Pharmacogenomics of Targeted Agents for Personalization of Colorectal Cancer Treatment


Abstract
:1. Introduction
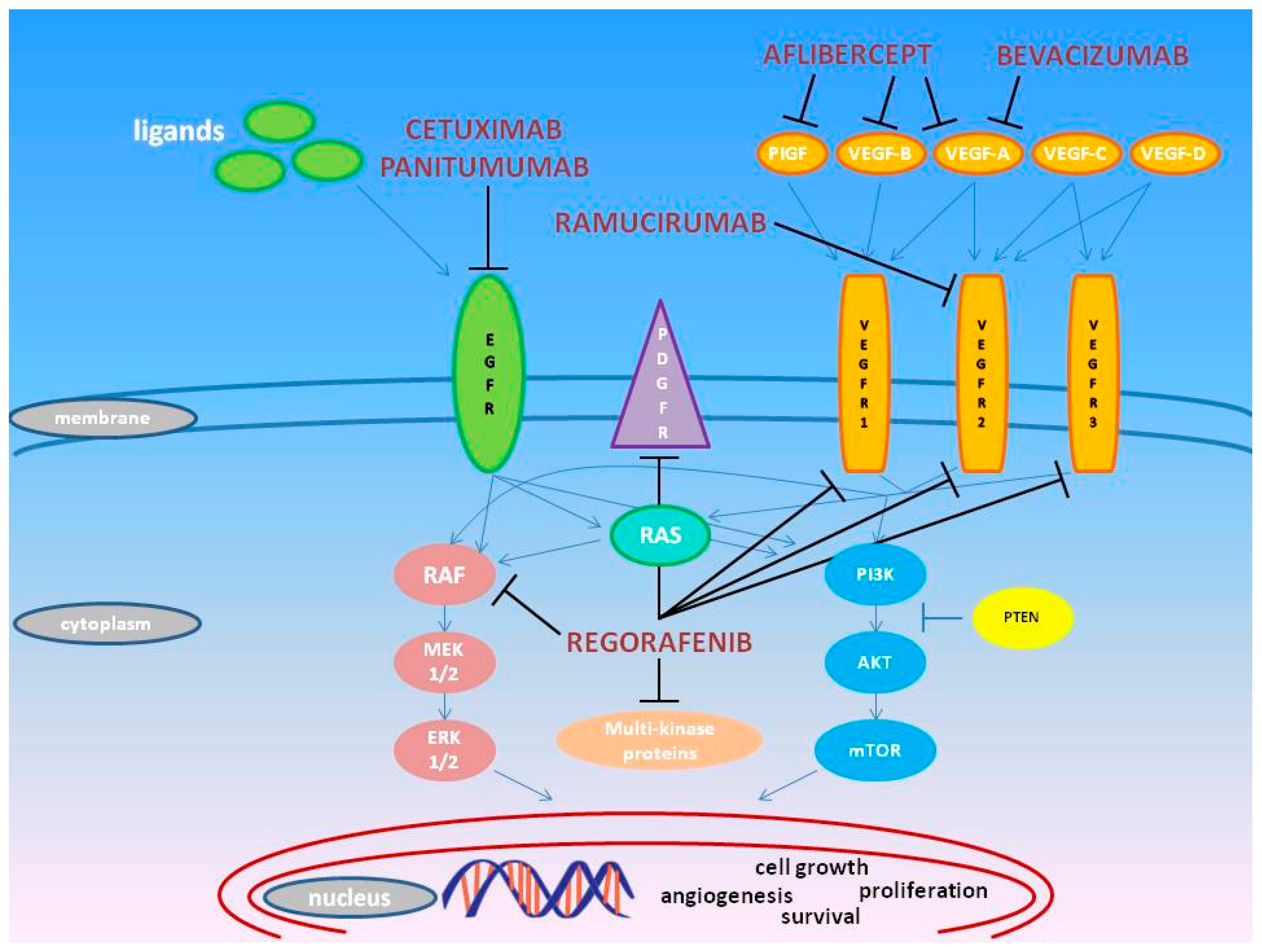
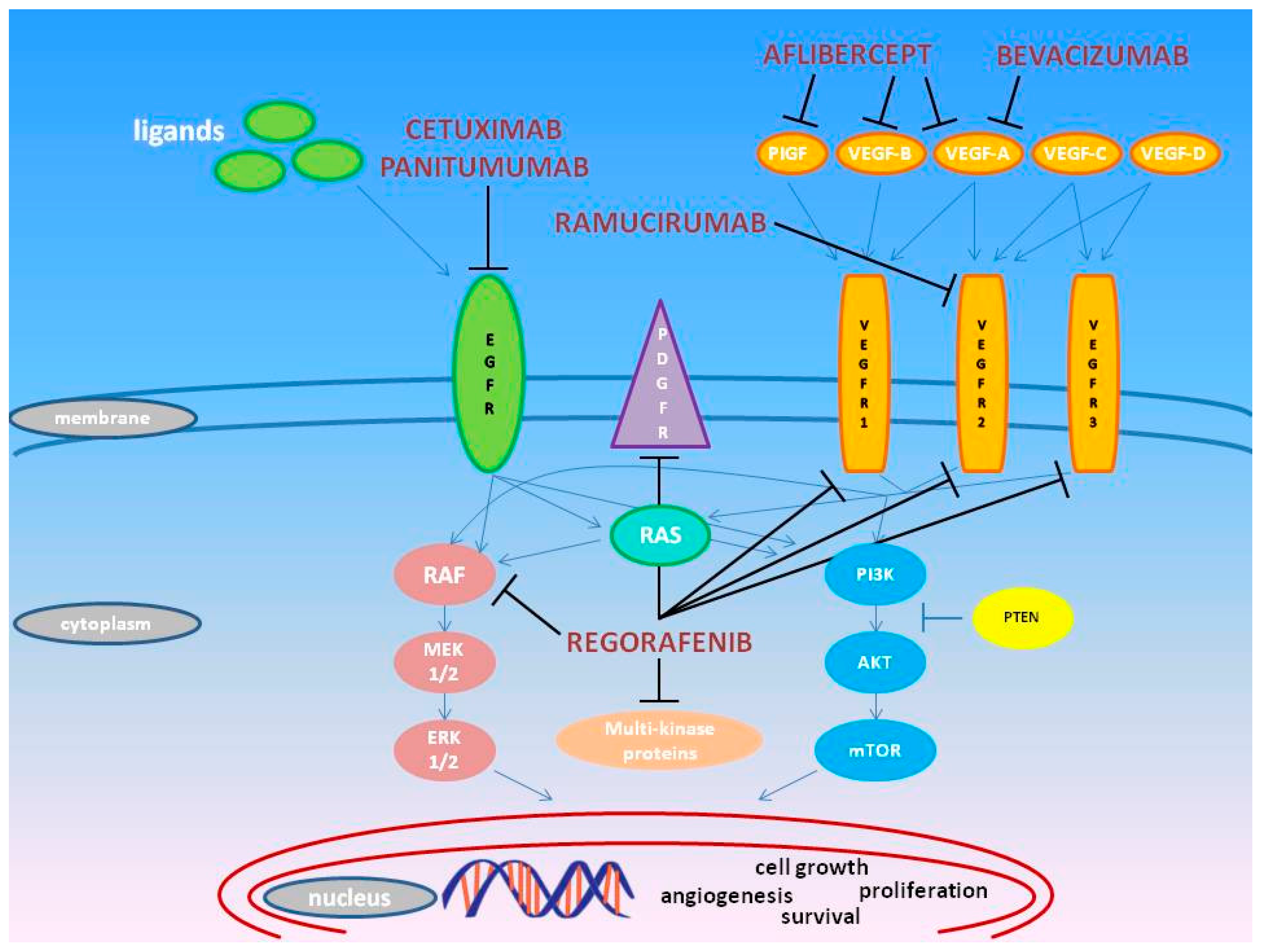
2. Pharmacogenomics of Approved Molecules Targeting the VEGF and EGF Pathways
2.1. Bevacizumab
2.2. Regorafenib
2.3. Ziv-Aflibercept
2.4. Ramucirumab
2.5. Cetuximab
2.6. Panitumumab
