Computational Investigation into the Interactions of Traditional Chinese Medicine Molecules of WenQingYin with GluR2

Abstract
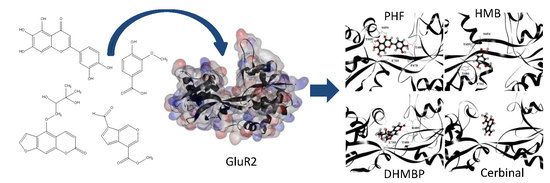
:
1. Introduction
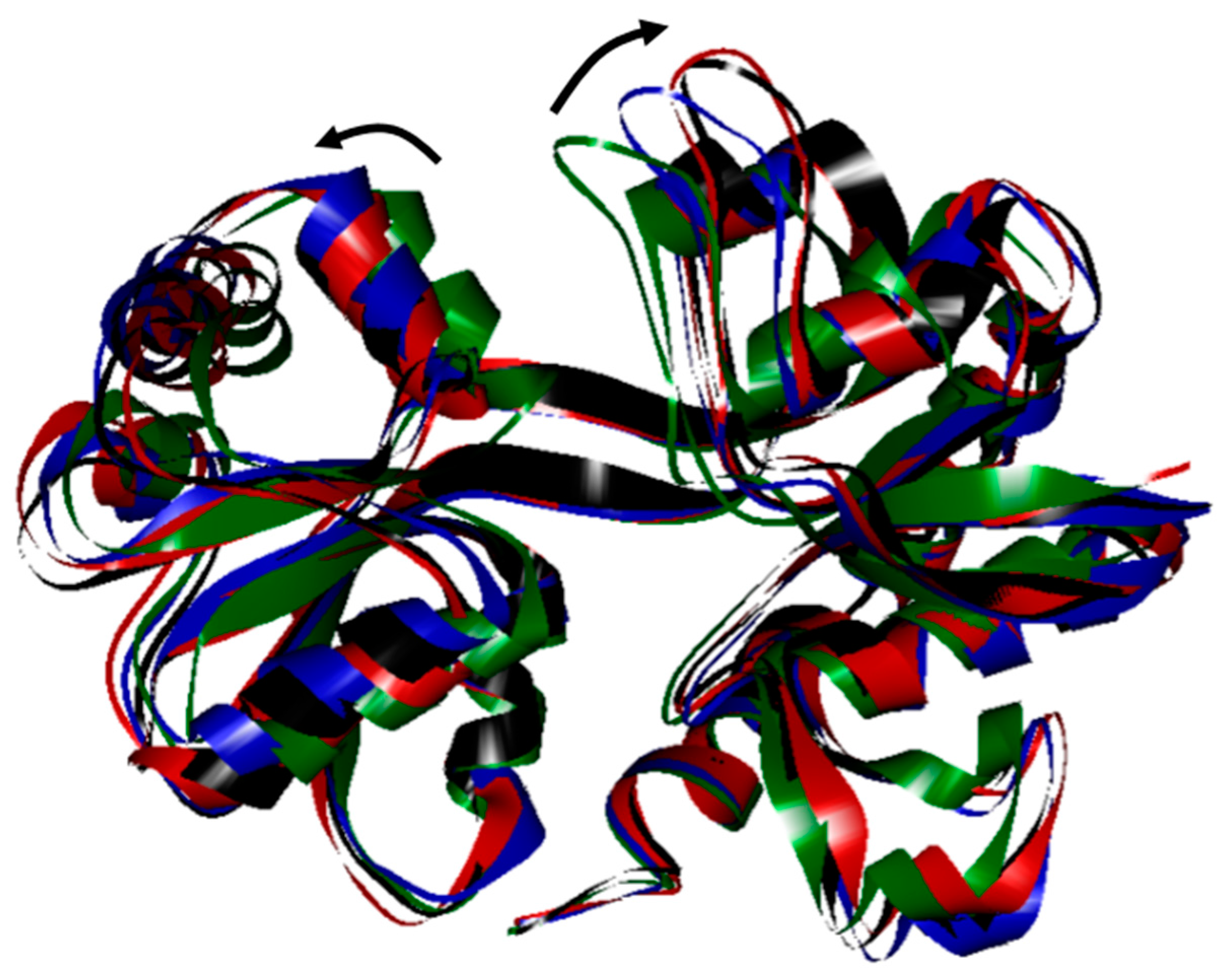
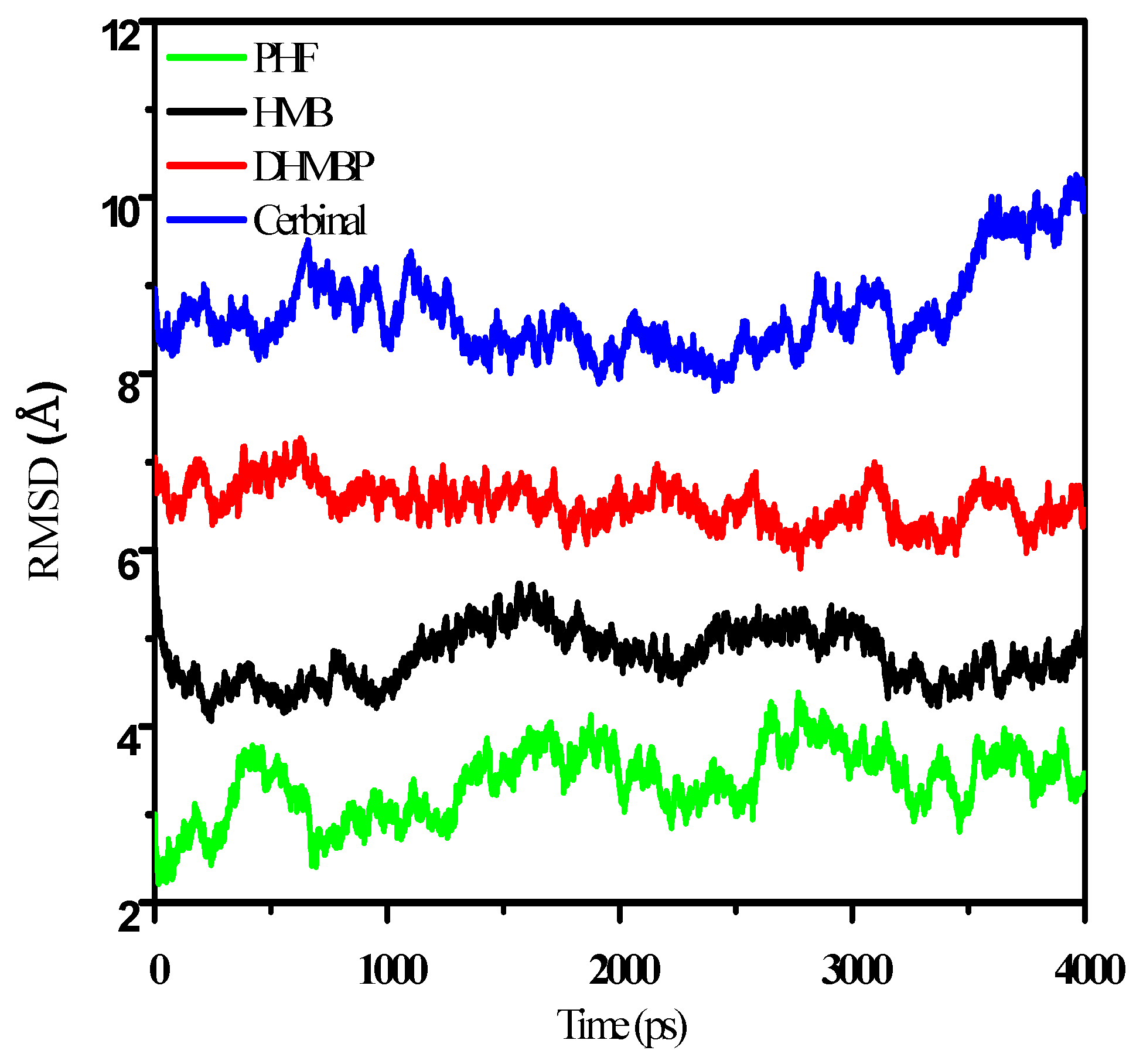
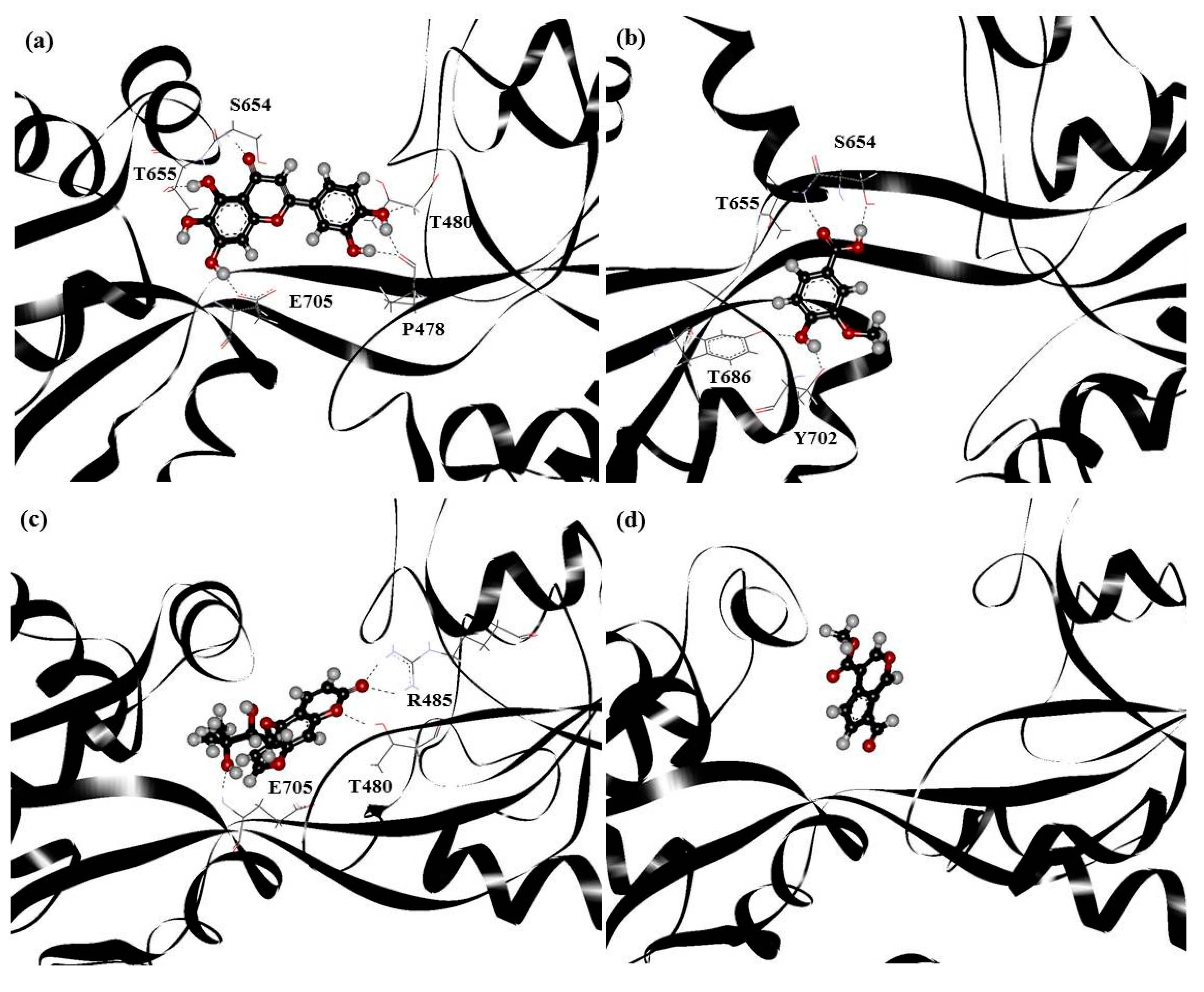
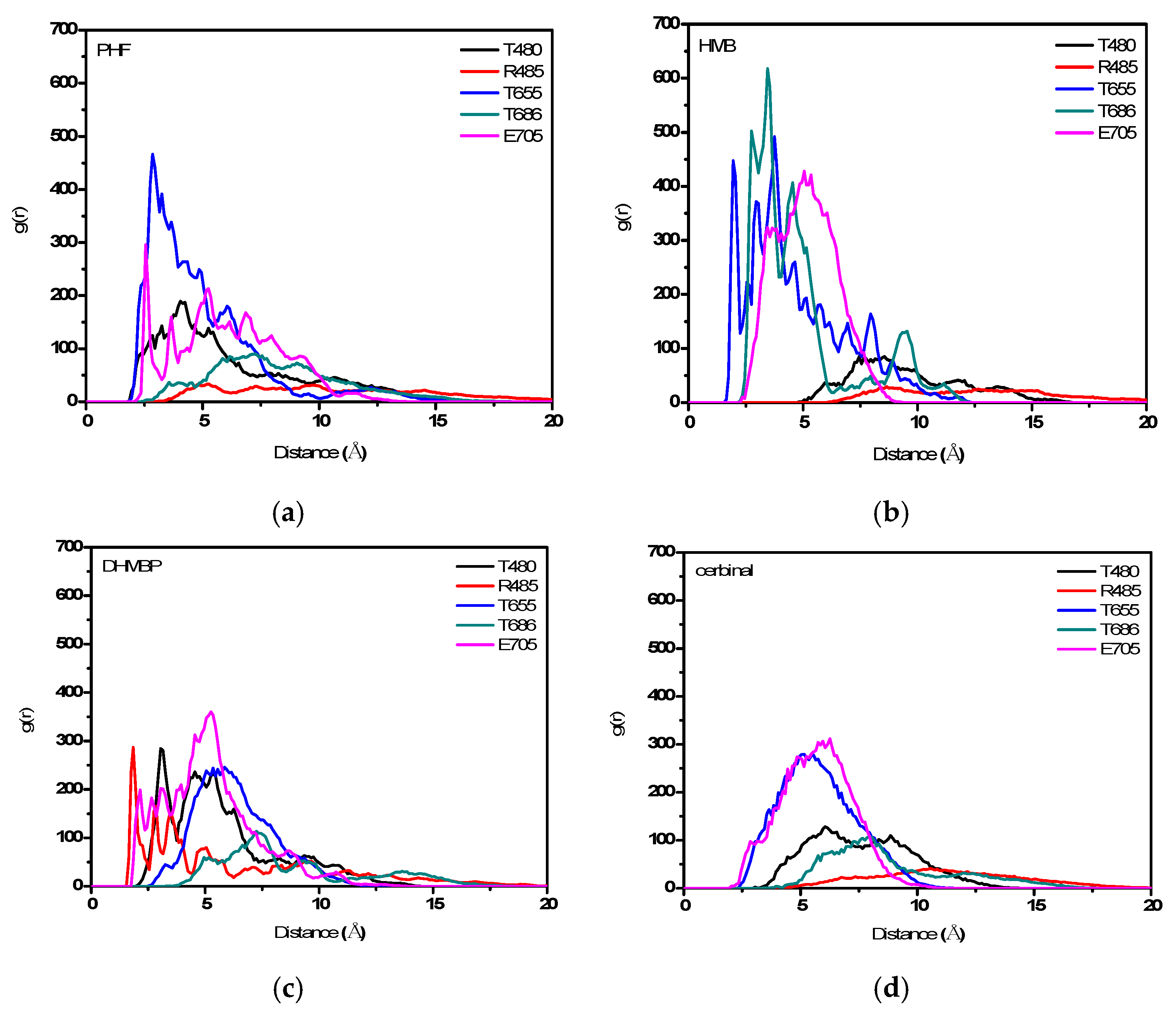
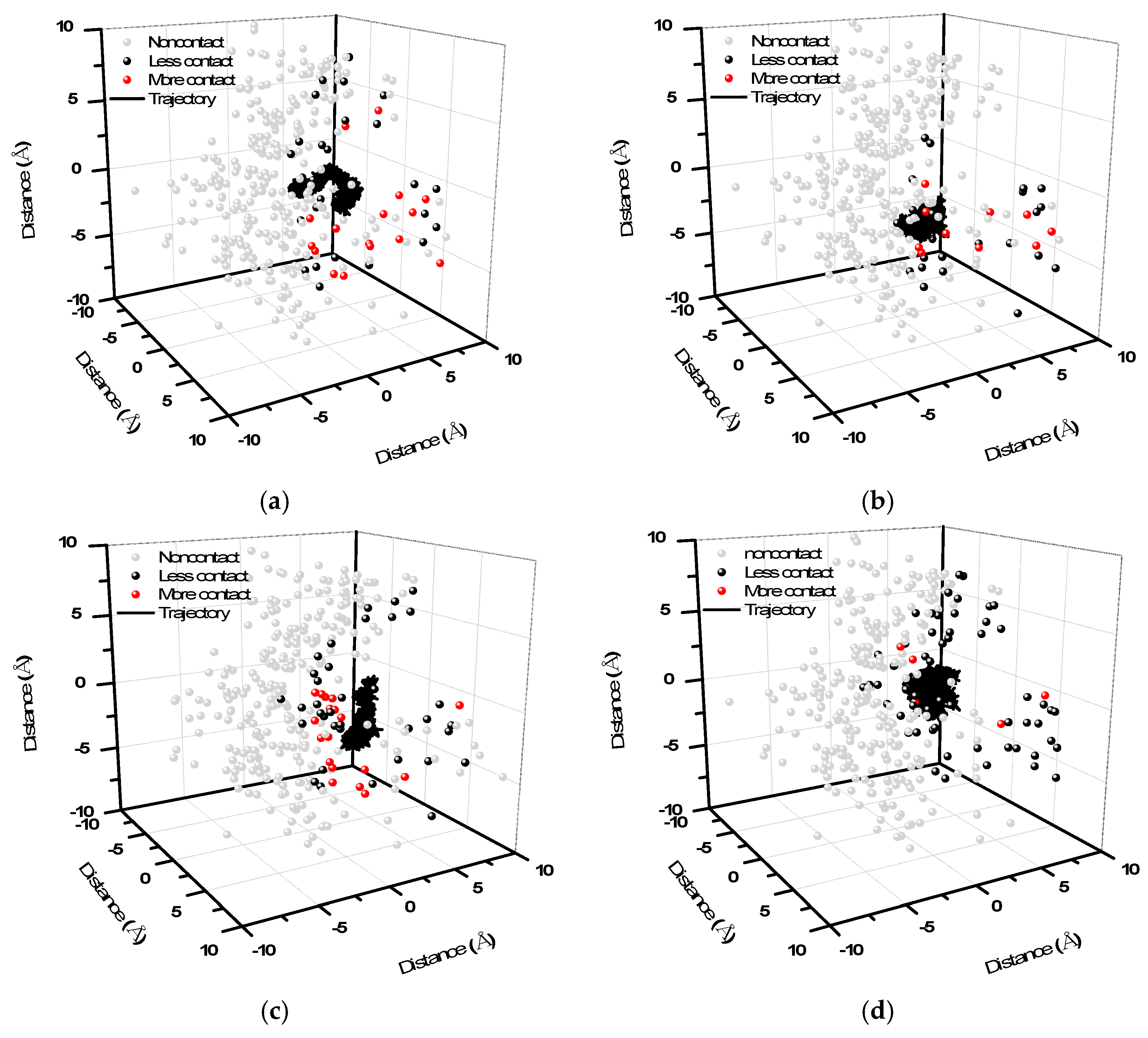
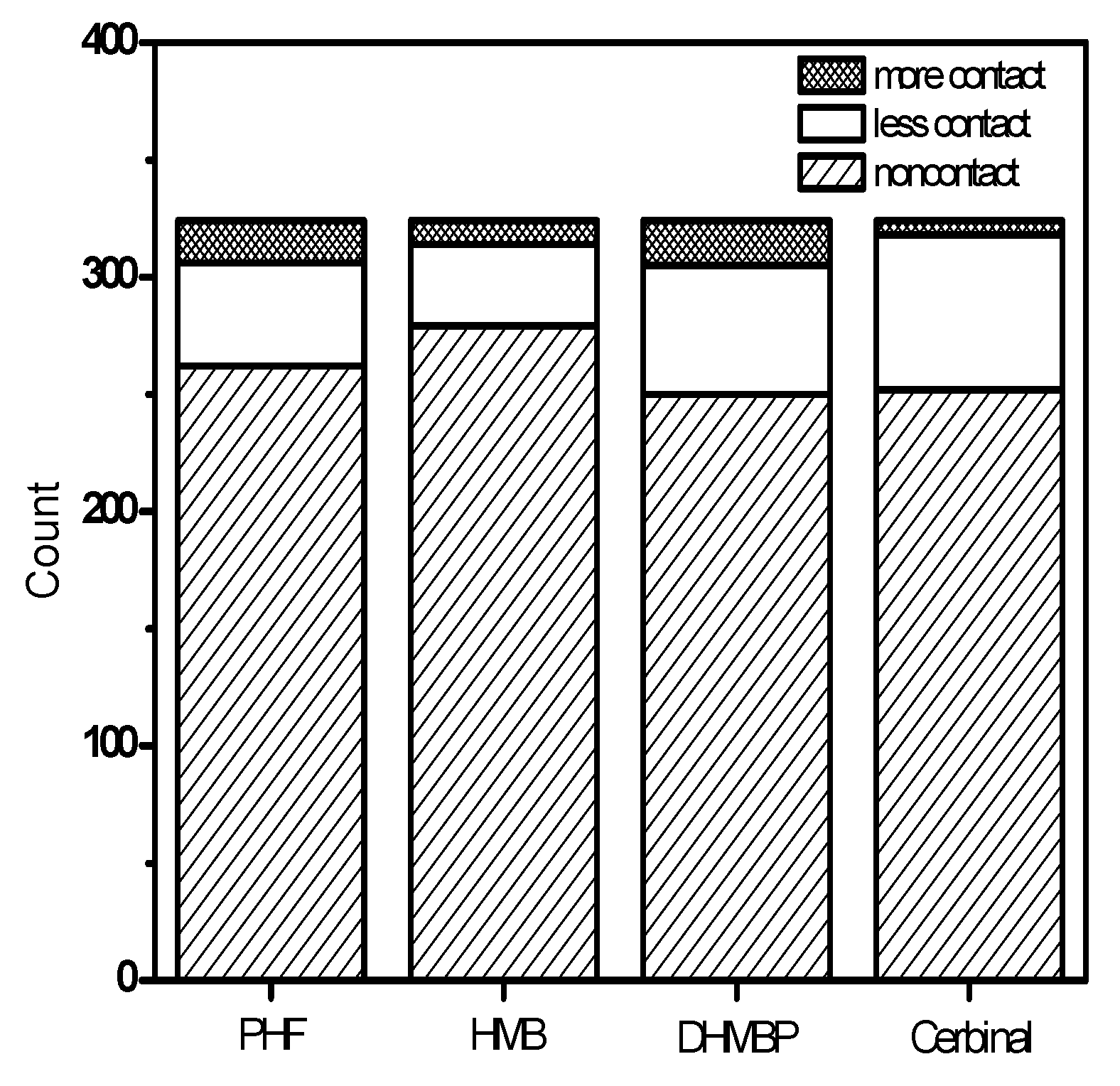
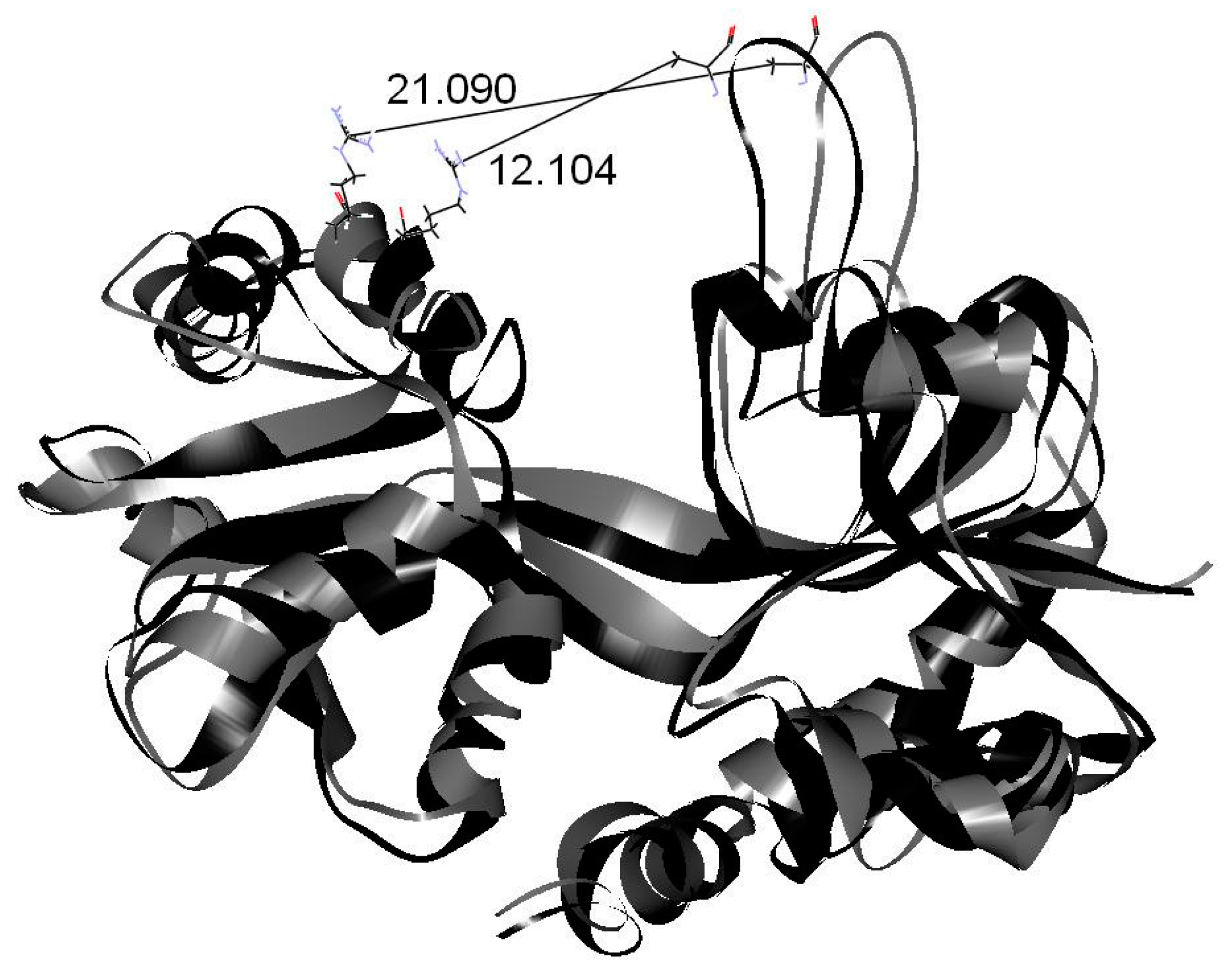
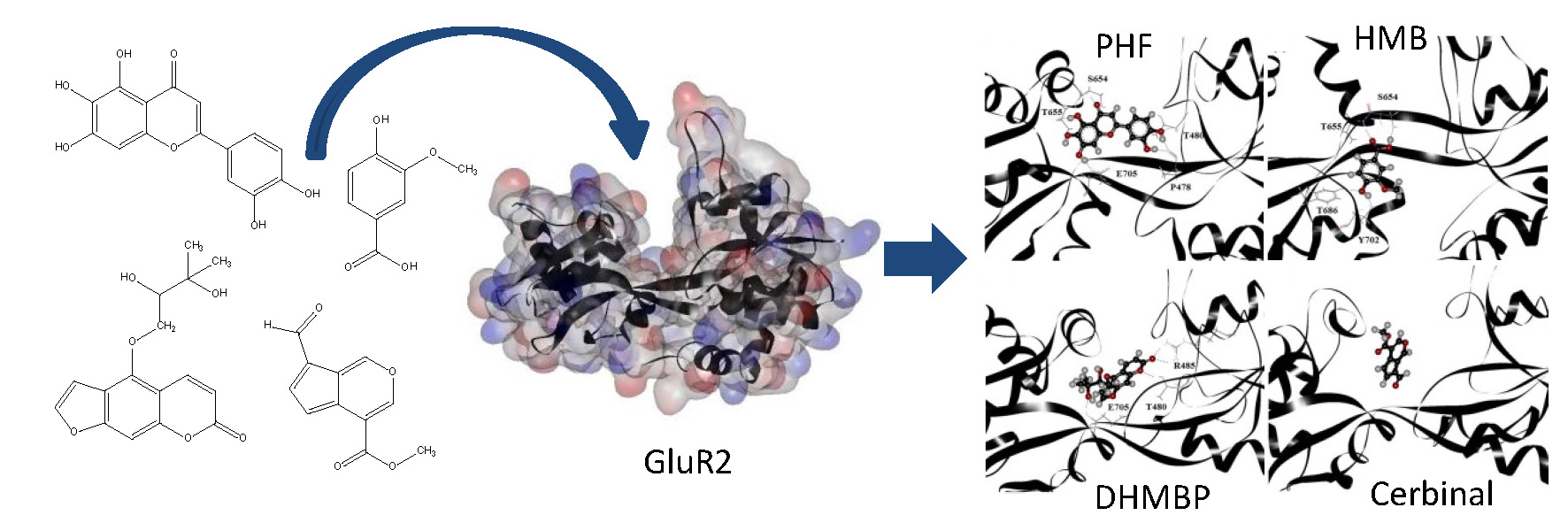
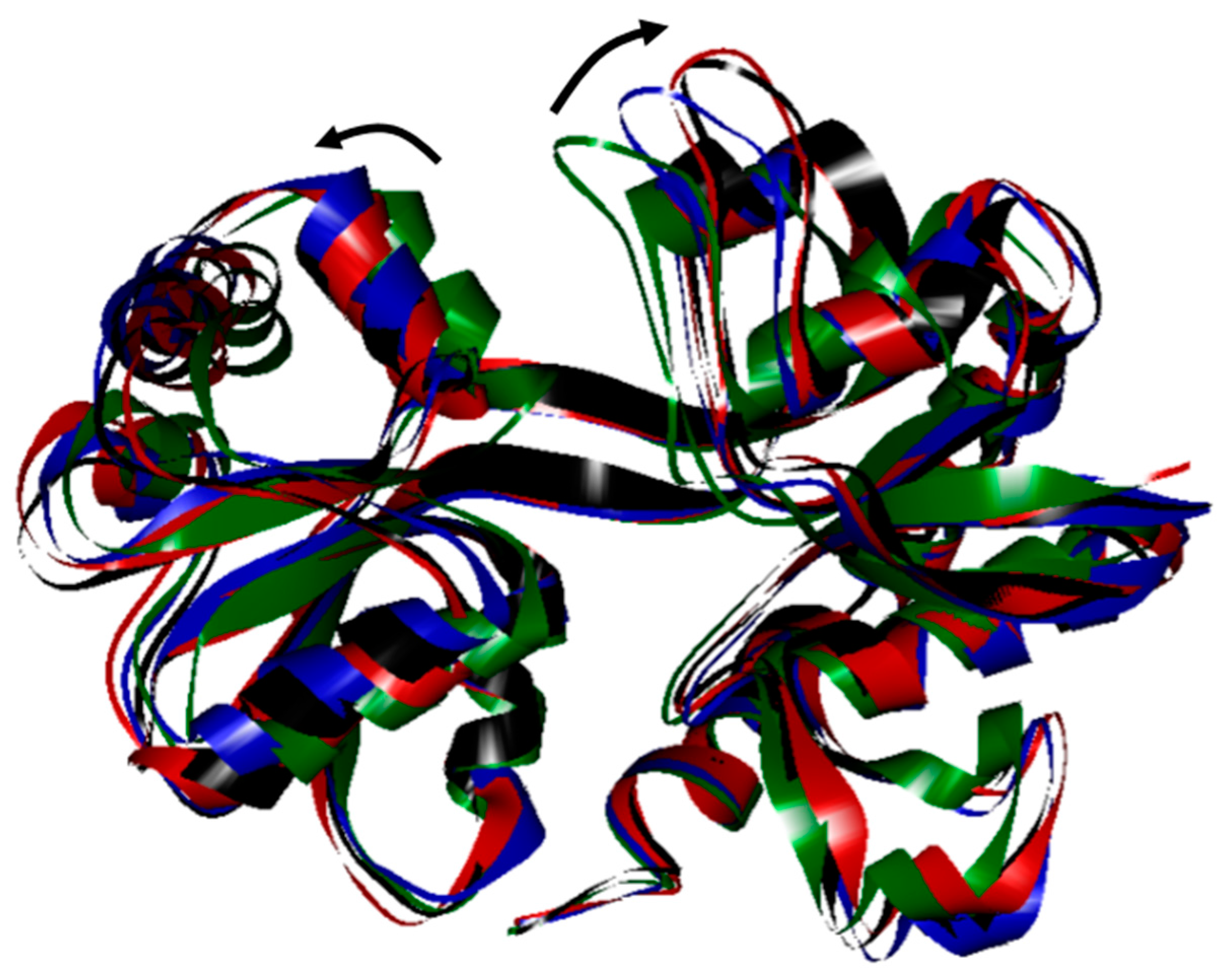
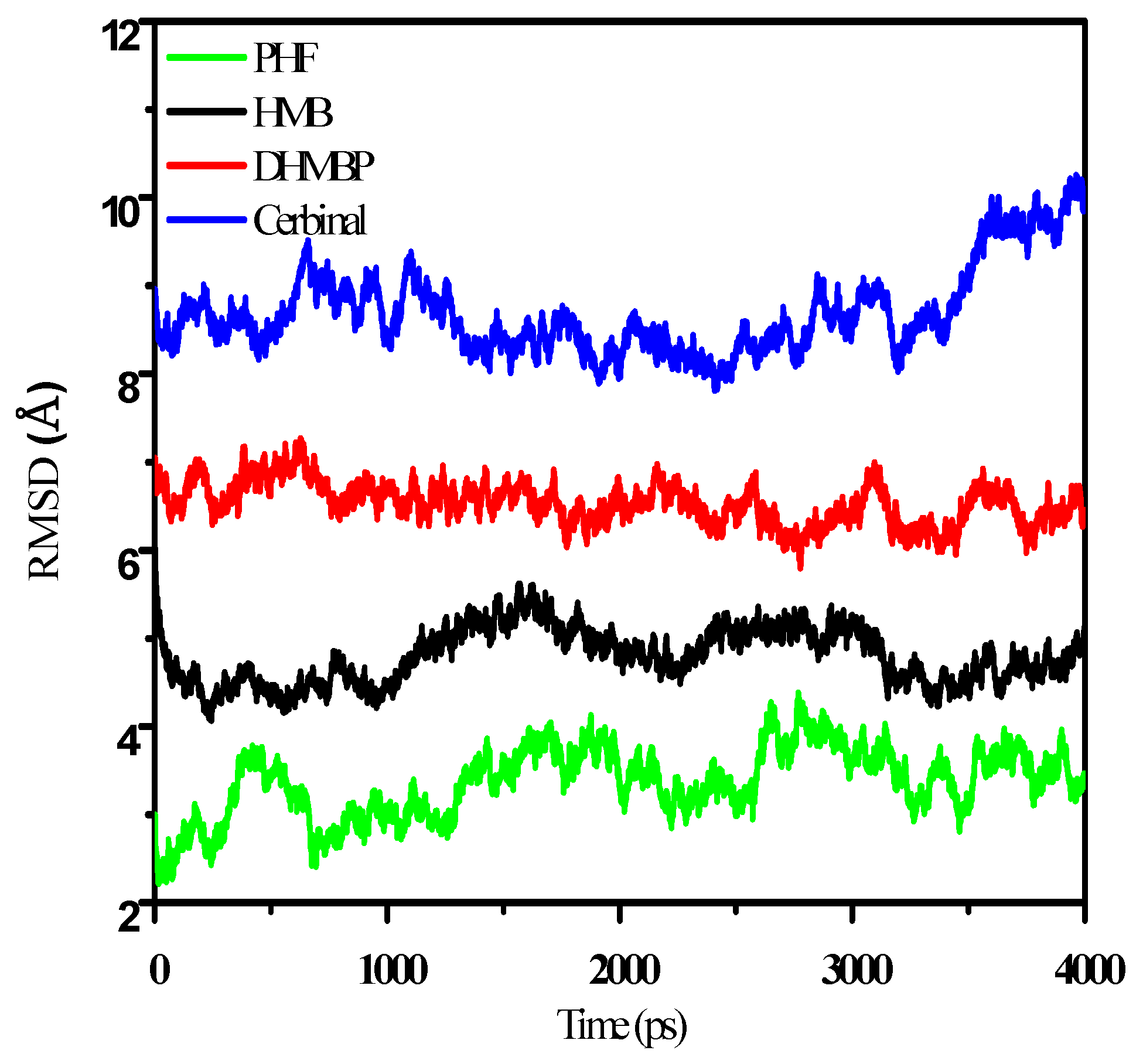
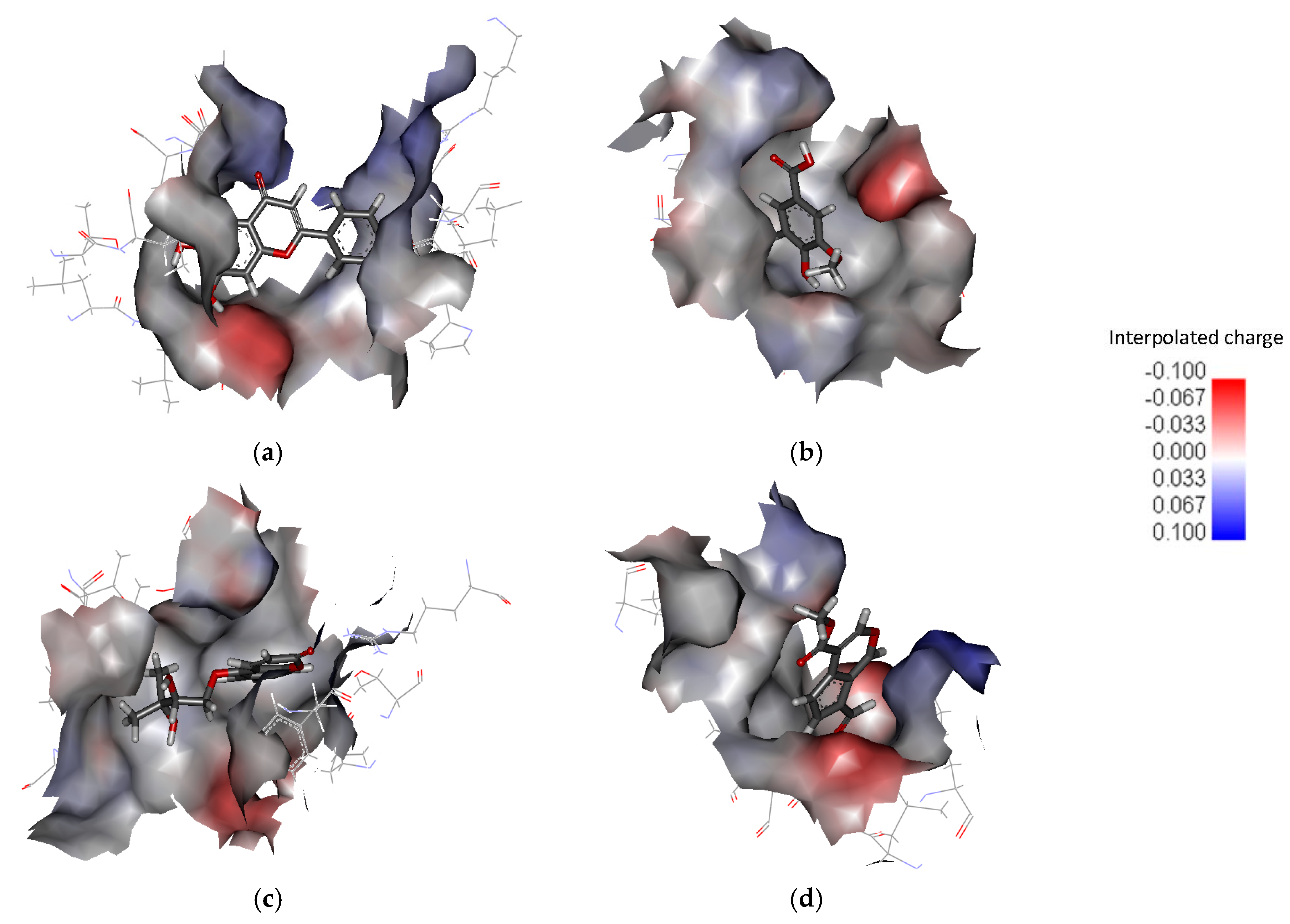
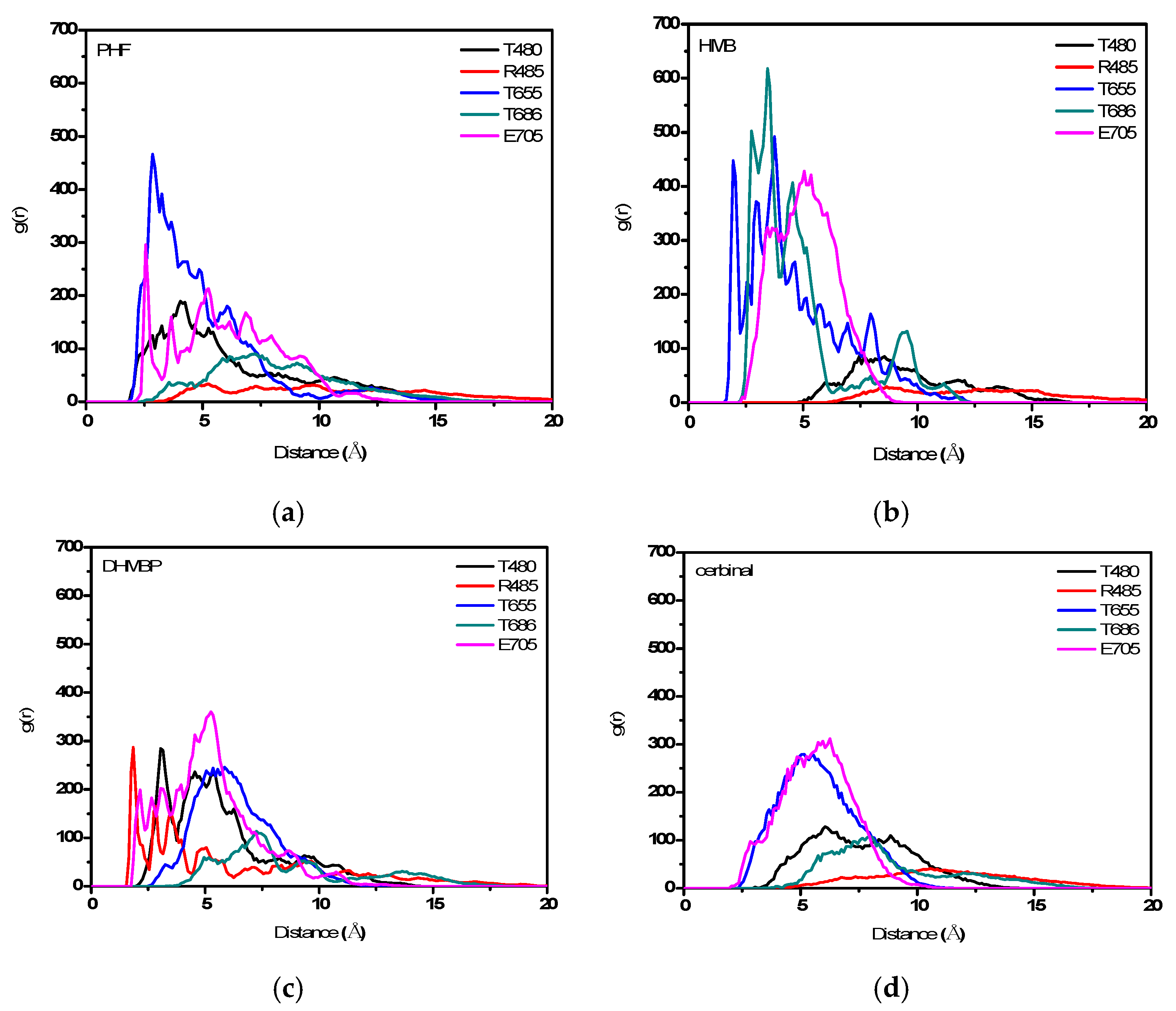
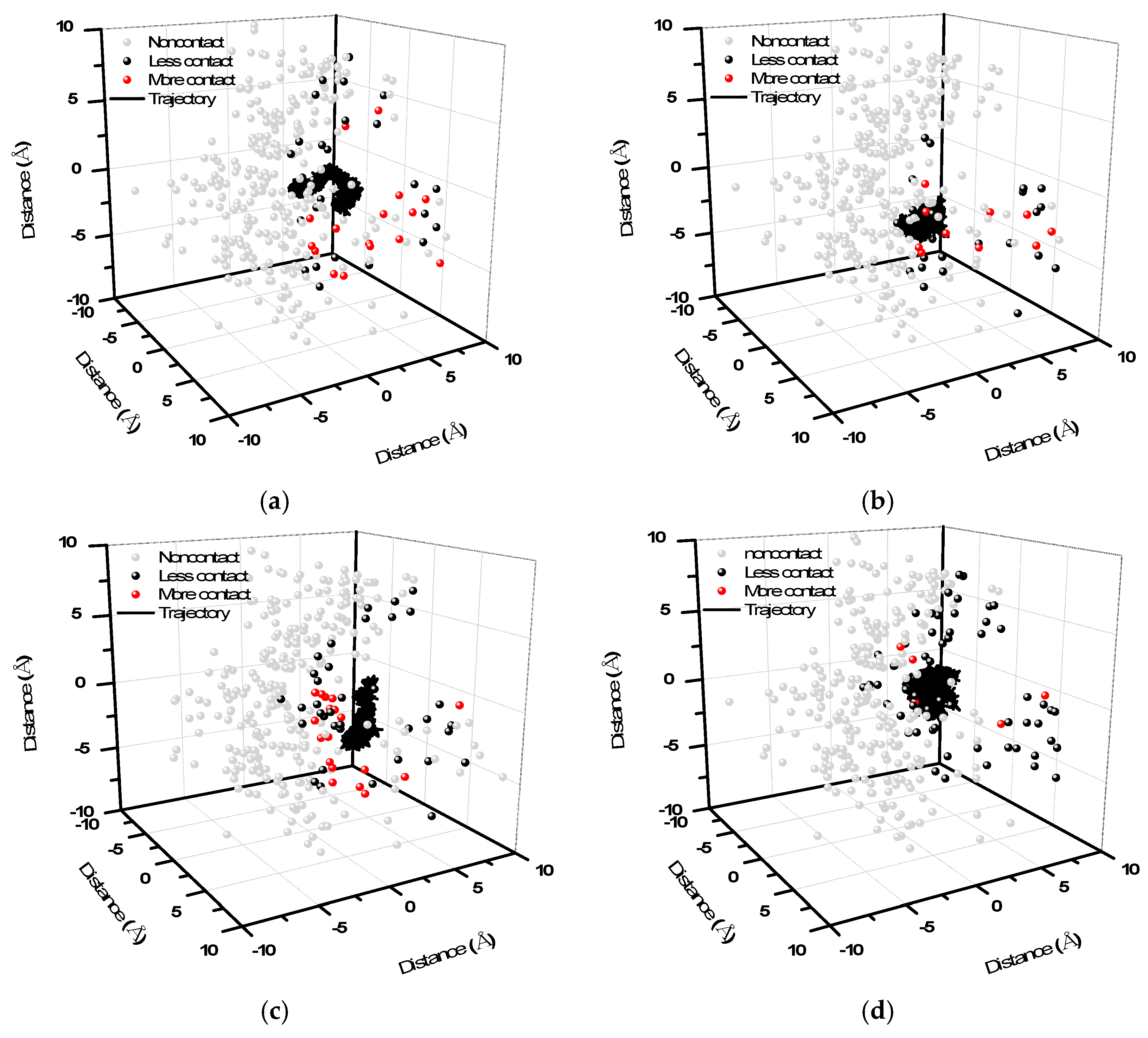
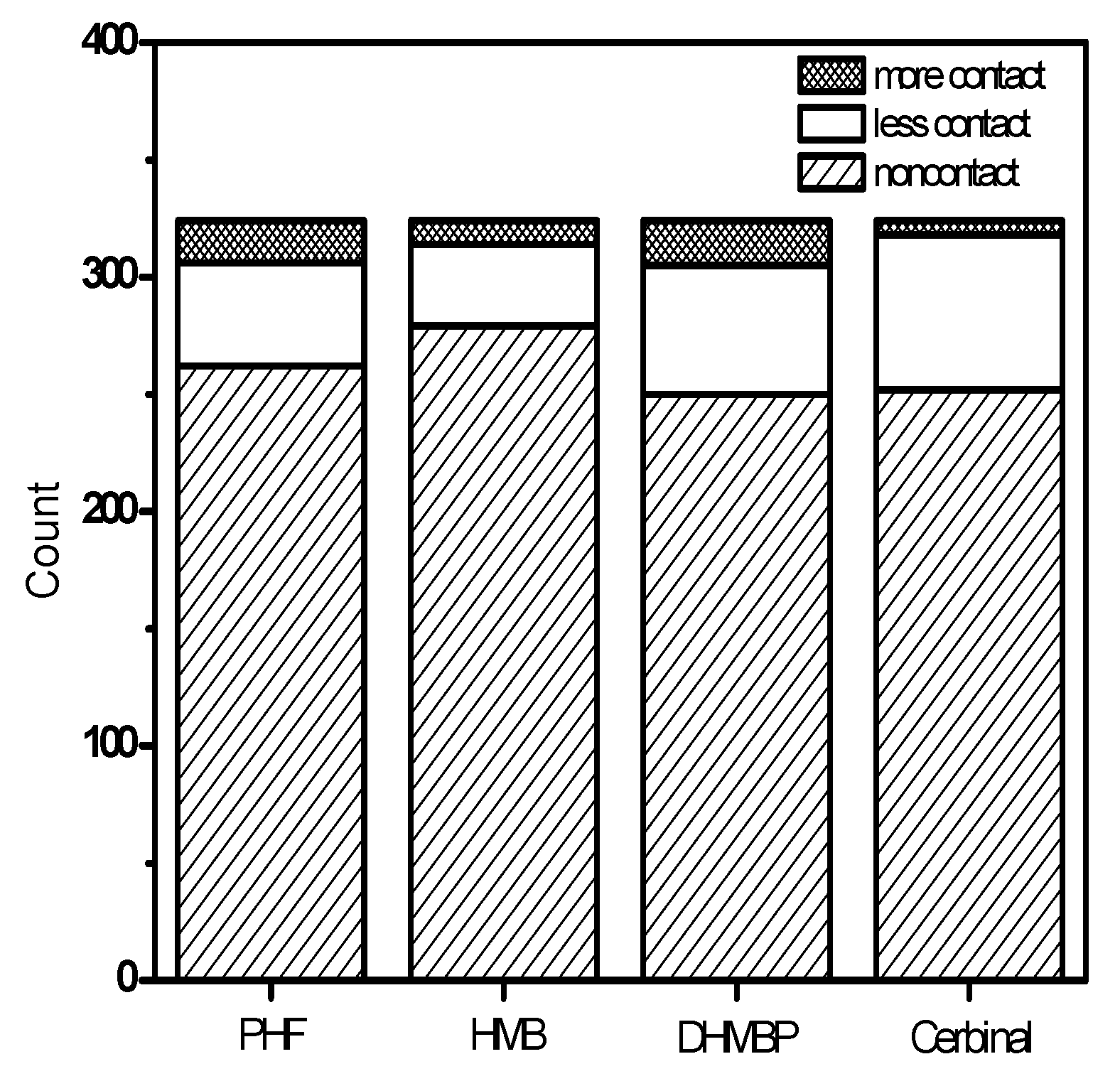
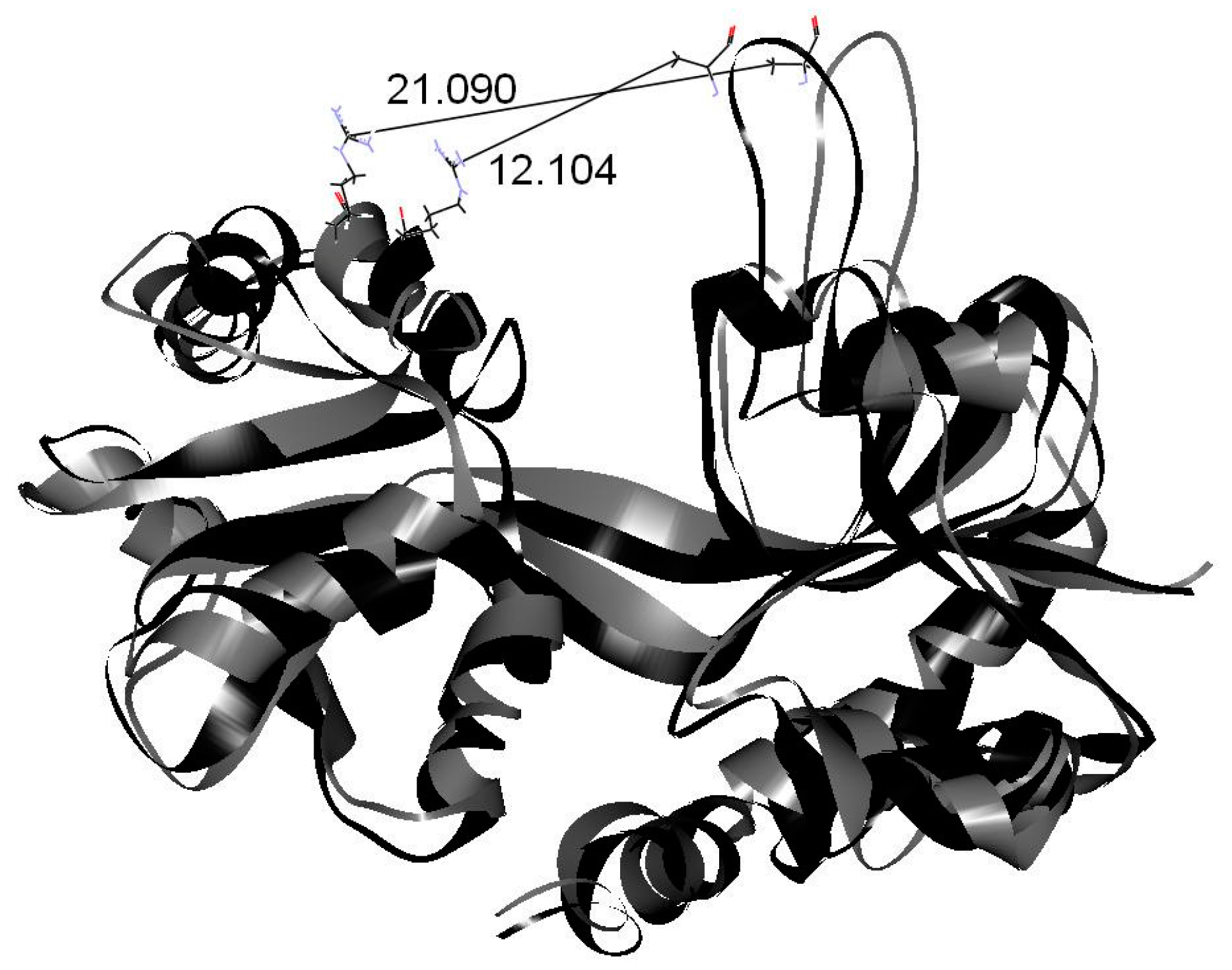
2. Results and Discussion
3. Methods
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yu, M.; Wang, Y.; Tian, L.; Wang, Y.Y.; Wang, X.Z.; Liang, W.G.; Yang, J.Y.; Yu, D.H.; Ma, T.H.; Fang, X.X. Safflomin a inhibits neuraminidase activity and influenza virus replication. RSC Adv. 2015, 5, 94053–94066. [Google Scholar] [CrossRef]
- Chen, K.C.; Sun, M.F.; Chen, H.Y.; Lee, C.C.; Chen, C.Y.C. Potential smoothened inhibitor from traditional Chinese medicine against the disease of diabetes, obesity, and cancer. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.C.; Chang, S.S.; Tsai, F.J.; Chen, C.Y.C. Han ethnicity-specific type 2 diabetic treatment from traditional Chinese medicine? J. Biomol. Struct. Dyn. 2013, 31, 1219–1235. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Chen, C.Y.C. Insights into designing the dual-targeted HER2/HSP90 inhibitors. J. Mol. Graph. 