Ac2-26, an Annexin A1 Peptide, Attenuates Ischemia-Reperfusion-Induced Acute Lung Injury

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
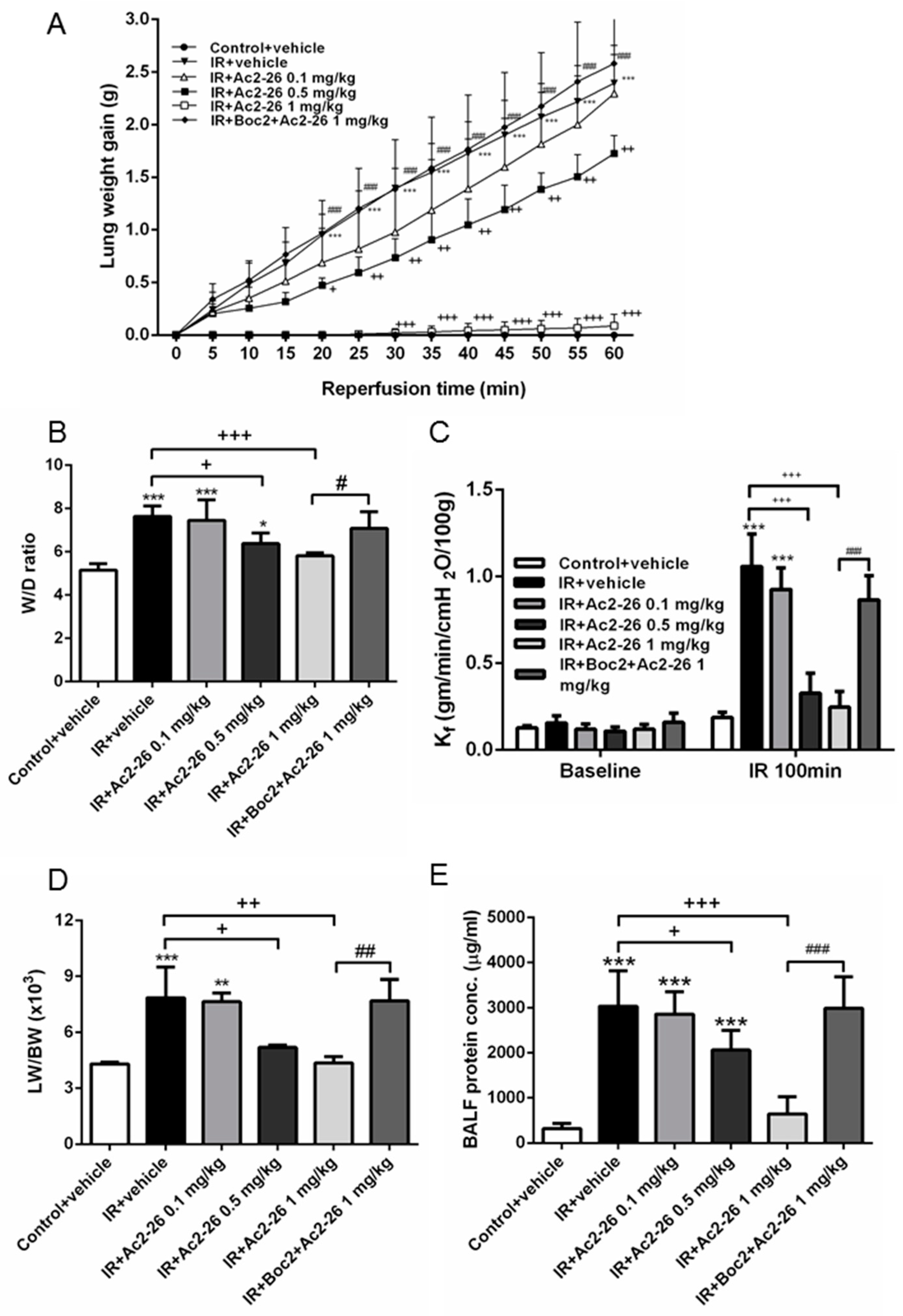
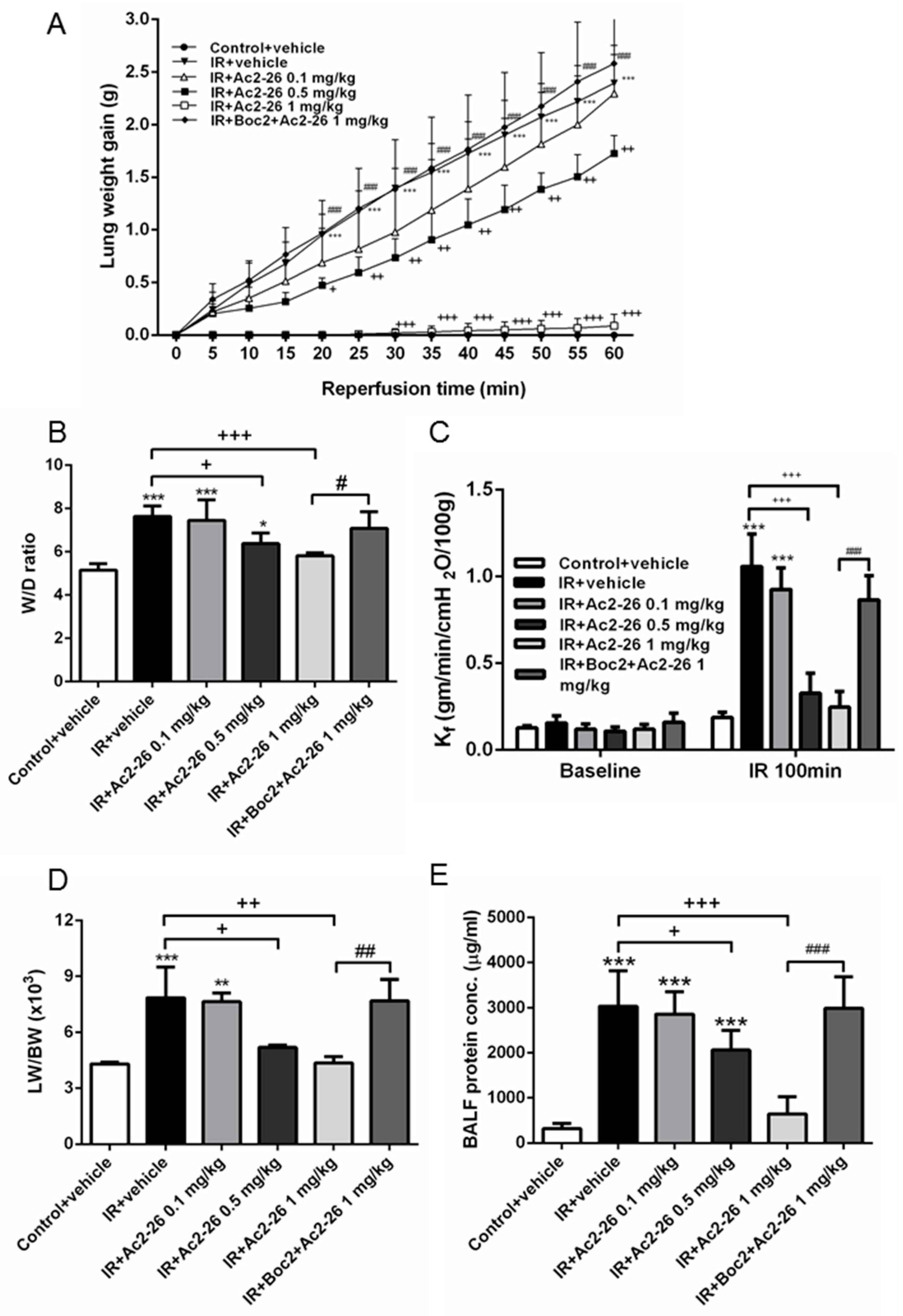
2.1. Effect of Ac-AMVSEFLKQAWFIENEEQEYVQTVK (Ac2-26) on Indices of Lung Edema
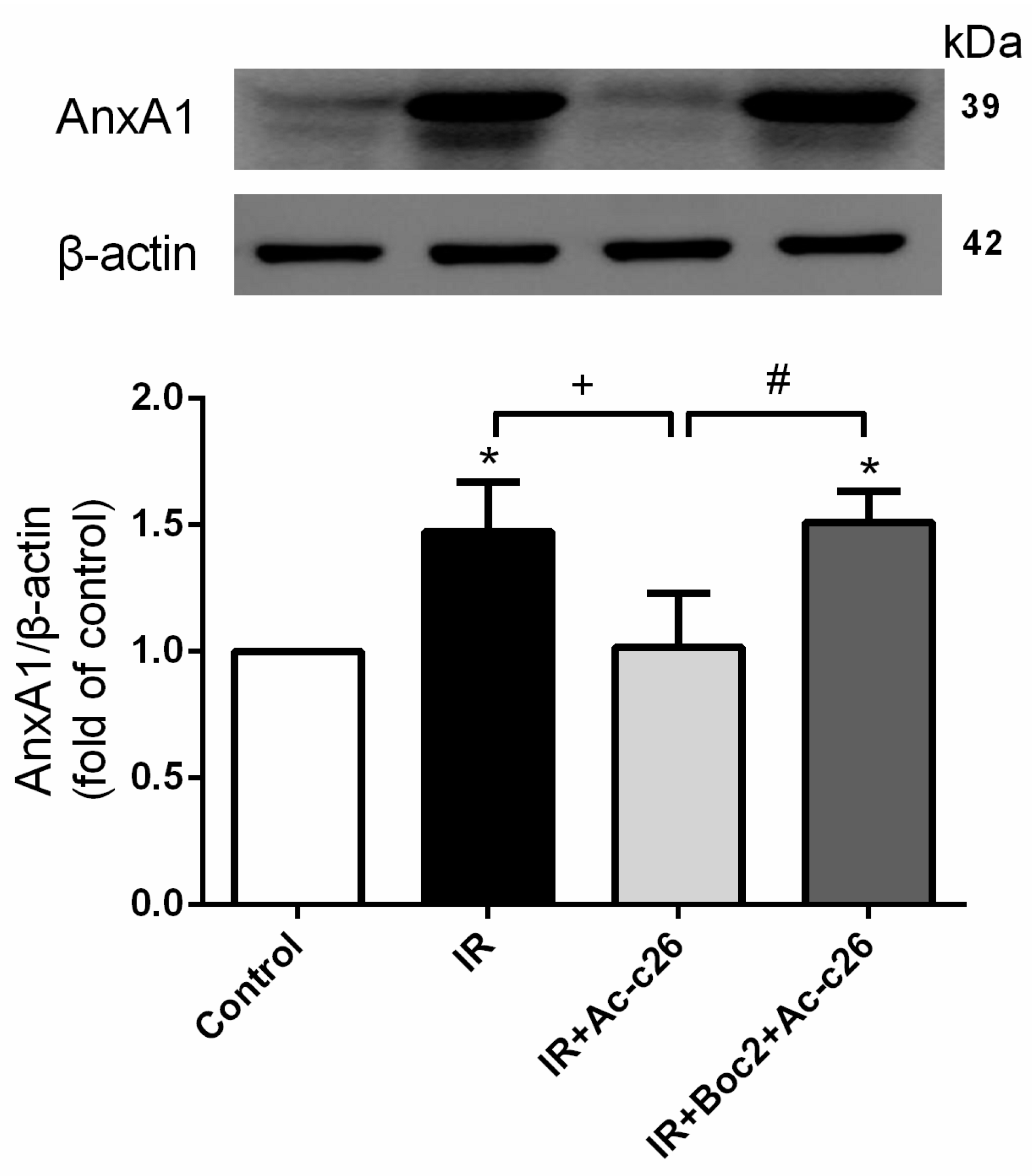
2.2. Effect of Ac2-26 on AnxA1 Protein Expression in Lung Tissue
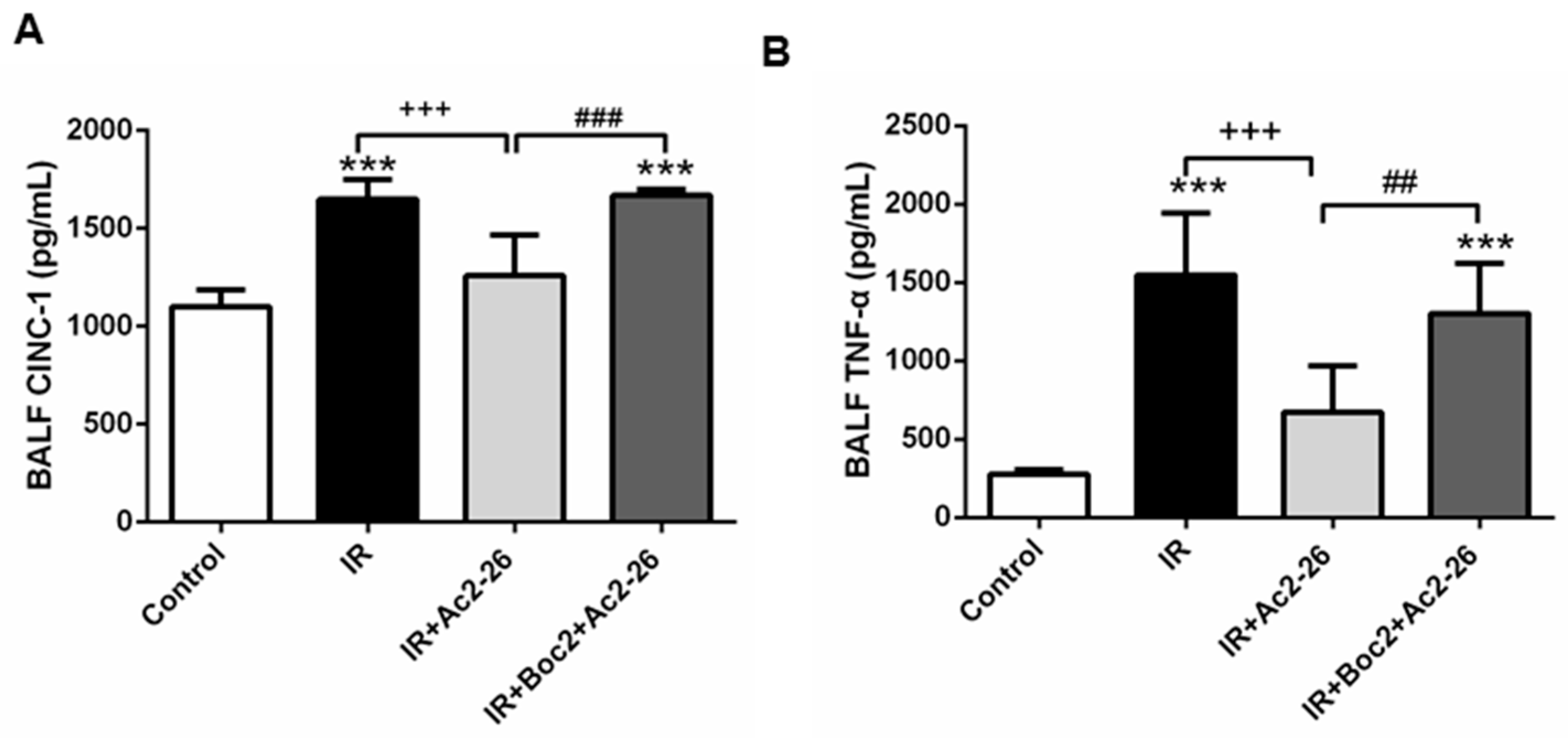
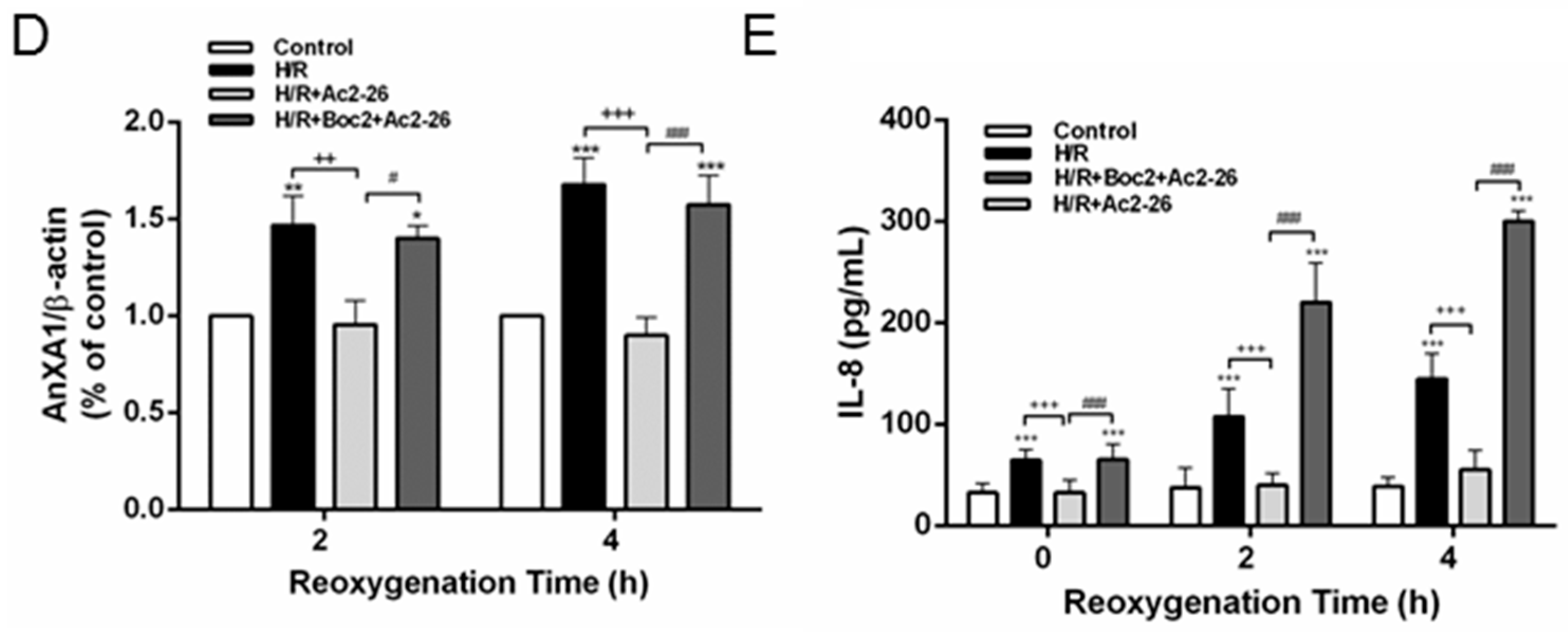
2.3. Effect of Ac2-26 on Cytokine-Induced Neutrophil Chemoattractant-1 (CINC-1) and TNF-α Concentrations in Bronchoalveolar Lavage Fluid (BALF)
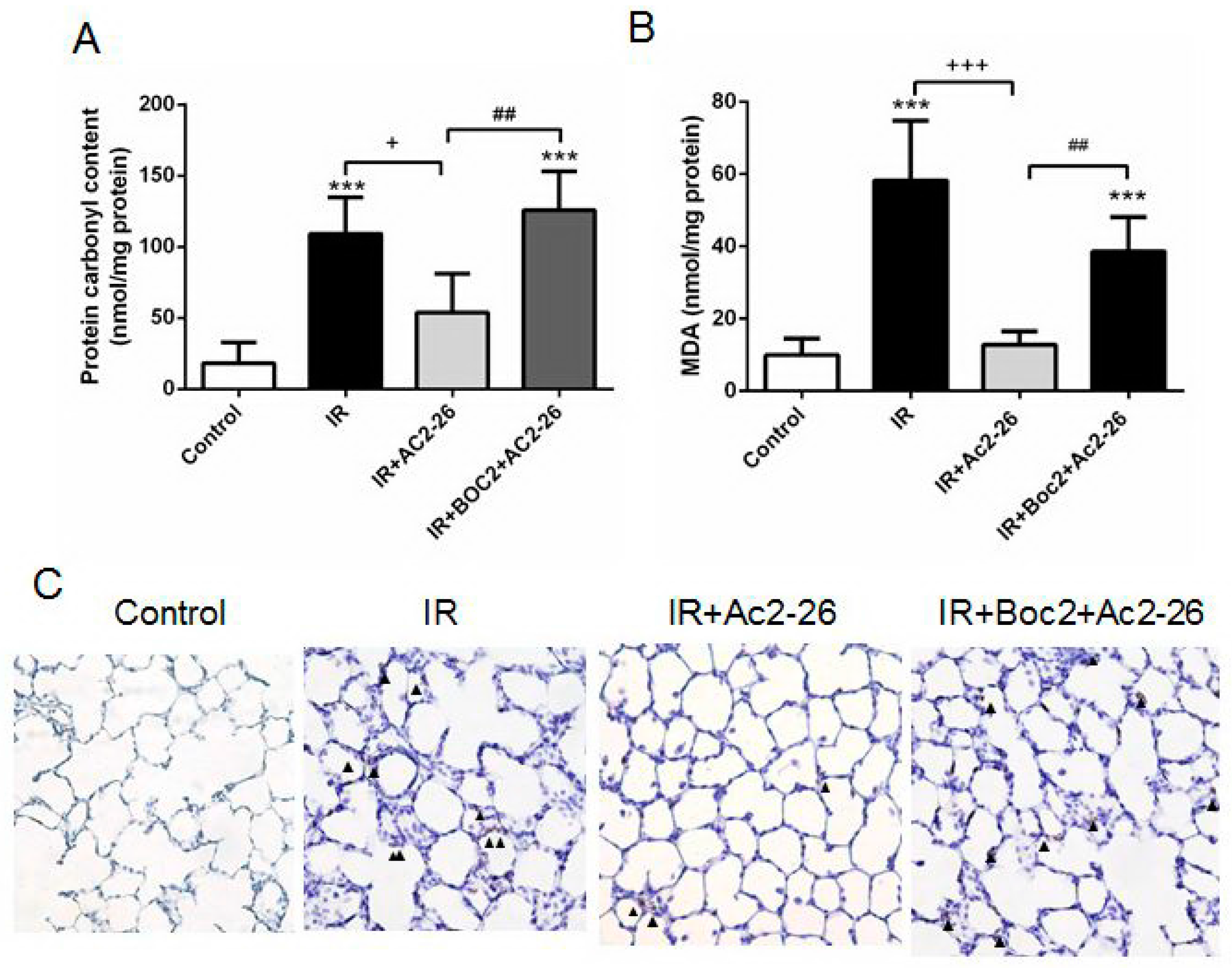
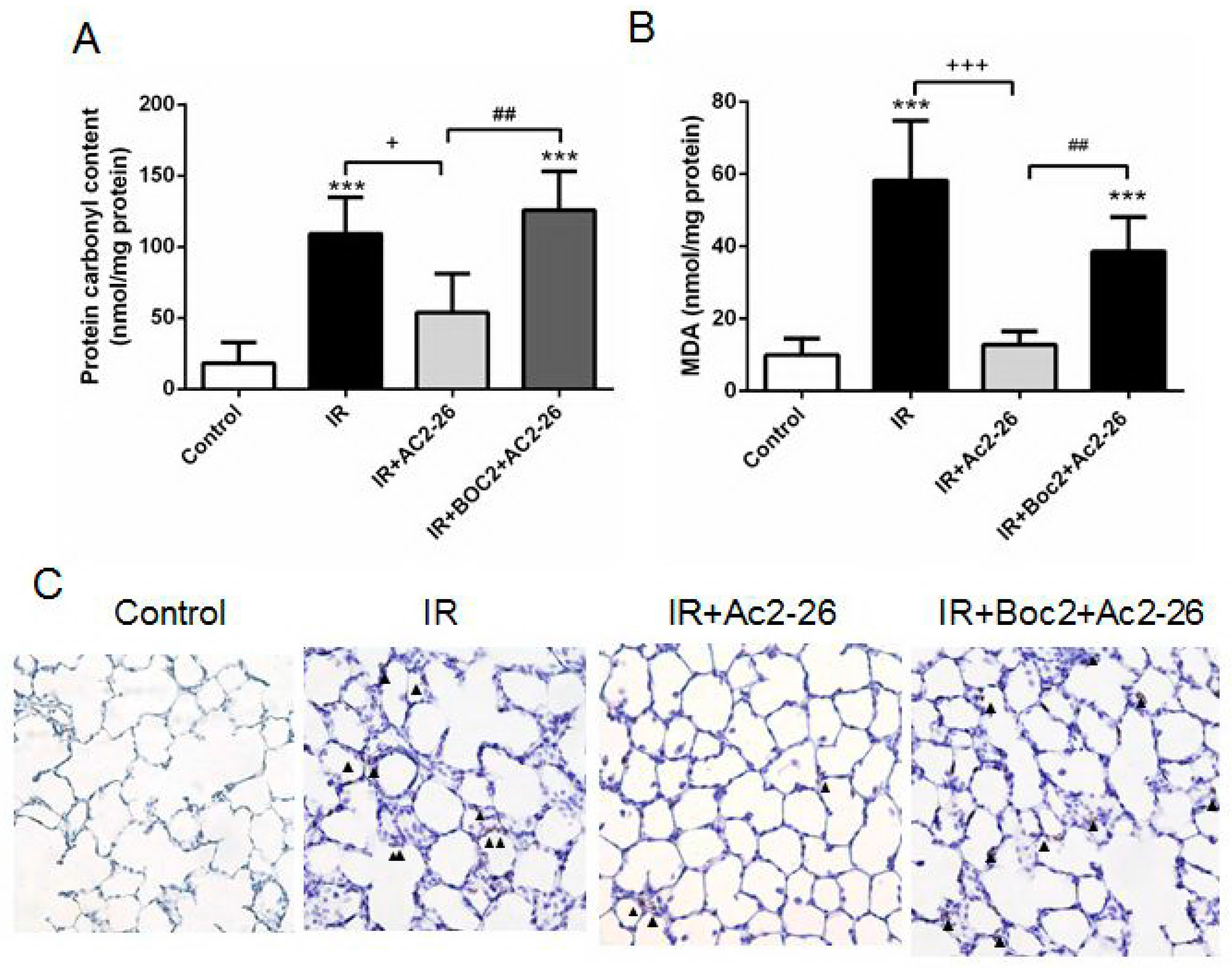
2.4. Effect of Ac2-26 on Carbonyl Content, Malondialdehyde (MDA) Level, and Myeloperoxidase (MPO)-Positive Cells in Lung Tissue
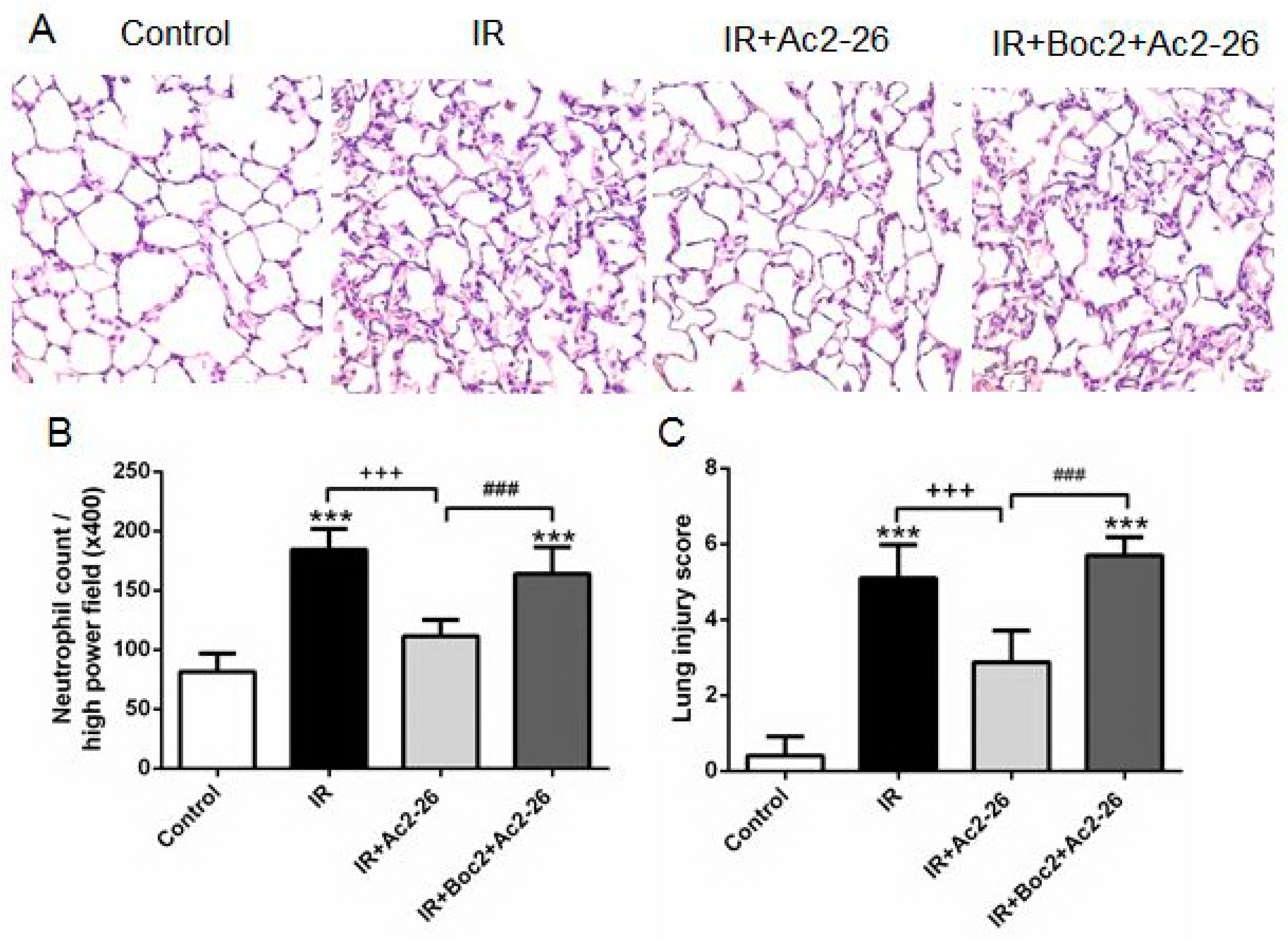
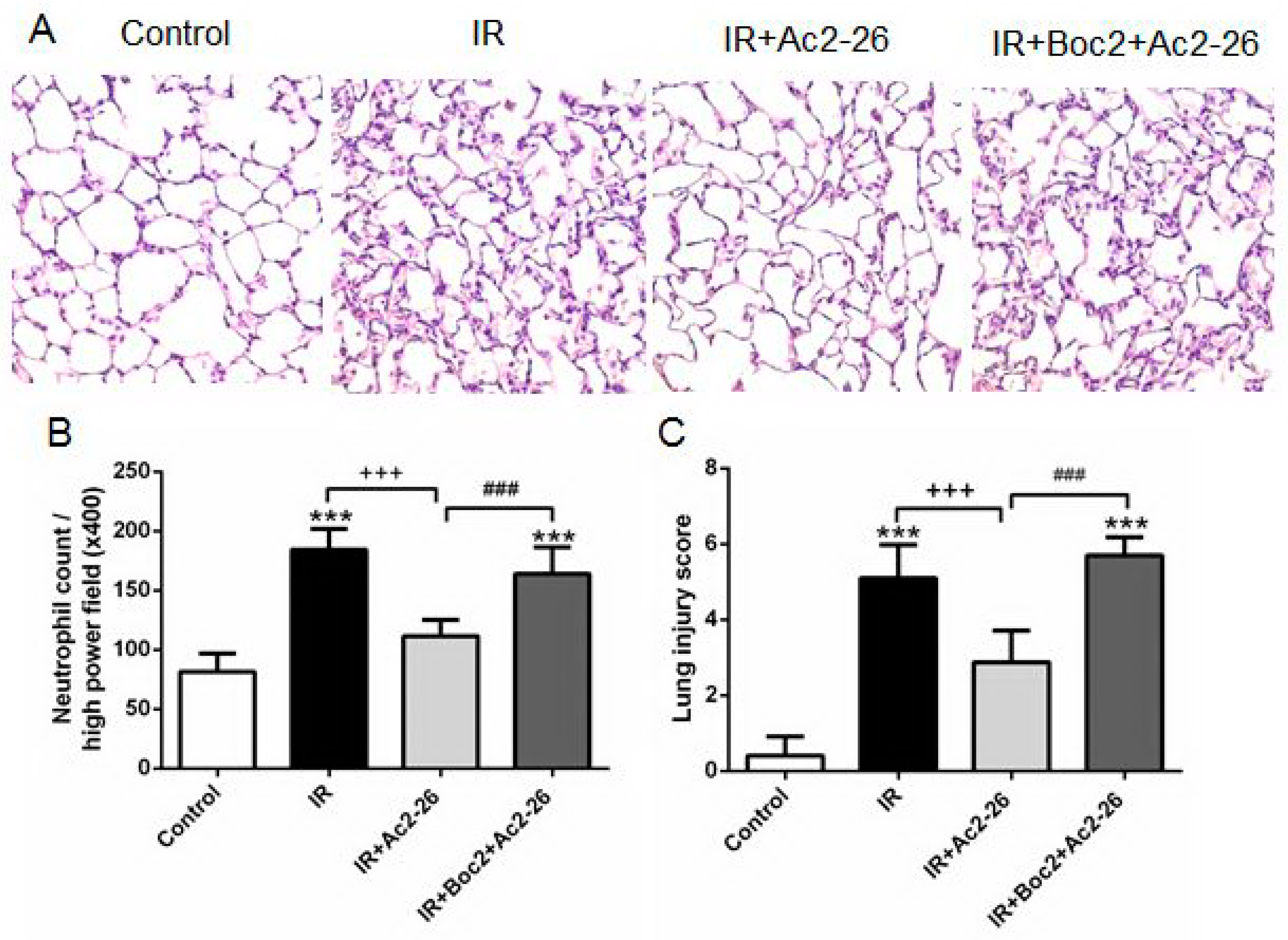
2.5. Effect of Ac2-26 on Lung Pathology
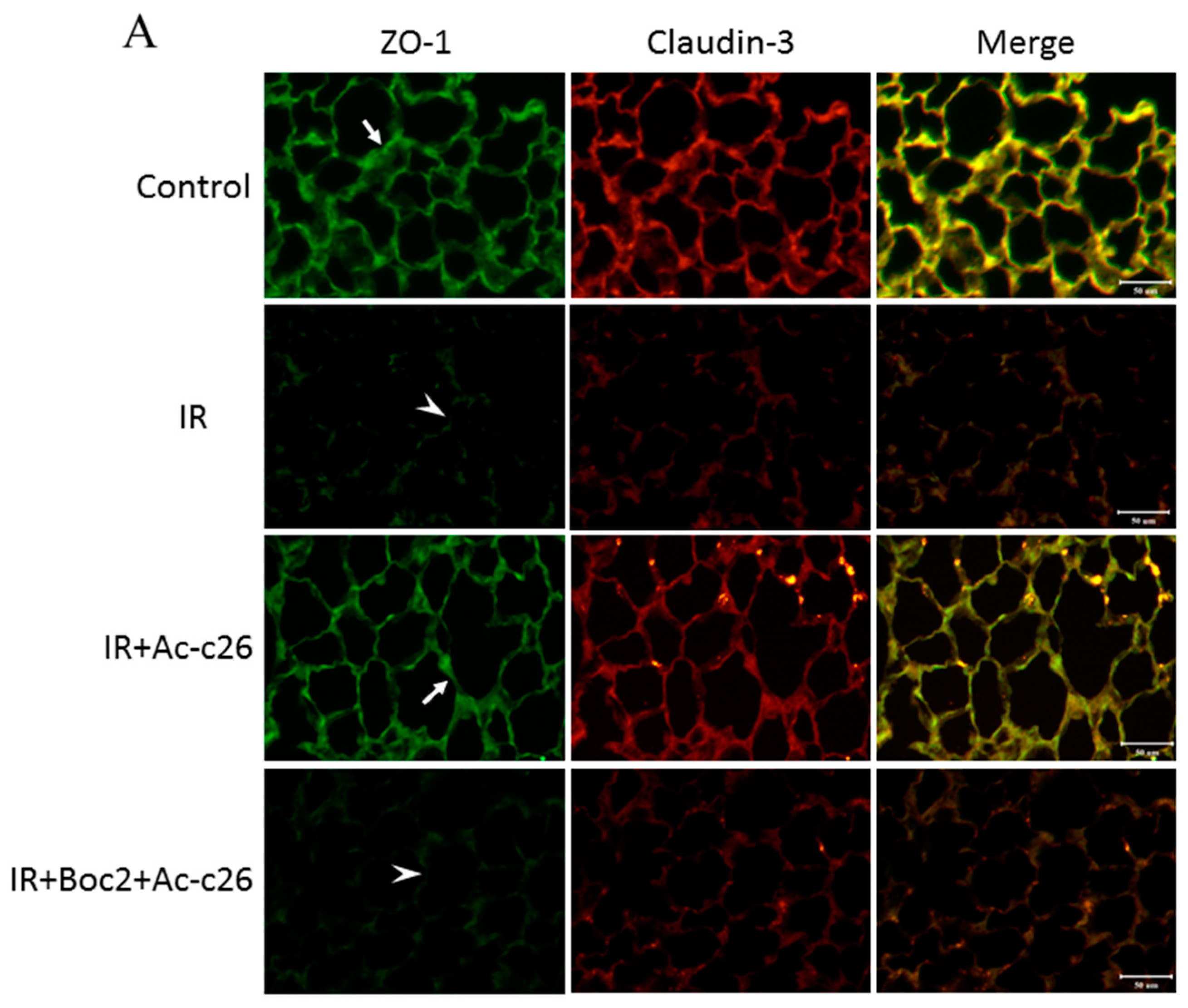
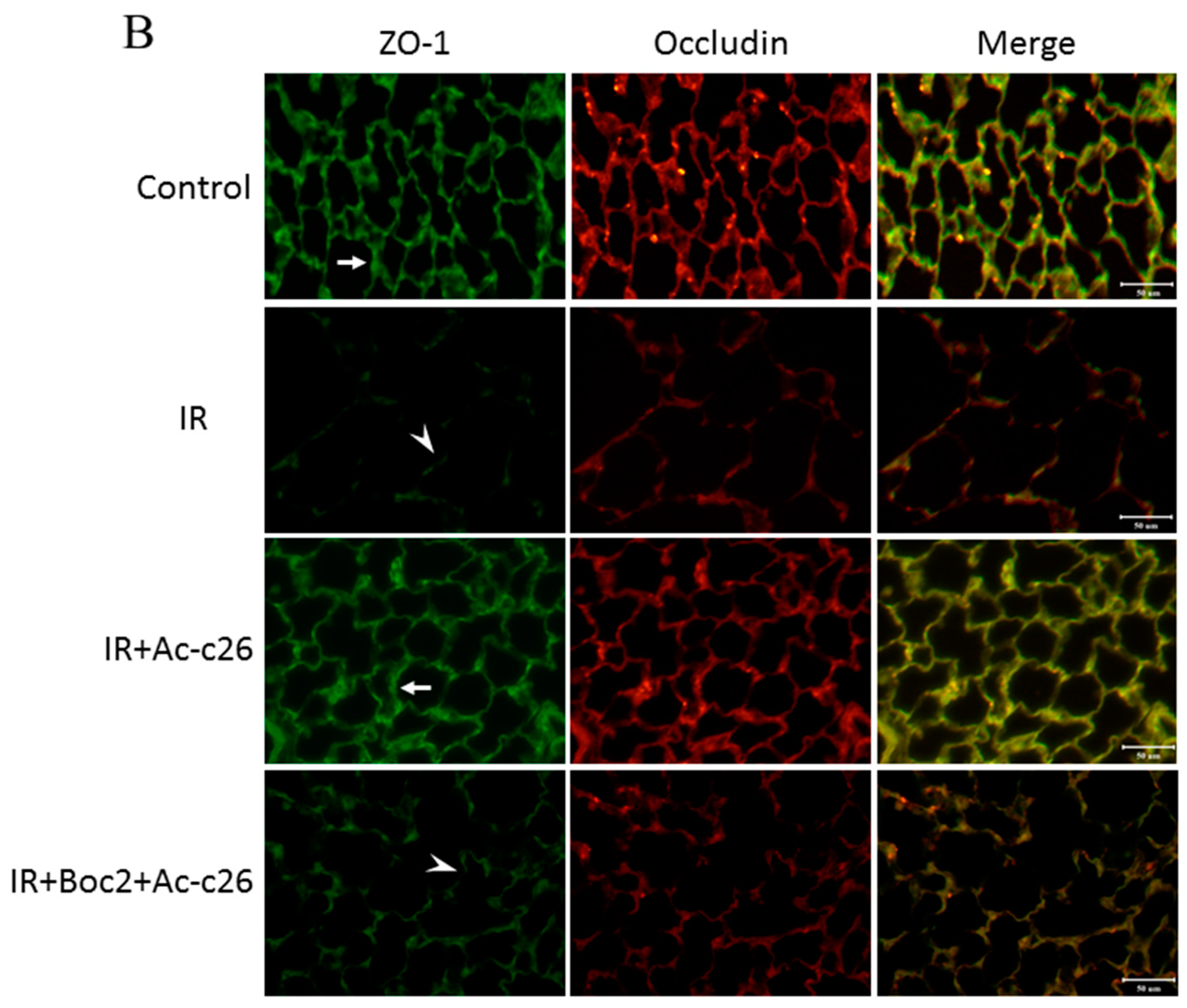
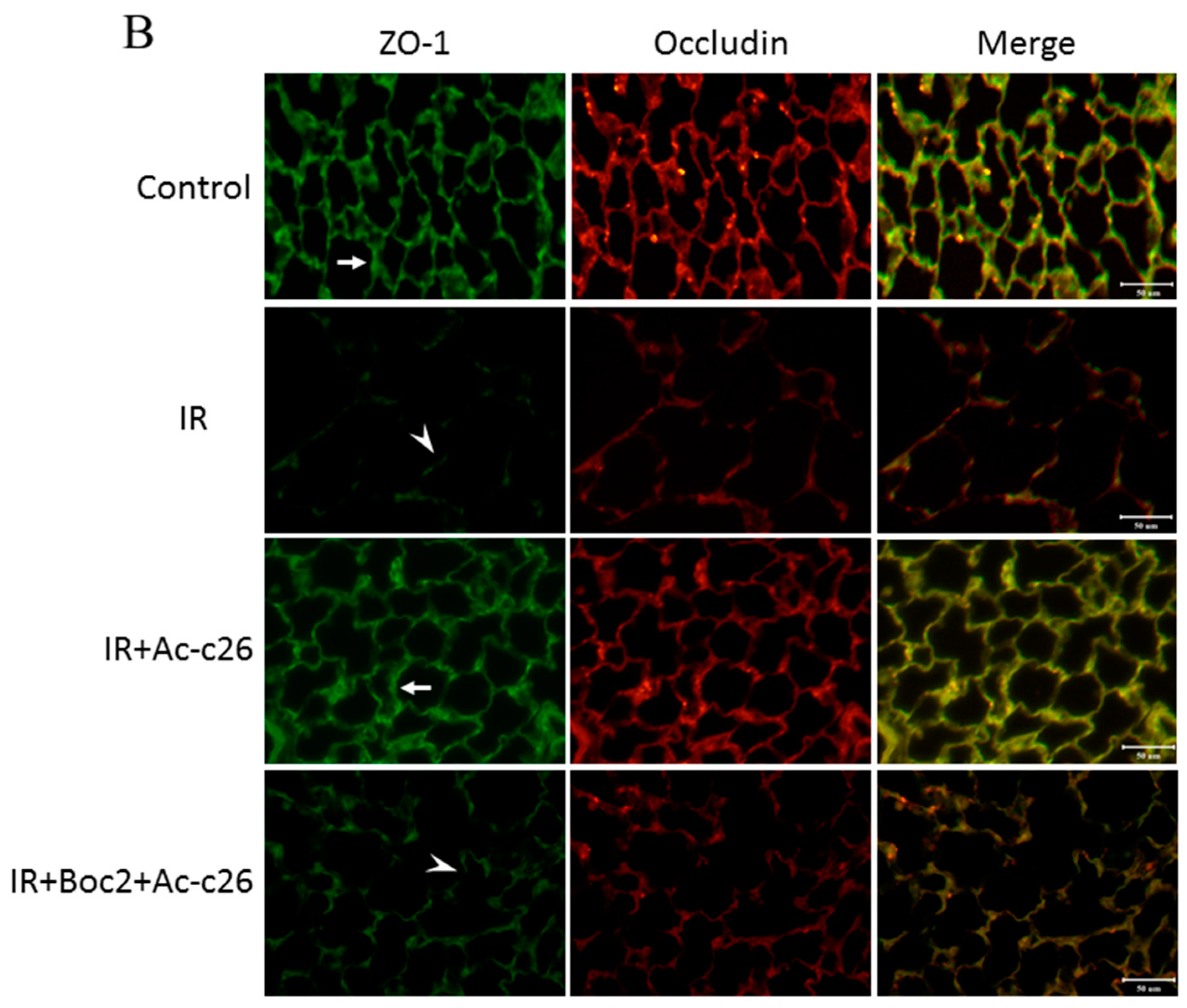
2.6. Effect of Ac2-26 on Claudin-3, Occludin, and Zonula Occludens-1 (ZO-1) Protein Expression in Lung Tissue
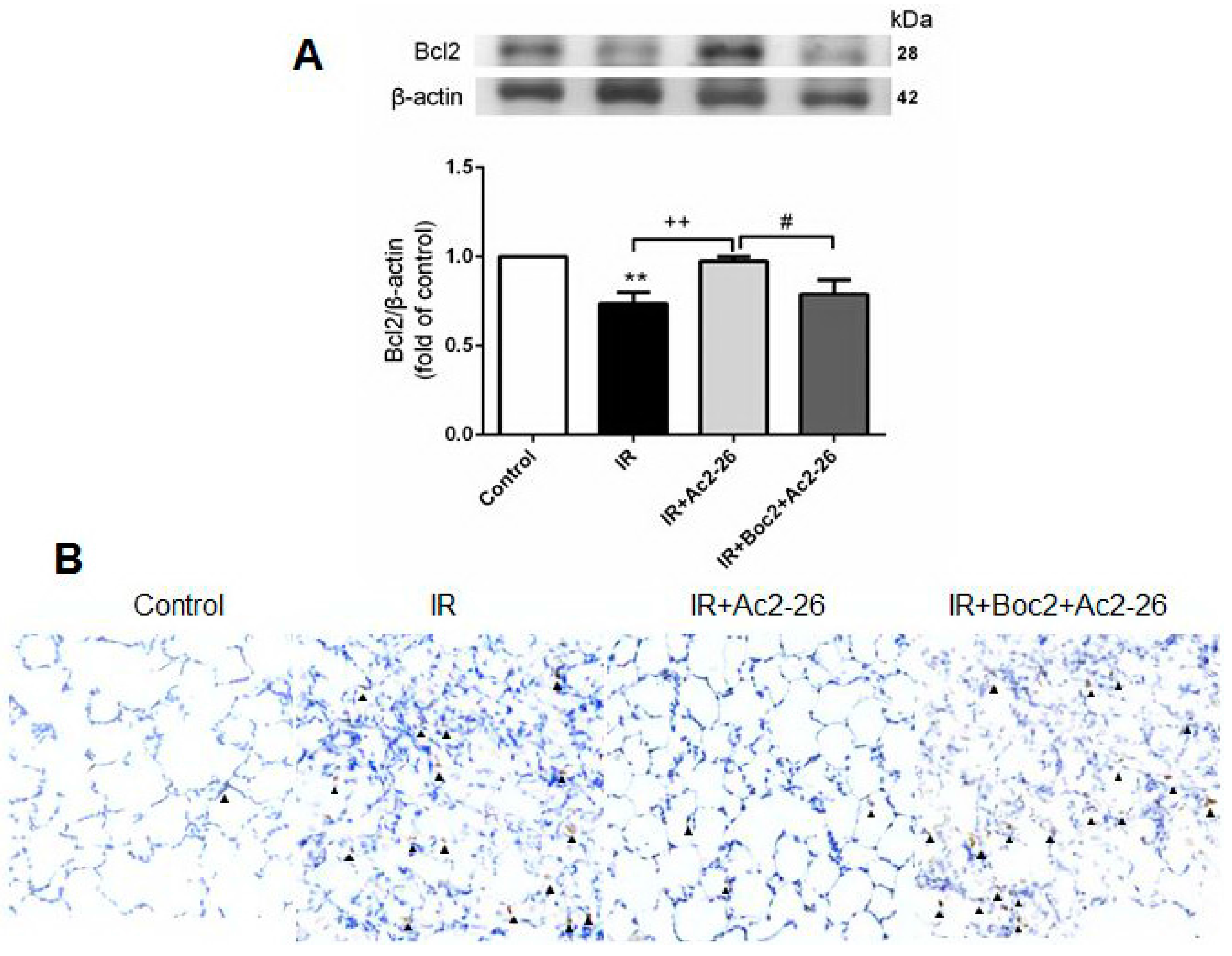
2.7. Effects of Ac2-26 on B-Cell Lymphoma (Bcl)-2 and Caspase-3 Protein Expression in Lung Tissue
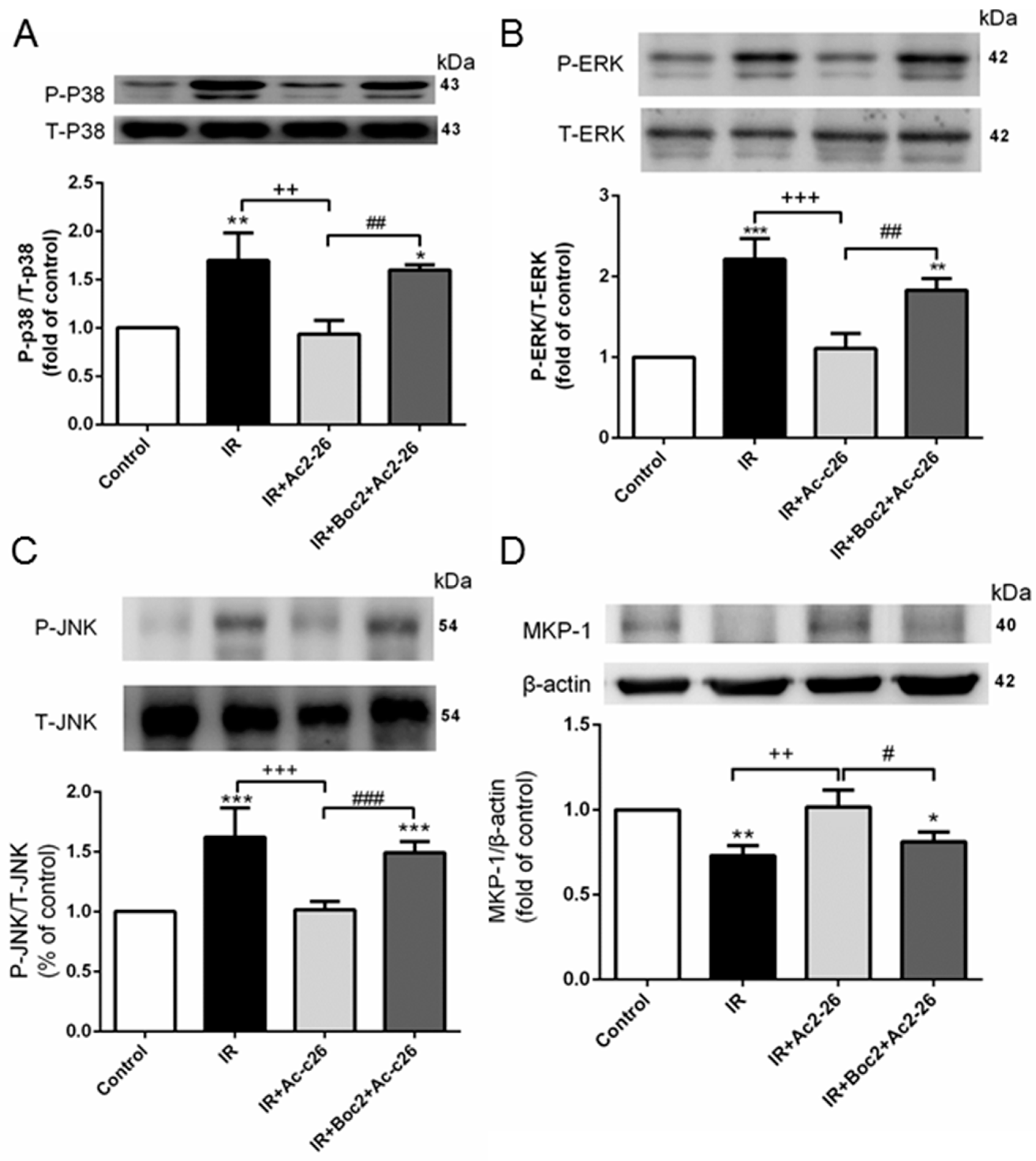
2.8. Effect of Ac2-26 on the Mitogen-Activated Protein Kinase (MAPK) Signaling Pathway and Mitogen-Activated Protein Kinase Phoshphotases-1 (MKP-1) Induction in Lung Tissue