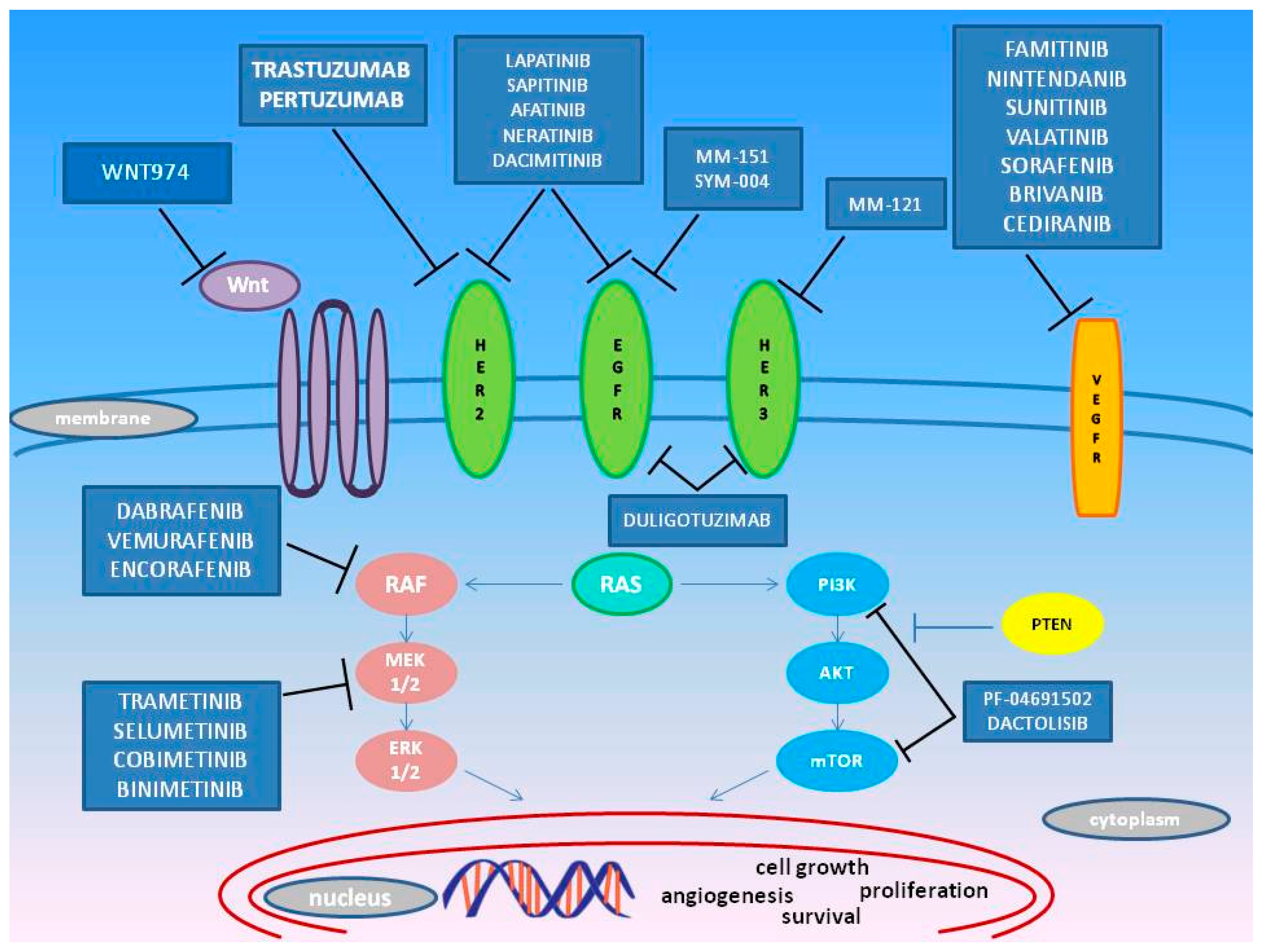
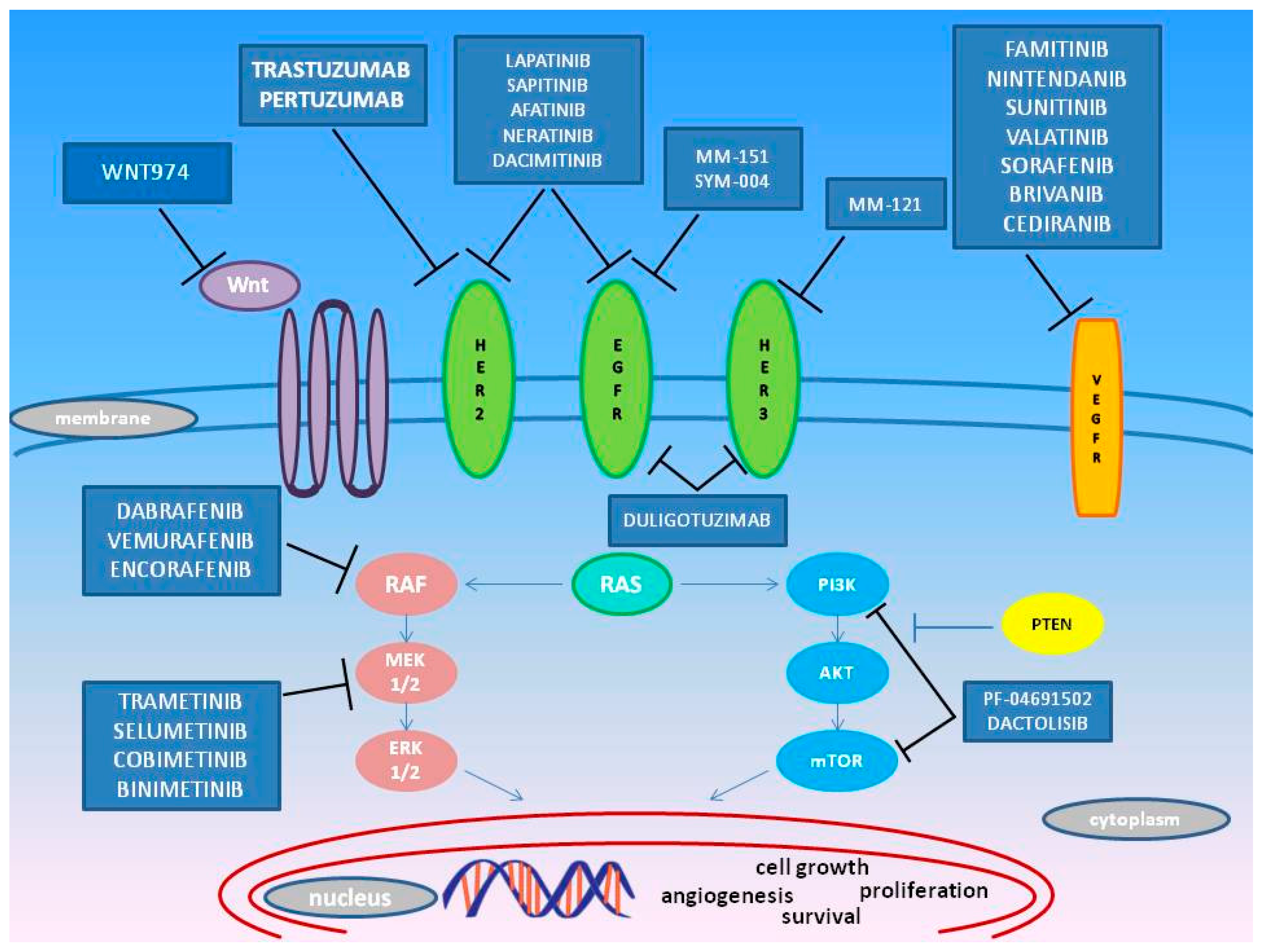
3. Emerging Target Molecules in mCRC Treatment
3.1. HER
3.2. MEK
3.3. BRAF
3.4. PI3K
3.5. VEGFR
4. Conclusions and Future Directions
Acknowledgments
Conflicts of Interest
References
- International Association of Cancer Registries. Available online: http://iicc.iarc.fr/ (accessed on 29 May 2015).