2010, 29, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.C.; Chang, S.S.; Chen, H.Y.; Chen, C.Y.C. Identification of potent EGFR inhibitors from TCM Database@Taiwan. PLoS Comput. Biol. 2011, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Tsou, Y.A.; Chen, K.C.; Lin, H.C.; Chang, S.S.; Chen, C.Y.C. Uroporphyrinogen decarboxylase as a potential target for specific components of traditional Chinese medicine: A virtual screening and molecular dynamics study. PLoS ONE 2012, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Tsou, Y.A.; Chen, K.C.; Chang, S.S.; Wen, Y.R.; Chen, C.Y.C. A possible strategy against head and neck cancer: In silico investigation of three-in-one inhibitors. J. Biomol. Struct. Dyn. 2013, 31, 1358–1369. [Google Scholar] [CrossRef] [PubMed]
- Kum, W.F.; Durairajan, S.S.K.; Bian, Z.X.; Man, S.C.; Lam, Y.C.; Xie, L.X.; Lu, J.H.; Wang, Y.; Huang, X.Z.; Li, M. Treatment of idiopathic Parkinson’s disease with traditional Chinese herbal medicine: A randomized placebo-controlled pilot clinical study. Evid. Based Complement. Altern. Med. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.C.; Lee, W.Y.; Chen, K.B.; Chan, Y.C.; Lee, C.C.; Chen, C.Y.C. In silico investigation of traditional Chinese medicine compounds to inhibit human histone deacetylase 2 for patients with Alzheimer’s disease. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Dingledine, R.; Borges, K.; Bowie, D.; Traynelis, S.F. The glutamate receptor ion channels. Pharmacol. Rev. 1999, 51, 7–61. [Google Scholar] [PubMed]
