Ankyrin Repeat Domain 1 Protein: A Functionally Pleiotropic Protein with Cardiac Biomarker Potential

 ,
,
Abstract
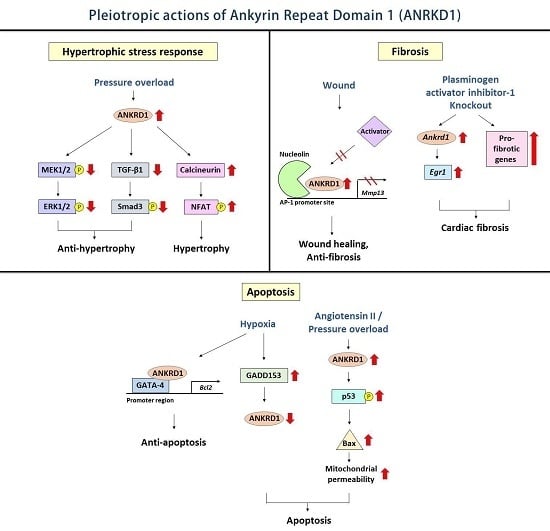
:
1. Introduction
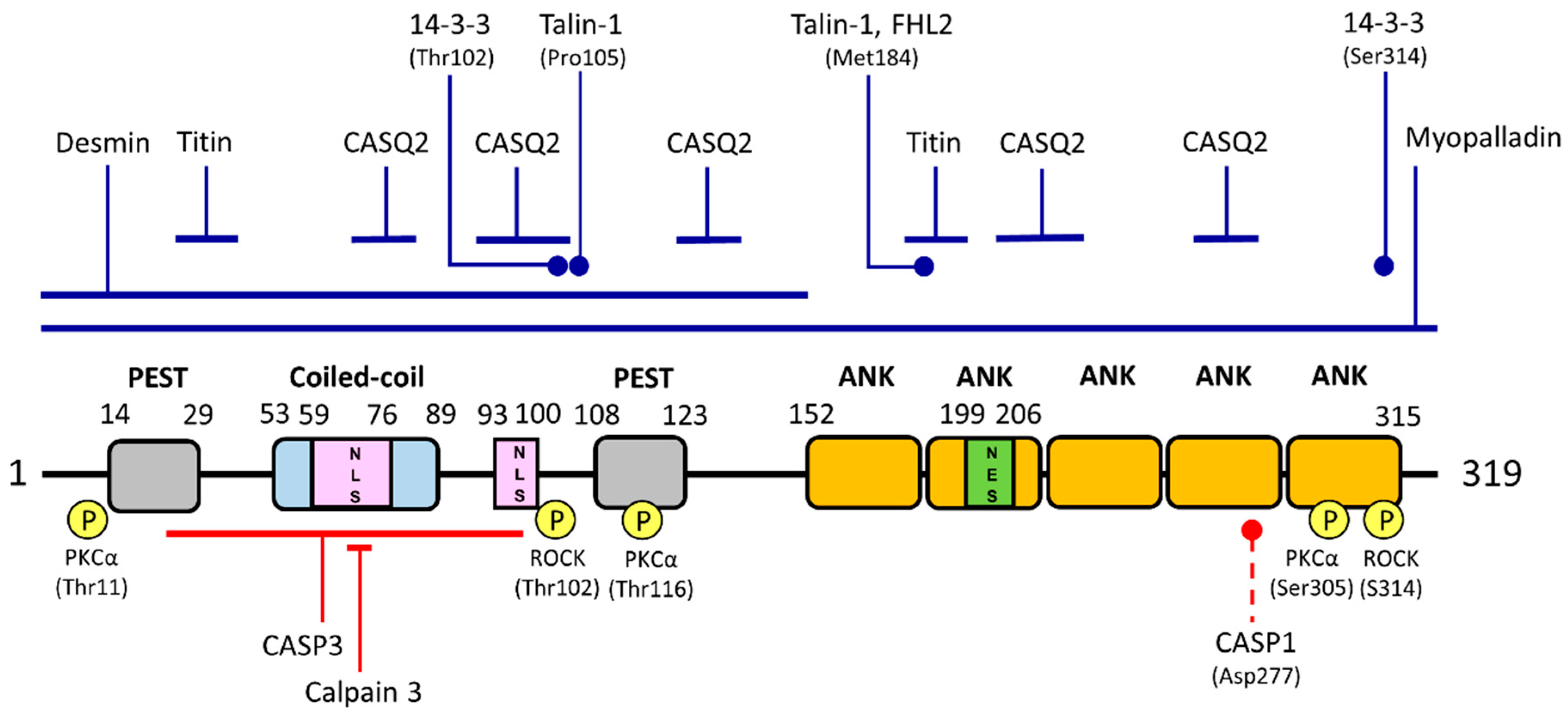
2. Gene and Protein Characteristics
3. Ankyrin Repeat Domain 1 Localization, Interacting Partners and Related Functions
3.1. Localization
3.2. Interaction with Transcription Factors: Regulation of Gene Expression
3.2.1. Y-Box Binding Protein 1 (YB-1)
3.2.2. Nuclear Factor κ-Light-Chain-Enhancer of Activated B Cells (NF-κB)
3.2.3. Nucleolin
3.2.4. Tumor Suppressor p53
3.3. Interaction with Structural Components of the Sarcomere: Maintenance of Sarcomere Integrity and Mechano-Sensing Functions