- Marley, A.R.; Nan, H. Epidemiology of colorectal cancer. Int. J. Mol. Epidemiol. Genet. 2016, 7, 105–114. [Google Scholar] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Van Loon, K.; Venook, A.P. Adjuvant treatment of colon cancer: What is next? Curr. Opin. Oncol. 2011, 23, 403–409. [Google Scholar]
- Price, T.J.; Segelov, E.; Burge, M.; Haller, D.G.; Ackland, S.P.; Tebbutt, N.C.; Karapetis, C.S.; Pavlakis, N.; Sobrero, A.F.; Cunningham, D.; et al. Current opinion on optimal treatment for colorectal cancer. Expert. Rev. Anticancer Ther. 2013, 13, 597–611. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Garcia, E.; Grasselli, J.; Argiles, G.; Elez, M.E.; Tabernero, J. Current and advancing treatments for metastatic colorectal cancer. Expert. Opin. Biol. Ther. 2016, 16, 93–110. [Google Scholar] [CrossRef] [PubMed]
- Marcus, L.; Lemery, S.J.; Khasar, S.; Wearne, E.; Helms, W.S.; Yuan, W.; He, K.; Cao, X.; Yu, J.; Zhao, H.; et al. FDA Approval Summary: TAS-102. Clin. Cancer Res. 2017, 23, 2924–2927. [Google Scholar] [CrossRef] [PubMed]
- Longo-Munoz, F.; Argiles, G.; Tabernero, J.; Cervantes, A.; Gravalos, C.; Pericay, C.; Gil-Calle, S.; Mizuguchi, H.; Carrato-Mena, A.; Limon, M.L.; et al. Efficacy of trifluridine and tipiracil (TAS-102) versus placebo, with supportive care, in a randomized, controlled trial of patients with metastatic colorectal cancer from Spain: Results of a subgroup analysis of the phase 3 RECOURSE trial. Clin. Transl. Oncol. 2017, 19, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.; Murphy, A. The emerging role of immunotherapy in colorectal cancer. Ann. Transl. Med. 2016, 4, 305. [Google Scholar] [CrossRef] [PubMed]
- De Mattia, E.; Cecchin, E.; Toffoli, G. Pharmacogenomics of intrinsic and acquired pharmacoresistance in colorectal cancer: Toward targeted personalized therapy. Drug Resist. Update 2015, 20, 39–70. [Google Scholar] [CrossRef] [PubMed]
- Fiala, O.; Buchler, T.; Mohelnikova-Duchonova, B.; Melichar, B.; Matejka, V.M.; Holubec, L.; Kulhankova, J.; Bortlicek, Z.; Bartouskova, M.; Liska, V.; et al. G12V and G12A KRAS mutations are associated with poor outcome in patients with metastatic colorectal cancer treated with bevacizumab. Tumour. Biol. 2016, 37, 6823–6830. [Google Scholar] [CrossRef] [PubMed]
- Cremolini, C.; Loupakis, F.; Antoniotti, C.; Lupi, C.; Sensi, E.; Lonardi, S.; Mezi, S.; Tomasello, G.; Ronzoni, M.; Zaniboni, A.; et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: Updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol. 2015, 16, 1306–1315. [Google Scholar] [CrossRef]
- Nakayama, I.; Shinozaki, E.; Matsushima, T.; Wakatsuki, T.; Ogura, M.; Ichimura, T.; Ozaka, M.; Takahari, D.; Suenaga, M.; Chin, K.; et al. Retrospective study of RAS/PIK3CA/BRAF tumor mutations as predictors of response to first-line chemotherapy with bevacizumab in metastatic colorectal cancer patients. BMC Cancer 2017, 17, 38. [Google Scholar] [CrossRef] [PubMed]
- Garde, N.J.; Jantus-Lewintre, E.; Gil-Raga, M.; Evgenyeva, E.; Macia, E.S.; Llombart-Cussac, A.; Camps, H.C. Role of RAS mutation status as a prognostic factor for patients with advanced colorectal cancer treated with first-line chemotherapy based on fluoropyrimidines and oxaliplatin, with or without bevavizumab: A retrospective analysis. Mol. Clin. Oncol. 2017, 6, 403–408. [Google Scholar]
- Renaud, S.; Schaeffer, M.; Falcoz, P.E.; Seitlinger, J.; Romain, B.; Voegeli, A.C.; Legrain, M.; Reeb, J.; Santelmo, N.; Rohr, S.; et al. Perioperative bevacizumab improves survival following lung metastasectomy for colorectal cancer in patients harbouring v-Ki-ras2 Kirsten rat sarcoma viral oncogene homologue exon 2 codon 12 mutationsdagger. Eur. J. Cardiothorac. Surg. 2017, 51, 255–262. [Google Scholar] [PubMed]
- Tabernero, J.; Takayuki, Y.; Cohn, A.L. Correction to Lancet Oncol 2015; 16: 499–508. Ramucirumab versus placebo in combination with second-line FOLFIRI in patients with metastatic colorectal carcinoma that progressed during or after first-line therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine (RAISE): A randomised, double-blind, multicentre, phase 3 study. Lancet Oncol. 2015, 16, e262. [Google Scholar] [PubMed]
- Adenis, A.; de la Fouchardiere, C.; Paule, B.; Burtin, P.; Tougeron, D.; Wallet, J.; Dourthe, L.M.; Etienne, P.L.; Mineur, L.; Clisant, S.; et al. Survival, safety, and prognostic factors for outcome with Regorafenib in patients with metastatic colorectal cancer refractory to standard therapies: Results from a multicenter study (REBACCA) nested within a compassionate use program. BMC. Cancer 2016, 16, 412. [Google Scholar]
- Loaiza-Bonilla, A.; Jensen, C.E.; Shroff, S.; Furth, E.; Bonilla-Reyes, P.A.; Deik, A.F.; Morrissette, J. KDR Mutation as a Novel Predictive Biomarker of Exceptional Response to Regorafenib in Metastatic Colorectal Cancer. Cureus 2016, 8, e478. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, D.; Thienpont, B.; Thuillier, V.; Sagaert, X.; Moisse, M.; Peuteman, G.; Pericay, C.; Folprecht, G.; Zalcberg, J.; Zilocchi, C.; et al. Evaluation of efficacy and safety markers in a phase II study of metastatic colorectal cancer treated with aflibercept in the first-line setting. Br. J. Cancer 2015, 113, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Obermannova, R.; Van Custem, E.; Yoshino, T.; Bodoky, G.; Prausova, J.; Garcia-Carbonero, R.; Ciuleanu, T.; Garcia, A.P.; Portnoy, D.; Cohn, A.; et al. Subgroup analysis in RAISE: A randomized, double-blind phase III study of irinotecan, folinic acid, and 5-fluorouracil (FOLFIRI) plus ramucirumab or placebo in patients with metastatic colorectal carcinoma progression. Ann. Oncol. 2016, 27, 2082–2090. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.H.; Odell, I.D.; Ko, C.J.; Choate, K.A. Somatic p.T771R KDR (VEGFR2) Mutation Arising in a Sporadic Angioma During Ramucirumab Therapy. JAMA Dermatol. 2015, 151, 1240–1243. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Aoyama, T.; Ishibashi, K.; Tsuji, A.; Takinishi, Y.; Shindo, Y.; Sakamoto, J.; Oba, K.; Mishima, H. Randomized phase II study of cetuximab versus irinotecan and cetuximab in patients with chemo-refractory KRAS codon G13D metastatic colorectal cancer (G13D-study). Cancer Chemother. Pharmacol. 2017, 79, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Segelov, E.; Waring, P.; Desai, J.; Wilson, K.; Gebski, V.; Thavaneswaran, S.; Elez, E.; Underhill, C.; Pavlakis, N.; Chantrill, L.; et al. ICECREAM: Randomised phase II study of cetuximab alone or in combination with irinotecan in patients with metastatic colorectal cancer with either KRAS, NRAS, BRAF and PI3KCA wild type, or G13D mutated tumours. BMC. Cancer 2016, 16, 339. [Google Scholar] [CrossRef] [PubMed]
