takami, S.; Minami, M.; Nagata, I.; Namura, S.; Satoh, M. Chemokine receptor antagonist peptide, viral MIP-II, protects the brain against focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 2001, 1430–1435. [Google Scholar] [CrossRef] [PubMed]
- Ghirnikar, R.S.; Lee, Y.L.; Eng, L.F. Chemokine antagonist infusion promotes axonal sparing after spinal cord contusion injury in rat. J. Neurosci. Res. 2001, 64, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Bedke, J.; Stojanovic, T.; Kiss, E.; Behnes, C.-L.; Proudfoot, A.E.; Gröne, H.-J. Viral macrophage inflammatory protein-II improves acute rejection in allogeneic rat kidney transplants. World J. Urol. 2010, 28, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L. Structural Study of the Interaction between Poxvirus-Encoded CC Chemokine Inhibitor VCCI and Human MIP-1β. Ph.D. Thesis, Texas A&M University, College Station, TX, USA, May 2008. [Google Scholar]
- DeRider, M.L.; Zhang, L.; LiWang, P.J. Resonance assignments and secondary structure of vCCI, a 26 kDa CC chemokine inhibitor from rabbitpox virus. J. Biomol. NMR 2006, 36, 22. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- PDBePISA (Proteins, Interfaces, Structures and Assemblies). Available online: http://www.ebi.ac.uk/pdbe/pisa/ (accessed on 12 July 2017).
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E. Crystal Contacts as Nature’s Docking Solutions. J. Comput. Chem. 2009, 28, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.C.; Feldmann, M. Anti-TNF biologic agents: Still the therapy of choice for rheumatoid arthritis. Nat. Rev. Rheumatol. 2009, 5, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Manickam, D.S.; Brynskikh, A.; Kabanov, A.V. Agile delivery of protein therapeutics to CNS. J. Control. Release 2014, 190, 637–663. [Google Scholar] [CrossRef] [PubMed]
- Boomker, J.M.; De Leij, L.F.M.H.; The, T.H.; Harmsen, M.C. Viral chemokine-modulatory proteins: Tools and targets. Cytokine Growth Factor Rev. 2005, 16, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Alcami, A.; Viejo-Borbolla, A. Identification and characterization of virus-encoded chemokine binding proteins. Methods Enzymol. 2009, 460, 173–191. [Google Scholar] [PubMed]
- Adcock, S.A.; McCammon, J.A. Molecular dynamics: Survey of methods for simulating the activity of proteins. Chem. Rev. 2006, 106, 1589–1615. [Google Scholar] [CrossRef] [PubMed]
- Shaw, D.E.; Maragakis, P.; Lindorff-Larsen, K.; Piana, S.; Shan, Y.; Wriggers, W. Atomic-level characterization of the structural dynamics of proteins. Science 2010, 330, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Dill, K.A.; MacCallum, J.L. The protein-folding problem, 50 years on. Science 2012, 338, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-G.; Kuo, N.-W.; Tseng, R.; LiWang, A. Flexibility of the C-terminal, or CII, ring of KaiC governs the rhythm of the circadian clock of cyanobacteria. Proc. Natl. Acad. Sci. USA 2011, 108, 14431–14436. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Bigam, C.G.; Yao, J.; Abildgaard, F.; Dyson, H.J.; Oldfield, E.; Markley, J.L.; Sykes, B.D. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomolec. NMR 1995, 6, 135–140. [Google Scholar] [CrossRef]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Garrett, D.S.; Powers, R.; Gronenborn, A.M.; Clore, G.M. A common sense approach to peak picking in two-, three-, and four-dimensional spectra using automatic computer analysis of contour diagrams. J. Magn. Reson. 1991, 95, 214–220. [Google Scholar] [CrossRef]
- Garrett, D.S.; Seok, Y.J.; Peterkofsky, A.; Clore, G.M.; Gronenborn, A.M. Identification by NMR of the binding surface for the histidine- containing phosphocarrier protein HPr on the N-terminal domain of enzyme I of the Escherichia coli phosphotransferase system. Biochemistry 1997, 36, 4393–4398. [Google Scholar] [CrossRef] [PubMed]
- Conlan, L.H.; Dupureur, C.M. Dissecting the metal ion dependence of DNA binding by PvuII endonuclease. Biochemistry 2002, 41, 1335–1342. [Google Scholar] [CrossRef] [PubMed]
- Reid, S.L.; Parry, D.; Liu, H.H.; Connolly, B.A. Binding and recognition of GATATC target sequences by the EcoRV restriction endonuclease: A study using fluorescent oligonucleotides and fluorescence polarization. Biochemistry 2001, 40, 2484–2494. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Pall, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Pronk, S.; Pall, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van Der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load balanced, and scalable molecular simulations. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinforma 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Shift Perturbation | Definition | vCCI Chemical Shift | vMIP-II Chemical Shift |
|---|---|---|---|
| 0 | No confirmable change | No peaks visible | No peaks visible |
| 1 | Less than or equal to average | Δδobs ≤ 0.045 | Δδobs ≤ 0.100 |
| 2 | Greater than average, up to one standard deviation above average | 0.045 < Δδobs ≤ 0.086 | 0.100 < Δδobs ≤ 0.178 |
| 3 | Greater than one standard deviation above average | Δδobs ≥ 0.086 | Δδobs ≥ 0.178 |
| Complex | vCCI Buried Surface Area (Å2) | Chemokine Buried Surface Area (Å2) |
|---|---|---|
| vCCI:vMIP-II | 1473 | 1528 |
| vCCI:MIP-1β wild type | 1355 | 1392 |
| vCCI:MIP-1β K45A/R46A/K48A (variant used in 2FFK structure determination) | 1020 | 1060 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, A.F.; Kuo, N.-W.; Showalter, L.J.; Ramos, R.; Dupureur, C.M.; Colvin, M.E.; LiWang, P.J. Biophysical and Computational Studies of the vCCI:vMIP-II Complex. Int. J. Mol. Sci. 2017, 18, 1778. https://doi.org/10.3390/ijms18081778
Nguyen AF, Kuo N-W, Showalter LJ, Ramos R, Dupureur CM, Colvin ME, LiWang PJ. Biophysical and Computational Studies of the vCCI:vMIP-II Complex. International Journal of Molecular Sciences. 2017; 18(8):1778. https://doi.org/10.3390/ijms18081778
Chicago/Turabian StyleNguyen, Anna F., Nai-Wei Kuo, Laura J. Showalter, Ricardo Ramos, Cynthia M. Dupureur, Michael E. Colvin, and Patricia J. LiWang. 2017. "Biophysical and Computational Studies of the vCCI:vMIP-II Complex" International Journal of Molecular Sciences 18, no. 8: 1778. https://doi.org/10.3390/ijms18081778