Glycosaminoglycans Regulate CXCR3 Ligands at Distinct Levels: Protection against Processing by Dipeptidyl Peptidase IV/CD26 and Interference with Receptor Signaling

 , and
, and
Abstract
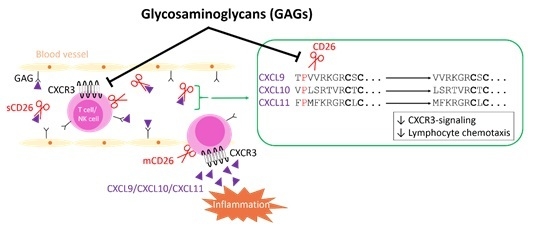
:
1. Introduction
2. Results
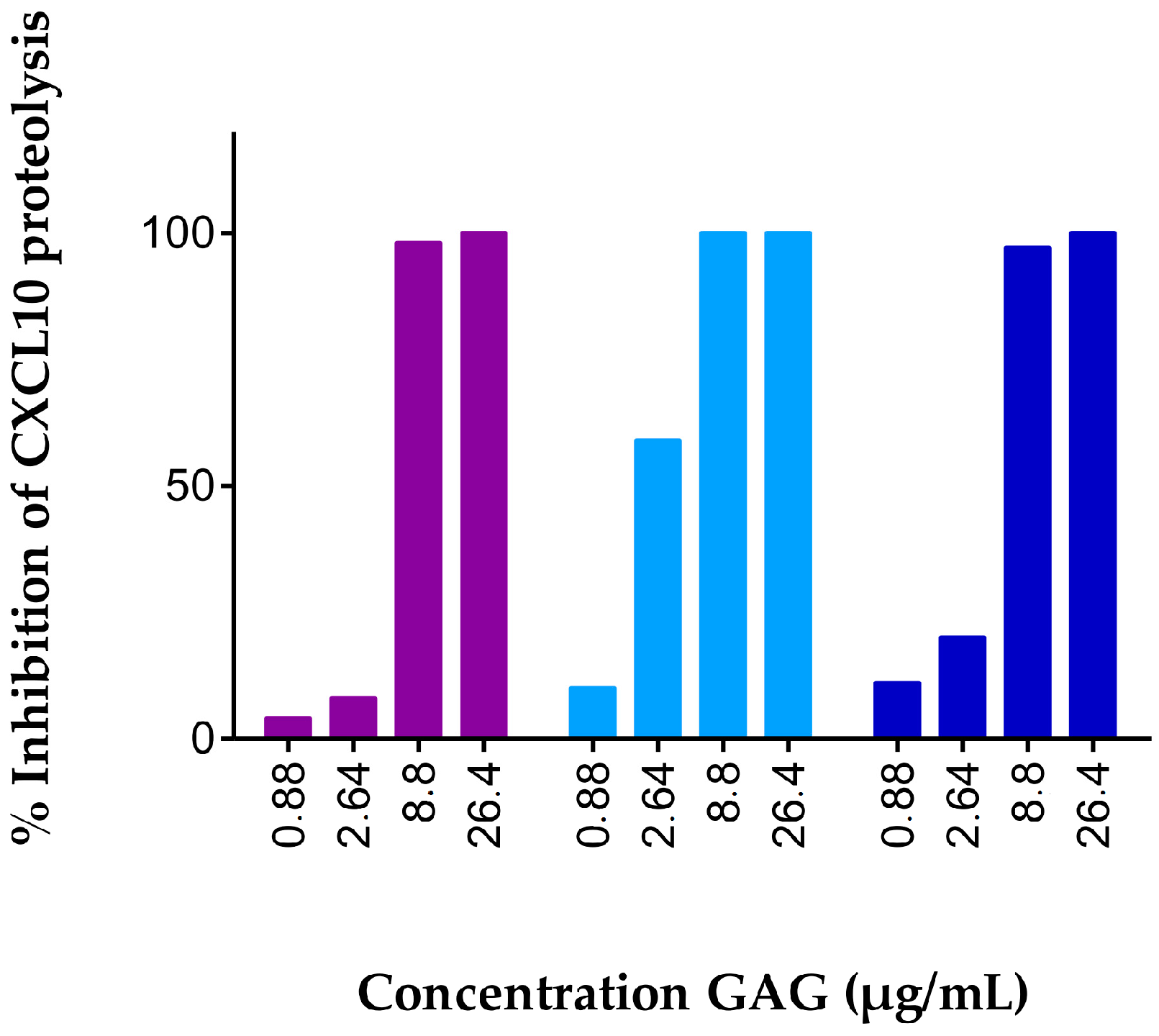
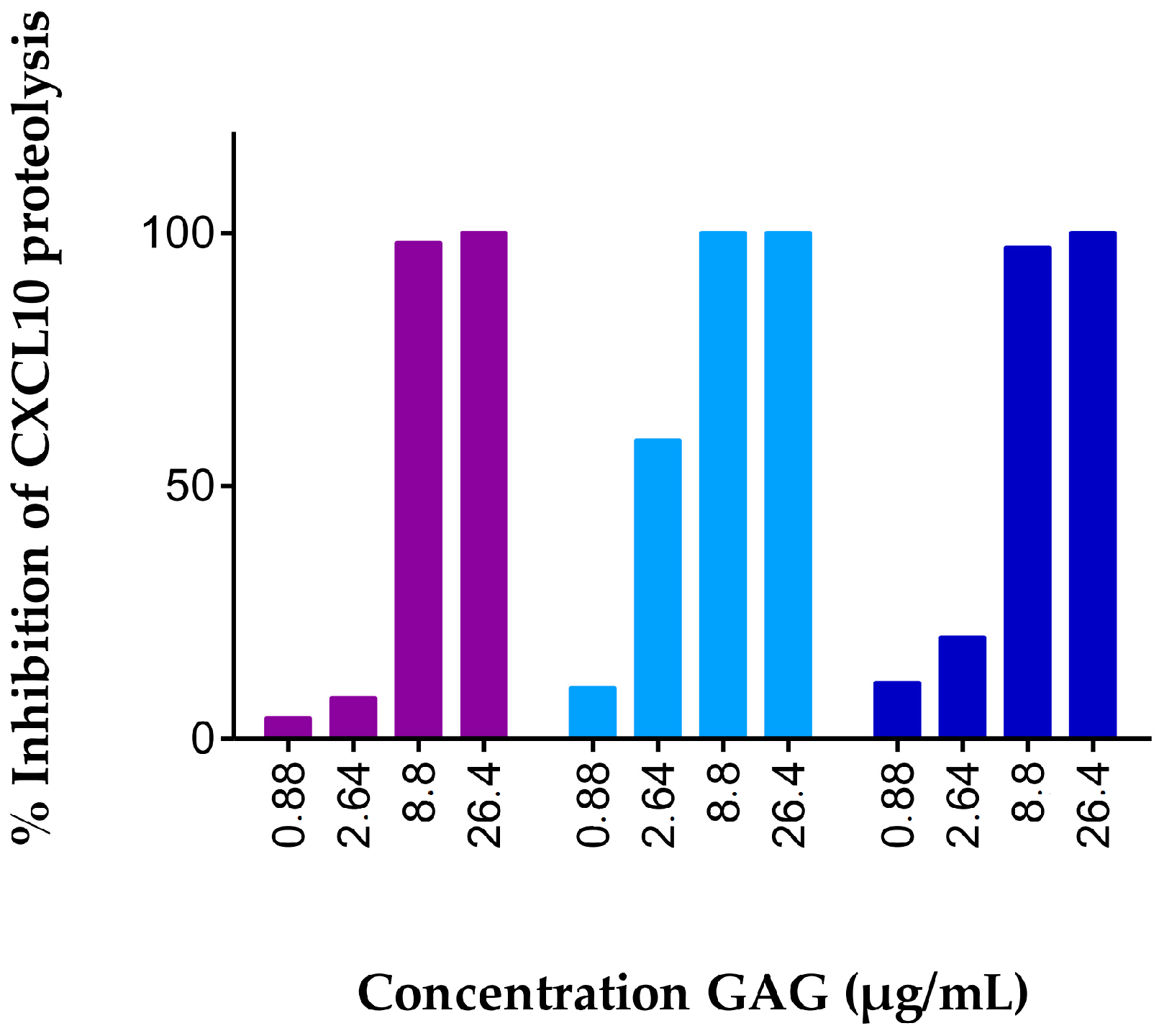
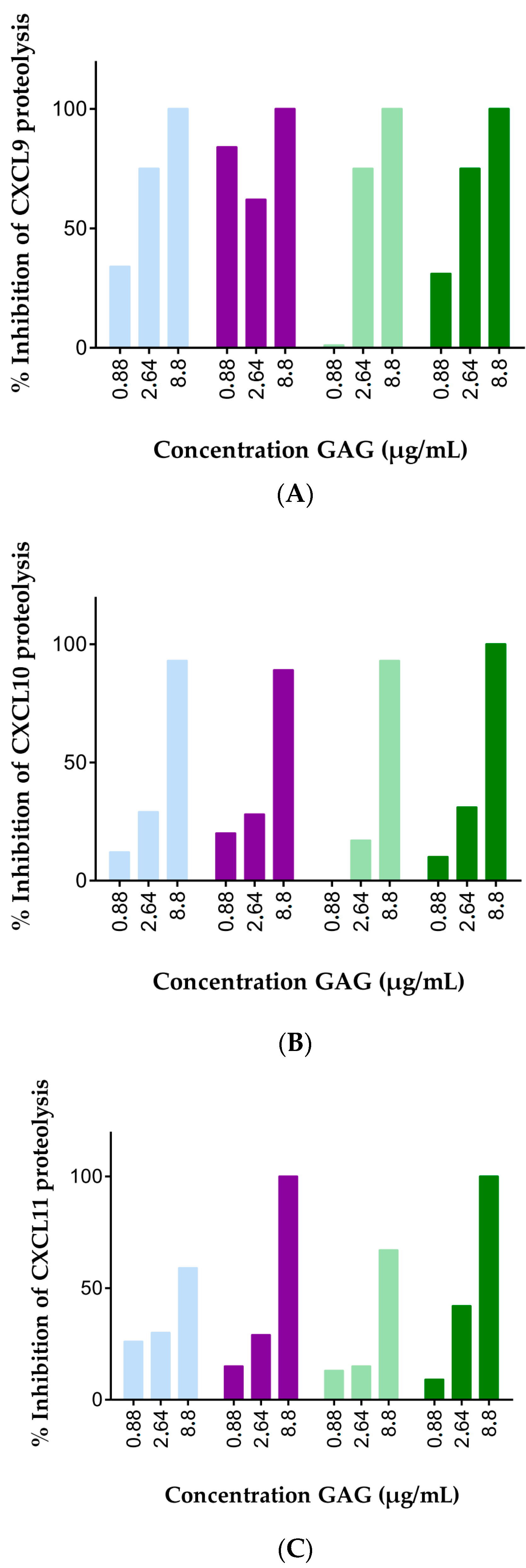
2.1. Soluble GAGs Protected CXCR3 Ligands against Truncation by Soluble CD26
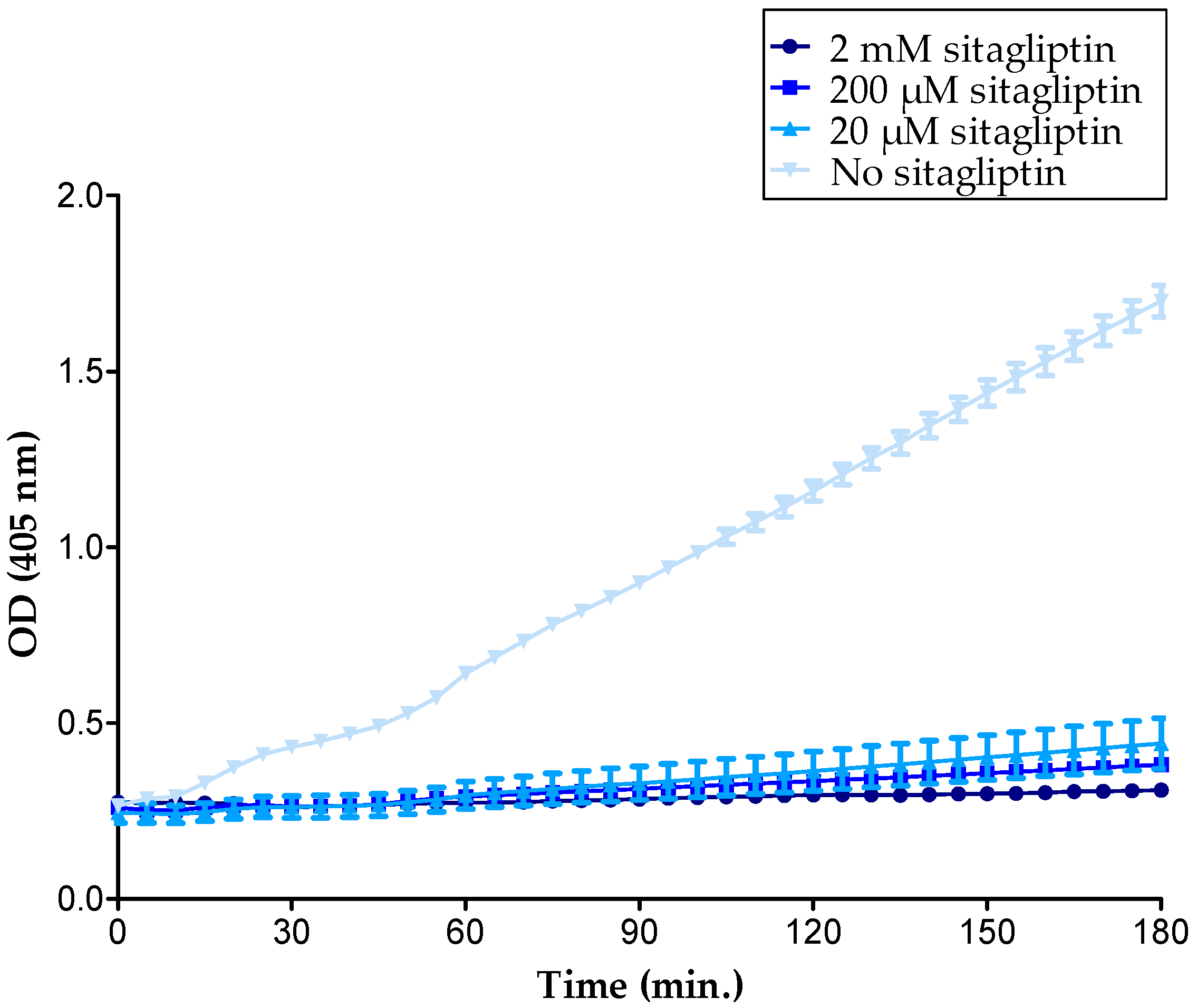
2.2. GAGs Did Not Inhibit the Enzymatic Activity of Soluble CD26 Directly
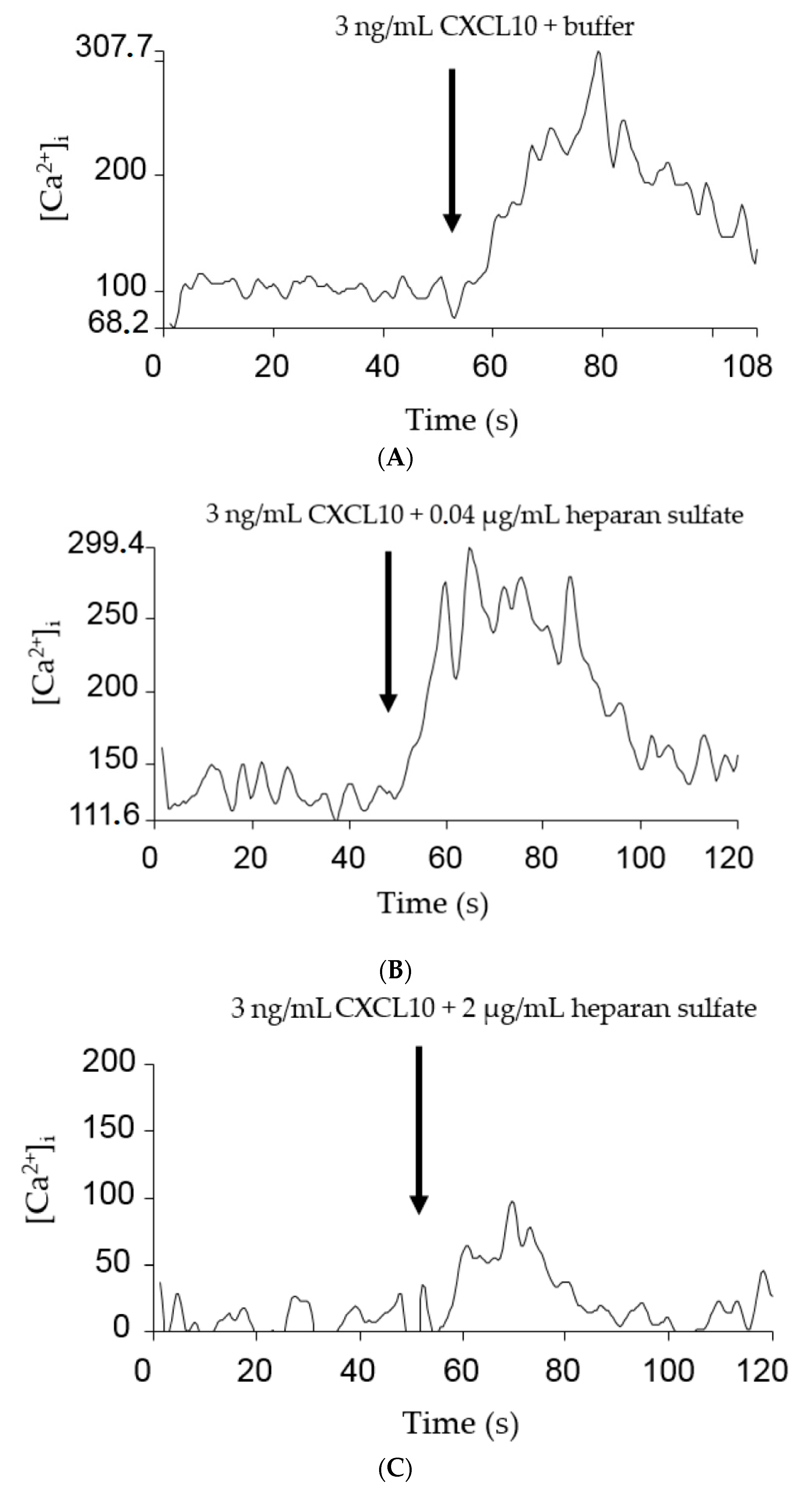
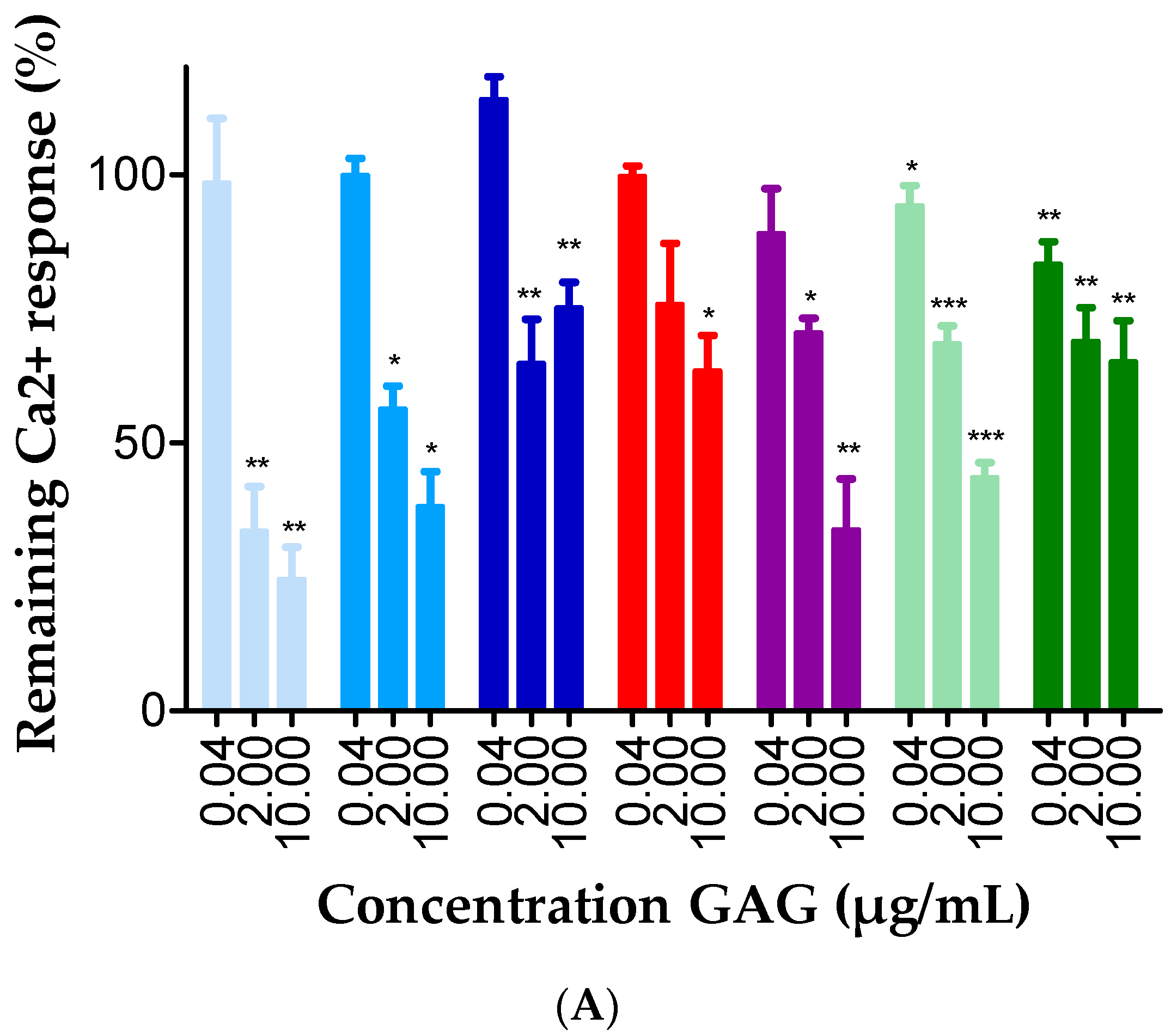
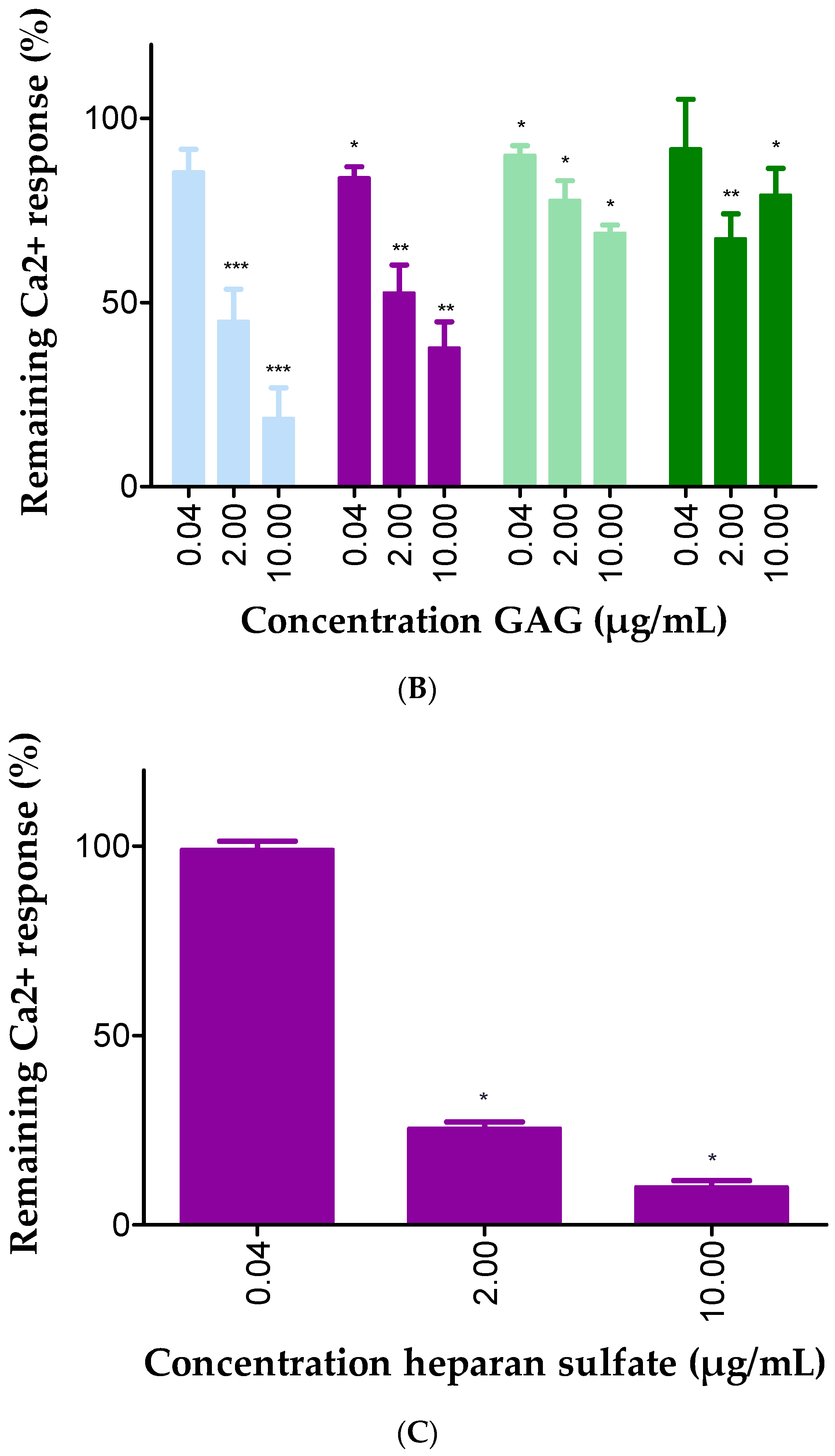
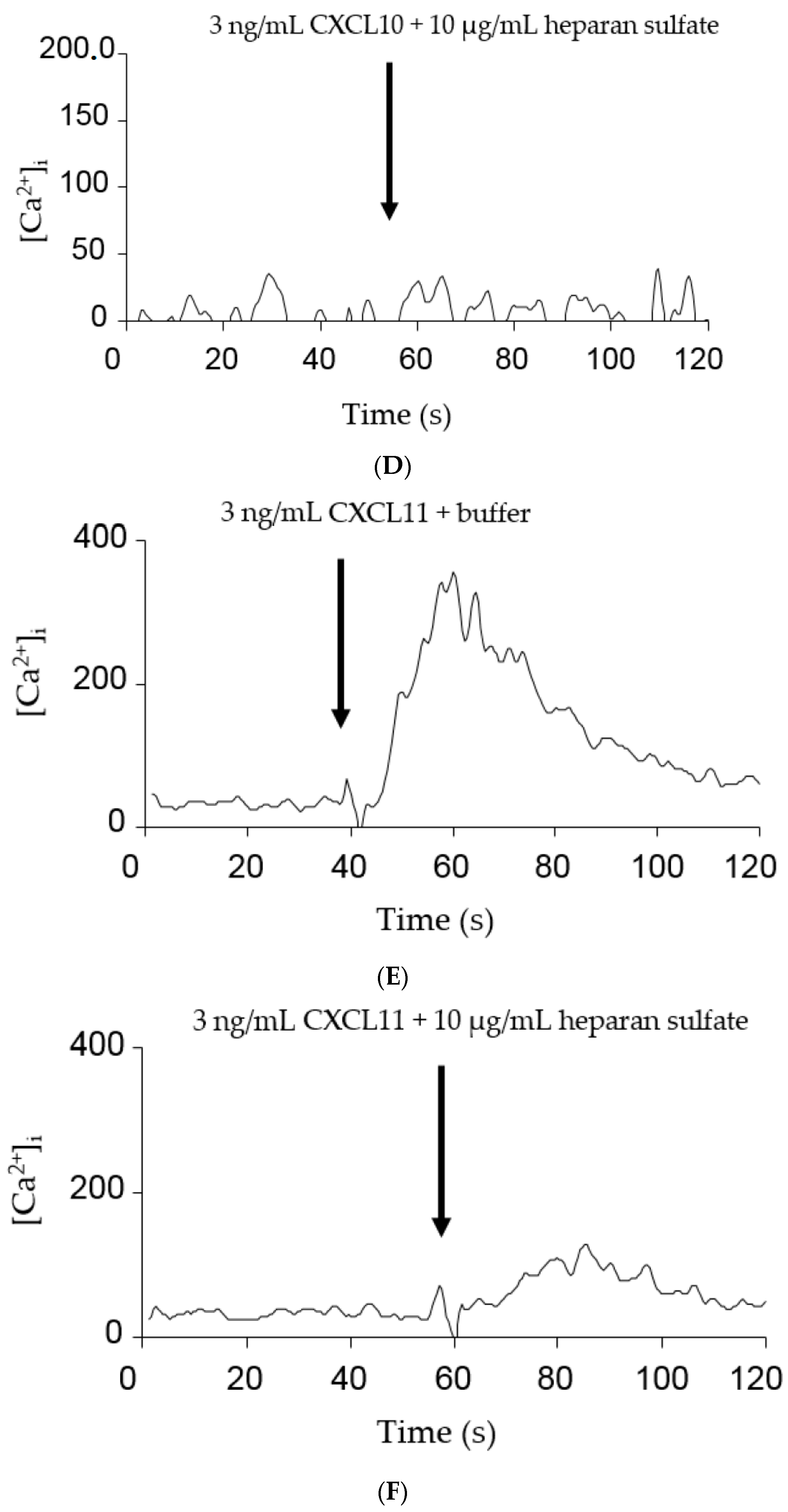
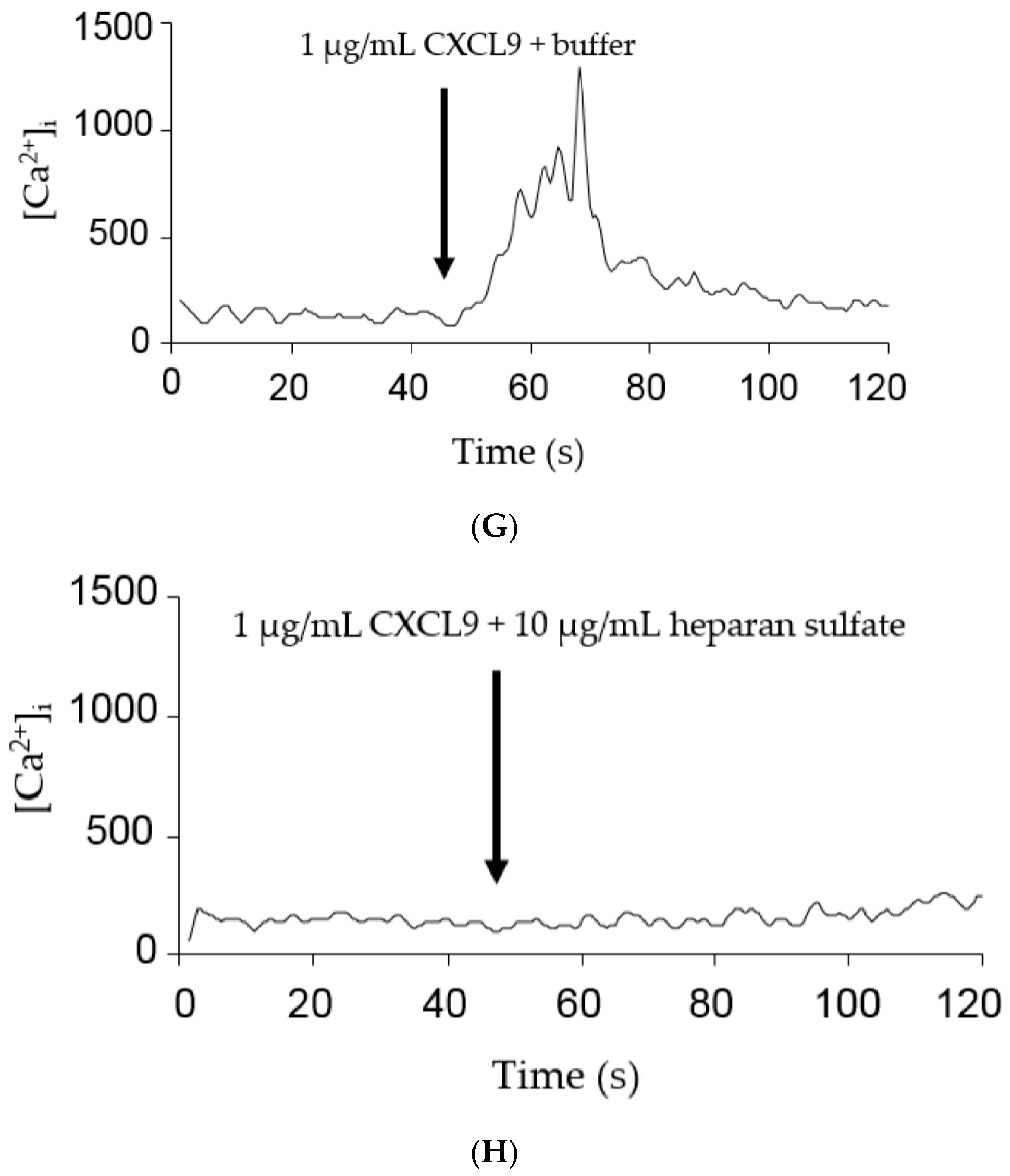
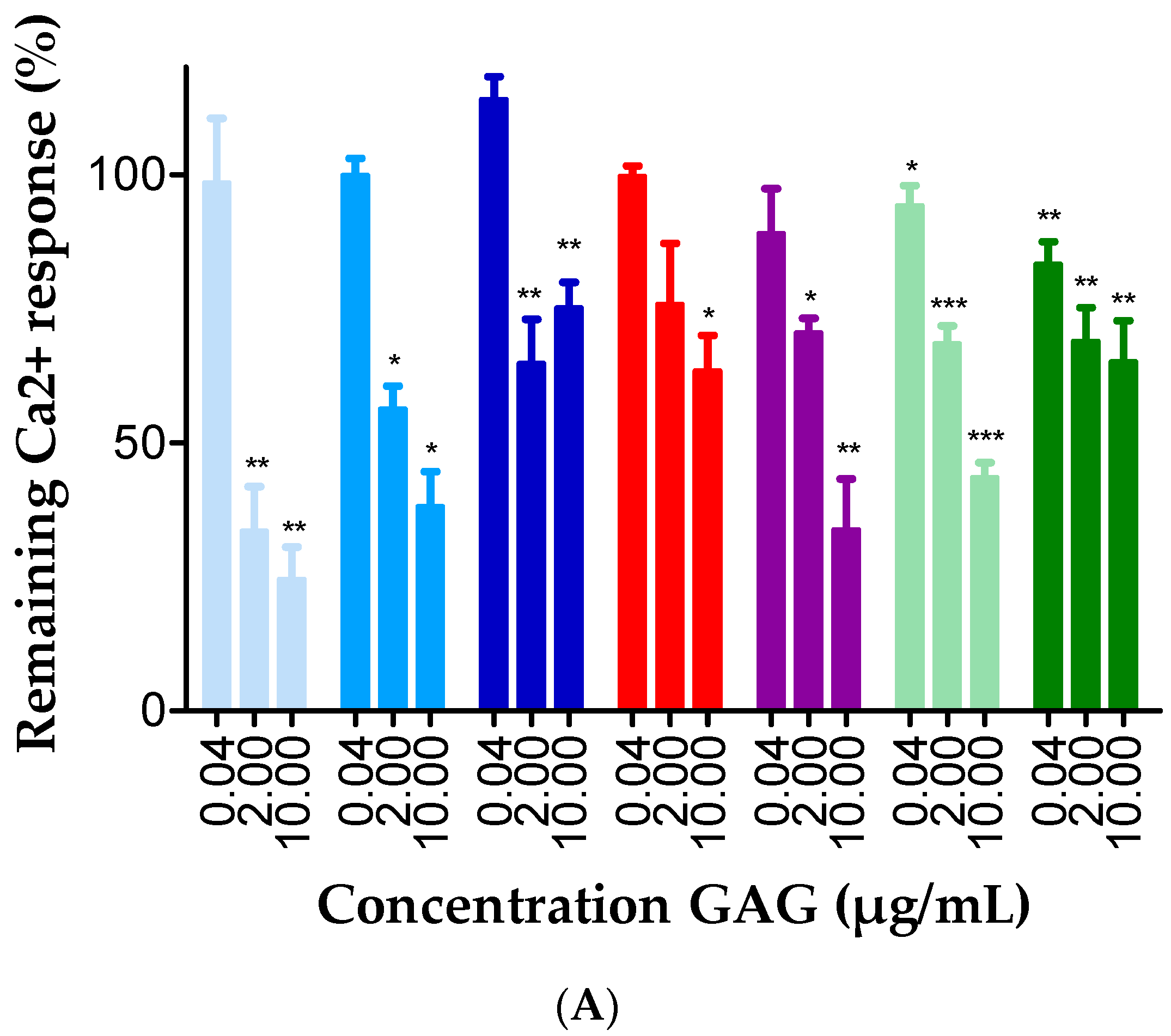
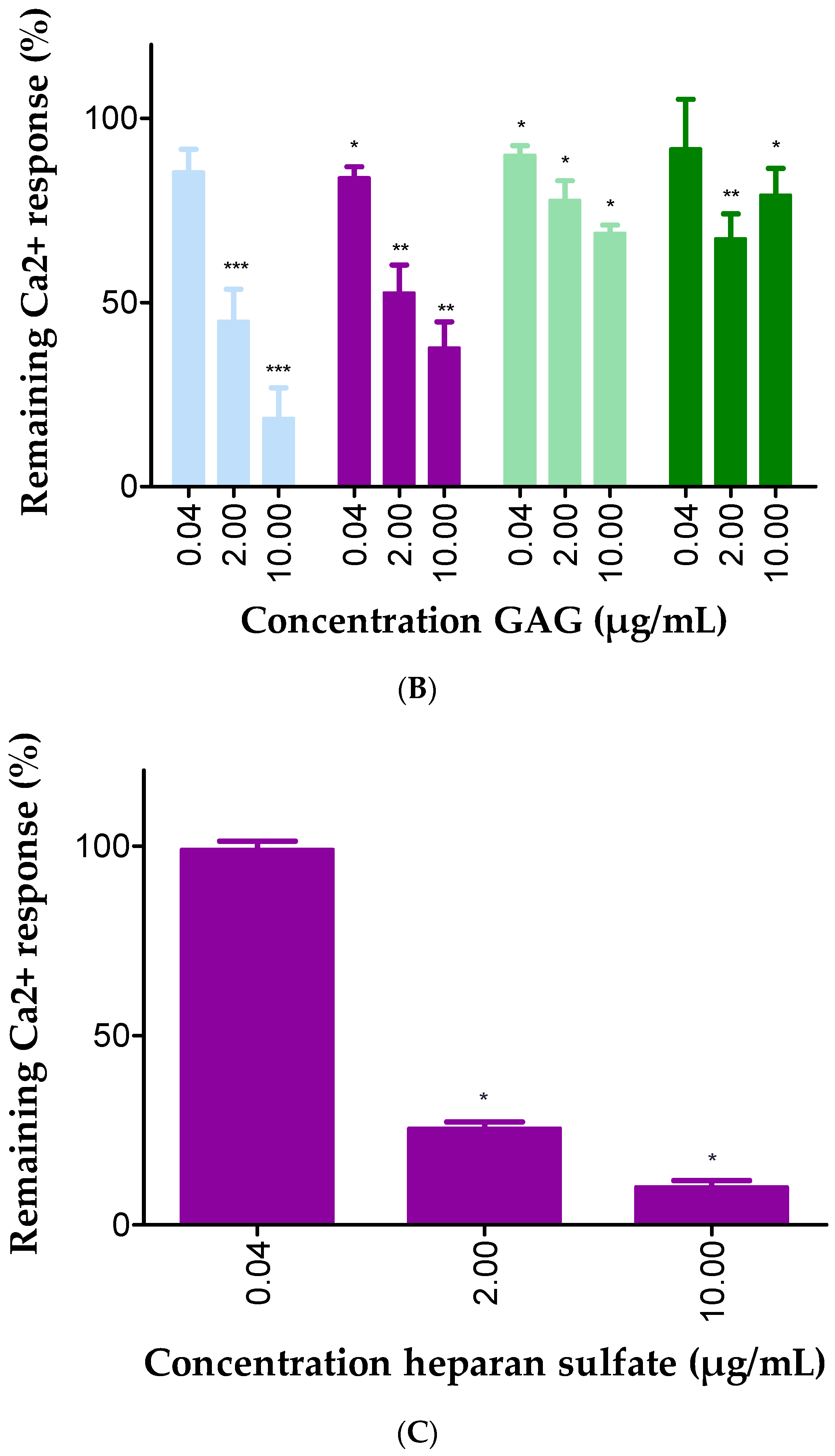
2.3. GAGs Interfered with Chemokine Signaling through CXCR3
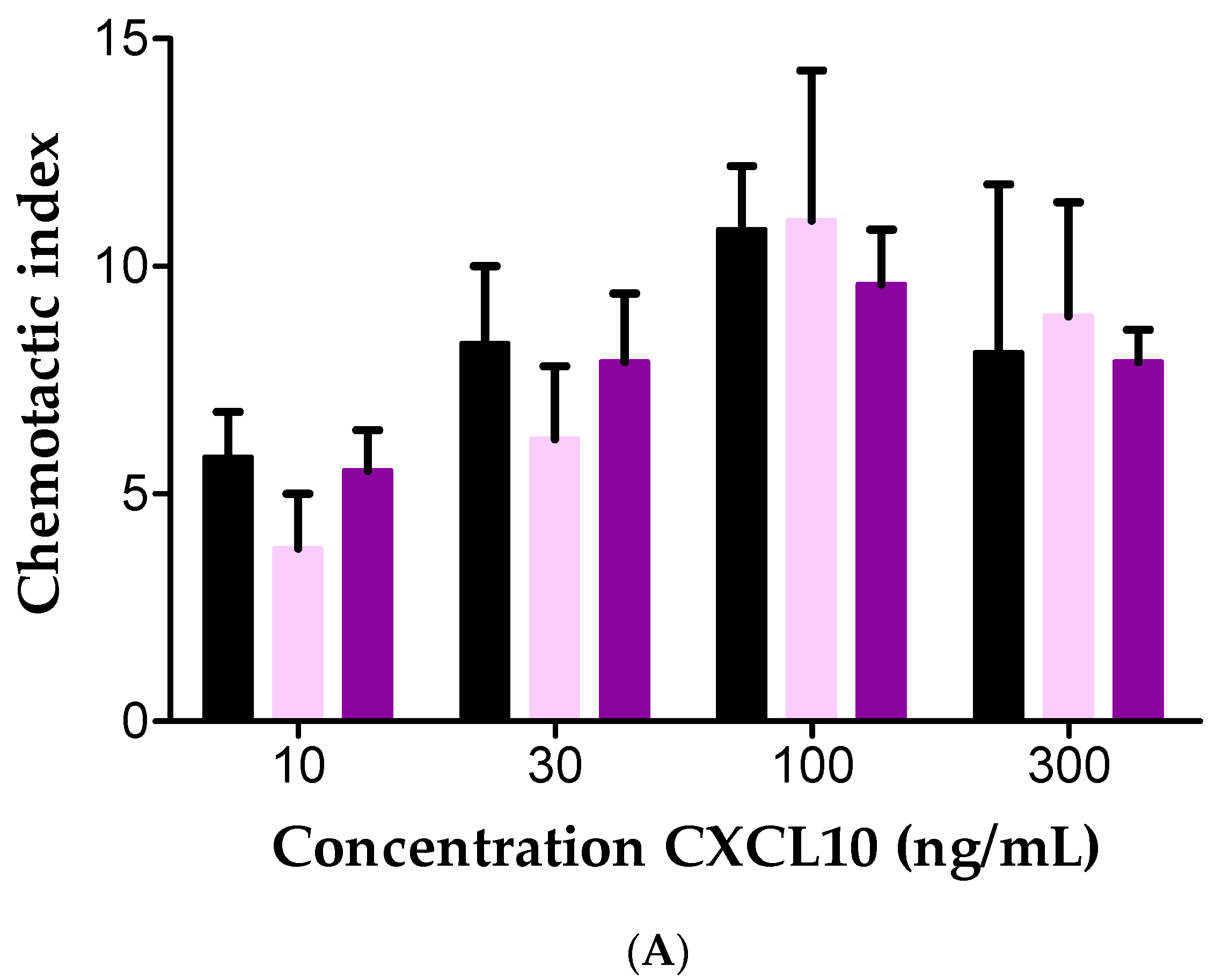
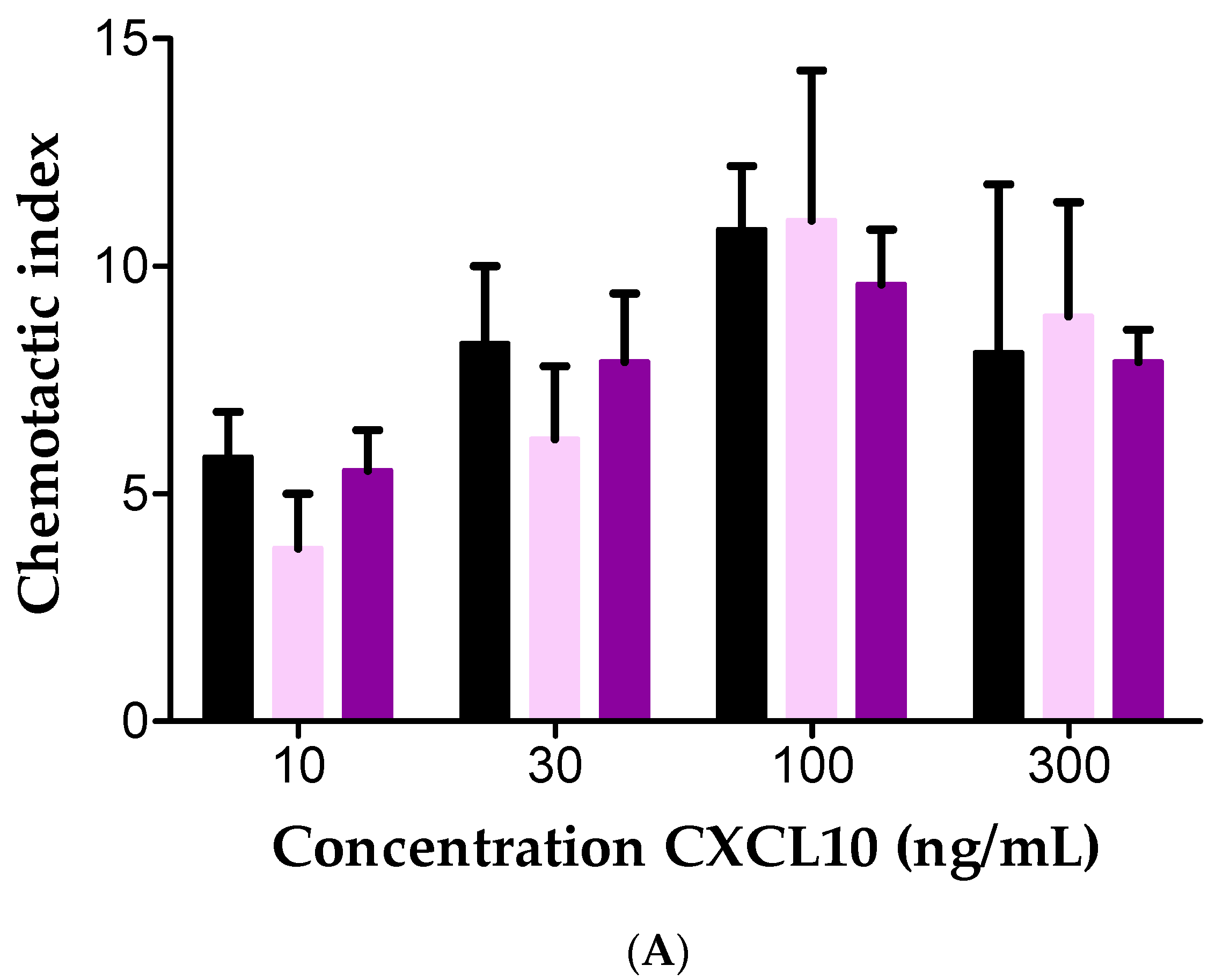
2.4. Effect of Soluble GAGs on CXCL10-Mediated CD26-Positive T Cell Chemotaxis In Vitro
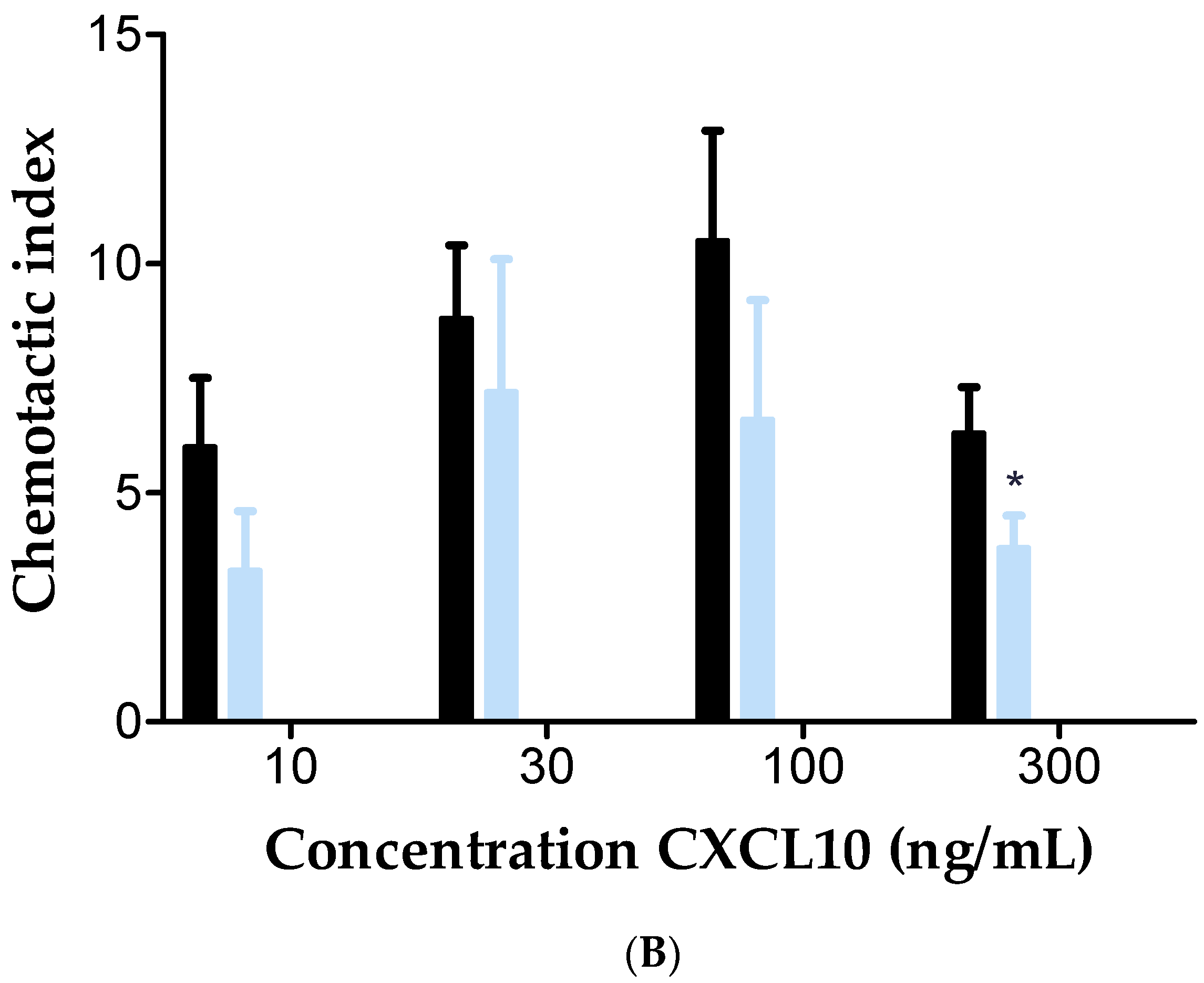
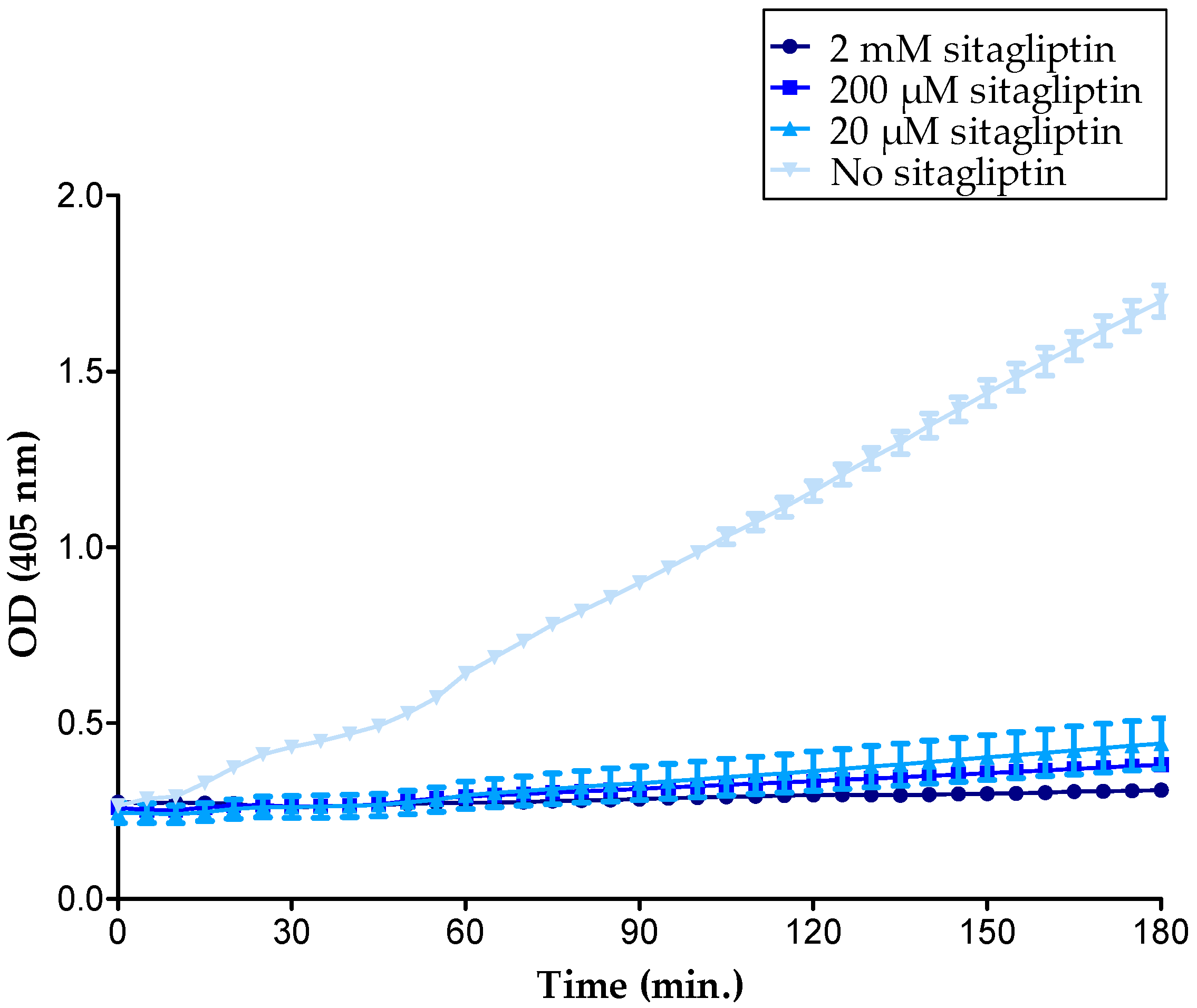
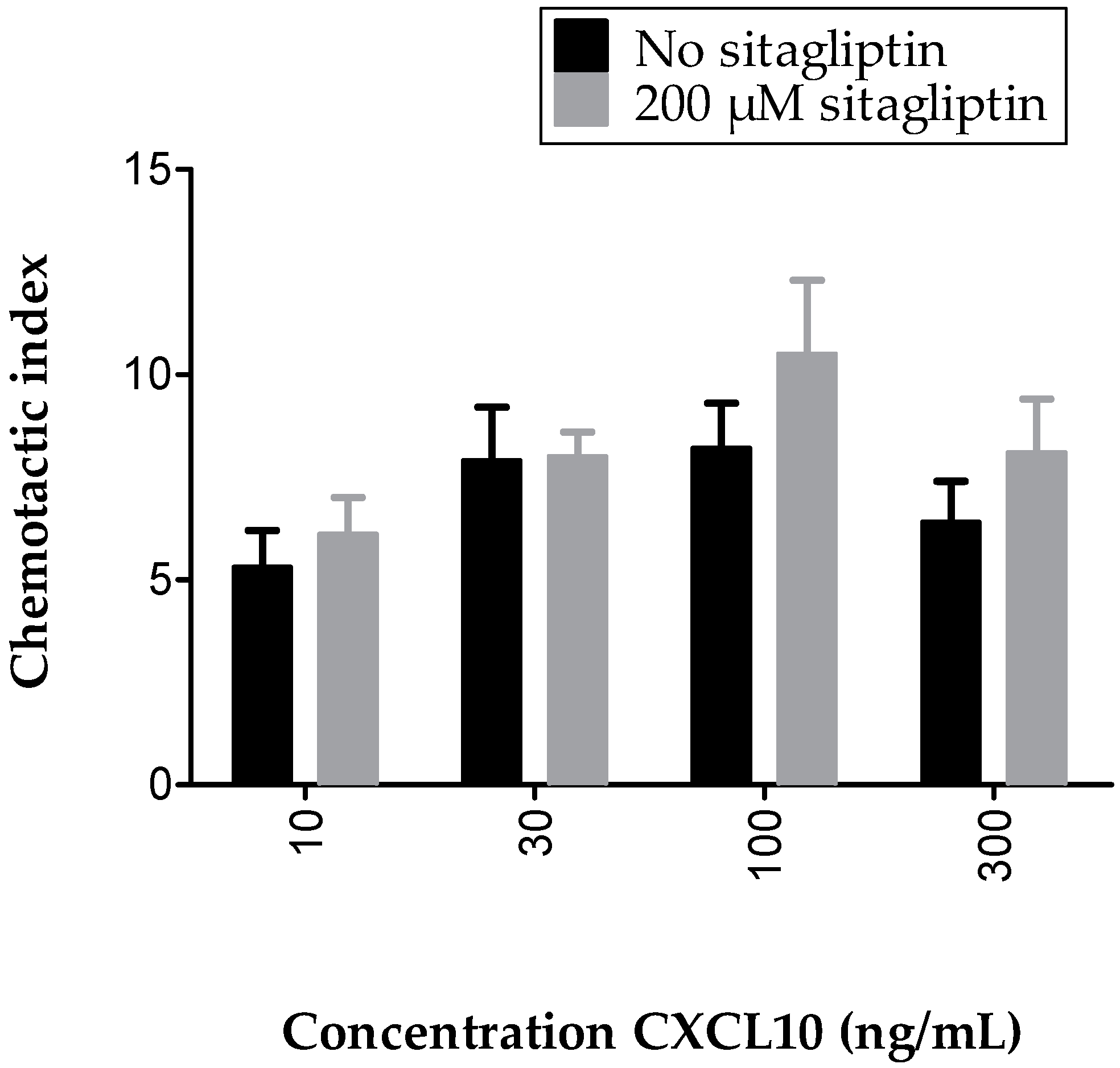
2.5. Inhibition of Membrane-Bound CD26 Did Not Affect CXCL10-Mediated T Cell Chemotaxis In Vitro
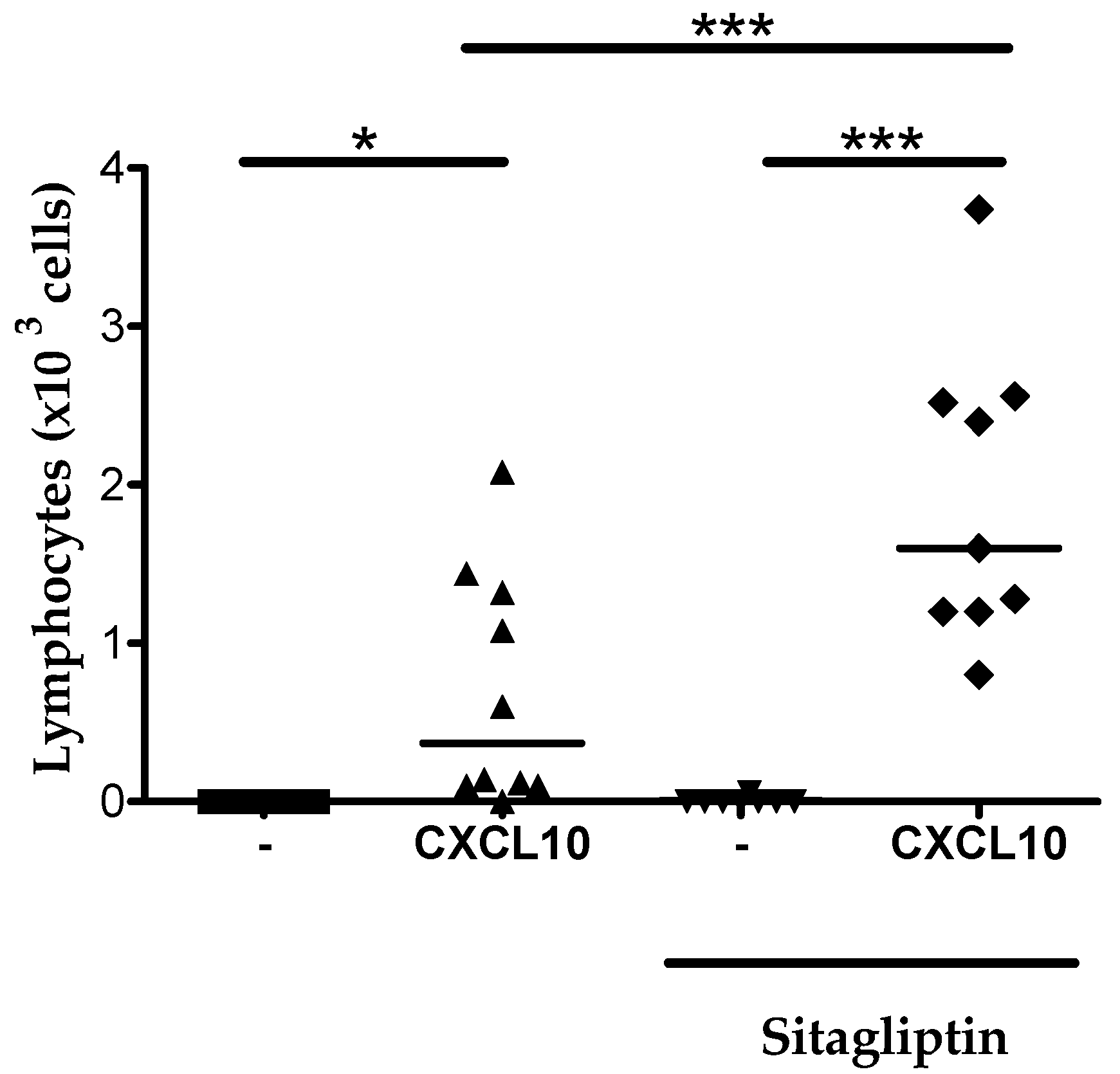
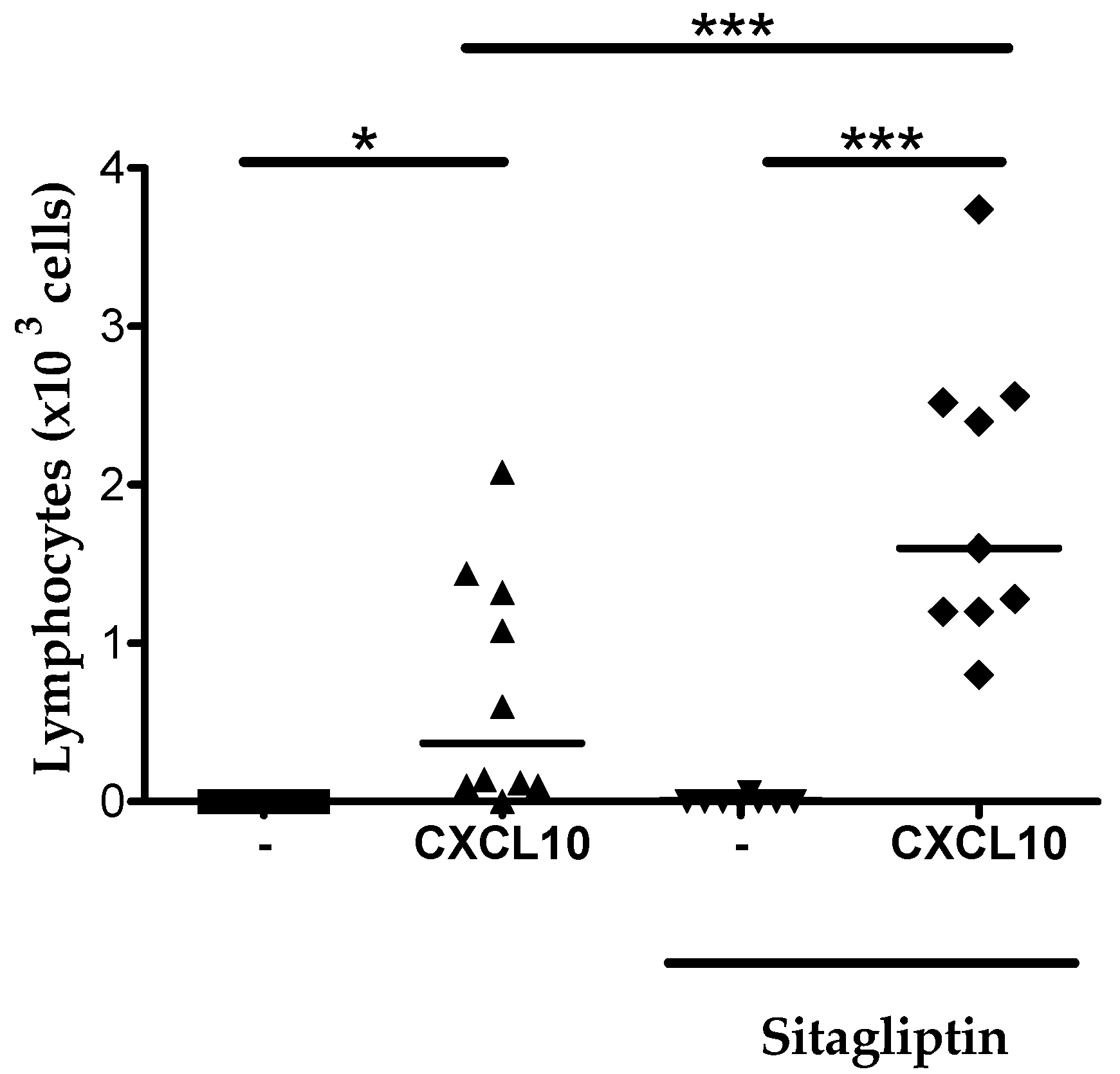
2.6. Inhibition of CD26 Significantly Increased CXCL10-Induced Lymphocyte Influx into the Joint In Vivo
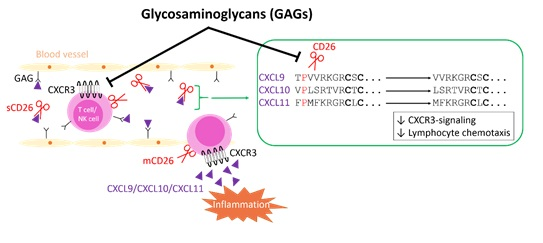
3. Discussion
4. Materials and Methods
4.1. Cells and Reagents
4.1.1. Chemokines and CD26
4.1.2. Cells
4.2. Proteolytic Processing of Chemokines by CD26 In Vitro
4.3. CD26 Activity Assays
4.4. Calcium-Mobilization Assays
4.5. In Vitro Chemotaxis Assays
4.6. In Vivo Cell Migration Assay
4.7. Flow Cytometry
4.8. Statistics
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| [Ca2+]i | Intracellular calcium concentration |
| GAG | glycosaminoglycan |
| GPCR | G protein-coupled receptor |
| Mr | Relative molecular mass |
References
- Zlotnik, A.; Yoshie, O. The chemokine superfamily revisited. Immunity 2012, 36, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Blanchet, X.; Langer, M.; Weber, C.; Koenen, R.R.; von Hundelshausen, P. Touch of chemokines. Front. Immunol. 2012, 3, 175. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, P.; Lasagni, L.; Annunziato, F.; Serio, M.; Romagnani, S. CXC chemokines: The regulatory link between inflammation and angiogenesis. Trends Immunol. 2004, 25, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Russo, R.C.; Garcia, C.C.; Teixeira, M.M.; Amaral, F.A. The CXCL8/IL-8 chemokine family and its receptors in inflammatory diseases. Expert. Rev. Clin. Immunol. 2014, 10, 593–619. [Google Scholar] [CrossRef] [PubMed]
- Keeley, E.C.; Mehrad, B.; Strieter, R.M. Chemokines as mediators of tumor angiogenesis and neovascularization. Exp. Cell Res. 2011, 317, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Bachelerie, F.; Ben-Baruch, A.; Burkhardt, A.M.; Combadiere, C.; Farber, J.M.; Graham, G.J.; Horuk, R.; Sparre-Ulrich, A.H.; Locati, M.; Luster, A.D.; et al. International union of basic and clinical pharmacology. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol. Rev. 2014, 66, 1–79. [Google Scholar] [CrossRef] [PubMed]
- Corsiero, E.; Nerviani, A.; Bombardieri, M.; Pitzalis, C. Ectopic lymphoid structures: Powerhouse of autoimmunity. Front. Immunol. 2016, 7, 430. [Google Scholar] [CrossRef] [PubMed]