3.3.1. Myopalladin and Titin
3.3.2. Desmin
3.3.3. Talin-1
3.4. Interaction with Signaling Molecules
3.4.1. Four-And-A-Half LIM Domains 2 (FHL2)
3.4.2. Calsequestrin 2 (CASQ2)
3.4.3. 14-3-3 Proteins
3.4.4. Protein Kinase C Alpha (PKCα)
3.5. Effects of Ankrd1 Knockout in Mice
4. Pathological Cardiac Functions of ANKRD1
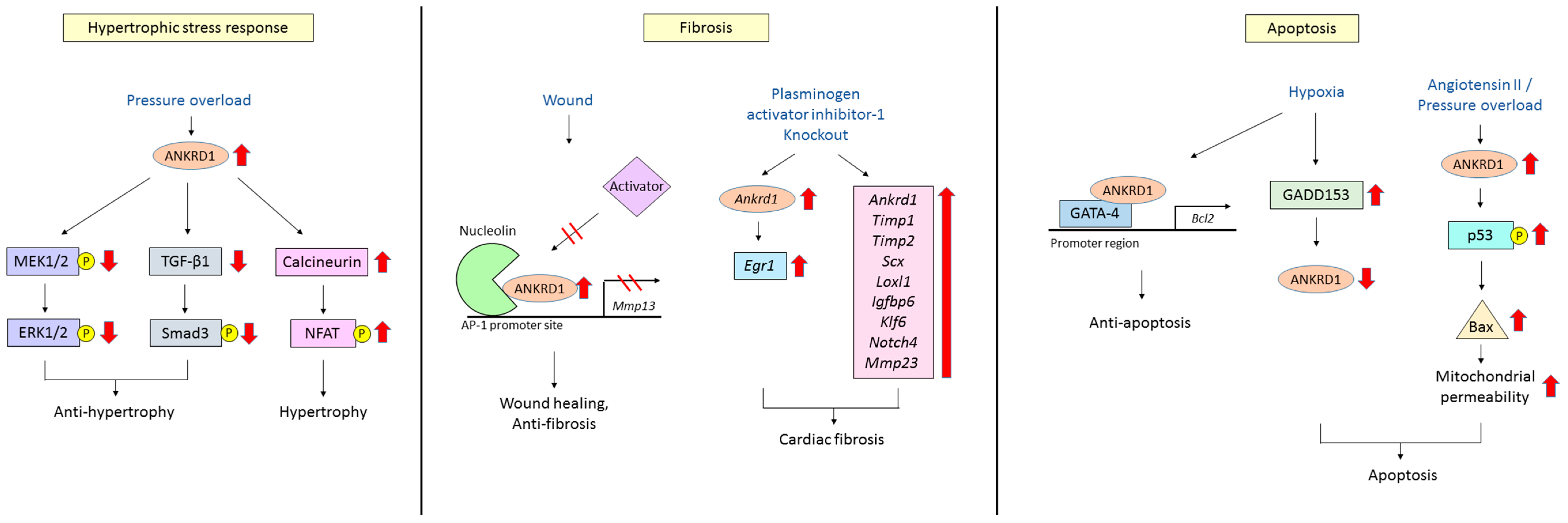
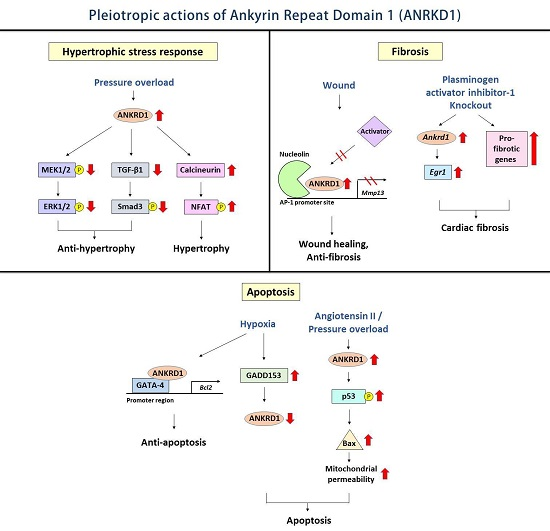
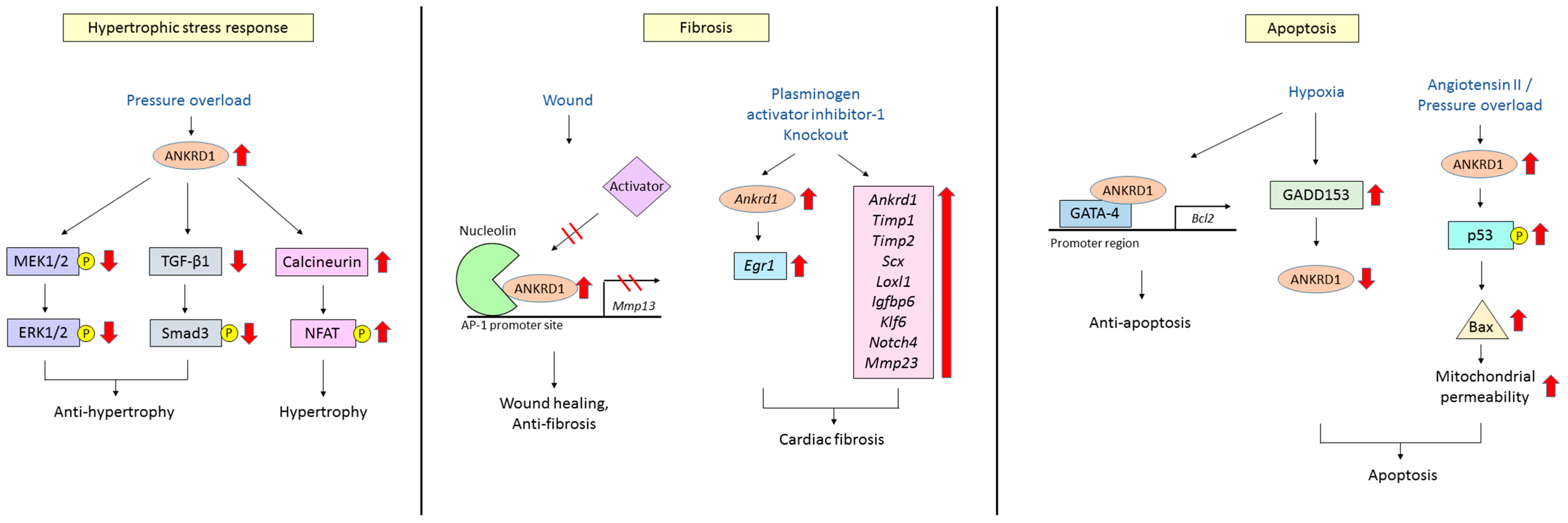
4.1. Hypertrophic Stress Responses
4.2. Cardiac Fibrosis
4.3. Cardiomyocyte Apoptosis
4.4. ANKRD1: Disease-Causing or Cardioprotective?
5. Regulation of ANKRD1
5.1. Regulation of ANKRD1 Promoter Activity
5.2. Post-Transcriptional Gene Silencing of ANKRD1
5.3. Proteolytic Processing
5.3.1. Calpain 3
5.3.2. Caspase 3
5.4. Degradation
26S Proteasome
6. ANKRD1 and Cardiovascular Diseases
6.1. Cardiomyopathy
6.2. Heart Failure
7. Potential as a Cardiac Biomarker
8. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Miller, M.K.; Bang, M.L.; Witt, C.C.; Labeit, D.; Trombitas, C.; Watanabe, K.; Granzier, H.; McElhinny, A.S.; Gregorio, C.C.; Labeit, S. The muscle ankyrin repeat proteins: CARP, ANKRD2/ARPP and DARP as a family of titin filament-based stress response molecules. J. Mol. Biol. 2003, 333, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Jeyaseelan, R.; Poizat, C.; Baker, R.K.; Abdishoo, S.; Isterabadi, L.B.; Lyons, G.E.; Kedes, L. A novel cardiac-restricted target for doxorubicin CARP, a nuclear modulator of gene expression in cardiac progenitor cells and cardiomyocytes. J. Biol. Chem. 1997, 272, 22800–22808. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, N.; Baba, T.; Ishida, T.; Takeuchi, K.; Osaki, M.; Araki, N.; Okada, E.; Takahashi, S.; Saito, M.; Watanabe, M.; et al. Carp, a cardiac ankyrin-repeated protein, and its new homologue, ARPP, are differentially expressed in heart, skeletal muscle, and rhabdomyosarcomas. Am. J. Pathol. 2002, 160, 1767–1778. [Google Scholar] [CrossRef]
- Ikeda, K.; Emoto, N.; Matsuo, M.; Yokoyama, M. Molecular identification and characterization of a novel nuclear protein whose expression is up-regulated in insulin-resistant animals. J. Biol. Chem. 2003, 278, 3514–3520. [Google Scholar] [CrossRef] [PubMed]
- Kuo, H.; Chen, J.; Ruiz-Lozano, P.; Zou, Y.; Nemer, M.; Chien, K.R. Control of segmental expression of the cardiac-restricted ankyrin repeat protein gene by distinct regulatory pathways in murine cardiogenesis. Development 1999, 126, 4223–4234. [Google Scholar] [PubMed]
- Aihara, Y.; Kurabayashi, M.; Saito, Y.; Ohyama, Y.; Tanaka, T.; Takeda, S.; Tomaru, K.; Sekiguchi, K.; Arai, M.; Nakamura, T.; et al. Cardiac ankyrin repeat protein is a novel marker of cardiac hypertrophy: Role of M-CAT element within the promoter. Hypertension 2000, 36, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Zolk, O.; Frohme, M.; Maurer, A.; Kluxen, F.W.; Hentsch, B.; Zubakov, D.; Hoheisel, J.D.; Zucker, I.H.; Pepe, S.; Eschenhagen, T. Cardiac ankyrin repeat protein, a negative regulator of cardiac gene expression, is augmented in human heart failure. Biochem. Biophys. Res. Commun. 2002, 293, 1377–1382. [Google Scholar] [CrossRef]
- Moulik, M.; Vatta, M.; Witt, S.H.; Arola, A.M.; Murphy, R.T.; McKenna, W.J.; Boriek, A.M.; Oka, K.; Labeit, S.; Bowles, N.E.; et al. ANKRD1, the gene encoding cardiac ankyrin repeat protein, is a novel dilated cardiomyopathy gene. J. Am. Coll. Cardiol. 2009, 54, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.; Burns, D.K.; Swerlick, R.A.; Presky, D.H. Identification and characterization of a novel cytokine-inducible nuclear protein from human endothelial cells. J. Biol. Chem. 1995, 270, 10236–10245. [Google Scholar] [CrossRef] [PubMed]