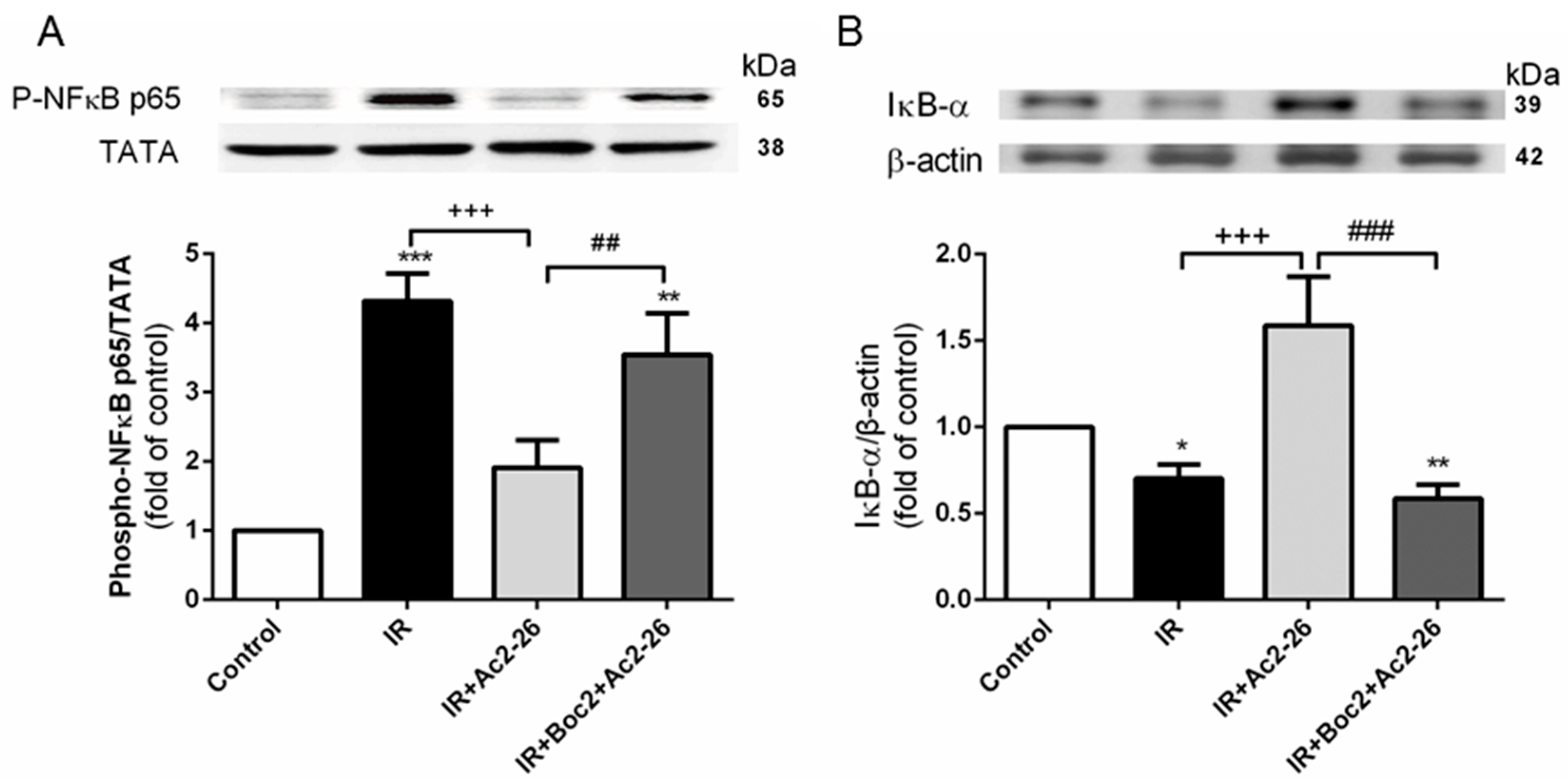
2.9. Effect of Ac2-26 on the Nuclear Factor (NF)-κB Signaling Pathway
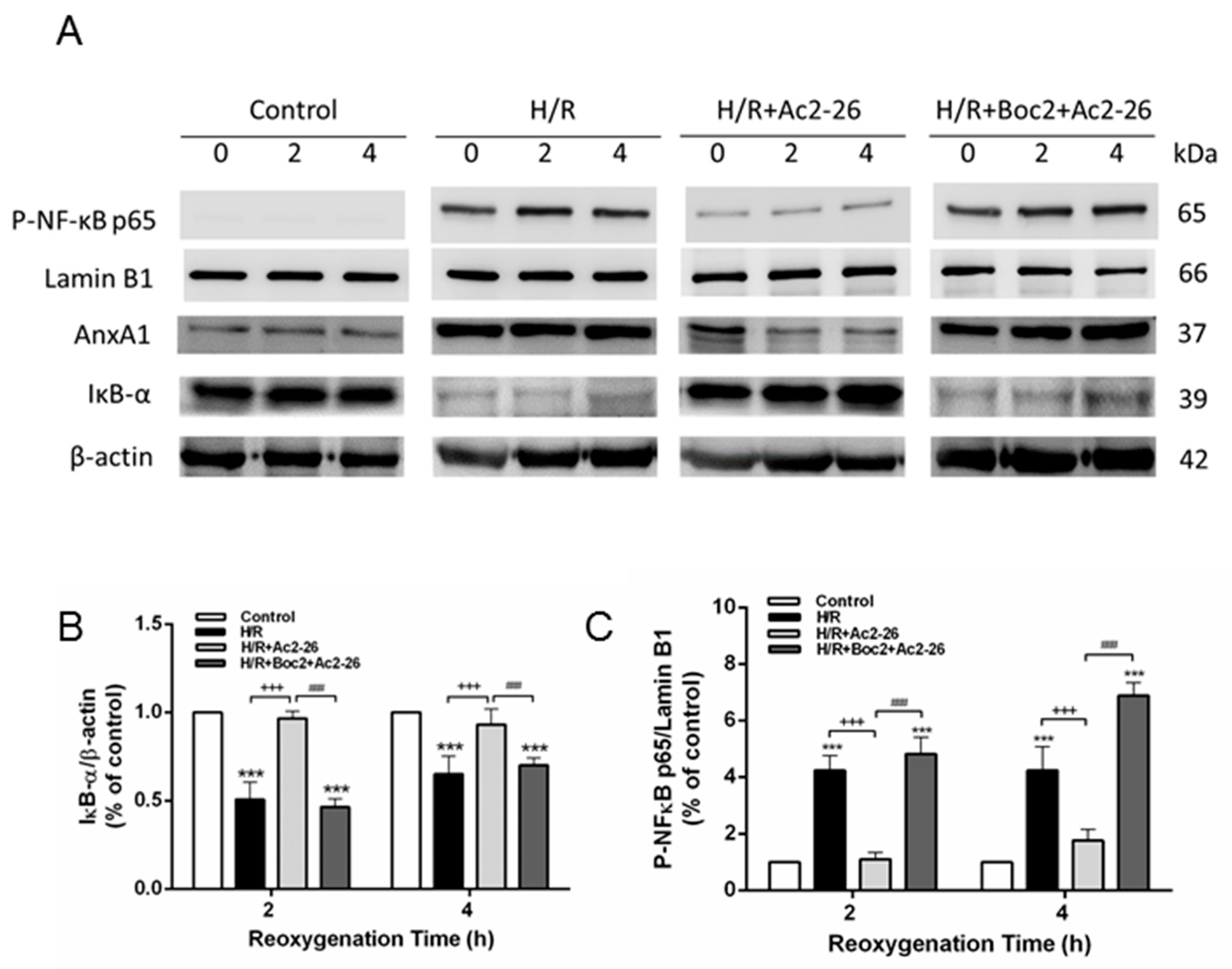
2.10. Effect of Ac2-26 in A549 Epithelial Cells Subjected to Hypoxia-Reoxygenation (H/R)
3. Discussion
4. Material and Methods
4.1. Isolated Perfused Lung Model in Rats
4.2. Vascular Filtration Coefficient
4.3. Lung Weight/Body Weight (LW/BW) and Wet/Dry (W/D) Weight Ratios
4.4. Assessment of Protein Concentration, CINC-1 and TNF-α Levels in BALF
4.5. Protein Carbonyl Content and MDA Level in Lung Tissue
4.6. Western Blotting
4.7. Immunohistochemical Analyses
4.8. Histopathology
4.9. Immunofluorescence Staining for Claudin-3, Occludin, and ZO-1
4.10. Experimental Design
4.11. Cell Culture and Induction of H/R
4.12. Data Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Perretti, M.; Dalli, J. Exploiting the Annexin A1 pathway for the development of novel anti-inflammatory therapeutics. Br. J. Pharmacol. 2009, 158, 936–946. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.H.; Morand, E.; Leech, M. Annexin A1: Potential for glucocorticoid sparing in RA. Nat. Rev. Rheumatol. 2013, 9, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Ernst, S.; Lange, C.; Wilbers, A.; Goebeler, V.; Gerke, V.; Rescher, U. An annexin 1 N-terminal peptide activates leukocytes by triggering different members of the formyl peptide receptor family. J. Immunol. 2004, 172, 7669–7676. [Google Scholar] [CrossRef] [PubMed]