- Opdenakker, G.; Proost, P.; Van Damme, J. Microbiomic and posttranslational modifications as preludes to autoimmune diseases. Trends Mol. Med. 2016, 22, 746–757. [Google Scholar] [CrossRef] [PubMed]
- Rot, A.; von Andrian, U.H. Chemokines in innate and adaptive host defense: Basic chemokinese grammar for immune cells. Annu. Rev. Immunol. 2004, 22, 891–928. [Google Scholar] [CrossRef] [PubMed]
- Groom, J.R.; Luster, A.D. CXCR3 ligands: Redundant, collaborative and antagonistic functions. Immunol. Cell Biol. 2011, 89, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Van Raemdonck, K.; Van den Steen, P.E.; Liekens, S.; Van Damme, J.; Struyf, S. CXCR3 ligands in disease and therapy. Cytokine Growth Factor Rev. 2015, 26, 311–327. [Google Scholar] [CrossRef] [PubMed]
- Groom, J.R.; Luster, A.D. CXCR3 in T cell function. Exp. Cell Res. 2011, 317, 620–631. [Google Scholar] [CrossRef] [PubMed]
- Lasagni, L.; Francalanci, M.; Annunziato, F.; Lazzeri, F.; Giannini, S.; Cosmi, L.; Sagrinati, C.; Mazzinghi, B.; Orlando, C.; Maggi, E.; et al. An alternatively spliced variant of CXCR3 mediates the inhibition of endothelial cell growth induced by IP-10, Mig, and I-TAC, and acts as functional receptor for platelet factor 4. J. Exp. Med. 2003, 197, 1537–1549. [Google Scholar] [CrossRef] [PubMed]
- Naumann, U.; Cameroni, E.; Pruenster, M.; Mahabaleshwar, H.; Raz, E.; Zerwes, H.G.; Rot, A.; Thelen, M. CXCR7 functions as a scavenger for CXCL12 and CXCL11. PLoS ONE 2010, 5, e9175. [Google Scholar] [CrossRef] [PubMed]
- Girard, M.; Rhainds, D.; St-Onge, G. Mutational analysis of atypical chemokine receptor 3 (ACKR3/CXCR7) interaction with its chemokine ligands CXCL11 and CXCL12. J. Biol. Chem. 2017, 292, 31–42. [Google Scholar]
- Burns, J.M.; Summers, B.C.; Wang, Y.; Melikian, A.; Berahovich, R.; Miao, Z.; Penfold, M.E.; Sunshine, M.J.; Littman, D.R.; Kuo, C.J.; et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J. Exp. Med. 2006, 203, 2201–2213. [Google Scholar] [CrossRef] [PubMed]
- Vanheule, V.; Janssens, R.; Boff, D.; Kitic, N.; Berghmans, N.; Ronsse, I.; Kungl, A.J.; Amaral, F.A.; Teixeira, M.M.; Van Damme, J.; et al. The Positively charged COOH-terminal glycosaminoglycan-binding CXCL9(74–103) peptide inhibits CXCL8-induced neutrophil extravasation and monosodium urate crystal-induced gout in mice. J. Biol. Chem. 2015, 290, 21292–21304. [Google Scholar] [CrossRef] [PubMed]
- Farber, J.M. Mig and IP-10: CXC chemokines that target lymphocytes. J. Leukoc. Biol. 1997, 61, 246–257. [Google Scholar] [PubMed]
- Ohmori, Y.; Schreiber, R.D.; Hamilton, T.A. Synergy between interferon-gamma and tumor necrosis factor-α in transcriptional activation is mediated by cooperation between signal transducer and activator of transcription 1 and nuclear factor κB. J. Biol. Chem. 1997, 272, 14899–14907. [Google Scholar] [CrossRef] [PubMed]
- Ohmori, Y.; Wyner, L.; Narumi, S.; Armstrong, D.; Stoler, M.; Hamilton, T.A. Tumor necrosis factoralpha induces cell type and tissue-specific expression of chemoattractant cytokines in vivo. Am. J. Pathol. 1993, 142, 861–870. [Google Scholar] [PubMed]
- Rani, M.R.; Foster, G.R.; Leung, S.; Leaman, D.; Stark, G.R.; Ransohoff, R.M. Characterization of β-R1, a gene that is selectively induced by interferon β (IFN-β) compared with IFN-α. J. Biol. Chem. 1996, 271, 22878–22884. [Google Scholar] [CrossRef] [PubMed]