- Torrado, M.; Iglesias, R.; Nespereira, B.; Centeno, A.; Lopez, E.; Mikhailov, A.T. Intron retention generates ANKRD1 splice variants that are co-regulated with the main transcript in normal and failing myocardium. Gene 2009, 440, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Lupas, A.; van Dyke, M.; Stock, J. Predicting coiled coils from protein sequences. Science 1991, 252, 1162–1164. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.M.; Arndt, K.M. Coiled coil domains: Stability, specificity, and biological implications. Chembiochem 2004, 5, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Witt, S.H.; Labeit, D.; Granzier, H.; Labeit, S.; Witt, C.C. Dimerization of the cardiac ankyrin protein CARP: Implications for MARP titin-based signaling. J. Muscle Res. Cell Motil. 2005, 26, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Lun, A.S.; Chen, J.; Lange, S. Probing muscle ankyrin-repeat protein (MARP) structure and function. Anat. Rec. 2014, 297, 1615–1629. [Google Scholar] [CrossRef] [PubMed]
- Mosavi, L.K.; Cammett, T.J.; Desrosiers, D.C.; Peng, Z.Y. The ankyrin repeat as molecular architecture for protein recognition. Protein Sci. 2004, 13, 1435–1448. [Google Scholar] [CrossRef] [PubMed]
- Rubtsov, A.M.; Lopina, O.D. Ankyrins. FEBS Lett. 2000, 482, 1–5. [Google Scholar] [CrossRef]
- Laure, L.; Daniele, N.; Suel, L.; Marchand, S.; Aubert, S.; Bourg, N.; Roudaut, C.; Duguez, S.; Bartoli, M.; Richard, I. A new pathway encompassing calpain 3 and its newly identified substrate cardiac ankyrin repeat protein is involved in the regulation of the nuclear factor-κB pathway in skeletal muscle. FEBS J. 2010, 277, 4322–4337. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Evans, S.; Chen, J.; Kuo, H.C.; Harvey, R.P.; Chien, K.R. CARP, a cardiac ankyrin repeat protein, is downstream in the Nkx2-5 homeobox gene pathway. Development 1997, 124, 793–804. [Google Scholar] [PubMed]
- La Cour, T.; Kiemer, L.; Molgaard, A.; Gupta, R.; Skriver, K.; Brunak, S. Analysis and prediction of leucine-rich nuclear export signals. Protein Eng. Des. Sel. 2004, 17, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Rechsteiner, M.; Rogers, S.W. PEST sequences and regulation by proteolysis. Trends Biochem. Sci. 1996, 21, 267–271. [Google Scholar] [CrossRef]
- Yura, Y.; Amano, M.; Takefuji, M.; Bando, T.; Suzuki, K.; Kato, K.; Hamaguchi, T.; Hasanuzzaman Shohag, M.; Takano, T.; Funahashi, Y.; et al. Focused proteomics revealed a novel Rho-kinase signaling pathway in the heart. Cell Struct. Funct. 2016, 41, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Brunak, S. Prediction of glycosylation across the human proteome and the correlation to protein function. Pac. Symp. Biocomput. 2002, 7, 310–322. [Google Scholar]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Bang, M.L.; Mudry, R.E.; McElhinny, A.S.; Trombitas, K.; Geach, A.J.; Yamasaki, R.; Sorimachi, H.; Granzier, H.; Gregorio, C.C.; Labeit, S. Myopalladin, a novel 145-kilodalton sarcomeric protein with multiple roles in Z-disc and I-band protein assemblies. J. Cell Biol. 2001, 153, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Chiusa, M.; Cadar, A.G.; Lin, A.; Samaras, S.; Davidson, J.M.; Lim, C.C. Targeted inhibition of ANKRD1 disrupts sarcomeric ERK-GATA4 signal transduction and abrogates phenylephrine-induced cardiomyocyte hypertrophy. Cardiovasc. Res. 2015, 106, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Mikhailov, A.T.; Torrado, M. The enigmatic role of the ankyrin repeat domain 1 gene in heart development and disease. Int. J. Dev. Biol. 2008, 52, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.H.; Bauman, W.A.; Cardozo, C. ANKRD1 modulates inflammatory responses in C2C12 myoblasts through feedback inhibition of NF-κB signaling activity. Biochem. Biophys. Res. Commun. 2015, 464, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Almodovar-Garcia, K.; Kwon, M.; Samaras, S.E.; Davidson, J.M. ANKRD1 acts as a transcriptional repressor of MMP13 via the AP-1 site. Mol. Cell. Biol. 2014, 34, 1500–1511. [Google Scholar] [CrossRef] [PubMed]
- Kojic, S.; Nestorovic, A.; Rakicevic, L.; Belgrano, A.; Stankovic, M.; Divac, A.; Faulkner, G. A novel role for cardiac ankyrin repeat protein ANKRD1/CARP as a co-activator of the p53 tumor suppressor protein. Arch. Biochem. Biophys. 2010, 502, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Ye, F.; Zhu, W.; Hu, D.; Xiao, C.; Nan, J.; Su, S.; Wang, Y.; Liu, M.; Gao, K.; et al. Cardiac ankyrin repeat protein attenuates cardiomyocyte apoptosis by upregulation of bcl-2 expression. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 3040–3049. [Google Scholar] [CrossRef] [PubMed]