- Trentin, P.G.; Ferreira, T.P.; Arantes, A.C.; Ciambarella, B.T.; Cordeiro, R.S.; Flower, R.J.; Perretti, M.; Martins, M.A.; Silva, P.M. Annexin A1 mimetic peptide controls the inflammatory and fibrotic effects of silica particles in mice. Br. J. Pharmacol. 2015, 172, 3058–3071. [Google Scholar] [CrossRef] [PubMed]
- Perretti, M.; D’Acquisto, F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat. Rev. Immunol. 2009, 9, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Gavins, F.N. Are formyl peptide receptors novel targets for therapeutic intervention in ischaemia-reperfusion injury? Trends Pharmacol. Sci. 2010, 31, 266–276. [Google Scholar] [CrossRef] [PubMed]
- De Perrot, M.; Liu, M.; Waddell, T.K.; Keshavjee, S. Ischemia-reperfusion-induced lung injury. Am. J. Respir. Crit. Care Med. 2003, 167, 490–511. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Zimmerman, G.A. Acute lung injury and the acute respiratory distress syndrome: Four decades of inquiry into pathogenesis and rational management. Am. J. Respir. Cell Mol. Biol. 2005, 33, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Wu, C.P.; Kang, B.H.; Li, M.H.; Chu, S.J.; Huang, K.L. Hypercapnic acidosis attenuates reperfusion injury in isolated and perfused rat lungs. Crit. Care Med. 2012, 40, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Guido, B.C.; Zanatelli, M.; Tavares-de-Lima, W.; Oliani, S.M.; Damazo, A.S. Annexin-A1 peptide down-regulates the leukocyte recruitment and up-regulates interleukin-10 release into lung after intestinal ischemia-reperfusion in mice. J. Inflamm. 