- Proost, P.; Struyf, S.; Loos, T.; Gouwy, M.; Schutyser, E.; Conings, R.; Ronsse, I.; Parmentier, M.; Grillet, B.; Opdenakker, G.; et al. Coexpression and interaction of CXCL10 and CD26 in mesenchymal cells by synergising inflammatory cytokines: CXCL8 and CXCL10 are discriminative markers for autoimmune arthropathies. Arthritis Res. Ther. 2006, 8, R107. [Google Scholar] [CrossRef] [PubMed]
- Proost, P.; Vynckier, A.K.; Mahieu, F.; Put, W.; Grillet, B.; Struyf, S.; Wuyts, A.; Opdenakker, G.; Van Damme, J. Microbial Toll-like receptor ligands differentially regulate CXCL10/IP-10 expression in fibroblasts and mononuclear leukocytes in synergy with IFN-γ and provide a mechanism for enhanced synovial chemokine levels in septic arthritis. Eur. J. Immunol. 2003, 33, 3146–3153. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.F.; Lai, S.L.; Chen, J.P.; Sung, J.M.; Lin, Y.L.; Wu-Hsieh, B.A.; Gerard, C.; Luster, A.; Liao, F. Both CXCR3 and CXCL10/IFN-inducible protein 10 are required for resistance to primary infection by dengue virus. J. Immunol. 2006, 177, 1855–1863. [Google Scholar] [CrossRef] [PubMed]
- Christen, U.; McGavern, D.B.; Luster, A.D.; von Herrath, M.G.; Oldstone, M.B. Among CXCR3 chemokines, IFN-γ-inducible protein of 10 kDa (CXC chemokine ligand (CXCL) 10) but not monokine induced by IFN-γ (CXCL9) imprints a pattern for the subsequent development of autoimmune disease. J. Immunol. 2003, 171, 6838–6845. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Xu, W.; Xu, L.; Jiang, Z.; Wen, Z.; Li, K.; Xiong, S. I-TAC is a dominant chemokine in controlling skin intragraft inflammation via recruiting CXCR3+ cells into the graft. Cell. Immunol. 2010, 260, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.S.; Lin, E.; Zhang, B.; Luster, A.D.; Tollett, J.; Samuel, M.A.; Engle, M.; Diamond, M.S. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J. Virol. 2005, 79, 11457–11466. [Google Scholar] [CrossRef] [PubMed]
- Campanella, G.S.; Tager, A.M.; EI Khoury, J.K.; Thomas, S.Y.; Abrazinski, T.A.; Manice, L.A.; Colvin, R.A.; Luster, A.D. Chemokine receptor CXCR3 and its ligands CXCL9 and CXCL10 are required for the development of murine cerebral malaria. Proc. Natl. Acad. Sci. USA 2008, 105, 4814–4819. [Google Scholar] [CrossRef] [PubMed]
- Thapa, M.; Welner, R.S.; Pelayo, R.; Carr, D.J. CXCL9 and CXCL10 expression are critical for control of genital herpes simplex virus type 2 infection through mobilization of HSV-specific CTL and NK cells to the nervous system. J. Immunol. 2008, 180, 1098–1106. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, J.M.; Shimoda, N.; Schenk, A.D.; Zhang, H.; Kish, D.D.; Keslar, K.; Farber, J.M.; Fairchild, R.L. CXC chemokine ligand (CXCL) 9 and CXCL10 are antagonistic costimulation molecules during the priming of alloreactive T cell effectors. J. Immunol. 2010, 184, 3450–3460. [Google Scholar] [CrossRef] [PubMed]
- Sierro, F.; Biben, C.; Martínez-Muñoz, L.; Mellado, M.; Ransohoff, R.M.; Li, M.; Woehl, B.; Leung, H.; Groom, J.; Batten, M.; et al. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc. Natl. Acad. Sci. USA 2007, 104, 14759–14764. [Google Scholar] [CrossRef] [PubMed]