- Arimura, T.; Bos, J.M.; Sato, A.; Kubo, T.; Okamoto, H.; Nishi, H.; Harada, H.; Koga, Y.; Moulik, M.; Doi, Y.L.; et al. Cardiac ankyrin repeat protein gene (ANKRD1) mutations in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2009, 54, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Torrado, M.; Nespereira, B.; Lopez, E.; Centeno, A.; Castro-Beiras, A.; Mikhailov, A.T. ANKRD1 specifically binds CASQ2 in heart extracts and both proteins are co-enriched in piglet cardiac Purkinje cells. J. Mol. Cell. Cardiol. 2005, 38, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Lange, S.; Gehmlich, K.; Lun, A.S.; Blondelle, J.; Hooper, C.; Dalton, N.D.; Alvarez, E.A.; Zhang, X.; Bang, M.L.; Abassi, Y.A.; et al. MLP and CARP are linked to chronic PKCα signalling in dilated cardiomyopathy. Nat. Commun. 2016, 7, 12120. [Google Scholar] [CrossRef] [PubMed]
- Eliseeva, I.A.; Kim, E.R.; Guryanov, S.G.; Ovchinnikov, L.P.; Lyabin, D.N. Y-box-binding protein 1 (YB-1) and its functions. Biochemistry 2011, 76, 1402–1433. [Google Scholar] [CrossRef] [PubMed]
- Nagasupriya, A.; Rao, D.B.; Ravikanth, M.; Kumar, N.G.; Ramachandran, C.R.; Saraswathi, T.R. Immunohistochemical expression of matrix metalloproteinase 13 in chronic periodontitis. Int. J. Periodontics Restor. Dent. 2014, 34, e79–e84. [Google Scholar] [CrossRef] [PubMed]
- Vincent-Chong, V.K.; Salahshourifar, I.; Karen-Ng, L.P.; Siow, M.Y.; Kallarakkal, T.G.; Ramanathan, A.; Yang, Y.H.; Khor, G.H.; Rahman, Z.A.; Ismail, S.M.; et al. Overexpression of MMP13 is associated with clinical outcomes and poor prognosis in oral squamous cell carcinoma. Sci. World J. 2014, 2014, 897523. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Reitmaier, B.; Regenbogen, J.; Slowey, R.M.; Opalenik, S.R.; Wolf, E.; Goppelt, A.; Davidson, J.M. CARP, a cardiac ankyrin repeat protein, is up-regulated during wound healing and induces angiogenesis in experimental granulation tissue. Am. J. Pathol. 2005, 166, 303–312. [Google Scholar] [CrossRef]
- Samaras, S.E.; Almodovar-Garcia, K.; Wu, N.; Yu, F.; Davidson, J.M. Global deletion of Ankrd1 results in a wound-healing phenotype associated with dermal fibroblast dysfunction. Am. J. Pathol. 2015, 185, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zhong, L.; Roush, S.F.; Pentassuglia, L.; Peng, X.; Samaras, S.; Davidson, J.M.; Sawyer, D.B.; Lim, C.C. Disruption of a GATA4/ANKRD1 signaling axis in cardiomyocytes leads to sarcomere disarray: Implications for anthracycline cardiomyopathy. PLoS ONE 2012, 7, e35743. [Google Scholar] [CrossRef] [PubMed]
- Granzier, H.L.; Labeit, S. The giant protein titin: A major player in myocardial mechanics, signaling, and disease. Circ. Res. 2004, 94, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Paulin, D.; Li, Z. Desmin: A major intermediate filament protein essential for the structural integrity and function of muscle. Exp. Cell Res. 2004, 301, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Belkin, A.M.; Zhidkova, N.I.; Koteliansky, V.E. Localization of talin in skeletal and cardiac muscles. FEBS Lett. 1986, 200, 32–36. [Google Scholar] [CrossRef]
- Critchley, D.R. Biochemical and structural properties of the integrin-associated cytoskeletal protein talin. Annu. Rev. Biophys. 2009, 38, 235–254. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, M.; Moller, S.; Hansen, T.; Moens, U.; Van Ghelue, M. The multifunctional roles of the four-and-a-half-LIM only protein FHL2. Cell. Mol. Life Sci. 2006, 63, 268–284. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.M.; Sunde, M. Zinc fingers—Folds for many occasions. IUBMB Life 2002, 54, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Samson, T.; Smyth, N.; Janetzky, S.; Wendler, O.; Muller, J.M.; Schule, R.; von der Mark, H.; von der Mark, K.; Wixler, V. The LIM-only proteins FHL2 and FHL3 interact with α- and β-subunits of the muscle α7β1 integrin receptor. J. Biol. Chem. 2004, 279, 28641–28652. [Google Scholar] [CrossRef] [PubMed]
