
Pathogenesis of Lafora Disease: Transition of Soluble Glycogen to Insoluble Polyglucosan

Abstract
: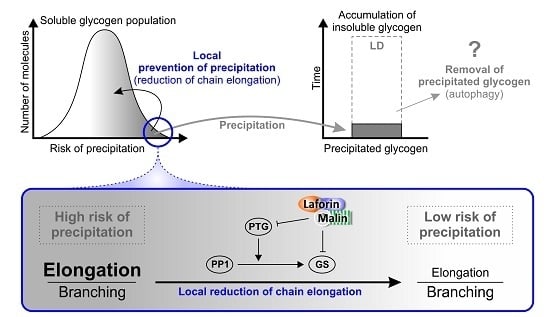
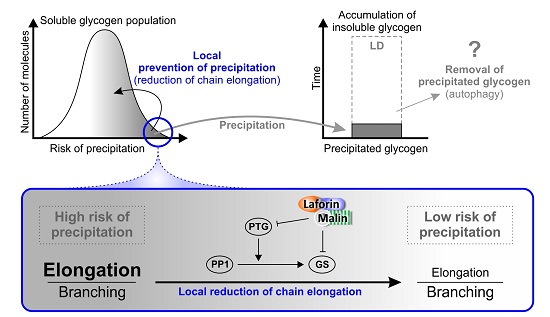{kind=link}
{kind=link}
{kind=link}
{kind=link}
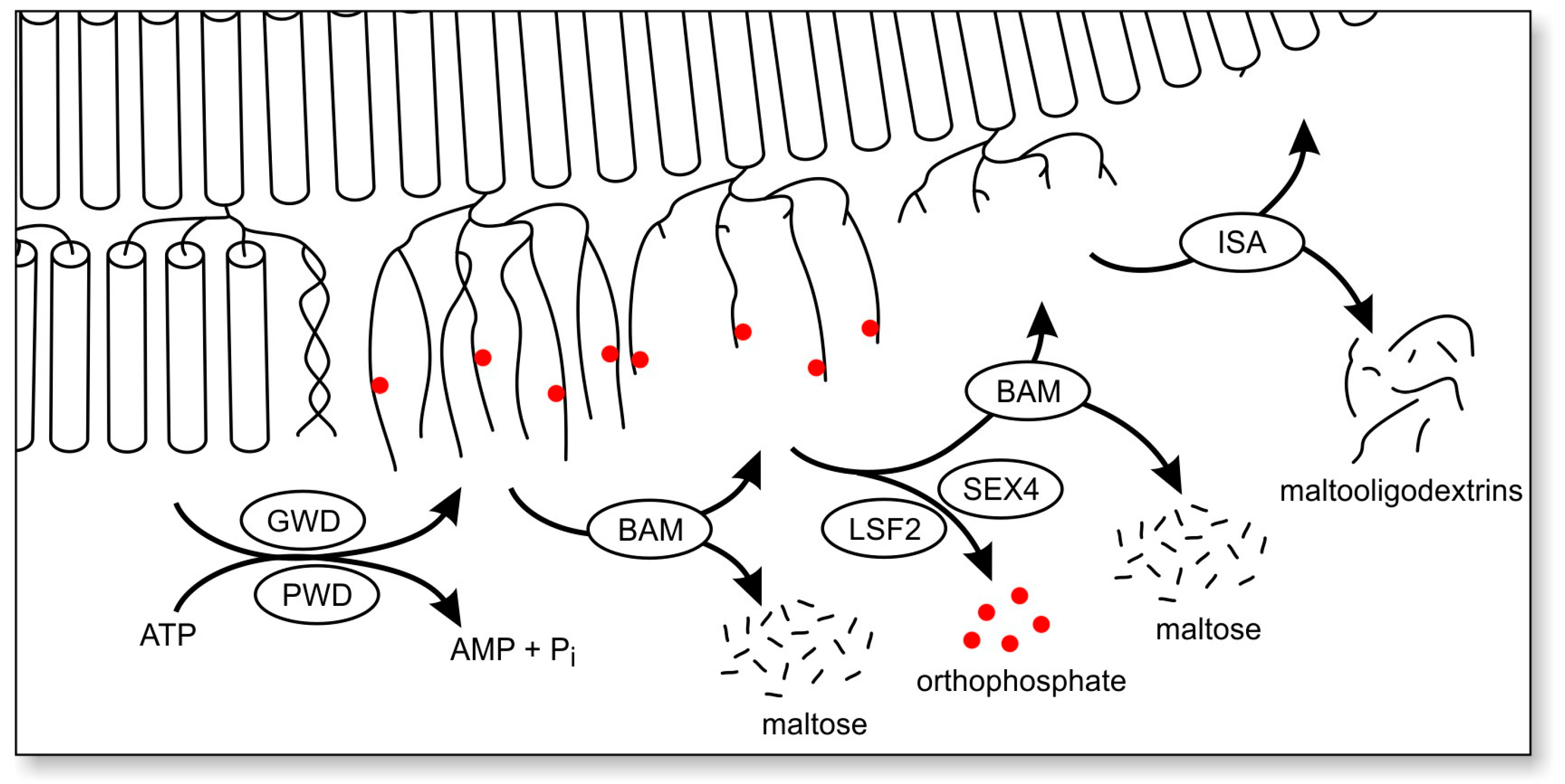{kind=link}
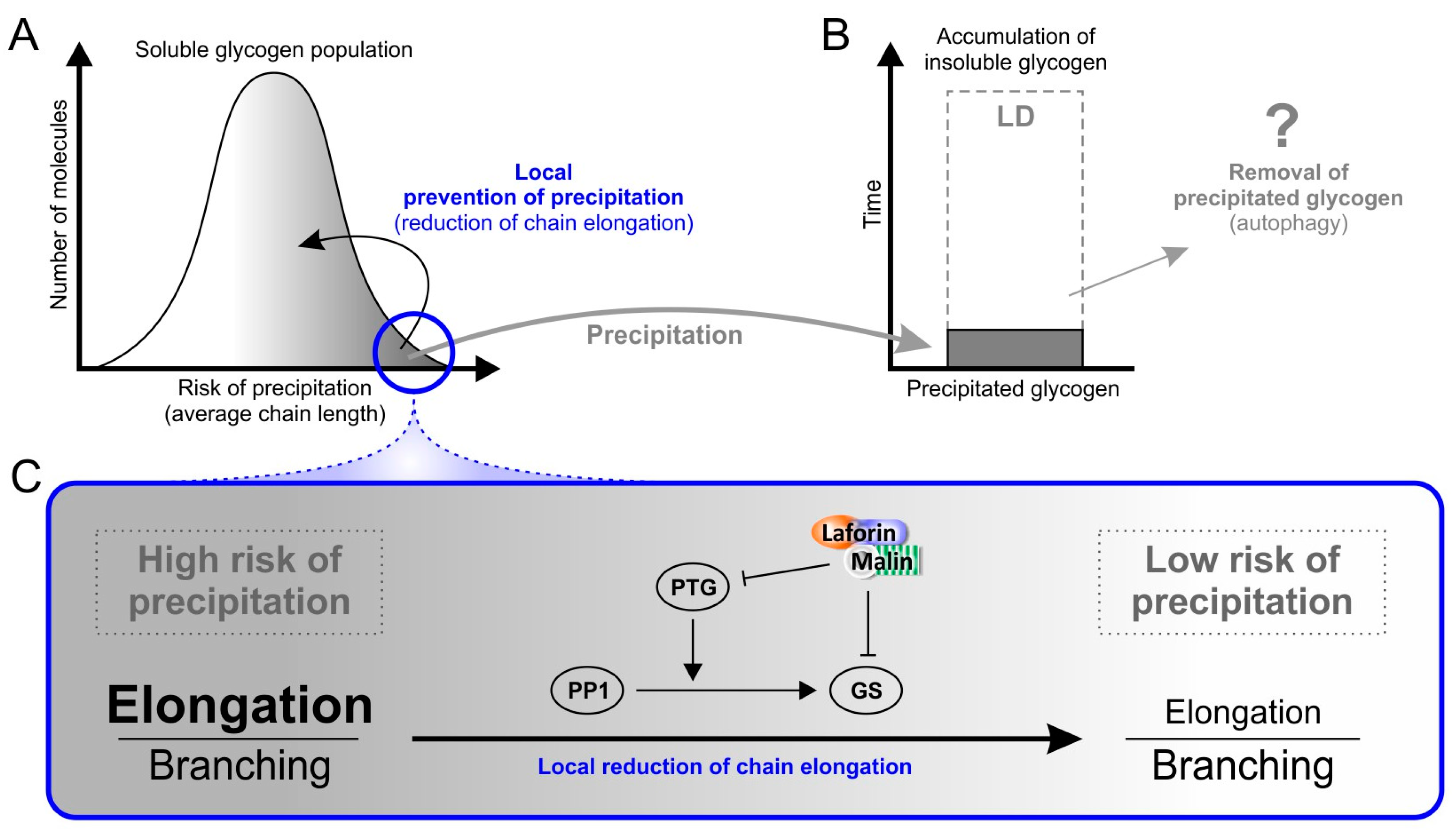{kind=link}
1. Introduction
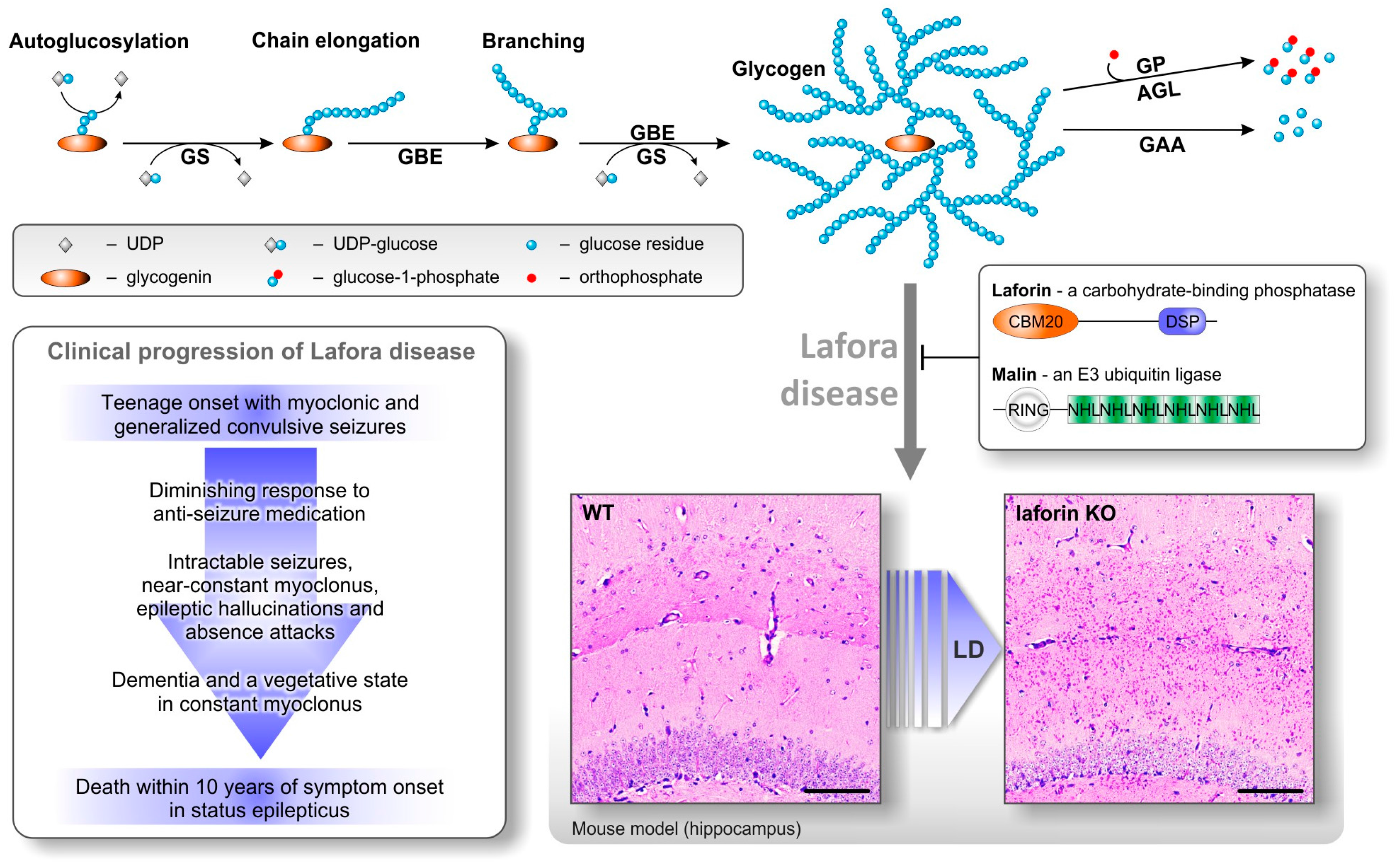
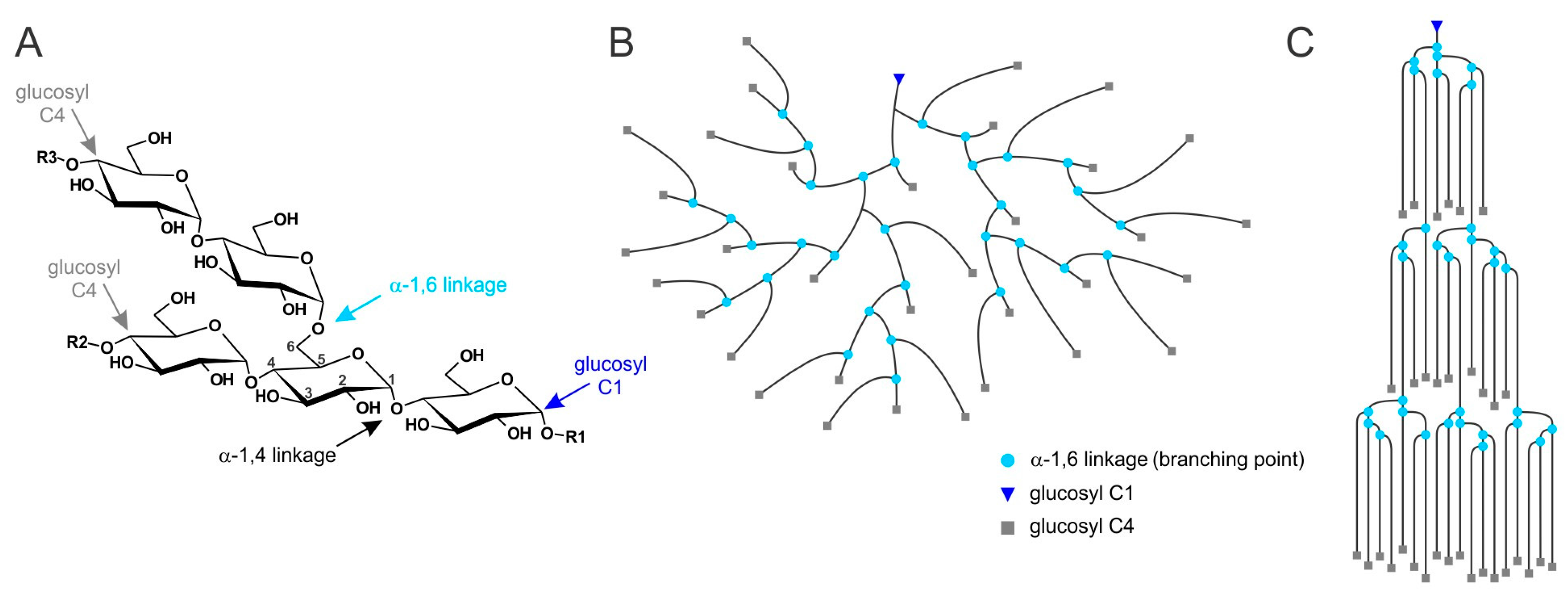
2. Lafora Bodies Put Lafora Disease in the Context of Polyglucan Metabolism
3. Lafora Bodies—Cause or Effect of Lafora Disease?
4. Laforin—A Carbohydrate-Binding Dual Specificity Phosphatase
5. Malin—An E3 Ubiquitin Ligase
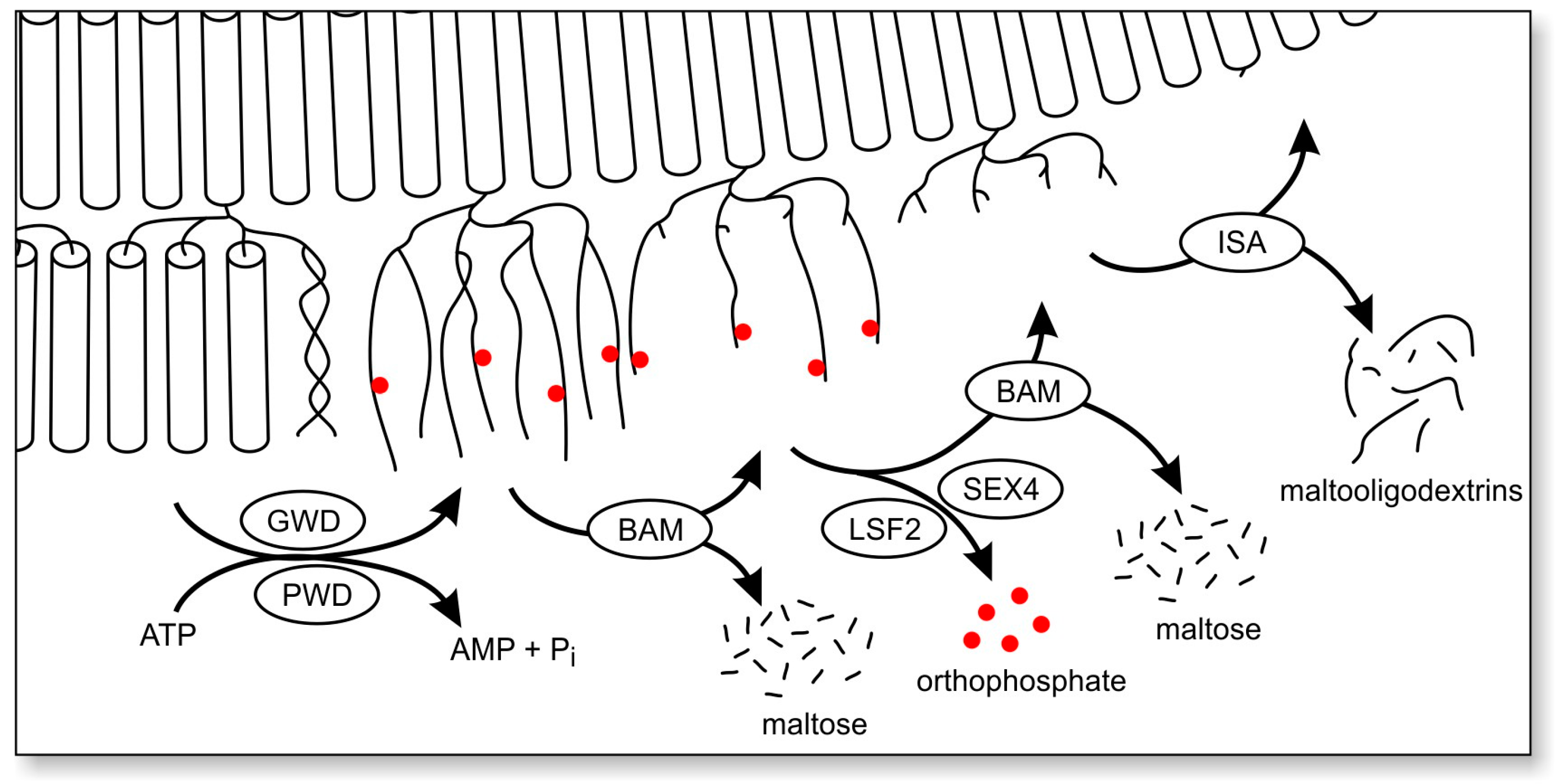
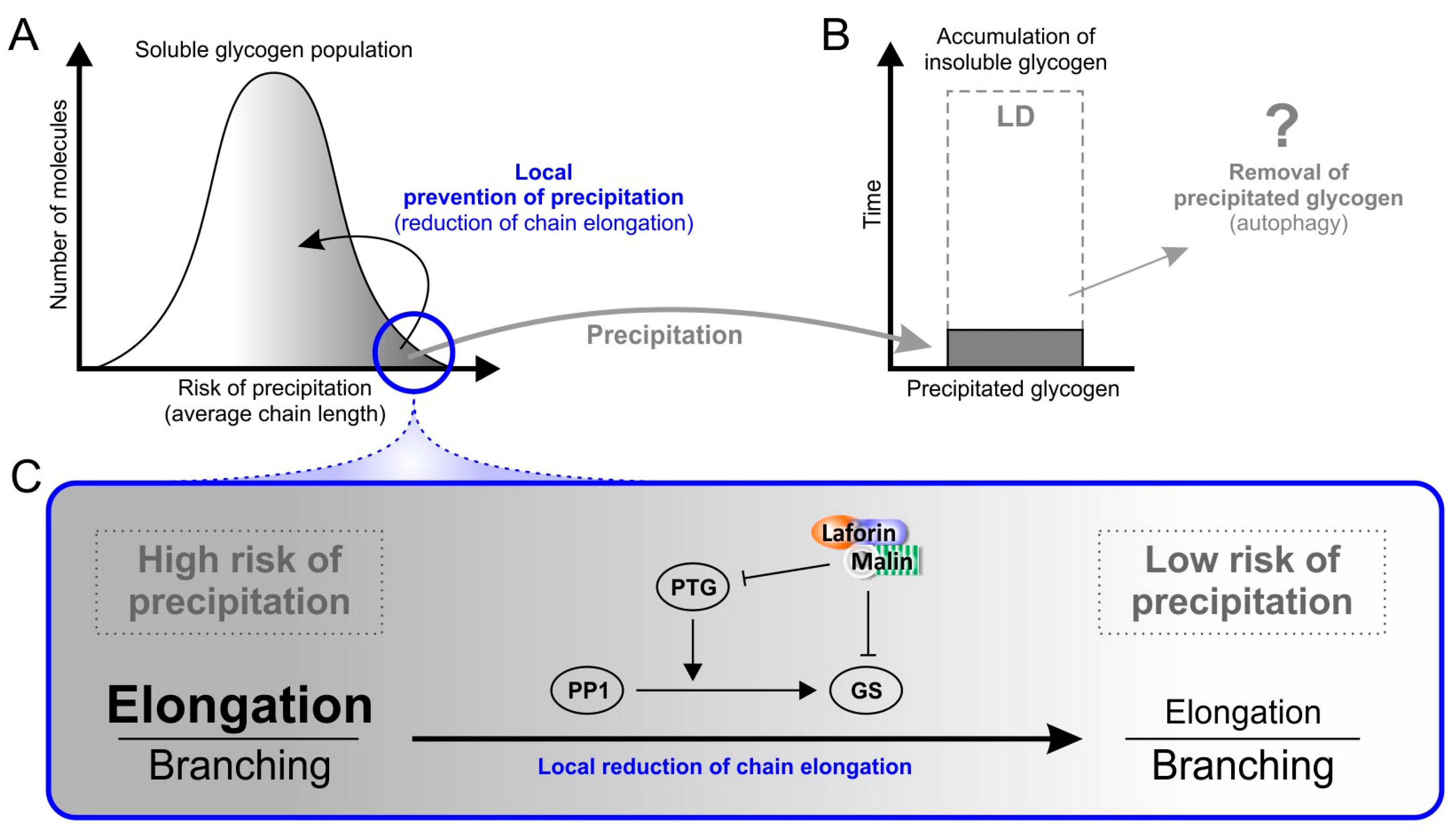
6. Factors That May Cause Glycogen Insolubility
7. Why Does Insoluble Glycogen Accumulate in Lafora Disease?
Acknowledgments
Conflicts of Interest
Abbreviations
| AGL | Glycogen debranching enzyme |
| AMPK | AMP-activated protein kinase |
| CBM | Carbohydrate-binding module |
| CLD | Chain length distribution |
| DSP | Dual specificity phosphatase |
| G6P | Glucose-6-phosphate |
| GBE | Glycogen branching enzyme |
| GP | Glycogen phosphorylase |
| GS | Glycogen synthase |
| GSK3 | Glycogen synthase kinase 3 |
| KO | Knockout |
| LB | Lafora body |
| LD | Lafora disease |
| PP1 | Protein phosphatase 1 |
| PTG | Protein targeting to glycogen – regulatory subunit of PP1 |
| WT | Wild-type |
References
- Minassian, B.A.; Lee, J.R.; Herbrick, J.A.; Huizenga, J.; Soder, S.; Mungall, A.J.; Dunham, I.; Gardner, R.; Fong, C.G.; Carpenter, S.; et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat. Genet. 1998, 20, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Serratosa, J.M.; Gomez-Garre, P.; Gallardo, M.E.; Anta, B.; de Bernabe, D.B.; Lindhout, D.; Augustijn, P.B.; Tassinari, C.A.; Malafosse, R.M.; Topcu, M.; et al. A novel protein tyrosine phosphatase gene is mutated in progressive myoclonus epilepsy of the lafora type (EPM2). Hum. Mol. Genet. 1999, 8, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.M.; Bulman, D.E.; Paterson, A.D.; Turnbull, J.; Andermann, E.; Andermann, F.; Rouleau, G.A.; Delgado-Escueta, A.V.; Scherer, S.W.; Minassian, B.A. Genetic mapping of a new lafora progressive myoclonus epilepsy locus (EPM2B) on 6p22. J. Med. Genet. 2003, 40, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Ganesh, S. Lafora progressive myoclonus epilepsy: A meta-analysis of reported mutations in the first decade following the discovery of the EPM2A and NHLRC1 genes. Hum. Mutat. 2009, 30, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Lesca, G.; Boutry-Kryza, N.; de Toffol, B.; Milh, M.; Steschenko, D.; Lemesle-Martin, M.; Maillard, L.; Foletti, G.; Rudolf, G.; Nielsen, J.E.; et al. Novel mutations in EPM2A and NHLRC1 widen the spectrum of lafora disease. Epilepsia 2010, 51, 1691–1698. [Google Scholar] [CrossRef] [PubMed]
- Poyrazoglu, H.G.; Karaca, E.; Per, H.; Gumus, H.; Onay, H.; Canpolat, M.; Canoz, O.; Ozkinay, F.; Kumandas, S. Three patients with lafora disease: Different clinical presentations and a novel mutation. J. Child. Neurol. 2015, 30, 777–781. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.M.; Omer, S.; Ahmed, M.; Bridges, L.R.; Bennett, C.; Scherer, S.W.; Minassian, B.A. Progressive myoclonus epilepsy with polyglucosans (lafora disease)—Evidence for a third locus. Neurology 2004, 63, 565–567. [Google Scholar] [CrossRef] [PubMed]