- Sarris, M.; Masson, J.B.; Maurin, D.; Van der Aa, L.M.; Boudinot, P.; Lortat-Jacob, H.; Herbomel, P. Inflammatory chemokines direct and restrict leukocyte migration within live tissues as glycan-bound gradients. Curr. Biol. 2012, 22, 2375–2382. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E.; Handel, T.M.; Johnson, Z.; Lau, E.K.; LiWang, P.; Clark-Lewis, I.; Borlat, F.; Wells, T.N.; Kosco-Vilbois, M.H. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl. Acad. Sci. USA 2003, 100, 1885–1890. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Fuster, M.; Sriramarao, P.; Esko, J.D. Endothelial heparan sulfate deficiency impairs l-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat. Immunol. 2005, 6, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Severin, I.C.; Gaudry, J.P.; Johnson, Z.; Kungl, A.; Jansma, A.; Gesslbauer, B.; Mulloy, B.; Power, C.; Proudfoot, A.E.; Handel, T. Characterization of the chemokine CXCL11-heparin interaction suggests two different affinities for glycosaminoglycans. J. Biol. Chem. 2010, 285, 17713–17724. [Google Scholar] [CrossRef] [PubMed]
- Campanella, G.S.; Grimm, J.; Manice, L.A.; Colvin, R.A.; Medoff, B.D.; Wojtkiewicz, G.R.; Weissleder, R.; Luster, A.D. Oligomerization of CXCL10 is necessary for endothelial cell presentation and in vivo activity. J. Immunol. 2006, 177, 6991–6998. [Google Scholar] [CrossRef] [PubMed]
- Massena, S.; Christoffersson, G.; Hjertström, E.; Zcharia, E.; Vlodavsky, I.; Ausmees, N.; Rolny, C.; Li, J.P.; Phillipson, M. A chemotactic gradient sequestered on endothelial heparan sulfate induces directional intraluminal crawling of neutrophils. Blood 2010, 116, 1924–1931. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Moseman, E.A.; Saito, H.; Petryanik, B.; Thiriot, A.; Hatakeyama, S.; Ito, Y.; Kawashima, H.; Yamaguchi, Y.; Lowe, J.B.; et al. Endothelial heparan sulfate controls chemokine presentation in recruitment of lymphocytes and dendritic cells to lymph nodes. Immunity 2010, 33, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.V.; Katakam, S.K.; Urbanowitz, A.K.; Gotte, M. Heparan sulfate as a regulator of leukocyte recruitment in inflammation. Curr. Protein Pept. Sci. 2015, 16, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Esko, J.D. Demystifying heparan sulfate-protein interactions. Annu. Rev. Biochem. 2014, 83, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Vanheule, V.; Vervaeke, P.; Mortier, A.; Noppen, S.; Gouwy, M.; Snoeck, R.; Andrei, G.; Van Damme, J.; Liekens, S.; Proost, P. Basic chemokine-derived glycosaminoglycan binding peptides exert antiviral properties against dengue virus serotype 2, herpes simplex virus-1 and respiratory syncytial virus. Biochem. Pharmacol. 2016, 100, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Baggiolini, M. CXCL8—The First Chemokine. Front. Immunol. 2015, 6, 285. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, J.; Van Beeumen, J.; Conings, R.; Decock, B.; Billiau, A. Purification of granulocyte chemotactic peptide/interleukin-8 reveals N-terminal sequence heterogeneity similar to that of beta-thromboglobulin. Eur. J. Biochem. 1989, 181, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Mortier, A.; Van Damme, J.; Proost, P. Overview of the mechanisms regulating chemokine activity and availability. Immunol. Lett. 2012, 145, 2–9. [Google Scholar]
- Krohn, S.C.; Bonvin, P.; Proudfoot, A.E. CCL18 exhibits a regulatory role through inhibition of receptor and glycosaminoglycan binding. PLoS ONE 2013, 8, e72321. [Google Scholar] [CrossRef] [PubMed]
- Sadir, R.; Imberty, A.; Baleux, F.; Lortat-Jacob, H. Heparan sulfate/heparin oligosaccharides protect stromal cell-derived factor-1 (SDF-1)/CXCL12 against proteolysis induced by CD26/dipeptidyl peptidase IV. J. Biol. Chem. 2004, 279, 43854–43860. [Google Scholar] [CrossRef] [PubMed]