- Beard, N.A.; Laver, D.R.; Dulhunty, A.F. Calsequestrin and the calcium release channel of skeletal and cardiac muscle. Prog. Biophys. Mol. Biol. 2004, 85, 33–69. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Subramanian, R.R.; Masters, S.C. 14-3-3 proteins: Structure, function, and regulation. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 617–647. [Google Scholar] [CrossRef] [PubMed]
- Bang, M.L.; Gu, Y.; Dalton, N.D.; Peterson, K.L.; Chien, K.R.; Chen, J. The muscle ankyrin repeat proteins CARP, ANKRD2, and DARP are not essential for normal cardiac development and function at basal conditions and in response to pressure overload. PLoS ONE 2014, 9, e93638. [Google Scholar] [CrossRef] [PubMed]
- Brunori, M. Nitric oxide moves myoglobin centre stage. Trends Biochem. Sci. 2001, 26, 209–210. [Google Scholar] [CrossRef]
- Garry, D.J.; Ordway, G.A.; Lorenz, J.N.; Radford, N.B.; Chin, E.R.; Grange, R.W.; Bassel-Duby, R.; Williams, R.S. Mice without myoglobin. Nature 1998, 395, 905–908. [Google Scholar] [PubMed]
- Schlieper, G.; Kim, J.H.; Molojavyi, A.; Jacoby, C.; Laussmann, T.; Flögel, U.; Gödecke, A.; Schrader, J. Adaptation of the myoglobin knockout mouse to hypoxic stress. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Molojavyi, A.; Lindecke, A.; Raupach, A.; Moellendorf, S.; Köhrer, K.; Gödecke, A. Myoglobin-deficient mice activate a distinct cardiac gene expression program in response to isoproterenol-induced hypertrophy. Physiol. Genom. 2010, 41, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Meeson, A.P.; Radford, N.; Shelton, J.M.; Mammen, P.P.; DiMaio, J.M.; Hutcheson, K.; Kong, Y.; Elterman, J.; Williams, R.S.; Garry, D.J. Adaptive mechanisms that preserve cardiac function in mice without myoglobin. Circ. Res. 2001, 88, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Gödecke, A.; Flögel, U.; Zanger, K.; Ding, Z.; Hirchenhain, J.; Decking, U.K.; Schrader, J. Disruption of myoglobin in mice induces multiple compensatory mechanisms. Proc. Natl. Acad. Sci. USA 1999, 96, 10495–10500. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Xu, J.; Li, Y.; Jia, C.; Ma, X.; Zhang, L.; Xie, X.; Zhang, Y.; Gao, X.; Zhu, D. Cardiac ankyrin repeat protein attenuates cardiac hypertrophy by inhibition of ERK1/2 and TGF-β signaling pathways. PLoS ONE 2012, 7, e50436. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Shen, L.; Cao, S.; Li, X.; Xuan, W.; Zhang, J.; Huang, X.; Bin, J.; Xu, D.; Li, G.; et al. Cytosolic CARP promotes angiotensin II- or pressure overload-induced cardiomyocyte hypertrophy through calcineurin accumulation. PLoS ONE 2014, 9, e104040. [Google Scholar] [CrossRef] [PubMed]
- Molkentin, J.D.; Lu, J.R.; Antos, C.L.; Markham, B.; Richardson, J.; Robbins, J.; Grant, S.R.; Olson, E.N. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 1998, 93, 215–228. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Murphy, S.B.; Kishore, R.; Vaughan, D.E. Global gene expression profiling in PAI-1 knockout murine heart and kidney: Molecular basis of cardiac-selective fibrosis. PLoS ONE 2013, 8, e63825. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Pipp, F.; Fernandez, B.; Ziegelhoeffer, T.; Schaper, W.; Deindl, E. Arteriogenesis is associated with an induction of the cardiac ankyrin repeat protein (CARP). Cardiovasc. Res. 2003, 59, 573–581. [Google Scholar] [CrossRef]