- Baykan, B.; Striano, P.; Gianotti, S.; Bebek, N.; Gennaro, E.; Gurses, C.; Zara, F. Late-onset and slow-progressing lafora disease in four siblings with EPM2B mutation. Epilepsia 2005, 46, 1695–1697. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Abad, C.; Gomez-Garre, P.; Gutierrez-Delicado, E.; Saygi, S.; Michelucci, R.; Tassinari, C.A.; de Cordoba, S.R.; Serratosa, J.M. Lafora disease due to EPM2B mutations—A clinical and genetic study. Neurology 2005, 64, 982–986. [Google Scholar] [CrossRef] [PubMed]
- Franceschetti, S.; Gambardella, A.; Canafoglia, L.; Striano, P.; Lohi, H.; Gennaro, E.; Ianzano, L.; Veggiotti, P.; Sofia, V.; Biondi, R.; et al. Clinical and genetic findings in 26 Italian patients with lafora disease. Epilepsia 2006, 47, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Jara-Prado, A.; Ochoa, A.; Alonso, M.E.; Lima Villeda, G.A.; Fernandez-Valverde, F.; Ruano-Calderon, L.; Vargas-Canas, S.; Duron, R.M.; Delgado-Escueta, A.V.; Martinez-Juarez, I.E. Late onset lafora disease and novel EPM2A mutations: Breaking paradigms. Epilepsy Res. 2014, 108, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Minassian, B.A. Lafora’s disease: Towards a clinical, pathologic, and molecular synthesis. Pediatr. Neurol. 2001, 25, 21–29. [Google Scholar] [CrossRef]
- Carpenter, S.; Karpati, G. Sweat gland duct cells in lafora disease: Diagnosis by skin biopsy. Neurology 1981, 31, 1564–1568. [Google Scholar] [CrossRef] [PubMed]
- Duran, J.; Gruart, A.; Garcia-Rocha, M.; Delgado-Garcia, J.M.; Guinovart, J.J. Glycogen accumulation underlies neurodegeneration and autophagy impairment in lafora disease. Hum. Mol. Genet. 2014, 23, 3147–3156. [Google Scholar] [CrossRef] [PubMed]
- Striano, P.; Zara, F.; Turnbull, J.; Girard, J.M.; Ackerley, C.A.; Cervasio, M.; de Rosa, G.; del Basso-de Caro, M.L.; Striano, S.; Minassian, B.A. Typical progression of myoclonic epilepsy of the lafora type: A case report. Nat. Clin. Pract. Neurol. 2008, 4, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, S.; Austin, J.; Witmer, F.; Sakai, M. Studies in myoclonus epilepsy (lafora body form): I. Isolation and preliminary characterization of lafora bodies in two cases. Arch. Neurol. 1968, 19, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Sakai, M.; Austin, J.; Witmer, F.; Trueb, L. Studies in myoclonus epilepsy (lafora body form). II. Polyglucosans in systemic deposits of myoclonus epilepsy and in corpora-amylacea. Neurology 1970, 20, 160. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.A.; Vilaplana, F.; Cave, R.A.; Stapleton, D.; Gray-Weale, A.A.; Gilbert, R.G. Nature of α and β particles in glycogen using molecular size distributions. Biomacromolecules 2010, 11, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
- Besford, Q.A.; Sullivan, M.A.; Zheng, L.; Gilbert, R.G.; Stapleton, D.; Gray-Weale, A. The structure of cardiac glycogen in healthy mice. Int. J. Biol. Macromol. 2012, 51, 887–891. [Google Scholar] [CrossRef] [PubMed]