- Ellyard, J.I.; Simson, L.; Bezos, A.; Johnston, K.; Freeman, C.; Parish, C.R. Eotaxin selectively binds heparin. An interaction that protects eotaxin from proteolysis and potentiates chemotactic activity in vivo. J. Biol. Chem. 2007, 282, 15238–15247. [Google Scholar] [CrossRef] [PubMed]
- Mortier, A.; Gouwy, M.; Van Damme, J.; Proost, P. Effect of posttranslational processing on the in vitro and in vivo activity of chemokines. Exp. Cell Res. 2011, 317, 642–654. [Google Scholar] [CrossRef] [PubMed]
- Moelants, E.A.; Mortier, A.; Van Damme, J.; Proost, P. In vivo regulation of chemokine activity by post-translational modification. Immunol. Cell Biol. 2013, 91, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Metzemaekers, M.; Van Damme, J.; Mortier, A.; Proost, P. Regulation of chemokine activity—A focus on the role of dipeptidyl peptidase IV/CD26. Front. Immunol. 2016, 7, 483. [Google Scholar] [CrossRef] [PubMed]
- Janssens, R.; Mortier, A.; Boff, D.; Ruytinx, P.; Gouwy, M.; Vantilt, B.; Larsen, O.; Daugvilaite, V.; Rosenkilde, M.M.; Parmentier, M.; et al. Truncation of CXCL12 by CD26 reduces its CXC chemokine receptor 4- and atypical chemokine receptor 3-dependent activity on endothelial cells and lymphocytes. Biochem. Pharmacol. 2017, 132, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Mortier, A.; Gouwy, M.; Van Damme, J.; Proost, P.; Struyf, S. CD26/dipeptidylpeptidase IV-chemokine interactions: Double-edged regulation of inflammation and tumor biology. J. Leukoc. Biol. 2016, 99, 955–969. [Google Scholar] [CrossRef] [PubMed]
- Proost, P.; Schutyser, E.; Menten, P.; Struyf, S.; Wuyts, A.; Opdenakker, G.; Detheux, M.; Parmentier, M.; Durinx, C.; Lambeir, A.M.; et al. Amino-terminal truncation of CXCR3 agonists impairs receptor signaling and lymphocyte chemotaxis, while preserving antiangiogenic properties. Blood 2001, 98, 3554–3561. [Google Scholar] [CrossRef] [PubMed]
- Lambeir, A.M.; Durinx, C.; Scharpé, S.; De Meester, I. Dipeptidyl-peptidase IV from bench to bedside: An update on structural properties, functions, and clinical aspects of the enzyme DPP IV. Crit. Rev. Clin. Lab. Sci. 2003, 40, 209–294. [Google Scholar] [CrossRef] [PubMed]
- Klemann, C.; Wagner, L.; Stephan, M.; von Hörsten, S. Cut to the chase: A review of CD26/dipeptidyl peptidase-4’s (DPP4) entanglement in the immune system. Clin. Exp. Immunol. 2016, 185, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Wagner, L.; Klemann, C.; Stephan, M.; von Hörsten, S. Unravelling the immunological roles of dipeptidyl peptidase 4 (DPP4) activity and/or structure homologue (DASH) proteins. Clin. Exp. Immunol. 2016, 184, 265–283. [Google Scholar] [CrossRef] [PubMed]
- Boonacker, E.; Van Noorden, C.J. The multifunctional or moonlighting protein CD26/DPPIV. Eur. J. Cell Biol. 2003, 82, 53–73. [Google Scholar] [CrossRef] [PubMed]
- Proost, P.; De Wolf-Peeters, C.; Conings, R.; Opdenakker, G.; Billiau, A.; Van Damme, J. Identification of a novel granulocyte chemotactic protein (GCP-2) from human tumor cells. In vitro and in vivo comparison with natural forms of GRO, IP-10, and IL-8., IP-10, and IL-8. J. Immunol. 1993, 150, 1000–1010. [Google Scholar] [PubMed]
- Ludwig, A.; Schiemann, F.; Mentlein, R.; Lindner, B.; Brandt, E. Dipeptidyl peptidase IV (CD26) on T cells cleaves the CXC chemokine CXCL11 (I-TAC) and abolishes the stimulating but not the desensitizing potential of the chemokine. J. Leukoc. Biol. 2002, 72, 183–191. [Google Scholar] [PubMed]