2013, 10, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Cunha, E.E.; Oliani, S.M.; Damazo, A.S. Effect of annexin-A1 peptide treatment during lung inflammation induced by lipopolysaccharide. Pulm. Pharmacol. Ther. 2012, 25, 303–311. [Google Scholar] [CrossRef] [PubMed]
- La, M.; D’Amico, M.; Bandiera, S.; Di Filippo, C.; Oliani, S.M.; Gavins, F.N.; Flower, R.J.; Perretti, M. Annexin 1 peptides protect against experimental myocardial ischemia-reperfusion: Analysis of their mechanism of action. FASEB J. 2001, 15, 2247–2256. [Google Scholar] [CrossRef] [PubMed]
- Facio, F.N., Jr.; Sena, A.A.; Araujo, L.P.; Mendes, G.E.; Castro, I.; Luz, M.A.; Yu, L.; Oliani, S.M.; Burdmann, E.A. Annexin 1 mimetic peptide protects against renal ischemia/reperfusion injury in rats. J. Mol. Med. 2011, 89, 51–63. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, M.; Di Filippo, C.; La, M.; Solito, E.; McLean, P.G.; Flower, R.J.; Oliani, S.M.; Perretti, M. Lipocortin 1 reduces myocardial ischemia-reperfusion injury by affecting local leukocyte recruitment. FASEB J. 2000, 14, 1867–1869. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Li, M.H.; Ko, F.C.; Wu, G.C.; Huang, K.L.; Chu, S.J. Protective effect of hypercapnic acidosis in ischemia-reperfusion lung injury is attributable to upregulation of heme oxygenase-1. PLoS ONE 2013, 8, e74742. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Tang, S.E.; Ko, F.C.; Wu, G.C.; Huang, K.L.; Chu, S.J. Valproic acid attenuates acute lung injury induced by ischemia-reperfusion in rats. Anesthesiology 2015, 122, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Huang, L.; Zhao, W.; Rigas, B. Annexin 1 induced by anti-inflammatory drugs binds to NF-κB and inhibits its activation: Anticancer effects in vitro and in vivo. Cancer Res. 2010, 70, 2379–2388. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.S.; Mura, M.; Seth, R.; Liu, M. Acute lung injury and cell death: How many ways can cells die? Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L632–L641. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.; Flavell, R.A. Acetylation of MKP-1 and the control of inflammation. Sci. Signal. 2008, 1, pe44. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.H.; Wu, S.Y.; Tang, S.E.; Wu, G.C.; Li, M.H.; Huang, K.L.; Chu, S.J. Protection against reperfusion lung injury via aborgating multiple signaling cascades by trichostatin A. Int. Immunopharmacol. 2015, 25, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Kochanek, A.R.; Fukudome, E.Y.; Li, Y.; Smith, E.J.; Liu, B.; Velmahos, G.C.; de Moya, M.; King, D.; Alam, H.B. Histone deacetylase inhibitor treatment attenuates MAP kinase pathway activation and pulmonary inflammation following hemorrhagic shock in a rodent model. J. Surg. Res. 