- Osumi, H.; Shinozaki, E.; Osako, M.; Kawazoe, Y.; Oba, M.; Misaka, T.; Goto, T.; Kamo, H.; Suenaga, M.; Kumekawa, Y.; et al. Cetuximab treatment for metastatic colorectal cancer with KRAS p.G13D mutations improves progression-free survival. Mol. Clin. Oncol. 2015, 3, 1053–1057. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Elme, A.; Kusic, Z.; Park, J.O.; Udrea, A.A.; Kim, S.Y.; Ahn, J.B.; Valencia, R.V.; Krishnan, S.; Bilic, A.; et al. A phase 3 trial evaluating panitumumab plus best supportive care vs best supportive care in chemorefractory wild-type KRAS or RAS metastatic colorectal cancer. Br. J. Cancer 2016, 115, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- Pietrantonio, F.; Petrelli, F.; Coinu, A.; Di, B.M.; Borgonovo, K.; Maggi, C.; Cabiddu, M.; Iacovelli, R.; Bossi, I.; Lonati, V.; et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: A meta-analysis. Eur. J. Cancer 2015, 51, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Sorich, M.J.; Wiese, M.D.; Rowland, A.; Kichenadasse, G.; McKinnon, R.A.; Karapetis, C.S. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: A meta-analysis of randomized, controlled trials. Ann. Oncol. 2015, 26, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Therkildsen, C.; Bergmann, T.K.; Henrichsen-Schnack, T.; Ladelund, S.; Nilbert, M. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol. 2014, 53, 852–864. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Liu, Z.; Deng, D.; Tan, A.; Liao, M.; Mo, Z.; Yang, X. Anti-epidermal growth factor receptor monoclonal antibody-based therapy for metastatic colorectal cancer: A meta-analysis of the effect of PIK3CA mutations in KRAS wild-type patients. Arch. Med. Sci. 2014, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Peeters, M.; Oliner, K.S.; Price, T.J.; Cervantes, A.; Sobrero, A.F.; Ducreux, M.; Hotko, Y.; Andre, T.; Chan, E.; Lordick, F.; et al. Analysis of KRAS/NRAS Mutations in a Phase III Study of Panitumumab with FOLFIRI Compared with FOLFIRI Alone as Second-line Treatment for Metastatic Colorectal Cancer. Clin. Cancer Res. 2015, 21, 5469–5479. [Google Scholar] [CrossRef] [PubMed]
- Ohhara, Y.; Fukuda, N.; Takeuchi, S.; Honma, R.; Shimizu, Y.; Kinoshita, I.; Dosaka-Akita, H. Role of targeted therapy in metastatic colorectal cancer. World J. Gastrointest. Oncol. 2016, 8, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Yu, P.; Qu, J.; Chen, Y.; Zhou, Y.; Fu, L.; Zhang, J. Efficacy of Bevacizumab in the First-Line Treatment of Patients with RAS Mutations Metastatic Colorectal Cancer: A Systematic Review and Network Meta-Analysis. Cell. Physiol. Biochem. 2016, 40, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Chu, E. Sequencing of antiangiogenic agents in the treatment of metastatic colorectal cancer. Clin. Colorectal Cancer 2014, 13, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Grothey, A.; Van Custem, E.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouche, O.; Mineur, L.; Barone, C.; et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef]
- Li, J.; Qin, S.; Xu, R.; Yau, T.C.; Ma, B.; Pan, H.; Xu, J.; Bai, Y.; Chi, Y.; Wang, L.; et al. Regorafenib plus best supportive care versus placebo plus best supportive care in Asian patients with previously treated metastatic colorectal cancer (CONCUR): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2015, 16, 619–629. [Google Scholar] [CrossRef]
- Van Custem, E.; Tabernero, J.; Lakomy, R.; Prenen, H.; Prausova, J.; Macarulla, T.; Ruff, P.; van Hazel, G.A.; Moiseyenko, V.; Ferry, D.; et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J. Clin. Oncol. 2012, 30, 3499–3506. [Google Scholar]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Lievre, A.; Bachet, J.B.; Boige, V.; Cayre, A.; Le, C.D.; Buc, E.; Ychou, M.; Bouche, O.; Landi, B.; Louvet, C.; et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J. Clin. Oncol. 2008, 26, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Giusti, R.M.; Shastri, K.A.; Cohen, M.H.; Keegan, P.; Pazdur, R. FDA drug approval summary: Panitumumab (Vectibix). Oncologist 2007, 12, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Sickmier, E.A.; Kurzeja, R.J.; Michelsen, K.; Vazir, M.; Yang, E.; Tasker, A.S. The Panitumumab EGFR Complex Reveals a Binding Mechanism That Overcomes Cetuximab Induced Resistance. PLoS ONE. 2016, 11, e0163366. [Google Scholar] [CrossRef] [PubMed]
- Bertotti, A.; Papp, E.; Jones, S.; Adleff, V.; Anagnostou, V.; Lupo, B.; Sausen, M.; Phallen, J.; Hruban, C.A.; Tokheim, C.; et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature 2015, 526, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Leslie, M. Potential Therapy for Refractory Colon Cancer. Cancer Discov. 2016, 6, 336–337. [Google Scholar] [PubMed]
- Van Emburgh, B.O.; Arena, S.; Siravegna, G.; Lazzari, L.; Crisafulli, G.; Corti, G.; Mussolin, B.; Baldi, F.; Buscarino, M.; Bartolini, A.; et al. Acquired RAS or EGFR mutations and duration of response to EGFR blockade in colorectal cancer. Nat. Commun. 2016, 7, 13665. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Oddo, D.; Sennott, E.M.; Barault, L.; Valtorta, E.; Arena, S.; Cassingena, A.; Filiciotto, G.; Marzolla, G.; Elez, E.; van Geel, R.M.; et al. Molecular Landscape of Acquired Resistance to Targeted Therapy Combinations in BRAF-Mutant Colorectal Cancer. Cancer Res. 2016, 76, 4504–4515. [Google Scholar] [CrossRef] [PubMed]
- Richman, S.D.; Southward, K.; Chambers, P.; Cross, D.; Barrett, J.; Hemmings, G.; Taylor, M.; Wood, H.; Hutchins, G.; Foster, J.M.; et al. HER2 overexpression and amplification as a potential therapeutic target in colorectal cancer: Analysis of 3256 patients enrolled in the QUASAR, FOCUS and PICCOLO colorectal cancer trials. J. Pathol. 2016, 238, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Parikh, A.; Atreya, C.; Korn, W.M.; Venook, A.P. Prolonged Response to HER2-Directed Therapy in a Patient With HER2-Amplified, Rapidly Progressive Metastatic Colorectal Cancer. J. Natl. Compr. Canc. Netw. 2017, 15, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): A proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 738–746. [Google Scholar] [CrossRef]
- Rubinson, D.A.; Hochster, H.S.; Ryan, D.P.; Wolpin, B.M.; McCleary, N.J.; Abrams, T.A.; Chan, J.A.; Iqbal, S.; Lenz, H.J.; Lim, D.; et al. Multi-drug inhibition of the HER pathway in metastatic colorectal cancer: Results of a phase I study of pertuzumab plus cetuximab in cetuximab-refractory patients. Investig. New Drugs 2014, 32, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Kloth, M.; Ruesseler, V.; Engel, C.; Koenig, K.; Peifer, M.; Mariotti, E.; Kuenstlinger, H.; Florin, A.; Rommerscheidt-Fuss, U.; Koitzsch, U.; et al. Activating ERBB2/HER2 mutations indicate susceptibility to pan-HER inhibitors in Lynch and Lynch-like colorectal cancer. Gut 2016, 65, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Aung, K.L.; Stockley, T.L.; Serra, S.; Kamel-Reid, S.; Bedard, P.L.; Siu, L.L. Testing ERBB2 p.L755S kinase domain mutation as a druggable target in a patient with advanced colorectal cancer. Cold Spring Harb. Mol. Case. Stud. 2016, 2, a001016. [Google Scholar] [CrossRef] [PubMed]
- Schoeberl, B.; Faber, A.C.; Li, D.; Liang, M.C.; Crosby, K.; Onsum, M.; Burenkova, O.; Pace, E.; Walton, Z.; Nie, L.; et al. An ErbB3 antibody, MM-121, is active in cancers with ligand-dependent activation. Cancer Res. 2010, 70, 2485–2494. [Google Scholar] [CrossRef] [PubMed]
- Breuleux, M. Role of heregulin in human cancer. Cell. Mol. Life Sci. 2007, 64, 2358–2377. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Michael, M.; Zalcberg, J.R. An overview of experimental and investigational multikinase inhibitors for the treatment of metastatic colorectal cancer. Expert. Opin. Investig. Drugs 2015, 24, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 29 May 2015).