- Meyerson, J.R.; Kumar, J.; Chittori, S.; Rao, P.; Pierson, J.; Bartesaghi, A.; Mayer, M.L.; Subramaniam, S. Structural mechanism of glutamate receptor activation and desensitization. Nature 2014, 514, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Penn, A.C.; Williams, S.R.; Greger, I.H. Gating motions underlie AMPA receptor secretion from the endoplasmic reticulum. EMBO. J. 2008, 27, 3056–3068. [Google Scholar] [CrossRef] [PubMed]
- Asztely, F.; Gustafsson, B. Ionotropic glutamate receptors—Their possible role in the expression of hippocampal synaptic plasticity. Mol. Neurobiol. 1996, 12, 1–11. [Google Scholar] [PubMed]
- Armstrong, N.; Gouaux, E. Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: Crystal structures of the GluR2 ligand binding core. Neuron 2000, 28, 165–181. [Google Scholar] [CrossRef]
- Ahmed, A.H.; Thompson, M.D.; Fenwick, M.K.; Romero, B.; Loh, A.P.; Jane, D.E.; Sondermann, H.; Oswald, R.E. Mechanisms of antagonism of the GluR2 AMPA receptor: Structure and dynamics of the complex of two willardiine antagonists with the glutamate binding domain. Biochemistry 2009, 48, 3894–3903. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, N.; Mayer, M.; Gouaux, E. Tuning activation of the AMPA-sensitive GluR2 ion channel by genetic adjustment of agonist-induced conformational changes. Proc. Natl. Acad. Sci. USA 2003, 100, 5736–5741. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Jayaraman, V. Chemistry and conformation of the ligand-binding domain of GluR2 subtype of glutamate receptors. J. Biol. Chem. 2004, 279, 26346–26350. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.S.; Banke, T.G.; Mayer, M.L.; Traynelis, S.F.; Gouaux, E. Structural basis for partial agonist action at ionotropic glutamate receptors. Nat. Neurosci. 2003, 6, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.S.; Gouaux, E. Probing the function, conformational plasticity, and dimer-dimer contacts of the GluR2 ligand-binding core: Studies of 5-substituted willardiines and GluR2 S1S2 in the crystal. Biochemistry 2003, 42, 5201–5213. [Google Scholar] [CrossRef] [PubMed]
- Hogner, A.; Greenwood, J.R.; Liljefors, T.; Lunn, M.L.; Egebjerg, J.; Larsen, I.K.; Gouaux, E.; Kastrup, J.S. Competitive antagonism of AMPA receptors by ligands of different classes: Crystal structure of ATPO bound to the GluR2 ligand-binding core, in comparison with DNQX. J. Med. Chem. 2003, 46, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Menuz, K.; Stroud, R.M.; Nicoll, R.A.; Hays, F.A. Tarp auxiliary subunits switch AMPA receptor antagonists into partial agonists. Science 2007, 318, 815–817. [Google Scholar] [CrossRef] [PubMed]
- Kasper, C.; Pickering, D.S.; Mirza, O.; Olsen, L.; Kristensen, A.S.; Greenwood, J.R.; Liljefors, T.; Schousboe, A.; Watjen, F.; Gajhede, M.; et al. The structure of a mixed GluR2 ligand-binding core dimer in complex with (s)-glutamate and the antagonist (s)-NS1209. J. Mol. Biol. 2006, 357, 1184–1201. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Wang, J.H.; Liu, P. Effects of WenQingYin on serum MDA and SOD of rats with stress ulcer of stomach. Chin. J. Tradit. Med. Sci. Technol. 2009, 16, 174–175. [Google Scholar]