- Proost, P.; Mortier, A.; Loos, T.; Vandercappellen, J.; Gouwy, M.; Ronsse, I.; Schutyser, E.; Put, W.; Parmentier, M.; Struyf, S.; et al. Proteolytic processing of CXCL11 by CD13/aminopeptidase N impairs CXCR3 and CXCR7 binding and signaling and reduces lymphocyte and endothelial cell migration. Blood 2007, 110, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Hensbergen, P.J.; van der Raaij-Helmer, E.M.; Dijkman, R.; van der Schors, R.C.; Werner-Felmayer, G.; Boorsma, D.M.; Scheper, R.J.; Willemze, R.; Tensen, C.P. Processing of natural and recombinant CXCR3-targeting chemokines and implications for biological activity. Eur. J. Biochem. 2001, 268, 4992–4999. [Google Scholar] [CrossRef] [PubMed]
- Lambeir, A.M.; Proost, P.; Durinx, C.; Bal, G.; Senten, K.; Augustyns, K.; Scharpé, S.; Van Damme, J.; De Meester, I. Kinetic investigation of chemokine truncation by CD26/dipeptidyl peptidase IV reveals a striking selectivity within the chemokine family. J. Biol. Chem. 2001, 276, 29839–29845. [Google Scholar] [CrossRef] [PubMed]
- Barreira da Silva, R.; Laird, M.E.; Yatim, N.; Fiette, L.; Ingersoll, M.A.; Albert, M.L. Dipeptidylpeptidase 4 inhibition enhances lymphocyte trafficking, improving both naturally occurring tumor immunity and immunotherapy. Nat. Immunol. 2015, 16, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Bleul, C.C.; Fuhlbrigge, R.C.; Casasnovas, J.M.; Aiuti, A.; Springer, T.A. A highly efficacious lymphocyte chemoattractant, stromal cell-derived factor 1 (SDF-1). J. Exp. Med. 1996, 184, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.; Hyun, Y.M.; Lambert-Emo, K.; Capece, T.; Bae, S.; Miller, R.; Topham, D.J.; Kim, M. Neutrophil trails guide influenza-specific CD8(+) T cells in the airways. Science 2015, 349, aaa4352. [Google Scholar] [CrossRef] [PubMed]
- Möhle, R.; Bautz, F.; Rafii, S.; Moore, M.A.; Brugger, W.; Kanz, L. The chemokine receptor CXCR-4 is expressed on CD34+ hematopoietic progenitors and leukemic cells and mediates transendothelial migration induced by stromal cell-derived factor-1. Blood 1998, 91, 4523–4530. [Google Scholar] [PubMed]
- Aiuti, A.; Webb, I.J.; Bleul, C.C.; Springer, T.; Gutierrez-Ramos, J.C. The chemokine SDF-1 is a chemoattractant for human CD34+ hematopoietic progenitor cells and provides a new mechanism to explain the mobilization of CD34+ progenitors to peripheral blood. J. Exp. Med. 1997, 185, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Shioda, T.; Kato, H.; Ohnishi, Y.; Tashiro, K.; Ikegawa, M.; Nakayama, E.E.; Hu, H.; Kato, A.; Sakai, Y.; Liu, H.; et al. Anti-HIV-1 and chemotactic activities of human stromal cell-derived factor 1alpha (SDF-1α) and SDF-1β are abolished by CD26/dipeptidyl peptidase IV-mediated cleavage. Proc. Natl. Acad. Sci. USA 1998, 95, 6331–6336. [Google Scholar] [CrossRef] [PubMed]
- Proost, P.; Struyf, S.; Schols, D.; Durinx, C.; Wuyts, A.; Lenaerts, J.P.; De Clercq, E.; De Meester, I.; Van Damme, J. Processing by CD26/dipeptidyl-peptidase IV reduces the chemotactic and anti-HIV-1 activity of stromal-cell-derived factor-1α. FEBS Lett. 1998, 432, 73–76. [Google Scholar] [CrossRef]