- Dienstmann, R.; Patnaik, A.; Garcia-Carbonero, R.; Cervantes, A.; Benavent, M.; Rosello, S.; Tops, B.B.; van der Post, R.S.; Argiles, G.; Skartved, N.J.; et al. Safety and Activity of the First-in-Class Sym004 Anti-EGFR Antibody Mixture in Patients with Refractory Colorectal Cancer. Cancer Discov. 2015, 5, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Martin, F.J.; Bellosillo, B.; Gelabert-Baldrich, M.; Dalmases, A.; Canadas, I.; Vidal, J.; Martinez, A.; Argiles, G.; Siravegna, G.; Arena, S.; et al. The First-in-class Anti-EGFR Antibody Mixture Sym004 Overcomes Cetuximab Resistance Mediated by EGFR Extracellular Domain Mutations in Colorectal Cancer. Clin. Cancer Res. 2016, 22, 3260–3267. [Google Scholar] [CrossRef] [PubMed]
- Arena, S.; Siravegna, G.; Mussolin, B.; Kearns, J.D.; Wolf, B.B.; Misale, S.; Lazzari, L.; Bertotti, A.; Trusolino, L.; Adjei, A.A.; et al. MM-151 overcomes acquired resistance to cetuximab and panitumumab in colorectal cancers harboring EGFR extracellular domain mutations. Sci. Transl. Med. 2016, 8, 324ra14. [Google Scholar] [CrossRef] [PubMed]
- Kearns, J.D.; Bukhalid, R.; Sevecka, M.; Tan, G.; Gerami-Moayed, N.; Werner, S.L.; Kohli, N.; Burenkova, O.; Sloss, C.M.; King, A.M.; et al. Enhanced Targeting of the EGFR Network with MM-151, an Oligoclonal Anti-EGFR Antibody Therapeutic. Mol. Cancer Ther. 2015, 14, 1625–1636. [Google Scholar] [CrossRef] [PubMed]
- Deming, D.A.; Cavalcante, L.L.; Lubner, S.J.; Mulkerin, D.L.; LoConte, N.K.; Eickhoff, J.C.; Kolesar, J.M.; Fioravanti, S.; Greten, T.F.; Compton, K.; et al. A phase I study of selumetinib (AZD6244/ARRY-142866), a MEK1/2 inhibitor, in combination with cetuximab in refractory solid tumors and KRAS mutant colorectal cancer. Invest. New Drugs 2016, 34, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; Atreya, C.E.; Falchook, G.S.; Kwak, E.L.; Ryan, D.P.; Bendell, J.C.; Hamid, O.; Messersmith, W.A.; Daud, A.; Kurzrock, R.; et al. Combined BRAF and MEK Inhibition With Dabrafenib and Trametinib in BRAF V600-Mutant Colorectal Cancer. J. Clin. Oncol. 2015, 33, 4023–4031. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, A.; Calegari, A.; Inno, A.; Berenato, R.; Caporale, M.; Niger, M.; Bossi, I.; Di, B.M.; de, B.F.; Pietrantonio, F. BRAF in metastatic colorectal cancer: The future starts now. Pharmacogenomics. 2015, 16, 2069–2081. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.B.; McMahon, C.; Ali, S.M.; Abramovitz, M.; Williams, K.A.; Klein, J.; McKean, H.; Yelensky, R.; George, T.J., Jr.; Elvin, J.A.; et al. A metastatic colon adenocarcinoma harboring BRAF V600E has a durable major response to dabrafenib/trametinib and chemotherapy. Onco. Targets. Ther. 2015, 8, 3561–3564. [Google Scholar] [PubMed]
- Kopetz, S.; Desai, J.; Chan, E.; Hecht, J.R.; O’Dwyer, P.J.; Maru, D.; Morris, V.; Janku, F.; Dasari, A.; Chung, W.; et al. Phase II Pilot Study of Vemurafenib in Patients With Metastatic BRAF-Mutated Colorectal Cancer. J. Clin. Oncol. 2015, 33, 4032–4038. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Morris, V.K.; El, O.B.; Sorokin, A.V.; Janku, F.; Fu, S.; Overman, M.J.; Piha-Paul, S.; Subbiah, V.; Kee, B.; et al. Phase IB Study of Vemurafenib in Combination with Irinotecan and Cetuximab in Patients with Metastatic Colorectal Cancer with BRAFV600E Mutation. Cancer Discov. 2016, 6, 1352–1365. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Cercek, A.; O'Reilly, E.M.; Reidy, D.L.; Kemeny, N.; Wolinsky, T.; Capanu, M.; Gollub, M.J.; Rosen, N.; Berger, M.F.; et al. Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clin. Cancer Res. 2015, 21, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Van Geel, R.M.J.M.; Tabernero, J.; Elez, E.; Bendell, J.C.; Spreafico, A.; Schuler, M.; Yoshino, T.; Delord, J.P.; Yamada, Y.; Lolkema, M.P.; et al. A Phase Ib Dose-Escalation Study of Encorafenib and Cetuximab with or without Alpelisib in Metastatic BRAF-Mutant Colorectal Cancer. Cancer Discov. 2017, 7, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lu, B.; Liu, D.; Shen, R.; Yan, Y.; Yang, L.; Zhang, M.; Zhang, L.; Cao, G.; Cao, H.; et al. EBI-907, a novel BRAF(V600E) inhibitor, has potent oral anti-tumor activity and a broad kinase selectivity profile. Cancer Biol. Ther. 2016, 17, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Yuan, X.; Du, R.; Cheung, S.H.; Zhang, G.; Wei, J.; Zhao, Y.; Feng, Y.; Peng, H.; Zhang, Y.; et al. BGB-283, a Novel RAF Kinase and EGFR Inhibitor, Displays Potent Antitumor Activity in BRAF-Mutated Colorectal Cancers. Mol. Cancer Ther. 2015, 14, 2187–2197. [Google Scholar] [CrossRef] [PubMed]
- Dasari, A.; Messersmith, W.A. New strategies in colorectal cancer: Biomarkers of response to epidermal growth factor receptor monoclonal antibodies and potential therapeutic targets in phosphoinositide 3-kinase and mitogen-activated protein kinase pathways. Clin. Cancer Res. 2010, 16, 3811–3818. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Yang, Z.Y.; Hu, X.F.; Chen, Q.; Tang, J.L. PIK3CA exon 20 mutations as a potential biomarker for resistance to anti-EGFR monoclonal antibodies in KRAS wild-type metastatic colorectal cancer: A systematic review and meta-analysis. Ann. Oncol. 2012, 23, 1518–1525. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Brana, I.; Siu, L.L.; De Jonge, M.J.; Homji, N.; Mills, D.; Di, T.E.; Sarr, C.; Trandafir, L.; Massacesi, C.; et al. Phase I dose-escalation and -expansion study of buparlisib (BKM120), an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2014, 32, 670–681. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, M.S.; Melo, S.; Velho, S.; Carneiro, P.; Carneiro, F.; Seruca, R. Specific inhibition of p110alpha subunit of PI3K: Putative therapeutic strategy for KRAS mutant colorectal cancers. Oncotarget. 2016, 7, 68546–68558. [Google Scholar] [PubMed]
- Jifu, E.; Xing, J.; Gong, H.; He, J.; Zhang, W. Combine MEK inhibition with PI3K/mTOR inhibition exert inhibitory tumor growth effect on KRAS and PIK3CA mutation CRC xenografts due to reduced expression of VEGF and matrix metallopeptidase-9. Tumour. Biol. 2015, 36, 1091–1097. [Google Scholar]
- Fang, D.D.; Zhang, C.C.; Gu, Y.; Jani, J.P.; Cao, J.; Tsaparikos, K.; Yuan, J.; Thiel, M.; Jackson-Fisher, A.; Zong, Q.; et al. Antitumor Efficacy of the Dual PI3K/mTOR Inhibitor PF-04691502 in a Human Xenograft Tumor Model Derived from Colorectal Cancer Stem Cells Harboring a PIK3CA Mutation. PLoS ONE 2013, 8, e67258. [Google Scholar] [CrossRef] [PubMed]
- Hanna, D.L.; Lenz, H.J. Novel therapeutics in metastatic colorectal cancer: Molecular insights and pharmacogenomic implications. Expert Rev. Clin. Pharmacol. 2016, 9, 1091–1108. [Google Scholar] [CrossRef] [PubMed]