- Nitschke, F.; Wang, P.; Schmieder, P.; Girard, J.-M.; Awrey, D.E.; Wang, T.; Israelian, J.; Zhao, X.; Turnbull, J.; Heydenreich, M.; et al. Hyperphosphorylation of glucosyl C6 carbons and altered structure of glycogen in the neurodegenerative epilepsy lafora disease. Cell Metab. 2013, 17, 756–767. [Google Scholar] [CrossRef] [PubMed]
- Wanson, J.C.; Drochmans, P. Rabbit skeletal muscle glycogen: A morphological and biochemical study of glycogen beta-particles isolated by the precipitation-centrifugation method. J. Cell Biol. 1968, 38, 130. [Google Scholar] [CrossRef] [PubMed]
- Irimia, J.M.; Tagliabracci, V.S.; Meyer, C.M.; Segvich, D.M.; DePaoli-Roach, A.A.; Roach, P.J. Muscle glycogen remodeling and glycogen phosphate metabolism following exhaustive exercise of wild type and laforin knockout mice. J. Biol. Chem. 2015, 290, 22686–22698. [Google Scholar] [CrossRef] [PubMed]
- Jayachandran, K.N.; Takacs-Cox, A.; Brooks, D.E. Synthesis and characterization of polymer brushes of poly(n,n-dimethylacrylamide) from polystyrene latex by aqueous atom transfer radical polymerization. Macromolecules 2002, 35, 4247–4257. [Google Scholar] [CrossRef]
- Roach, P.J.; Depaoli-Roach, A.A.; Hurley, T.D.; Tagliabracci, V.S. Glycogen and its metabolism: Some new developments and old themes. Biochem. J. 2012, 441, 763–787. [Google Scholar] [CrossRef] [PubMed]
- Testoni, G.; Duran, J.; Garcia-Rocha, M.; Vilaplana, F.; Serrano, A.L.; Sebastian, D.; Lopez-Soldado, I.; Sullivan, M.A.; Slebe, F.; Vilaseca, M.; et al. Lack of glycogenin causes glycogen accumulation and muscle function impairment. Cell. Metab. 2017, 26, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Szydlowski, N.; Ragel, P.; Raynaud, S.; Lucas, M.M.; Roldán, I.; Montero, M.; Munoz, F.J.; Ovecka, M.; Bahaji, A.; Planchot, V.; et al. Starch granule initiation in arabidopsis requires the presence of either class IV or class III starch synthases. Plant Cell 2009, 21, 2443–2457. [Google Scholar] [CrossRef] [PubMed]
- Hunter, R.W.; Zeqiraj, E.; Morrice, N.; Sicheri, F.; Sakamoto, K. Expression and purification of functional human glycogen synthase-1:Glycogenin-1 complex in insect cells. Protein Expr. Purif. 2015, 108, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Doble, B.W.; MacAulay, K.; Sinclair, E.M.; Drucker, D.J.; Woodgett, J.R. Tissue-specific role of glycogen synthase kinase 3β in glucose homeostasis and insulin action. Mol. Cell Biol. 2008, 28, 6314–6328. [Google Scholar] [CrossRef] [PubMed]
- Fong, N.M.; Jensen, T.C.; Shah, A.S.; Parekh, N.N.; Saltiel, A.R.; Brady, M.J. Identification of binding sites on protein targeting to glycogen for enzymes of glycogen metabolism. J. Biol. Chem. 2000, 275, 35034–35039. [Google Scholar] [CrossRef] [PubMed]
- Worby, C.A.; Gentry, M.S.; Dixon, J.E. Malin decreases glycogen accumulation by promoting the degradation of protein targeting to glycogen (PTG). J. Biol. Chem. 2008, 283, 4069–4076. [Google Scholar] [CrossRef] [PubMed]
- Petty, H.R.; Worth, R.G.; Kindzelskii, A.L. Imaging sustained dissipative patterns in the metabolism of individual living cells. Phys. Rev. Lett. 2000, 84, 2754–2757. [Google Scholar] [CrossRef] [PubMed]
- Rickey Welch, G.; Easterby, J.S. Metabolic channeling versus free diffusion: Transition-time analysis. Trends Biochem. Sci. 1994, 19, 193–197. [Google Scholar] [CrossRef]