- Wang, J.; Gibbert, L.; Djudjaj, S.; Alidousty, C.; Rauen, T.; Kunter, U.; Rembiak, A.; Enders, D.; Jankowski, V.; Braun, G.S.; et al. Therapeutic nuclear shuttling of YB-1 reduces renal damage and fibrosis. Kidney Int. 2016, 90, 1226–1237. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Kwak, Y.K.; You, K.R.; Lee, B.H.; Kim, D.G. Involvement of GADD153 and cardiac ankyrin repeat protein in cardiac ischemia-reperfusion injury. Exp. Mol. Med. 2009, 41, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Han, X.J.; Chae, J.K.; Lee, M.J.; You, K.R.; Lee, B.H.; Kim, D.G. Involvement of GADD153 and cardiac ankyrin repeat protein in hypoxia-induced apoptosis of H9c2 cells. J. Biol. Chem. 2005, 280, 23122–23129. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Chen, C.; Wei, X.; Li, X.; Luo, G.; Zhang, J.; Bin, J.; Huang, X.; Cao, S.; Li, G.; et al. Overexpression of ankyrin repeat domain 1 enhances cardiomyocyte apoptosis by promoting p53 activation and mitochondrial dysfunction in rodents. Clin. Sci. 2015, 128, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Liu, L.; Kim, I.H.; Kim, J.H.; You, K.R.; Kim, D.G. Identification of the genes involved in enhanced fenretinide-induced apoptosis by parthenolide in human hepatoma cells. Cancer Res. 2005, 65, 2804–2814. [Google Scholar] [CrossRef] [PubMed]
- Torrado, M.; Nespereira, B.; Bouzamayor, Y.; Centeno, A.; Lopez, E.; Mikhailov, A.T. Differential atrial vs. ventricular ANKRD1 gene expression is oppositely regulated at diastolic heart failure. FEBS Lett. 2006, 580, 4182–4187. [Google Scholar] [CrossRef] [PubMed]
- Torrado, M.; Lopez, E.; Centeno, A.; Castro-Beiras, A.; Mikhailov, A.T. Left-right asymmetric ventricular expression of CARP in the piglet heart: Regional response to experimental heart failure. Eur. J. Heart Fail. 2004, 6, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Zolk, O.; Marx, M.; Jäckel, E.; El-Armouche, A.; Eschenhagen, T. β-adrenergic stimulation induces cardiac ankyrin repeat protein expression: Involvement of protein kinase A and calmodulin-dependent kinase. Cardiovasc. Res. 2003, 59, 563–572. [Google Scholar]
- Maeda, T.; Sepulveda, J.; Chen, H.H.; Stewart, A.F. α(1)-adrenergic activation of the cardiac ankyrin repeat protein gene in cardiac myocytes. Gene 2002, 297, 1–9. [Google Scholar] [CrossRef]
- Kanai, H.; Tanaka, T.; Aihara, Y.; Takeda, S.; Kawabata, M.; Miyazono, K.; Nagai, R.; Kurabayashi, M. Transforming growth factor-β/Smads signaling induces transcription of the cell type-restricted ankyrin repeat protein CARP gene through CAGA motif in vascular smooth muscle cells. Circ. Res. 2001, 88, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Lehti, M.; Kivela, R.; Komi, P.; Komulainen, J.; Kainulainen, H.; Kyrolainen, H. Effects of fatiguing jumping exercise on mRNA expression of titin-complex proteins and calpains. J. Appl. Physiol. 2009, 106, 1419–1424. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, A.; Arber, S.; Caroni, P. Accumulation of muscle ankyrin repeat protein transcript reveals local activation of primary myotube endcompartments during muscle morphogenesis. J. Cell Biol. 1997, 139, 1231–1242. [Google Scholar] [CrossRef] [PubMed]
- Petrie, M.A.; Kimball, A.L.; McHenry, C.L.; Suneja, M.; Yen, C.L.; Sharma, A.; Shields, R.K. Distinct skeletal muscle gene regulation from active contraction, passive vibration, and whole body heat stress in humans. PLoS ONE 2016, 11, e0160594. [Google Scholar] [CrossRef] [PubMed]
- Thuerauf, D.J.; Glembotski, C.C. Differential effects of protein kinase C, Ras, and Raf-1 kinase on the induction of the cardiac B-type natriuretic peptide gene through a critical promoter-proximal M-CAT element. J. Biol. Chem. 1997, 272, 7464–7472. [Google Scholar] [CrossRef] [PubMed]
- Van Loo, P.F.; Mahtab, E.A.; Wisse, L.J.; Hou, J.; Grosveld, F.; Suske, G.; Philipsen, S.; Gittenberger-de Groot, A.C. Transcription factor SP3 knockout mice display serious cardiac malformations. Mol. Cell. Biol. 2007, 27, 8571–8582. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Olson, E.N. MicroRNAs: Powerful new regulators of heart disease and provocative therapeutic targets. J. Clin. Investig. 2007, 117, 2369–2376. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar] [CrossRef] [PubMed]