- Gasiorowski, K.; Lamer-Zarawska, E.; Leszek, J.; Parvathaneni, K.; Yendluri, B.B.; Blach-Olszewska, Z.; Aliev, G. Flavones from root of Scutellaria baicalensis Georgi: Drugs of the future in neurodegeneration? CNS Neurol. Disord. Drug Targets 2011, 10, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.S.; Kim, M.H.; Gwak, N.G.; Im, Y.S.; Lee, K.Y.; Sohn, Y.; Choi, H.; Yang, W.M. Antiallergic effects of Scutellaria baicalensis on inflammation in vivo and in vitro. J. Ethnopharmacol. 2012, 141, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Ma, J.; Ma, Z.; Wang, Y.; Tan, H.; Xiao, C.; Liu, M.; Lu, B.; Zhang, B.; Gao, Y. Chemical comparison of dried rehmannia root and prepared rehmannia root by UPLC-TOF MS and HPLC-ELSD with multivariate statistical analysis. Acta Pharm. Sin. B 2013, 3, 55–64. [Google Scholar] [CrossRef]
- Sung, Y.Y.; Lee, A.Y.; Kim, H.K. The Gardenia jasminoides extract and its constituent, geniposide, elicit anti-allergic effects on atopic dermatitis by inhibiting histamine in vitro and in vivo. J. Ethnopharmacol. 2014, 156, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Xue, R.; Fang, Z.; Zhang, M.; Yi, Z.; Wen, C.; Shi, T. TCMID: Traditional Chinese medicine integrative database for herb molecular mechanism analysis. Nucleic Acids Res. 2013, 41, D1089–D1095. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.C. TCM database@taiwan: The world’s largest traditional Chinese medicine database for drug screening in silico. PLoS ONE 2011, 6, e15939. [Google Scholar] [CrossRef] [PubMed]