- Aw, T.Y. Intracellular compartmentation of organelles and gradients of low molecular weight species. Int. Rev. Cytol. 1999, 192, 223–253. [Google Scholar]
- Tagliabracci, V.S.; Turnbull, J.; Wang, W.; Girard, J.-M.; Zhao, X.; Skurat, A.V.; Delgado-Escueta, A.V.; Minassian, B.A.; DePaoli-Roach, A.A.; Roach, P.J. Laforin is a glycogen phosphatase, deficiency of which leads to elevated phosphorylation of glycogen in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 19262–19266. [Google Scholar] [CrossRef] [PubMed]
- Tagliabracci, V.S.; Girard, J.M.; Segvich, D.; Meyer, C.; Turnbull, J.; Zhao, X.C.; Minassian, B.A.; DePaoli-Roach, A.A.; Roach, P.J. Abnormal metabolism of glycogen phosphate as a cause for lafora disease. J. Biol. Chem. 2008, 283, 33816–33825. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, J.; DePaoli-Roach, A.A.; Zhao, X.C.; Cortez, M.A.; Pencea, N.; Tiberia, E.; Piliguian, M.; Roach, P.J.; Wang, P.X.; Ackerley, C.A.; et al. PTG depletion removes lafora bodies and rescues the fatal epilepsy of lafora disease. PLoS Genet. 2011, 7, e1002037. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, J.; Epp, J.R.; Goldsmith, D.; Zhao, X.C.; Pencea, N.; Wang, P.X.; Frankland, P.W.; Ackerley, C.A.; Minassian, B.A. PTG protein depletion rescues malin-deficient lafora disease in mouse. Ann. Neurol. 2014, 75, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Gayarre, J.; Duran-Trio, L.; Garcia, O.C.; Aguado, C.; Juana-Lopez, L.; Crespo, I.; Knecht, E.; Bovolenta, P.; de Cordoba, S.R. The phosphatase activity of laforin is dispensable to rescue Epm2a−/− mice from lafora disease. Brain 2014, 137, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Duran, J.; Tevy, M.F.; Garcia-Rocha, M.; Calbo, J.; Milan, M.; Guinovart, J.J. Deleterious effects of neuronal accumulation of glycogen in flies and mice. EMBO Mol. Med. 2012, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- McMahon, J.; Huang, X.X.; Yang, J.; Komatsu, M.; Yue, Z.Y.; Qian, J.; Zhu, X.J.; Huang, Y.F. Impaired autophagy in neurons after disinhibition of mammalian target of rapamycin and its contribution to epileptogenesis. J. Neurosci. 2012, 32, 15704–15714. [Google Scholar] [CrossRef] [PubMed]
- Gentry, M.S.; Roma-Mateo, C.; Sanz, P. Laforin, a protein with many faces: Glucan phosphatase, adapter protein, et alii. FEBS J. 2013, 280, 525–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubey, D.; Parihar, R.; Ganesh, S. Identification and characterization of novel splice variants of the human EPM2A gene mutated in lafora progressive myoclonus epilepsy. Genomics 2012, 99, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Pulido, R.; Hooft van Huijsduijnen, R. Protein tyrosine phosphatases: Dual-specificity phosphatases in health and disease. FEBS J. 2008, 275, 848–866. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Stuckey, J.A.; Wishart, M.J.; Dixon, J.E. A unique carbohydrate binding domain targets the lafora disease phosphatase to glycogen. J. Biol. Chem. 2002, 277, 2377–2380. [Google Scholar] [CrossRef] [PubMed]
- Emanuelle, S.; Brewer, M.K.; Meekins, D.A.; Gentry, M.S. Unique carbohydrate binding platforms employed by the glucan phosphatases. Cell. Mol. Life Sci. 2016, 73, 2765–2778. [Google Scholar] [CrossRef] [PubMed]