- Castoldi, G.; di Gioia, C.R.; Bombardi, C.; Catalucci, D.; Corradi, B.; Gualazzi, M.G.; Leopizzi, M.; Mancini, M.; Zerbini, G.; Condorelli, G.; et al. miR-133a regulates collagen 1A1: Potential role of miR-133a in myocardial fibrosis in angiotensin II-dependent hypertension. J. Cell. Physiol. 2012, 227, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Kim, I.K.; Kumar, S.; Jayasinghe, S.; Hong, N.; Castoldi, G.; Catalucci, D.; Jones, W.K.; Gupta, S. NF-κB mediated miR-26a regulation in cardiac fibrosis. J. Cell. Physiol. 2013, 228, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.; Wang, X. miRDB: An online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015, 43, D146–D152. [Google Scholar] [CrossRef] [PubMed]
- Betel, D.; Koppal, A.; Agius, P.; Sander, C.; Leslie, C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010, 11, R90. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.L.; Wang, J.; Liew, O.W.; Richards, A.M.; Chen, Y.T. MicroRNA and heart failure. Int. J. Mol. Sci. 2016, 17, 502. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.B.; Sanderson, J.E.; Izzat, M.B.; Yu, C.M. Micro-RNA and mRNA myocardial tissue expression in biopsy specimen from patients with heart failure. Int. J. Cardiol. 2015, 199, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Rane, S.; He, M.; Sayed, D.; Vashistha, H.; Malhotra, A.; Sadoshima, J.; Vatner, D.E.; Vatner, S.F.; Abdellatif, M. Downregulation of miR-199a derepresses hypoxia-inducible factor-1α and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ. Res. 2009, 104, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Rane, S.; He, M.; Sayed, D.; Yan, L.; Vatner, D.; Abdellatif, M. An antagonism between the AKT and β-adrenergic signaling pathways mediated through their reciprocal effects on miR-199a-5p. Cell. Signal. 2010, 22, 1054–1062. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Song, Y.; Liu, L.; Hou, N.; An, X.; Zhan, D.; Li, Y.; Zhou, L.; Li, P.; Yu, L.; et al. miR-199a impairs autophagy and induces cardiac hypertrophy through mTOR activation. Cell Death Differ. 2015. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Cheng, H.W.; Qiu, Y.; Dupee, D.; Noonan, M.; Lin, Y.D.; Fisch, S.; Unno, K.; Sereti, K.I.; Liao, R. MicroRNA-34a plays a key role in cardiac repair and regeneration following myocardial infarction. Circ. Res. 2015, 117, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Qi, Y.; Du, J.Q.; Zhang, D.F. MicroRNA-34a regulates cardiac fibrosis after myocardial infarction by targeting Smad4. Expert Opin. Ther. Targets 2014, 18, 1355–1365. [Google Scholar] [CrossRef] [PubMed]
- Goren, Y.; Kushnir, M.; Zafrir, B.; Tabak, S.; Lewis, B.S.; Amir, O. Serum levels of microRNAs in patients with heart failure. Eur. J. Heart Fail. 2012, 14, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Tijsen, A.J.; Creemers, E.E.; Moerland, P.D.; de Windt, L.J.; van der Wal, A.C.; Kok, W.E.; Pinto, Y.M. miR423–5p as a circulating biomarker for heart failure. Circ. Res. 2010, 106, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Voellenkle, C.; van Rooij, J.; Cappuzzello, C.; Greco, S.; Arcelli, D.; di Vito, L.; Melillo, G.; Rigolini, R.; Costa, E.; Crea, F.; et al. MicroRNA signatures in peripheral blood mononuclear cells of chronic heart failure patients. Physiol. Genom. 2010, 42, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.L.; Armugam, A.; Sepramaniam, S.; Karolina, D.S.; Lim, K.Y.; Lim, J.Y.; Chong, J.P.; Ng, J.Y.; Chen, Y.T.; Chan, M.M.; et al. Circulating microRNAs in heart failure with reduced and preserved left ventricular ejection fraction. Eur. J. Heart Fail. 2015, 17, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Sakata, Y.; Suna, S.; Nakatani, D.; Usami, M.; Hara, M.; Kitamura, T.; Hamasaki, T.; Nanto, S.; Kawahara, Y.; et al. Circulating p53-responsive microRNAs are predictive indicators of heart failure after acute myocardial infarction. Circ. Res. 2013, 113, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Spengler, R.M.; Zhang, X.; Cheng, C.; McLendon, J.M.; Skeie, J.M.; Johnson, F.L.; Davidson, B.L.; Boudreau, R.L. Elucidation of transcriptome-wide microRNA binding sites in human cardiac tissues by Ago2 HITS-CLIP. Nucleic Acids Res. 2016, 44, 7120–7131. [Google Scholar] [CrossRef] [PubMed]