- Chuang, P.H.; Gu, Q.R.; Tseng, Y.H.; Chen, C.L. Estimation of electron transfer properties of ferrocenyl–dicholesteryl-peptide in liquid and gel. J. Colloid Interface Sci. 2014, 417, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Chuang, P.H.; Tseng, Y.H.; Gu, Q.R.; Chen, C.L. Phase behavior and electron transfer properties of ferrocenyl cholesteryl N-formanidoformamide gelator: A computational study. Colloid Polym. Sci. 2015, 293, 2113–2119. [Google Scholar] [CrossRef]
- Werder, T.; Walther, J.H.; Jaffe, R.L.; Halicioglu, T.; Koumoutsakos, P. On the water-carbon interaction for use in molecular dynamics simulations of graphite and carbon nanotubes. J. Phys. Chem. B 2003, 107, 1345–1352. [Google Scholar] [CrossRef]
- Skoulidas, A.I.; Sholl, D.S. Self-diffusion and transport diffusion of light gases in metal-organic framework materials assessed using molecular dynamics simulations. J. Phys. Chem. B 2005, 109, 15760–15768. [Google Scholar] [CrossRef] [PubMed]
- Beck, D.A.C.; Daggett, V. Methods for molecular dynamics simulations of protein folding/unfolding in solution. Methods 2004, 34, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wade, R.C. Implicit solvent models for flexible protein-protein docking by molecular dynamics simulation. Proteins 2003, 50, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.Y.; Nussinov, R. Simulations as analytical tools to understand protein aggregation and predict amyloid conformation. Curr. Opin. Chem. Biol. 2006, 10, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Cichero, E.; D’Ursi, P.; Moscatelli, M.; Bruno, O.; Orro, A.; Rotolo, C.; Milanesi, L.; Fossa, P. Homology modeling, docking studies and molecular dynamic simulations using graphical processing unit architecture to probe the type-11 phosphodiesterase catalytic site: A computational approach for the rational design of selective inhibitors. Chem. Biol. Drug Des. 2013, 82, 718–731. [Google Scholar] [CrossRef] [PubMed]