- Konda, B.; Shum, H.; Rajdev, L. Anti-angiogenic agents in metastatic colorectal cancer. World J. Gastrointest. Oncol. 2015, 7, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.S.L.; Wang, K.; Wu, G.; Shi, C.; Ding, K.; Lin, L.; Wang, J.; Xiong, J.; Wu, C.; Li, J.; et al. A randomized, double-blind, parallel-group, placebo-controlled, multicenter, phase II clinical study of famitinib in the treatment of advanced metastatic colorectal cancer. J. Clin. Oncol. 2015, 33, 513. [Google Scholar] [CrossRef]
- Van Custem, E.; Prenen, H.; D'Haens, G.; Bennouna, J.; Carrato, A.; Ducreux, M.; Bouche, O.; Sobrero, A.; Latini, L.; Staines, H.; et al. A phase I/II, open-label, randomised study of nintedanib plus mFOLFOX6 versus bevacizumab plus mFOLFOX6 in first-line metastatic colorectal cancer patients. Ann. Oncol. 2015, 26, 2085–2091. [Google Scholar]
- Van Custem, E.; Yoshino, T.; Hocke, J.; Oum’Hamed, Z.; Studeny, M.; Tabernero, J. Rationale and Design for the LUME-Colon 1 Study: A Randomized, Double-Blind, Placebo-Controlled Phase III Trial of Nintedanib Plus Best Supportive Care Versus Placebo Plus Best Supportive Care in Patients With Advanced Colorectal Cancer Refractory to Standard Treatment. Clin. Colorectal Cancer 2016, 15, 91–94. [Google Scholar]
- Hecht, J.R.; Trarbach, T.; Hainsworth, J.D.; Major, P.; Jager, E.; Wolff, R.A.; Lloyd-Salvant, K.; Bodoky, G.; Pendergrass, K.; Berg, W.; et al. Randomized, placebo-controlled, phase III study of first-line oxaliplatin-based chemotherapy plus PTK787/ZK 222584, an oral vascular endothelial growth factor receptor inhibitor, in patients with metastatic colorectal adenocarcinoma. J. Clin. Oncol. 2011, 29, 1997–2003. [Google Scholar] [CrossRef] [PubMed]
- Van Custem, E.; Bajetta, E.; Valle, J.; Kohne, C.H.; Hecht, J.R.; Moore, M.; Germond, C.; Berg, W.; Chen, B.L.; Jalava, T.; et al. Randomized, placebo-controlled, phase III study of oxaliplatin, fluorouracil, and leucovorin with or without PTK787/ZK 222584 in patients with previously treated metastatic colorectal adenocarcinoma. J. Clin. Oncol. 2011, 29, 2004–2010. [Google Scholar]
- Carrato, A.; Swieboda-Sadlej, A.; Staszewska-Skurczynska, M.; Lim, R.; Roman, L.; Shparyk, Y.; Bondarenko, I.; Jonker, D.J.; Sun, Y.; De la Cruz, J.A.; et al. Fluorouracil, leucovorin, and irinotecan plus either sunitinib or placebo in metastatic colorectal cancer: A randomized, phase III trial. J. Clin. Oncol. 2013, 31, 1341–1347. [Google Scholar] [CrossRef] [PubMed]
- Samalin, E.; Bouche, O.; Thezenas, S.; Francois, E.; Adenis, A.; Bennouna, J.; Taieb, J.; Desseigne, F.; Seitz, J.F.; Conroy, T.; et al. Sorafenib and irinotecan (NEXIRI) as second- or later-line treatment for patients with metastatic colorectal cancer and KRAS-mutated tumours: A multicentre Phase I/II trial. Br. J. Cancer 2014, 110, 1148–1154. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Garcia-Carbonero, R.; Cassidy, J.; Sobrero, A.; Van Custem, E.; Kohne, C.H.; Tejpar, S.; Gladkov, O.; Davidenko, I.; Salazar, R.; et al. Sorafenib in combination with oxaliplatin, leucovorin, and fluorouracil (modified FOLFOX6) as first-line treatment of metastatic colorectal cancer: The RESPECT trial. Clin. Cancer Res. 2013, 19, 2541–2550. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.A., Jr.; Bardelli, A. Liquid biopsies: Genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Diehl, F.; Li, M.; Dressman, D.; He, Y.; Shen, D.; Szabo, S.; Diaz, L.A., Jr.; Goodman, S.N.; David, K.A.; Juhl, H.; et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc. Natl. Acad. Sci. USA 2005, 102, 16368–16373. [Google Scholar] [CrossRef] [PubMed]
- Higgins, M.J.; Jelovac, D.; Barnathan, E.; Blair, B.; Slater, S.; Powers, P.; Zorzi, J.; Jeter, S.C.; Oliver, G.R.; Fetting, J.; et al. Detection of tumor PIK3CA status in metastatic breast cancer using peripheral blood. Clin. Cancer Res. 2012, 18, 3462–3469. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.W.; Balaj, L.; Liau, L.M.; Samuels, M.L.; Kotsopoulos, S.K.; Maguire, C.A.; Loguidice, L.; Soto, H.; Garrett, M.; Zhu, L.D.; et al. BEAMing and Droplet Digital PCR Analysis of Mutant IDH1 mRNA in Glioma Patient Serum and Cerebrospinal Fluid Extracellular Vesicles. Mol. Ther. Nucleic Acids 2013, 2, e109. [Google Scholar] [CrossRef] [PubMed]
- Kinde, I.; Wu, J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 9530–9535. [Google Scholar] [CrossRef] [PubMed]
- El Massaoudi, S.; Mouliere, F.; Du, M.S.; Bascoul-Mollevi, C.; Gillet, B.; Nouaille, M.; Fiess, C.; Crapez, E.; Bibeau, F.; Theillet, C.; et al. Circulating DNA as a Strong Multimarker Prognostic Tool for Metastatic Colorectal Cancer Patient Management Care. Clin. Cancer Res. 2016, 22, 3067–3077. [Google Scholar] [CrossRef] [PubMed]
- Spindler, K.L.; Pallisgaard, N.; Andersen, R.F.; Brandslund, I.; Jakobsen, A. Circulating free DNA as biomarker and source for mutation detection in metastatic colorectal cancer. PLoS ONE. 2015, 10, e0108247. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Kinde, I.; Wang, Y.; Wong, H.L.; Roebert, J.; Christie, M.; Tacey, M.; Wong, R.; Singh, M.; Karapetis, C.S.; et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann. Oncol. 2015, 26, 1715–1722. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, M.; Dawson, S.J.; Tsui, D.W.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.F.; Kingsbury, Z.; Wong, A.S.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, L. Gastrointestinal cancer: A step closer to combating acquired resistance in CRC. Nat. Rev. Clin. Oncol. 2012, 9, 428. [Google Scholar] [CrossRef] [PubMed]
- McLeod, H.L. Cancer pharmacogenomics: Early promise, but concerted effort needed. Science 2013, 339, 1563–1566. [Google Scholar] [CrossRef] [PubMed]
- Sadanandam, A.; Lyssiotis, C.A.; Homicsko, K.; Collisson, E.A.; Gibb, W.J.; Wullschleger, S.; Ostos, L.C.; Lannon, W.A.; Grotzinger, C.; Del, R.M.; et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat. Med. 2013, 19, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Mlecnik, B.; Bindea, G.; Angell, H.K.; Maby, P.; Angelova, M.; Tougeron, D.; Church, S.E.; Lafontaine, L.; Fischer, M.; Fredriksen, T.; et al. Integrative Analyses of Colorectal Cancer Show Immunoscore Is a Stronger Predictor of Patient Survival Than Microsatellite Instability. Immunity 2016, 44, 698–711. [Google Scholar] [CrossRef] [PubMed]
- U.S. FOOD & DRUG Administration. Available online: http://www.fda.gov/drugs/scienceresearch/researchareas/pharmacogenetics/ucm083378.htm (accessed on 29 May 2015).