2012, 176, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.M.; Guo, G.F.; Huang, X.H.; Duan, W.L.; Zeng, Z.L. Isotetrandrine protects against lipopolysaccharide-induced acute lung injury by suppression of mitogen-activated protein kinase and nuclear factor-κB. J. Surg. Res. 2014, 187, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kim, D.W.; Kim, H.R.; Woo, S.J.; Kim, S.M.; Jo, H.S.; Jeon, S.G.; Cho, S.W.; Park, J.H.; Won, M.H. Anti-inflammatory effects of Tat-Annexin protein on ovalbumin-induced airway inflammation in a mouse model of asthma. Biochem. Biophys. Res. Commun. 2012, 417, 1024–1029. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kim, D.W.; Back, S.S.; Hwang, H.S.; Park, E.Y.; Kang, T.C.; Kwon, O.S.; Park, J.H.; Cho, S.W.; Han, K.H. Transduced Tat-Annexin protein suppresses inflammation-associated gene expression in lipopolysaccharide (LPS)-stimulated Raw 264.7 cells. BMB Rep. 2011, 44, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Wang, X.; Nelin, L.D.; Yao, Y.; Matta, R.; Manson, M.E.; Baliga, R.S.; Meng, X.; Smith, C.V.; Bauer, J.A. MAP kinase phosphatase 1 controls innate immune responses and suppresses endotoxic shock. J. Exp. Med. 2006, 203, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.H.; Toh, M.L.; Clyne, C.D.; Leech, M.; Aeberli, D.; Xue, J.; Dacumos, A.; Sharma, L.; Morand, E.F. Annexin 1 negatively regulates IL-6 expression via effects on p38 MAPK and MAPK phosphatase-1. J. Immunol. 2006, 177, 8148–8153. [Google Scholar] [CrossRef] [PubMed]