- D’Ursi, P.; Guariento, S.; Trombetti, G.; Orro, A.; Cichero, E.; Milanesi, L.; Fossa, P.; Bruno, O. Further insights in the binding mode of selective inhibitors to human PDE4D enzyme combining docking and molecular dynamics. Mol. Inform. 2016, 35, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Speranskiy, K.; Kurnikova, M. On the binding determinants of the glutamate agonist with the glutamate receptor ligand binding domain. Biochemistry 2005, 44, 11508–11517. [Google Scholar] [CrossRef] [PubMed]
- Mamonova, T.; Yonkunas, M.J.; Kurnikova, M.G. Energetics of the cleft closing transition and the role of electrostatic interactions in conformational rearrangements of the glutamate receptor ligand binding domain. Biochemistry 2008, 47, 11077–11085. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, N.; Sun, Y.; Chen, G.Q.; Gouaux, E. Structure of a glutamate-receptor ligand-binding core in complex with kainate. Nature 1998, 395, 913–917. [Google Scholar] [PubMed]
- Mendieta, J.; Gago, F.; Ramirez, G. Binding of 5′-GMP to the GluR2 AMPA receptor: Insight from targeted molecular dynamics simulations. Biochemistry 2005, 44, 14470–14476. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.E.; Wyllie, D.J.A. Pharmacological insights obtained from structure-function studies of ionotropic glutamate receptors. Br. J. Pharmacol. 2006, 147, 839–853. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
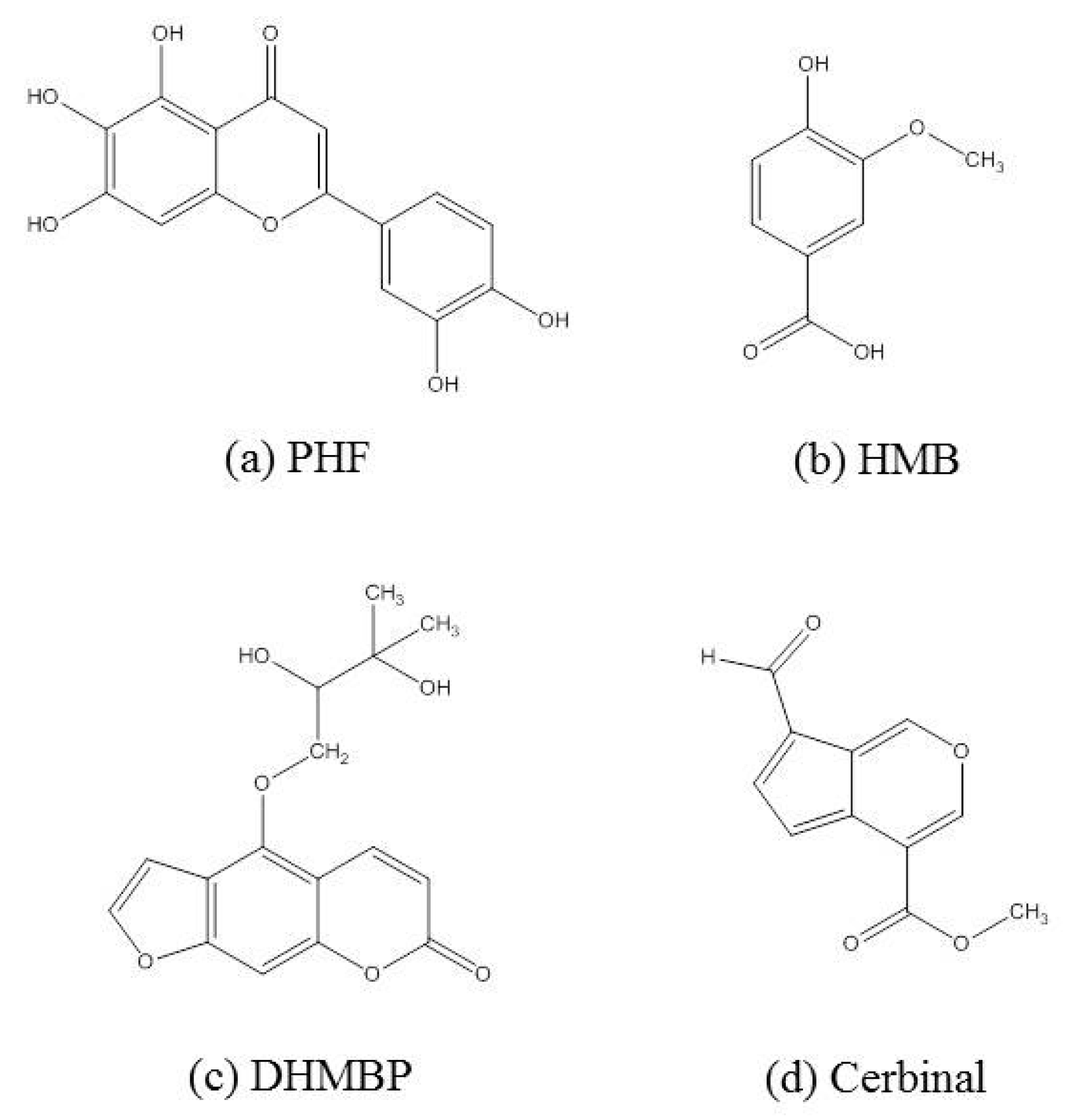
| Drugs | Binding Energy (kcal/mol) |
|---|---|