- Lohi, H.; Ianzano, L.; Zhao, X.C.; Chan, E.M.; Turnbull, J.; Scherer, S.W.; Ackerley, C.A.; Minassian, B.A. Novel glycogen synthase kinase 3 and ubiquitination pathways in progressive myoclonus epilepsy. Hum. Mol. Genet. 2005, 14, 2727–2736. [Google Scholar] [CrossRef] [PubMed]
- Worby, C.A.; Gentry, M.S.; Dixon, J.E. Laforin, a dual specificity phosphatase that dephosphorylates complex carbohydrates. J. Biol. Chem. 2006, 281, 30412–30418. [Google Scholar] [CrossRef] [PubMed]
- Roach, P.J. Glycogen phosphorylation and lafora disease. Mol. Asp. Med. 2015, 46, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Nitschke, F.; Sullivan, M.A.; Wang, P.; Zhao, X.; Chown, E.E.; Perri, A.M.; Israelian, L.; Juana-Lopez, L.; Bovolenta, P.; Rodriguez de Cordoba, S.; et al. Abnormal glycogen chain length pattern, not hyperphosphorylation, is critical in lafora disease. EMBO Mol. Med. 2017, 9, 906–917. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Sanchez, M.E.; Criado-Garcia, O.; Heath, K.E.; Garcia-Fojeda, B.; Medrano-Fernandez, I.; Gomez-Garre, P.; Sanz, P.; Serratosa, J.M.; de Cordoba, S.R. Laforin, the dual-phosphatase responsible for lafora disease, interacts with R5 (PTG), a regulatory subunit of protein phosphatase-1 that enhances glycogen accumulation. Hum. Mol. Genet. 2003, 12, 3161–3171. [Google Scholar] [CrossRef] [PubMed]
- Solaz-Fuster, M.C.; Gimeno-Alcaniz, J.V.; Ros, S.; Fernandez-Sanchez, M.E.; Garcia-Fojeda, B.; Garcia, O.C.; Vilchez, D.; Dominguez, J.; Garcia-Rocha, M.; Sanchez-Piris, M.; et al. Regulation of glycogen synthesis by the laforin-malin complex is modulated by the amp-activated protein kinase pathway. Hum. Mol. Genet. 2008, 17, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Puri, R.; Suzuki, T.; Yamakawa, K.; Ganesh, S. Hyperphosphorylation and aggregation of tau in laforin-deficient mice, an animal model for lafora disease. J. Biol. Chem. 2009, 284, 22657–22663. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, S.; Tsurutani, N.; Suzuki, T.; Ueda, K.; Agarwala, K.L.; Osada, H.; Delgado-Escueta, A.V.; Yamakawa, K. The lafora disease gene product laforin interacts with HIRIP5, a phylogenetically conserved protein containing a NifU-like domain. Hum. Mol. Genet. 2003, 12, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Ianzano, L.; Zhao, X.C.; Minassian, B.A.; Scherer, S.W. Identification of a novel protein interacting with laforin, the EPM2A progressive myoclonus epilepsy gene product. Genomics 2003, 81, 579–587. [Google Scholar] [CrossRef]
- Sanchez-Martin, P.; Roma-Mateo, C.; Viana, R.; Sanz, P. Ubiquitin conjugating enzyme E2-N and sequestosome-1 (p62) are components of the ubiquitination process mediated by the malin-laforin E3-ubiquitin ligase complex. Int. J. Biochem. Cell Biol. 2015, 69, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Vilchez, D.; Ros, S.; Cifuentes, D.; Pujadas, L.; Valles, J.; Garcia-Fojeda, B.; Criado-Garcia, O.; Fernandez-Sanchez, E.; Medrano-Fernandez, I.; Dominguez, J.; et al. Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat. Neurosci. 2007, 10, 1407–1413. [Google Scholar] [CrossRef] [PubMed]
- Freemont, P.S. Ubiquitination: Ring for destruction? Curr. Biol. 2000, 10, R84–R87. [Google Scholar] [CrossRef]