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Gene | Rs Code | Nucleotide Change and/or Location | Therapy | Setting | Patients Population | Ethnicity | Clinical End-Points | Main Effect | Citation |
|---|---|---|---|---|---|---|---|---|---|
| Bevacizumab | |||||||||
| KRAS | n.a. | G12A/V (exon 2) | BV-based therapy (combination with FOLFOX, FOLFIRI, FUFA, XELOX, XELIRI, XELODA, CAMPTO or OXALIPLATIN) | Fisrt-line and greater | 404 mCRC | Caucasian | PFS OS | At the multivariable analysis KRAS G12V and G12A mutations showed a lower PSF (HR = 2.18, p < 0.001) and OS (HR = 2.58, p < 0.001). | [11] |
| KRAS | n.a. | codons 12, 13 (exon 2) 59, 61 (exon 3) 117 and 146 (exon 4) | FOLFOXIRI plus BV vs FOLFIRI plus BV (TRIBE) | First-line | 508 mCRC | Caucasian | OS PFS | RAS (HR = 1.49) and BRAF mutation (HR = 2.79) subgroups have shorter median OS compared with RAS/ BRAF wt subgroup (p < 0.0001) RAS (HR = 1.23) and BRAF (HR = 2.27) mutation subgroups have shorter median PFS compared with RAS and BRAF wt subgroup (p = 0.002) Comparing the two regimes, treatment effect was not significantly different across molecular subgroups | [12] |
| NRAS | n.a. | codons 12, 13 (exon 2) 59, 61 (exon 3) 117 and 146 (exon 4) | |||||||
| BRAF | rs113488022 | NM_004333.4:c.1799T > A (V600E) | |||||||
| KRAS | n.a. | codons 12, 13 (exon 2) 61 (exon 3) and 146 (exon 4) | FOLFOX6 or CapeOX or FOLFIRI plus BV | First-line | 90 mCRC | Japanese | ORR PFS | Even if not statistically significant, ORR was higher for patients with wt tumors (64.3%) compared to those with tumors that were only wt with respect to KRAS exon 2 (54.8%); the differences in ORR between patients with wt and mutant-type tumors were greater when considering only KRAS exon 2 mutations (6.8%) rather than RAS/PIK3CA/BRAF mutations (18.4%). At the multivariate analysis liver metastasis, unresectable primary tumor, RAS and BRAF tumor mutations resulted predictive for early progression. | [13] |
| NRAS | n.a. | codons 12, 13 (exon 2) and 61 (exon 3) | |||||||
| BRAF | rs113488022 | NM_004333.4:c.1799T > A (V600E) | |||||||
| PIK3CA | n.a. | codons 542, 545, 546 (exon 9) and 1047 (exon 20) | |||||||
| KRAS | n.a. | codons 12, 13 (exon 2) 59, 61 (exon 3) 117 and 146 (exon 4) | FOLFOX or XELOX plus BV vs. FOLFOX or XELOX | First-line | 93 mCRC | Caucasian | ORR PFS OS | RAS (KRAS or NRAS) mutations are not a prognostic marker for RR, PFS and OS | [14] |
| NRAS | n.a. | codons 12, 13 (exon 2) 59, 61 (exon 3) 117 and 146 (exon 4) | |||||||
| KRAS | n.a. | codons 12, 13 | 5-FU alone or in combination with oxaliplatin (FOLFOX/XELOX) and/or irinotecan (FOLFIRI/FOLFOXIRI) ± BV | Preoperative chemotherapy | 167 mCRC underwent pulmonary metastasectomy | Caucasian | LRRFS OS | For patients with KRAS exon 2 codon 12 mutations , perioperative BV was associated with a significant improvement in both LRRFS (p < 0.001) and OS (p < 0.001) | [15] |
| BRAF | rs113488022 | NM_004333.4:c.1799T > A (V600E) | |||||||
| Regorafenib | |||||||||
| KRAS | rs121913529 | NM_004985.4:c.35G > A (G12D) | Regorafenib plus BSC vs. placebo plus BSC (phase III CORRECT trial) | Salvage-line CT | 760 mCRC (505 regorafenib arm vs. 253 placebo arm) | Caucasian, Asian, North American, Australian | OS PFS | PFS and OS were higher in the Regorafenib arm irrespective of KRAS and PIK3CA mutational status. | [16] |
| NM_004985.4:c.35G > T (G12V) | |||||||||
| NM_004985.4:c.35G > C (G12A) | |||||||||
| rs122193530 | NM_004985.4:c.34G > T (G12C) | ||||||||
| NM_004985.4:c.34G > A (G12S) | |||||||||
| NM_004985.4:c.34G > C (G12R) | |||||||||
| rs112445441 | NM_004985.4:c.38G > A (G13D) | ||||||||
| rs17851045 | NM_004985.4:c.183A > C (Q61H) | ||||||||
| rs121913527 | NM_004985.4:c.436G > A (A146T) | ||||||||
| PIK3CA | rs121913273 | NM_006218.3:c.1624G > A (E542K) | |||||||
| rs104886003 | NM_006218.3:c.1633G > A (E545K) | ||||||||
| rs121913274 | NM_006218.3:c.1634A > G (E545G) | ||||||||
| rs121913279 | NM_006218.3:c.3140A > G (H1047R) | ||||||||
| NM_006218.3:c.3140A > T (H1047L) | |||||||||
| rs121913281 | NM_006218.3:c.3139C > T (H1047Y) | ||||||||
| KRAS | n.a. | Exons 2,3 and 4 | Regorafenib (REBECCA observational trial) | French compassionate program | 654 mCRC | Mostly caucasian | OS | In the multivariate analysis KRAS mutations were associated with shorter OS (HR:1.25; p = 0.016) in an independently manner from PS, number of metastatic sites and time of initial diagnosis | [17] |
| KDR | rs80338758 | NM_005120.2:c.2881C > T (R961W) | Regorafenib monotherapy | Second-line | 1 case report mCRC | American | RR | The patient with KDR c.2881C > T mutation experienced excellent tolerance and response to Regorafenib administration. | [18] |
| Aflibercept | |||||||||
| KRAS | n.a. | 9 mutation in codons 12, 13 (Exon 2) | Ziv-aflibercept plus mFOLFOX6 vs mFOLFOX6 (phase II AFFIRM trial) | First-line | 93 mCRC (47 treated with Aflibercept) | Asian/oriental, Black, Caucasian/white | PFS (primary endpoint) | Only patients with mutations in KRAS showed a not statistically significant trend to worse PFS when treated with mFOLFOX6 rather than mFOLFOX6 plus Aflibercept. Mutations in KRAS, NRAS and BRAF did not influenced PFS in both arms. | [19] |
| n.a. | 6 mutation in codon 61 (Exon 3) | ||||||||
| BRAF | rs113488022 | NM_004333.4:c.1799T > A (V600E) | |||||||
| rs121913338 | NM_004333.4:c.1781A > G (D594G) | ||||||||
| NRAS | n.a. | 12 mutations in codons 12,13 (exon 2) | |||||||
| n.a. | 5 mutations in codon 61 (exon 3) | ||||||||
| PTEN | n.a. | 2 deletions (codons 267,323) | |||||||
| n.a. | 5 mutations (codons 85, 173, 233, 130) | ||||||||
| PIK3CA | n.a. | 22 mutations (codons 345,38,420,539,542,545,546,88,901,1043,1047,1049,106,118) | |||||||
| PIK3R2 | n.a. | C1546G > A(R345Q) | |||||||
| PIK3R1 | n.a. | 17 mutations (codons 162,285,348,358,376,455,461,527,543,564,565,574,576,642,649,666,682) 5 insertions (codons 376,448,459,562–563,668–669) 2 deletions (codons 447,601) | |||||||
| EGFR | n.a. | 6 mutations (codons 289,858,719,790)2 deletions (codons 746–750, 746–750) | |||||||
| Ramucirumab | |||||||||
| KRAS | n.a. | Codon 12,13 (Exon 2) | RAM plus FOLFIRI vs. placebo plus FOLFIRI (RAISE trial) | Second-line | 1072 mCRC | mostly White and Asian | OS | KRAS wt patients showed a trend to longer OS (HR = 0.82, p = 0.049) in RAM-arm compared to placebo arm; KRAS mutated patients showed a trend to longer OS (HR = 0.89, p = 0.263) in RAM-arm compared to placebo arm. | [20] |
| KDR | n.a. | c.2312C > G (T771R) | RAM plus CTX plus CPT11 | Second-line | 1 case report of mCRC KRAS wt | American | n.a. | c.2312C > G variant is probably an activating mutation in response to Ramucirumab treatment | [21] |
| Cetuximab | |||||||||