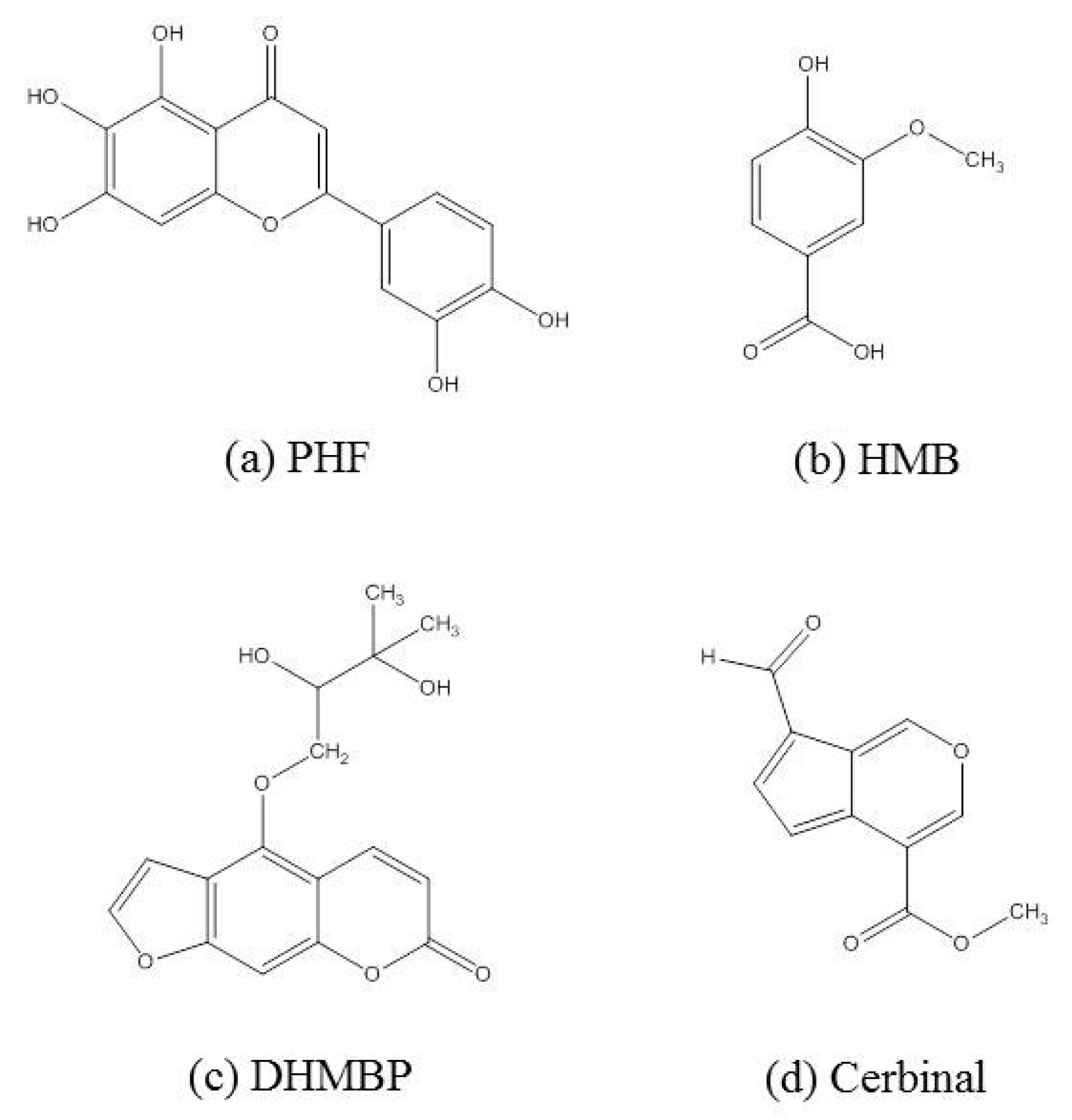
| 2-(3,4-dihydroxyphenyl)-5,6,7-trihydroxy-4H-chromen-4-one (PHF) | −7.69 |
| 4-hydroxy-3-methoxybenzoic acid (HMB) | −5.26 |
| 4-(2,3-dihydroxy-3-methylbutoxy)-7H-furo[3,2-g]chromen-7-one (DHMBP) | −6.57 |
| Cerbinal | −5.53 |
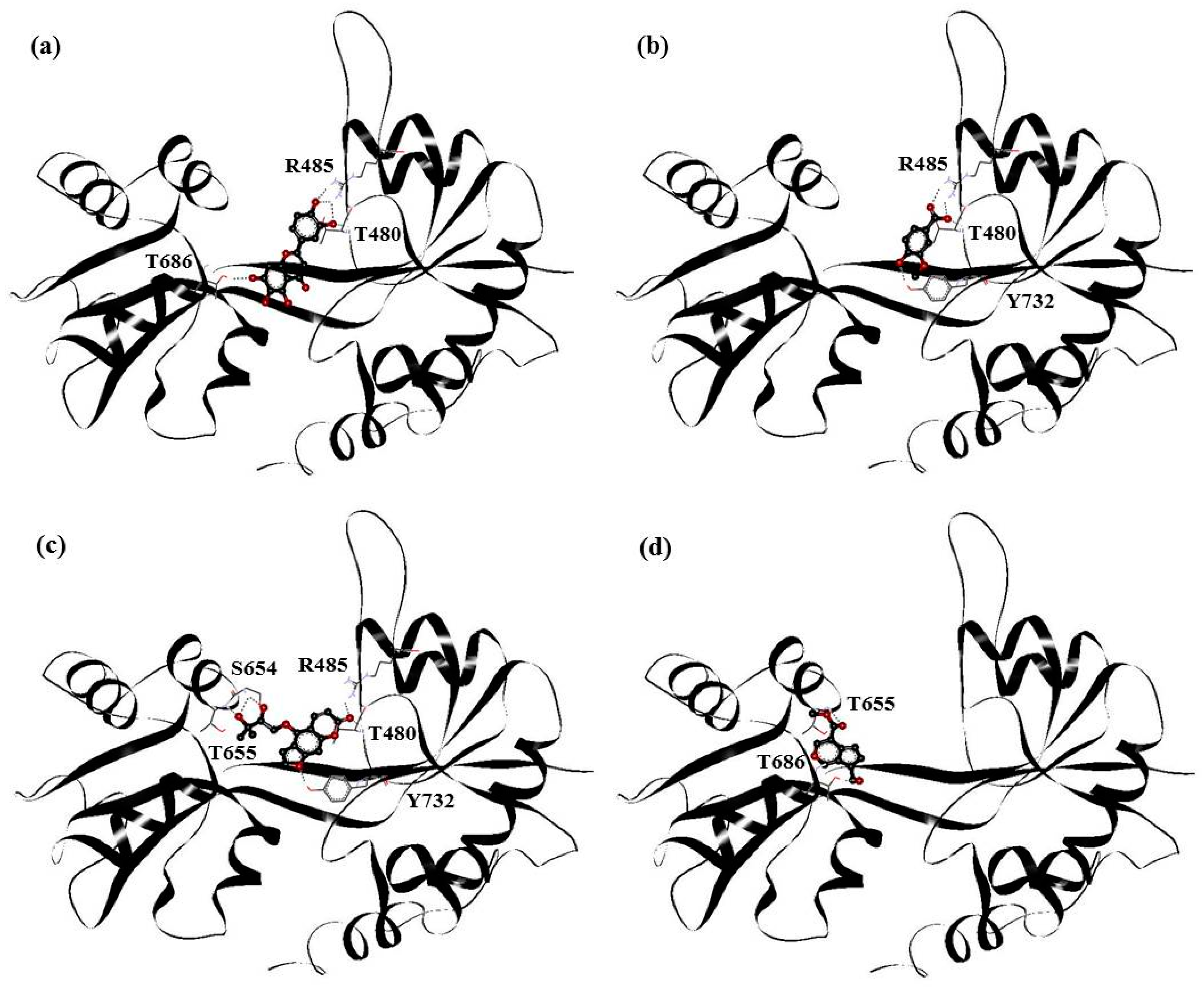
| Drugs | Residues |
|---|---|
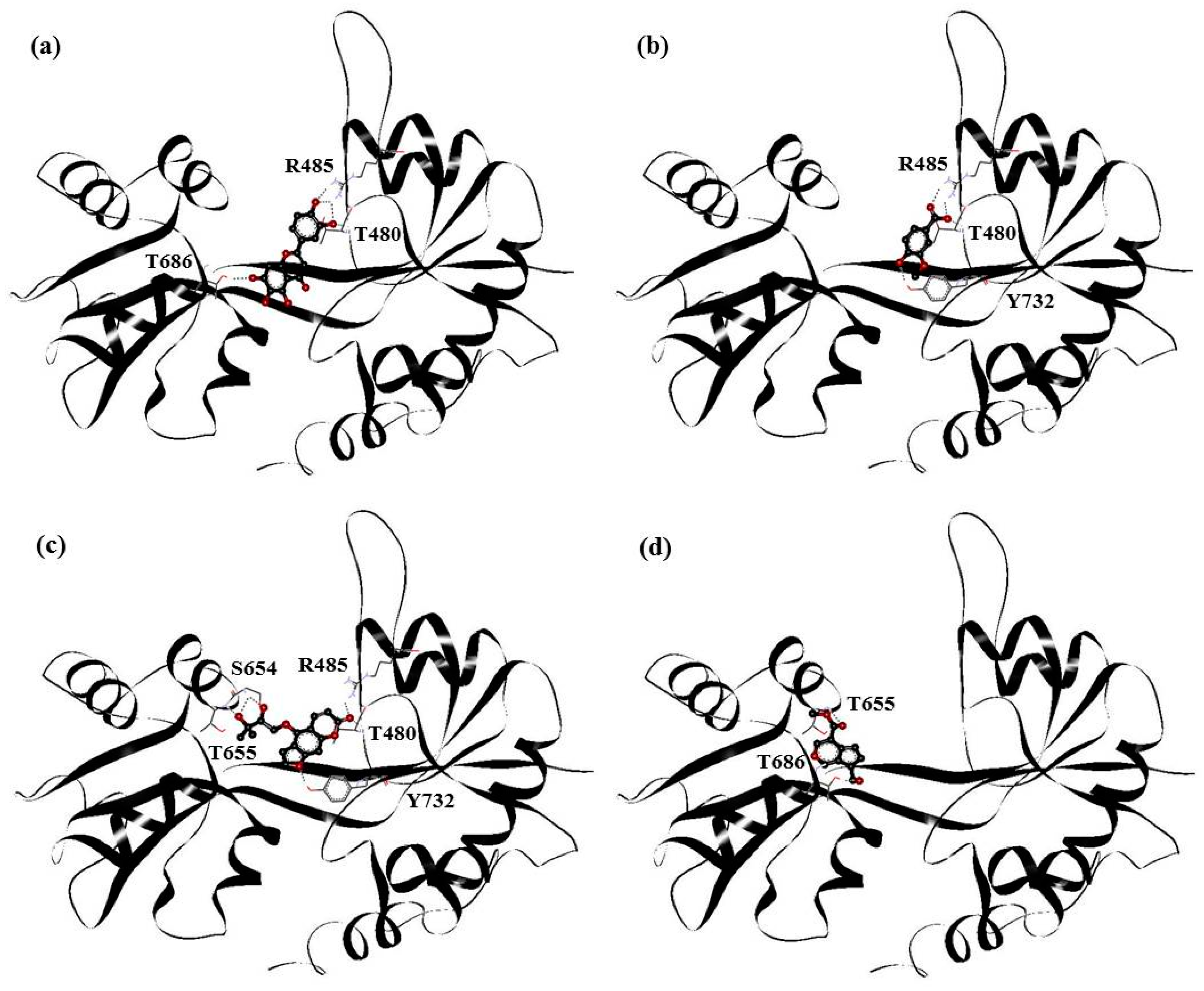
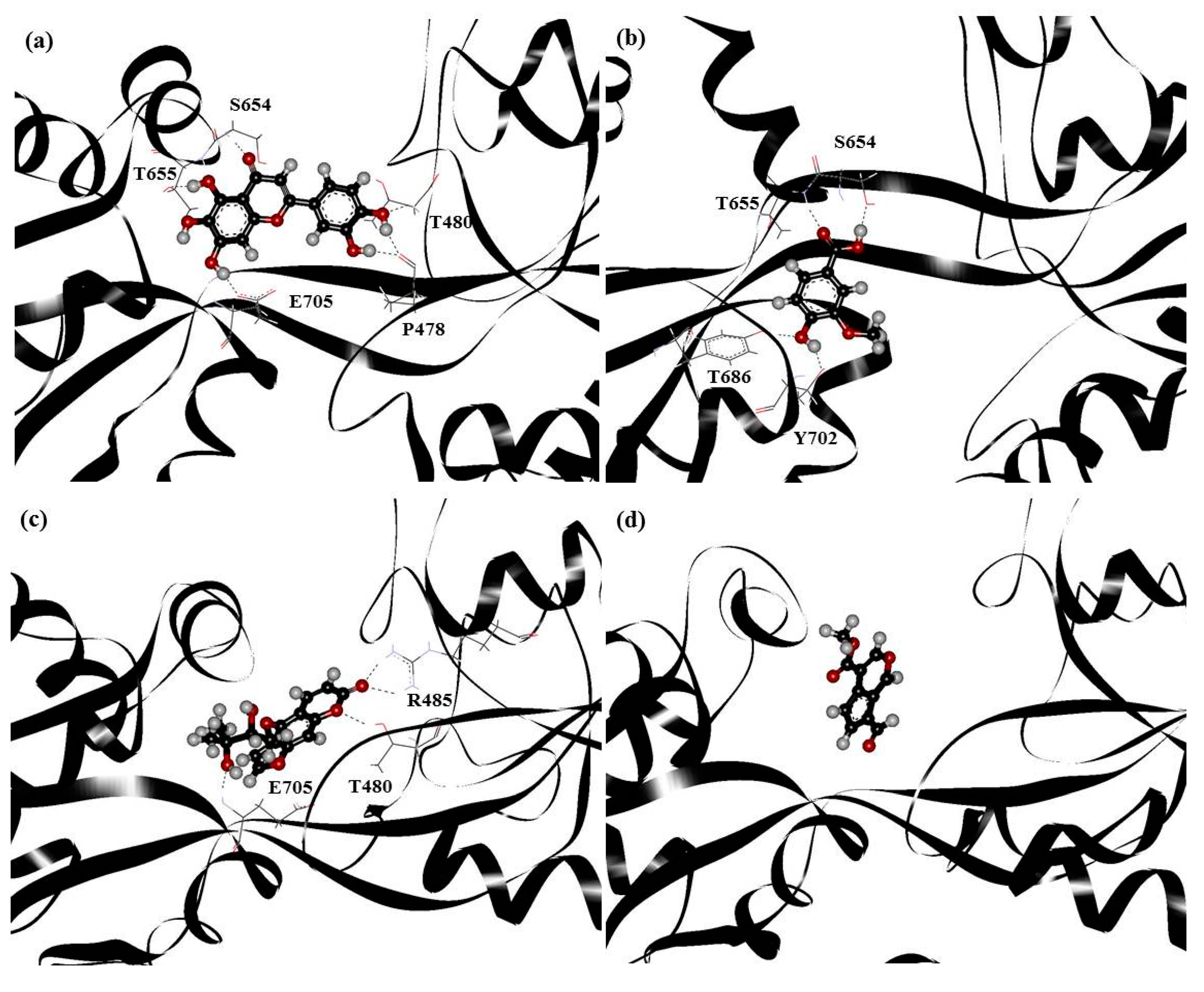
| PHF | Y450, P478, L479, T480, R485, T649, L650, G653, S654, T655, L703, L704, E705, Y732 |
| HMB | T649, L650, G653, S654, T655, T686, Y702, L703, L704, E705, M708 |
| DHMBP | Y450, T480, R485, L498, G499, I500, T649, L650, S654, T655, T686, Y702, L703, L704, E705, S706 |
| Cerbinal | L498, G499, I500, L650, S654, T655, E705, D728, K730 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tseng, Y.-H.; Chuang, P.-H.; Huang, Y.-R.; Chen, C.-L. Computational Investigation into the Interactions of Traditional Chinese Medicine Molecules of WenQingYin with GluR2. Int. J. Mol. Sci. 2017, 18, 1443. https://doi.org/10.3390/ijms18071443
Tseng Y-H, Chuang P-H, Huang Y-R, Chen C-L. Computational Investigation into the Interactions of Traditional Chinese Medicine Molecules of WenQingYin with GluR2. International Journal of Molecular Sciences. 2017; 18(7):1443. https://doi.org/10.3390/ijms18071443
Chicago/Turabian StyleTseng, Yu-Hui, Po-Hsiang Chuang, Yu-Ren Huang, and Cheng-Lung Chen. 2017. "Computational Investigation into the Interactions of Traditional Chinese Medicine Molecules of WenQingYin with GluR2" International Journal of Molecular Sciences 18, no. 7: 1443. https://doi.org/10.3390/ijms18071443