- Chan, E.M.; Young, E.J.; Ianzano, L.; Munteanu, I.; Zhao, X.C.; Christopoulos, C.C.; Avanzini, G.; Elia, M.; Ackerley, C.A.; Jovic, N.J.; et al. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat. Genet. 2003, 35, 125–127. [Google Scholar] [CrossRef] [PubMed]
- Moreno, D.; Towler, M.C.; Hardie, D.G.; Knecht, E.; Sanz, P. The laforin-malin complex, involved in lafora disease, promotes the incorporation of K63-linked ubiquitin chains into amp-activated protein kinase beta subunits. Mol. Biol. Cell 2010, 21, 2578–2588. [Google Scholar] [CrossRef] [PubMed]
- Roma-Mateo, C.; Moreno, D.; Vernia, S.; Rubio, T.; Bridges, T.M.; Gentry, M.S.; Sanz, P. Lafora disease E3-ubiquitin ligase malin is related to TRIM32 at both the phylogenetic and functional level. BMC Evol. Biol. 2011, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubio-Villena, C.; Garcia-Gimeno, M.A.; Sanz, P. Glycogenic activity of R6, a protein phosphatase 1 regulatory subunit, is modulated by the laforin-malin complex. Int. J. Biochem. Cell Biol. 2013, 45, 1479–1488. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J.; Joazeiro, C.A.P. Ring domain E3 ubiquitin ligases. Ann. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.M.M.; Wong, E.S.P.; Kirkpatrick, D.S.; Pletnikova, O.; Ko, H.S.; Tay, S.P.; Ho, M.W.L.; Troncoso, J.; Gygi, S.P.; Lee, M.K.; et al. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum. Mol. Genet. 2008, 17, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Nathan, J.A.; Kim, H.T.; Ting, L.; Gygi, S.P.; Goldberg, A.L. Why do cell proteins linked to K63-polyubiquitin chains not associate with proteasomes? EMBO J. 2013, 32, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.; Mulherkar, S.; Mukherjee, D.; Jana, N.R. Malin regulates wnt signaling pathway through degradation of dishevelled2. J. Biol. Chem. 2012, 287, 6830–6839. [Google Scholar] [CrossRef] [PubMed]
- Gentry, M.S.; Worby, C.A.; Dixon, J.E. Insights into lafora disease: Malin is an E3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc. Natl. Acad. Sci. USA 2005, 102, 8501–8506. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Zhang, M.; Gentry, M.S.; Worby, C.A.; Dixon, J.E.; Saltiel, A.R. A role for AGL ubiquitination in the glycogen storage disorders of lafora and Cori’s disease. Genes Dev. 2007, 21, 2399–2409. [Google Scholar] [CrossRef] [PubMed]
- Viana, R.; Lujan, P.; Sanz, P. The laforin/malin E3-ubiquitin ligase complex ubiquitinates pyruvate kinase M1/M2. BMC Biochem. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- DePaoli-Roach, A.A.; Tagliabracci, V.S.; Segvich, D.M.; Meyer, C.M.; Irimia, J.M.; Roach, P.J. Genetic depletion of the malin E3 ubiquitin ligase in mice leads to lafora bodies and the accumulation of insoluble laforin. J. Biol. Chem. 2010, 285, 25372–25381. [Google Scholar] [CrossRef] [PubMed]
- Cenci, U.; Nitschke, F.; Steup, M.; Minassian, B.A.; Colleoni, C.; Ball, S.G. Transition from glycogen to starch metabolism in archaeplastida. Trends Plant Sci. 2014, 19, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Buleon, A.; Colonna, P.; Planchot, V.; Ball, S. Starch granules: Structure and biosynthesis. Int. J. Biol. Macromolecules 1998, 23, 85–112. [Google Scholar] [CrossRef]