- Ziarek, J.J.; Veldkamp, C.T.; Zhang, F.; Murray, N.J.; Kartz, G.A.; Liang, X.; Su, J.; Baker, J.E.; Linhardt, R.J.; Volkman, B.F. Heparin oligosaccharides inhibit chemokine (CXC motif) ligand 12 (CXCL12) cardioprotection by binding orthogonal to the dimerization interface, promoting oligomerization, and competing with the chemokine (CXC motif) receptor 4 (CXCR4) N terminus. J. Biol. Chem. 2013, 288, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Gangavarapu, P.; Rajagopalan, L.; Kolli, D.; Guerrero-Plata, A.; Garofalo, R.P.; Rajarathnam, K. The monomer-dimer equilibrium and glycosaminoglycan interactions of chemokine CXCL8 regulate tissue-specific neutrophil recruitment. J. Leukoc. Biol. 2012, 91, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Dyer, D.P.; Migliorini, E.; Salanga, C.L.; Thakar, D.; Handel, T.M.; Richter, R.P. Differential structural remodelling of heparan sulfate by chemokines: The role of chemokine oligomerization. Open Biol. 2017, 7, 160286. [Google Scholar] [CrossRef] [PubMed]
- Dyer, D.P.; Salanga, C.L.; Volkman, B.F.; Kawamura, T.; Handel, T.M. The dependence of chemokine-glycosaminoglycan interactions on chemokine oligomerization. Glycobiology. 2016, 26, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Egesten, A.; Eliasson, M.; Johansson, H.M.; Olin, A.I.; Morgelin, M.; Mueller, A.; Pease, J.E.; Frick, I.M.; Bjorck, L. The CXC chemokine MIG/CXCL9 is important in innate immunity against Streptococcus pyogenes. J. Infect. Dis. 2007, 195, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, G.J.; Holloway, D.E.; Colvin, R.A.; Campanella, G.K.; Papageorgiou, A.C.; Luster, A.D.; Acharya, K.R. Crystal structures of oligomeric forms of the IP-10/CXCL10 chemokine. Structure 2003, 11, 521–532. [Google Scholar] [CrossRef]
- Dufour, J.H.; Dziejman, M.; Liu, M.T.; Leung, J.H.; Lane, T.E.; Luster, A.D. IFN-γ-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J. Immunol. 2002, 168, 3195–3204. [Google Scholar] [CrossRef] [PubMed]
- Casrouge, A.; Decalf, J.; Ahloulay, M.; Lababidi, C.; Mansour, H.; Vallet-Pichard, A.; Mallet, V.; Mottez, E.; Mapes, J.; Fontanet, A.; et al. Evidence for an antagonist form of the chemokine CXCL10 in patients chronically infected with HCV. J. Clin. Investig. 2011, 121, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Decalf, J.; Tarbell, K.V.; Casrouge, A.; Price, J.D.; Linder, G.; Mottez, E.; Sultanik, P.; Mallet, V.; Pol, S.; Duffy, D.; et al. Inhibition of DPP4 activity in humans establishes its in vivo role in CXCL10 post-translational modification: Prospective placebo-controlled clinical studies. EMBO Mol. Med. 2016, 8, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Campanella, G.S.; Lee, E.M.; Sun, J.; Luster, A.D. CXCR3 and heparin binding sites of the chemokine IP-10 (CXCL10). J. Biol. Chem. 2003, 278, 17066–17074. [Google Scholar] [CrossRef] [PubMed]
- Loos, T.; Mortier, A.; Gouwy, M.; Ronsse, I.; Put, W.; Lenaerts, J.P.; Van Damme, J.; Proost, P. Citrullination of CXCL10 and CXCL11 by peptidylarginine deiminase: A naturally occurring posttranslational modification of chemokines and new dimension of immunoregulation. Blood 2008, 112, 2648–2656. [Google Scholar] [CrossRef] [PubMed]
- Kuschert, G.S.; Coulin, F.; Power, C.A.; Proudfoot, A.E.; Hubbard, R.E.; Hoogewerf, A.J.; Wells, T.N. Glycosaminoglycans interact selectively with chemokines and modulate receptor binding and cellular responses. Biochemistry 1999, 38, 12959–12968. [Google Scholar] [CrossRef] [PubMed]
- Gorrell, M.D.; Gysbers, V.; McCaughan, G.W. CD26: A multifunctional integral membrane and secreted protein of activated lymphocytes. Scand. J. Immunol. 2001, 54, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Waumans, Y.; Baerts, L.; Kehoe, K.; Lambeir, A.M.; De Meester, I. The Dipeptidyl Peptidase Family, Prolyl Oligopeptidase, and Prolyl Carboxypeptidase in the Immune System and Inflammatory Disease, Including Atherosclerosis. Front. Immunol. 2015, 6, 387. [Google Scholar] [CrossRef] [PubMed]