- Perretti, M.; Getting, S.J.; Solito, E.; Murphy, P.M.; Gao, J.L. Involvement of the receptor for formylated peptides in the in vivo anti-migratory actions of annexin 1 and its mimetics. Am. J. Pathol. 2001, 158, 1969–1973. [Google Scholar] [CrossRef]
- Girol, A.P.; Mimura, K.K.; Drewes, C.C.; Bolonheis, S.M.; Solito, E.; Farsky, S.H.; Gil, C.D.; Oliani, S.M. Anti-inflammatory mechanisms of the annexin A1 protein and its mimetic peptide Ac2-26 in models of ocular inflammation in vivo and in vitro. J. Immunol. 2013, 190, 5689–5701. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.R.; Perretti, M.; Flower, R.J.; Wallace, J.L. Annexin-1 modulates repair of gastric mucosal injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G764–G769. [Google Scholar] [CrossRef] [PubMed]
- Dufton, N.; Hannon, R.; Brancaleone, V.; Dalli, J.; Patel, H.B.; Gray, M.; D’Acquisto, F.; Buckingham, J.C.; Perretti, M.; Flower, R.J. Anti-inflammatory role of the murine formyl-peptide receptor 2: Ligand-specific effects on leukocyte responses and experimental inflammation. J. Immunol. 2010, 184, 2611–2619. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, R.H.; Gordon, J.M.; Woodman, O.L.; Cao, A.H.; Dusting, G.J. Annexin-1 peptide Anx-1(2–26) protects adult rat cardiac myocytes from cellular injury induced by simulated ischaemia. Br. J. Pharmacol. 