- Manners, D.J. Recent developments in our understanding of glycogen structure. Carbohydr. Polym. 1991, 16, 37–82. [Google Scholar] [CrossRef]
- Ritte, G.; Scharf, A.; Eckermann, N.; Haebel, S.; Steup, M. Phosphorylation of transitory starch is increased during degradation. Plant. Physiol. 2004, 135, 2068–2077. [Google Scholar] [CrossRef] [PubMed]
- Pfister, B.; Zeeman, S.C. Formation of starch in plant cells. Cell. Mol. Life Sci. 2016, 73, 2781–2807. [Google Scholar] [CrossRef] [PubMed]
- Hejazi, M.; Fettke, J.; Haebel, S.; Edner, C.; Paris, O.; Frohberg, C.; Steup, M.; Ritte, G. Glucan, water dikinase phosphorylates crystalline maltodextrins and thereby initiates solubilization. Plant J. 2008, 55, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Zeeman, S.C.; Kossmann, J.; Smith, A.M. Starch: Its metabolism, evolution, and biotechnological modification in plants. Annu. Rev. Plant Biol. 2010, 61, 209–234. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.B.; Williams, P.F.; Cooney, G.J.; Caterson, I.D. Diurnal rhythms of glycogen-metabolism in the liver and skeletal-muscle in gold thioglucose induced-obese mice with developing insulin resistance. Int. J. Obes. 1992, 16, 913–921. [Google Scholar]
- Saez, I.; Duran, J.; Sinadinos, C.; Beltran, A.; Yanes, O.; Tevy, M.F.; Martinez-Pons, C.; Milan, M.; Guinovart, J.J. Neurons have an active glycogen metabolism that contributes to tolerance to hypoxia. J. Cereb. Blood Flow Metab. 2014, 34, 945–955. [Google Scholar] [CrossRef] [PubMed]
- Bischof, S.; Umhang, M.; Eicke, S.; Streb, S.; Qi, W.; Zeeman, S.C. Cecropia peltata accumulates starch or soluble glycogen by differentially regulating starch biosynthetic genes. Plant Cell 2013, 25, 1400–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, N.; Hanashiro, I.; Suzuki, S.; Higuchi, T.; Toyosawa, Y.; Utsumi, Y.; Itoh, R.; Aihara, S.; Nakamura, Y. Elongated phytoglycogen chain length in transgenic rice endosperm expressing active starch synthase IIa affects the altered solubility and crystallinity of the storage α-glucan. J. Exp. Bot. 2012, 63, 5859–5872. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.G.; Wu, A.C.; Sullivan, M.A.; Sumarriva, G.E.; Ersch, N.; Hasjim, J. Improving human health through understanding the complex structure of glucose polymers. Anal. Bioanal. Chem. 2013, 405, 8969–8980. [Google Scholar] [CrossRef] [PubMed]
- Powell, P.O.; Sullivan, M.A.; Sweedman, M.C.; Stapleton, D.I.; Hasjim, J.; Gilbert, R.G. Extraction, isolation and characterisation of phytoglycogen from su-1 maize leaves and grain. Carbohyd. Polym. 2014, 101, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.C.; Gilbert, R.G. Molecular weight distributions of starch branches reveal genetic constraints on biosynthesis. Biomacromolecules 2010, 11, 3539–3547. [Google Scholar] [CrossRef] [PubMed]
- Umeki, K.; Kainuma, K. Fine structure of naegeli amylodextrin obtained by acid treatment of defatted waxy-maize starch—Structural evidence to support the double-helix hypothesis. Carbohydr. Res. 1981, 96, 143–159. [Google Scholar] [CrossRef]
- Gidley, M.J.; Bulpin, P.V. Chrystallization of maltoologosaccharides as models of the crystalline forms of starch-minimum chain-length requirement for the formation of double helices. Carbohydr. Res. 1987, 161, 291–300. [Google Scholar] [CrossRef]