- Loos, T.; Mortier, A.; Proost, P. Chapter 1. Isolation, identification, and production of posttranslationally modified chemokines. Methods Enzymol. 2009, 461, 3–29. [Google Scholar] [PubMed]
- De Meester, I.; Vanhoof, G.; Lambeir, A.M.; Scharpé, S. Use of immobilized adenosine deaminase for the rapid purification of native human CD26/dipeptidyl peptidase IV. J. Immunol. Methods 1996, 189, 99–105. [Google Scholar] [CrossRef]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [PubMed]
- Boyden, S. The chemotactic effect of mixtures of antibody and antigen on polymorphonuclear leucocytes. J. Exp. Med. 1962, 115, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Janssens, R.; Mortier, A.; Boff, D.; Vanheule, V.; Gouwy, M.; Franck, C.; Larsen, O.; Rosenkilde, M.M.; Van Damme, J.; Amaral, F.A.; et al. Natural nitration of CXCL12 reduces its signaling capacity and chemotactic activity in vitro and abrogates intra-articular lymphocyte recruitment in vivo. Oncotarget 2016, 7, 62439–62459. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration Heparin DP30 (µg/mL) | CD26 Activity (U/L) |
|---|---|
| 0 | 4.6 |
| 10 | 4.19 |
| 100 | 4.35 |
| GAG | Source | Company | Composition | Relative Molecular Mass Mr |
|---|---|---|---|---|
| Heparin | Porcine mucosa | Iduron | ∆HexA,2S–GlcNS,6S–(IdoUA,2S–GlcNS,6S)n | ±40 kDa a |
| Heparin DP30 | Porcine mucosa | Iduron | ∆HexA,2S–GlcNS,6S–(IdoUA,2S–GlcNS,6S)30 | >9 kDa b |
| Heparin DP8 | Porcine mucosa | Iduron | ∆HexA,2S–GlcNS,6S–(IdoUA,2S–GlcNS,6S)8 | ±2.4 kDa b |
| Heparan sulfate | Porcine mucosa | Iduron | GlcA-GlcNAc and IdoA/Glc-GlcNS (variable O-sulfation); contains both low and high sulfated heparan sulfates | ±40 kDa a |
| Dermatan sulfate | Porcine mucosa | Iduron | HexA-GalNAc,4S (88%); HexA-GalNAc (5%); HexA,2S-GalNAc,4S (7%) | ±41 kDa b |
| Chondroitin sulfate A | Bovine trachea | Sigma-Aldrich | Alternating Copoly β-glucuronic acid-(1→3)-N-acetyl-β-galactosamine-4-sulfate-(1→4) | ±40 kDa a |
| Chondroitin sulfate C | Shark cartilage | Sigma-Aldrich | Poly[β-glucuronic acid-(1→3)-N-acetyl-β-galactosamine-6-sulfate-(1→4)] alternating | ±40 kDa a |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Metzemaekers, M.; Mortier, A.; Janssens, R.; Boff, D.; Vanbrabant, L.; Lamoen, N.; Van Damme, J.; Teixeira, M.M.; De Meester, I.; Amaral, F.A.; et al. Glycosaminoglycans Regulate CXCR3 Ligands at Distinct Levels: Protection against Processing by Dipeptidyl Peptidase IV/CD26 and Interference with Receptor Signaling. Int. J. Mol. Sci. 2017, 18, 1513. https://doi.org/10.3390/ijms18071513
Metzemaekers M, Mortier A, Janssens R, Boff D, Vanbrabant L, Lamoen N, Van Damme J, Teixeira MM, De Meester I, Amaral FA, et al. Glycosaminoglycans Regulate CXCR3 Ligands at Distinct Levels: Protection against Processing by Dipeptidyl Peptidase IV/CD26 and Interference with Receptor Signaling. International Journal of Molecular Sciences. 2017; 18(7):1513. https://doi.org/10.3390/ijms18071513
Chicago/Turabian StyleMetzemaekers, Mieke, Anneleen Mortier, Rik Janssens, Daiane Boff, Lotte Vanbrabant, Nicole Lamoen, Jo Van Damme, Mauro M. Teixeira, Ingrid De Meester, Flávio A. Amaral, and et al. 2017. "Glycosaminoglycans Regulate CXCR3 Ligands at Distinct Levels: Protection against Processing by Dipeptidyl Peptidase IV/CD26 and Interference with Receptor Signaling" International Journal of Molecular Sciences 18, no. 7: 1513. https://doi.org/10.3390/ijms18071513