2005, 145, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Buxton, K.D.; Pepe, S.; Cao, A.H.; Venardos, K.; Love, J.E.; Kaye, D.M.; Yang, Y.H.; Morand, E.F.; Ritchie, R.H. Reperfusion-induced myocardial dysfunction is prevented by endogenous annexin-A1 and its N-terminal-derived peptide Ac-ANX-A1(2–26). Br. J. Pharmacol. 2013, 168, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.E.; Wu, C.P.; Wu, S.Y.; Peng, C.K.; Perng, W.C.; Kang, B.H.; Chu, S.J.; Huang, K.L. Stanniocalcin-1 ameliorates lipopolysaccharide-induced pulmonary oxidative stress, inflammation, and apoptosis in mice. Free Radic. Biol. Med. 2014, 71, 321–331. [Google Scholar] [CrossRef] [PubMed]







© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, W.-I.; Wu, S.-Y.; Wu, G.-C.; Pao, H.-P.; Tang, S.-E.; Huang, K.-L.; Chu, S.-J. Ac2-26, an Annexin A1 Peptide, Attenuates Ischemia-Reperfusion-Induced Acute Lung Injury. Int. J. Mol. Sci. 2017, 18, 1771. https://doi.org/10.3390/ijms18081771
Liao W-I, Wu S-Y, Wu G-C, Pao H-P, Tang S-E, Huang K-L, Chu S-J. Ac2-26, an Annexin A1 Peptide, Attenuates Ischemia-Reperfusion-Induced Acute Lung Injury. International Journal of Molecular Sciences. 2017; 18(8):1771. https://doi.org/10.3390/ijms18081771
Chicago/Turabian StyleLiao, Wen-I, Shu-Yu Wu, Geng-Chin Wu, Hsin-Ping Pao, Shih-En Tang, Kun-Lun Huang, and Shi-Jye Chu. 2017. "Ac2-26, an Annexin A1 Peptide, Attenuates Ischemia-Reperfusion-Induced Acute Lung Injury" International Journal of Molecular Sciences 18, no. 8: 1771. https://doi.org/10.3390/ijms18081771