- Wong, K.-S.; Kubo, A.; Jane, J.-L.; Harada, K.; Satoh, H.; Nakamura, Y. Structures and properties of amylopectin and phytoglycogen in the endosperm of sugary-1 mutants of rice. J. Cereal Sci. 2003, 37, 139–149. [Google Scholar] [CrossRef]
- Fujita, N.; Kubo, A.; Suh, D.S.; Wong, K.S.; Jane, J.L.; Ozawa, K.; Takaiwa, F.; Inaba, Y.; Nakamura, Y. Antisense inhibition of isoamylase alters the structure of amylopectin and the physicochemical properties of starch in rice endosperm. Plant Cell Physiol. 2003, 44, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Akman, H.O.; Sheiko, T.; Tay, S.K.H.; Finegold, M.J.; DiMauro, S.; Craigen, W.J. Generation of a novel mouse model that recapitulates early and adult onset glycogenosis type IV. Hum. Mol. Genet. 2011, 20, 4430–4439. [Google Scholar] [CrossRef] [PubMed]
- Raben, N.; Danon, M.; Lu, N.; Lee, E.; Shliselfeld, L.; Skurat, A.V.; Roach, P.J.; Lawrence, J.C., Jr.; Musumeci, O.; Shanske, S.; et al. Surprises of genetic engineering: A possible model of polyglucosan body disease. Neurology 2001, 56, 1739–1745. [Google Scholar] [CrossRef] [PubMed]
- Palmer, T.N.; Macaskie, L.E.; Grewal, K.K. Spatial-distribution of unit chains in glycogen. Carbohydr. Res. 1983, 115, 139–150. [Google Scholar] [CrossRef]
- Liu, Y.; Zeng, L.; Ma, K.L.; Baba, O.; Zheng, P.; Liu, Y.; Wang, Y. Laforin-malin complex degrades polyglucosan bodies in concert with glycogen debranching enzyme and brain isoform glycogen phosphorylase. Mol. Neurobiol. 2014, 49, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.M.; Ackerley, C.A.; Lohi, H.; Ianzano, L.; Cortez, M.A.; Shannon, P.; Scherer, S.W.; Minassian, B.A. Laforin preferentially binds the neurotoxic starch-like polyglucosans, which form in its absence in progressive myoclonus epilepsy. Hum. Mol. Genet. 2004, 13, 1117–1129. [Google Scholar] [CrossRef] [PubMed]
- Duran, J.; Guinovart, J.J. Brain glycogen in health and disease. Mol. Aspects Med. 2015, 46, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Israelian, L.; Xue, Y.; Song, S.; Attisano, L.; Minassian, B.A. SGK1 (glucose transport), dishevelled2 (wnt signaling), LC3/p62(autophagy) and p53 (apoptosis) proteins are unaltered in lafora disease. All Results J. Biol. 2016, 7, 28–33. [Google Scholar]
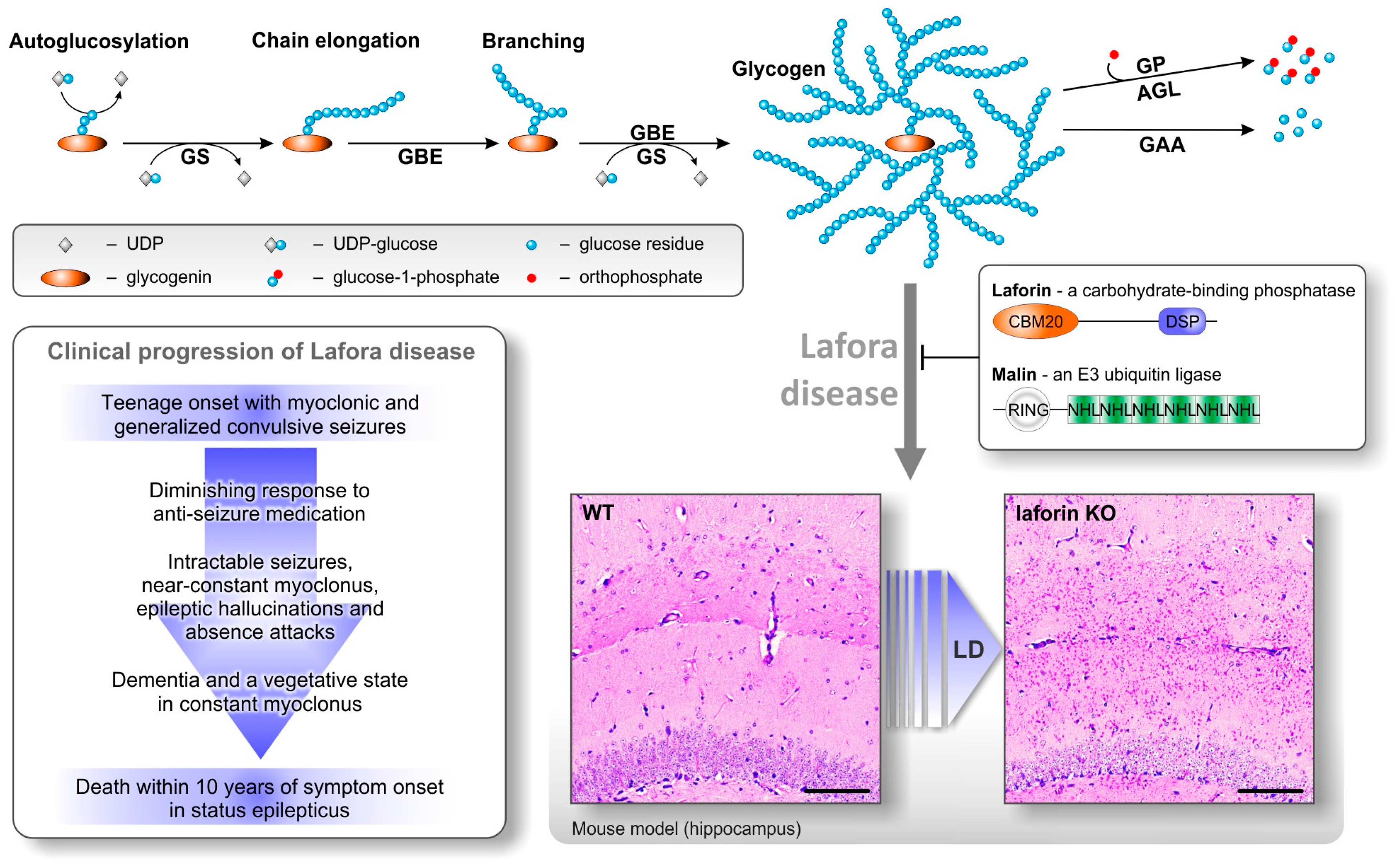

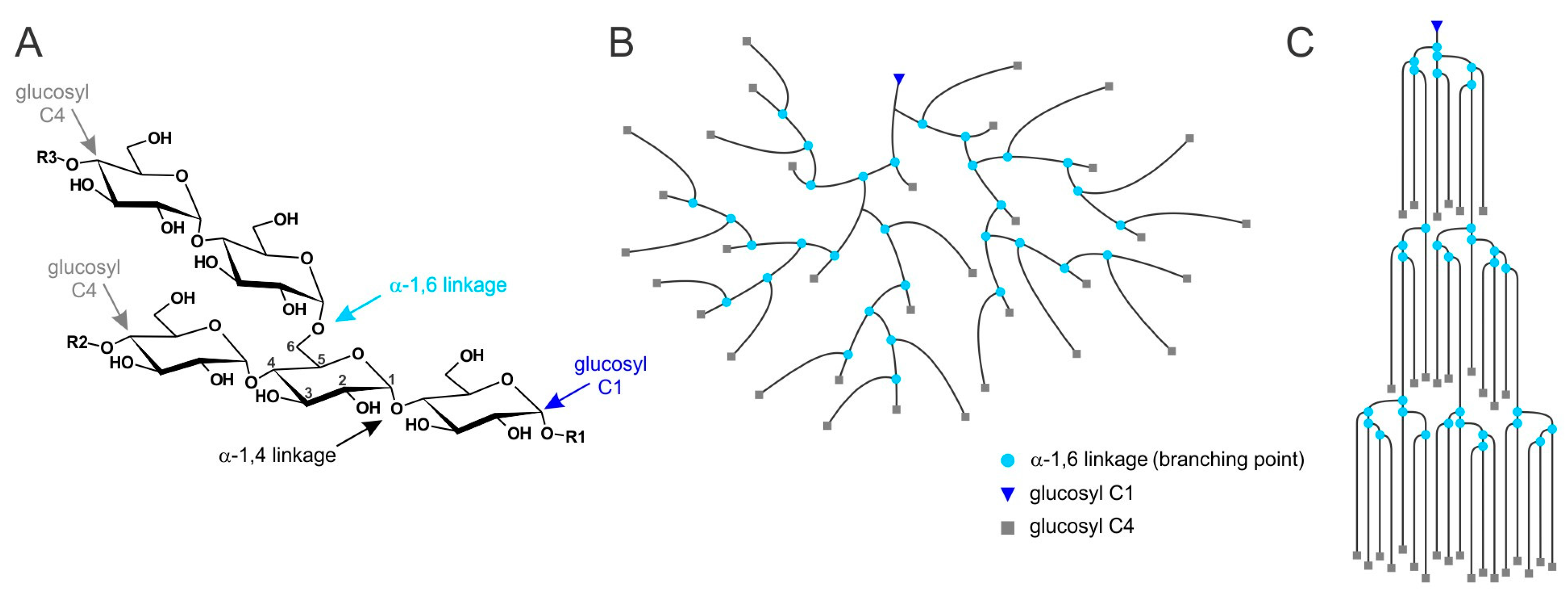
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sullivan, M.A.; Nitschke, S.; Steup, M.; Minassian, B.A.; Nitschke, F. Pathogenesis of Lafora Disease: Transition of Soluble Glycogen to Insoluble Polyglucosan. Int. J. Mol. Sci. 2017, 18, 1743. https://doi.org/10.3390/ijms18081743
Sullivan MA, Nitschke S, Steup M, Minassian BA, Nitschke F. Pathogenesis of Lafora Disease: Transition of Soluble Glycogen to Insoluble Polyglucosan. International Journal of Molecular Sciences. 2017; 18(8):1743. https://doi.org/10.3390/ijms18081743
Chicago/Turabian StyleSullivan, Mitchell A., Silvia Nitschke, Martin Steup, Berge A. Minassian, and Felix Nitschke. 2017. "Pathogenesis of Lafora Disease: Transition of Soluble Glycogen to Insoluble Polyglucosan" International Journal of Molecular Sciences 18, no. 8: 1743. https://doi.org/10.3390/ijms18081743