| KRAS | rs112445441 | NM_004985.4: c.38G > A (G13D) | CTX vs. CTX plus CPT11 (phase II trial) | Second-line and greater | 29 mCRC | Japanese | PFS OS | In the KRAS G13D mutant subgroup, PFS and OS were not statistically different between the two arms. CTX effectiveness was similar in KRAS wt and KRAS G13D mutated | [22] |
| KRAS | rs112445441 | NM_004985.4:c.38G > A (G13D) | CTX vs. CTX plus CPT11 (phase II trial ICECREAM) | Second-line | 50 mCRC quadruple RAS wt and 50 mCRC KRAS G13D mutated | Australian | PFS | Efficacy of CTX administered alone or in combinatorial regimens was similar in quadruple RAS wt (KRAS, NRAS, BRAF and PIK3CA wt) and in G13D KRAS mutated | [23] |
| n.a. | Exons 3,4 | ||||||||
| NRAS | n.a. | Exons 2,3,4 | |||||||
| BRAF | n.a. | Exon 15 | |||||||
| PIK3CA | n.a. | Exons 9,20 | |||||||
| KRAS | rs112445441 | NM_004985.4:c.38G>A (G13D) | CTX | n.a. | 98 mCRC | Japanese | OS PFS | At the multivariate analysis there was a trend to better PFS (HR = 0.29; p = 0.07) in KRAS G13D mutated patients compared to other KRAS mutations. | [24] |
| Panitumumab | |||||||||
| KRAS | n.a. | codons 12 and 13 (exon 2) | PAN plus BSC vs. BSC (Phase III trial) | Second line and greater | 377 mCRC KRAS exon 2 wt | Asian Caucasian and other ethnicities | OS PFS | OS was significantly longer in PAN arm in both wt KRAS exon 2 (HR = 0.73, p = 0.0096) subgroup and wt RAS subgroup (HR = 0.70, p = 0.0135). PFS was significantly longer in PAN arm in both wt KRAS exon 2 (HR = 0.51, p < 0.0001) and wt RAS subgroup (HR=0.46, p < 0.0001). | [25] |
| n.a. | codons 59 and 61 (exon 3) | ||||||||
| n.a. | codons 117 and 146 (exon 4) | ||||||||
| NRAS | n.a. | codons 12 and 13 (exon 2) | |||||||
| n.a. | codons 59 and 61 (exon 3) | ||||||||
| n.a. | codons 117 and 146 (exon 4) | ||||||||
| KRAS | n.a. | Exons 2,3,4 | PAN or CTX plus CT PAN or CTX plus BSC (Meta-analysis of 10 studies) | 6 first-line 2 second-line 2 BSC | 463 CRC KRAS wt | n.a. | OS PFS ORR | EGFR-Is combined with chemotherapy do not significantly increase OS (HR: 0.91 p = 0.63), PFS (HR: 0.88 p = 0.33) and ORR (RR = 1.31 p = 0.25) in patients with BRAF mutated CRC. | [26] |
| NRAS | n.a. | Exons 2,3,4 | |||||||
| BRAF | rs113488022 | NM_004333.4:c.1799T > A (V600E) | |||||||
| KRAS | n.a. | codons 12 and 13 (exon 2) | PAN or CTX plus 5-FU, CPT11, Oxaliplatin based CT or BSC (Meta-analysis of 9 studies ) | 6 first-line 2 second-line 1 third-line (BSC) | 5948 mCRC | n.a. | PFS OS | EGFR-Is efficacy was found to be significantly superior in terms of PFS (HR=0.60, p < 0.001) and OS (HR = 0.72, p = 0.008) in all RAS wt patients compared with RAS mutated patients (KRAS exon 3 and 4 and NRAS exon 2,3 and 4) No difference in terms of both PFS and OS was found between KRAS exon 2 mutated and new RAS mutated subgroups. | [27] |
| codons 59 and 61 (exon 3) | |||||||||
| codons 117 and 146 (exon 4) | |||||||||
| NRAS | n.a. | codons 12 and 13 (exon 2) | |||||||
| codons 59 and 61 (exon 3) | |||||||||
| codons 117 and 146 (exon 4) | |||||||||
| KRAS | n.a. | codons 59 and 61 (exon 3) | PAN or CTX plus CT (Meta-analysis: 22 studies) | first-line and greater | 2395 mCRC KRAS exon 2 wt | Caucasian, American, Asian, African, Australian | ORR PFS OS | From 5 studies: KRAS exons 3 and 4 mutations were significantly correlated with worse ORR (OR = 0.26) and shorter PFS (HR = 2.19) and OS (HR = 1.78) From 3 studies: NRAS exons 2,3 and 4 mutations showed a trend towards poor ORR (OR = 0.23), and significant worse PFS (HR = 2.30) and OS (HR = 1.85) From 17 studies: BRAF mutations were correlated with significant worse ORR (OR = 0.29) and shorter PFS (HR = 2.95) and OS (HR = 2.52) From 6 studies: PIK3CA exons 9 and 20 mutations were significant predictors of poor ORR (OR = 0.39) and were significantly associated with shorter OS (HR = 1.43); only a trend for worse PFS was detected. From 5 studies of primary tumor: PTEN mutations showed non-significant effect on ORR whereas PTEN mutations and/or reduced expression were significantly correlated with lower ORR (OR = 0.41) and shorter PFS (PFS = 2.6) and OS (HR = 1.77) | [28] |
| n.a. | codons 117 and 146 (exon 4) | ||||||||
| NRAS | n.a. | codons 12 and 13 (exon 2) | |||||||
| n.a. | codons 59 and 61 (exon 3) | ||||||||
| n.a. | codons 117 and 146 (exon 4) | ||||||||
| BRAF | rs113488022 | NM_004333.4:c.1799T > A (V600E) | |||||||
| PIK3CA | n.a. | exons 1,2,9,10,20 | |||||||
| PTEN | n.a. | exons 1,2,3,4,5,6,7,8,9 | |||||||
| PIK3CA | rs121913279 | NM_006218.3: c.3140A > G (H1047R) | anti-EGFR mAb (Meta-analysis: 11 studies) | n.a. | 864 mCRC KRAS wt patients | n.a. | ORR PFS OS | All PIK3CA mutations were associated with overall reduced ORR (OR = 0.42 , p = 0.003) and the result remained significant considering only exon 20 mutated subset (OR = 0.21, p = 0.04) PIK3CA mutations were associated with shorter PFS (HR = 1.54, p = 0.006) and OS (HR = 1.4 p = 0.036) | [29] |
| NM_006218.3: c.3140A > T (H1047L) | |||||||||
| rs104886003 | NM_006218.3: c.1633G > A (E545K) | ||||||||
| n.a. | codon 542 (exon 9) | ||||||||
| KRAS | n.a. | exon 3 and 4 | FOLFIRI plus PAN vs. FOLFIRI (phase III trial) NCT0039183 | second-line | 1186 mCRC KRAS wt exon 2 | n.a. | PFS OS ORR | PFS was significantly longer in the PAN-arm with respect to both the KRAS exon 2 wt patients (HR = 0.73, p = 0.004) and all RAS wt patients (HR = 0.70, p = 0.007); in the same analysis there was a trend to longer OS in the PAN-arm | [30] |
| NRAS | n.a. | exon 2,3 and 4 | |||||||
| Targeted Agent | Biological Target | Registration Trial for mCRC | Somatic Variant Approved or Mandatory | Somatic Variant Explorative |
|---|---|---|---|---|
| Bevacizumab | VEGF-A/VEGFR2 | TRIBE, CAIRO2, NO16966 | / | KRAS, NRAS (exon 2) |
| Regorafenib | VEGFR-1-2-3, FGFR, PDGFR, RET, TIE-2, DDR-2, RAF-1, BRAF | CORRECT, CONCUR | / | KRAS |
| Aflibercept | VEGF-A-B PIGF | VELOUR | / | / |
| Ramucirumab | VEGFR-2 | RAISE | / | / |
| Cetuximab | ED-EGFR | CRYSTAL, OPUS, PRIME, NORDIC, COIN | * KRAS, NRAS, HRAS (Exons 2,3,4) | PIK3CA, PTEN (all exons) |
| Panitumumab | ED-EGFR | PICCOLO, 20050181 trial | * KRAS, NRAS, HRAS, EGFR (Exons 2,3,4) | PIK3CA, PTEN (all exons) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bignucolo, A.; De Mattia, E.; Cecchin, E.; Roncato, R.; Toffoli, G. Pharmacogenomics of Targeted Agents for Personalization of Colorectal Cancer Treatment. Int. J. Mol. Sci. 2017, 18, 1522. https://doi.org/10.3390/ijms18071522
Bignucolo A, De Mattia E, Cecchin E, Roncato R, Toffoli G. Pharmacogenomics of Targeted Agents for Personalization of Colorectal Cancer Treatment. International Journal of Molecular Sciences. 2017; 18(7):1522. https://doi.org/10.3390/ijms18071522
Chicago/Turabian StyleBignucolo, Alessia, Elena De Mattia, Erika Cecchin, Rossana Roncato, and Giuseppe Toffoli. 2017. "Pharmacogenomics of Targeted Agents for Personalization of Colorectal Cancer Treatment" International Journal of Molecular Sciences 18, no. 7: 1522. https://doi.org/10.3390/ijms18071522