- Sucharov, C.; Bristow, M.R.; Port, J.D. miRNA expression in the failing human heart: Functional correlates. J. Mol. Cell. Cardiol. 2008, 45, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Soifer, H.S.; Rossi, J.J.; Saetrom, P. MicroRNAs in disease and potential therapeutic applications. Mol. Ther. 2007, 15, 2070–2079. [Google Scholar] [CrossRef] [PubMed]
- Sorimachi, H.; Ono, Y. Regulation and physiological roles of the calpain system in muscular disorders. Cardiovasc. Res. 2012, 96, 11–22. [Google Scholar] [CrossRef] [PubMed]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.; Valencia, C.A.; Pang, H.; Ke, Y.; Gao, W.; Dong, B.; Liu, R. Proteome-wide identification of family member-specific natural substrate repertoire of caspases. Proc. Natl. Acad. Sci. USA 2007, 104, 14294–14299. [Google Scholar] [CrossRef] [PubMed]
- Samaras, S.E.; Chen, B.; Koch, S.R.; Sawyer, D.B.; Lim, C.C.; Davidson, J.M. 26S proteasome regulation of ANKRD1/CARP in adult rat ventricular myocytes and human microvascular endothelial cells. Biochem. Biophys. Res. Commun. 2012, 425, 830–835. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.E.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Kumar, S.; van Raam, B.J.; Salvesen, G.S.; Cieplak, P. Caspase cleavage sites in the human proteome: CaspDB, a database of predicted substrates. PLoS ONE 2014, 9, e110539. [Google Scholar] [CrossRef] [PubMed]
- Schlossarek, S.; Frey, N.; Carrier, L. Ubiquitin-proteasome system and hereditary cardiomyopathies. J. Mol. Cell. Cardiol. 2014, 71, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Badi, I.; Cinquetti, R.; Frascoli, M.; Parolini, C.; Chiesa, G.; Taramelli, R.; Acquati, F. Intracellular ANKRD1 protein levels are regulated by 26S proteasome-mediated degradation. FEBS Lett. 2009, 583, 2486–2492. [Google Scholar] [CrossRef] [PubMed]
- Witt, C.C.; Witt, S.H.; Lerche, S.; Labeit, D.; Back, W.; Labeit, S. Cooperative control of striated muscle mass and metabolism by MuRF1 and MuRF2. EMBO J. 2008, 27, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Herrer, I.; Rosello-Lleti, E.; Rivera, M.; Molina-Navarro, M.M.; Tarazon, E.; Ortega, A.; Martinez-Dolz, L.; Trivino, J.C.; Lago, F.; Gonzalez-Juanatey, J.R.; et al. RNA-sequencing analysis reveals new alterations in cardiomyocyte cytoskeletal genes in patients with heart failure. Lab. Investig. 2014, 94, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.J.; Cui, C.J.; Huang, Y.X.; Zhang, X.L.; Zhang, H.; Hu, S.S. Upregulated expression of cardiac ankyrin repeat protein in human failing hearts due to arrhythmogenic right ventricular cardiomyopathy. Eur. J. Heart Fail. 2009, 11, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Duboscq-Bidot, L.; Charron, P.; Ruppert, V.; Fauchier, L.; Richter, A.; Tavazzi, L.; Arbustini, E.; Wichter, T.; Maisch, B.; Komajda, M.; et al. Mutations in the ANKRD1 gene encoding CARP are responsible for human dilated cardiomyopathy. Eur. Heart J. 2009, 30, 2128–2136. [Google Scholar] [CrossRef] [PubMed]
- Bogomolovas, J.; Brohm, K.; Celutkiene, J.; Balciunaite, G.; Bironaite, D.; Bukelskiene, V.; Daunoravicus, D.; Witt, C.C.; Fielitz, J.; Grabauskiene, V.; et al. Induction of ANKRD1 in dilated cardiomyopathy correlates with the heart failure progression. BioMed Res. Int. 2015, 2015, 273936. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards suite: From gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinform. 2016, 54, 1–30. [Google Scholar]
- Owens, A.T.; Jessup, M. The year in heart failure. J. Am. Coll. Cardiol. 2011, 57, 1573–1583. [Google Scholar] [CrossRef] [PubMed]
- Fathil, M.F.; Md Arshad, M.K.; Gopinath, S.C.; Hashim, U.; Adzhri, R.; Ayub, R.M.; Ruslinda, A.R.; Nuzaihan, M.N.M.; Azman, A.H.; Zaki, M.; et al. Diagnostics on acute myocardial infarction: Cardiac troponin biomarkers. Biosens. Bioelectron. 2015, 70, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Gyongyosi, M.; Winkler, J.; Ramos, I.; Do, Q.T.; Firat, H.; McDonald, K.; Gonzalez, A.; Thum, T.; Diez, J.; Jaisser, F.; et al. Myocardial fibrosis: Biomedical research from bench to bedside. Eur. J. Heart Fail. 2017, 19, 177–191. [Google Scholar] [CrossRef] [PubMed]
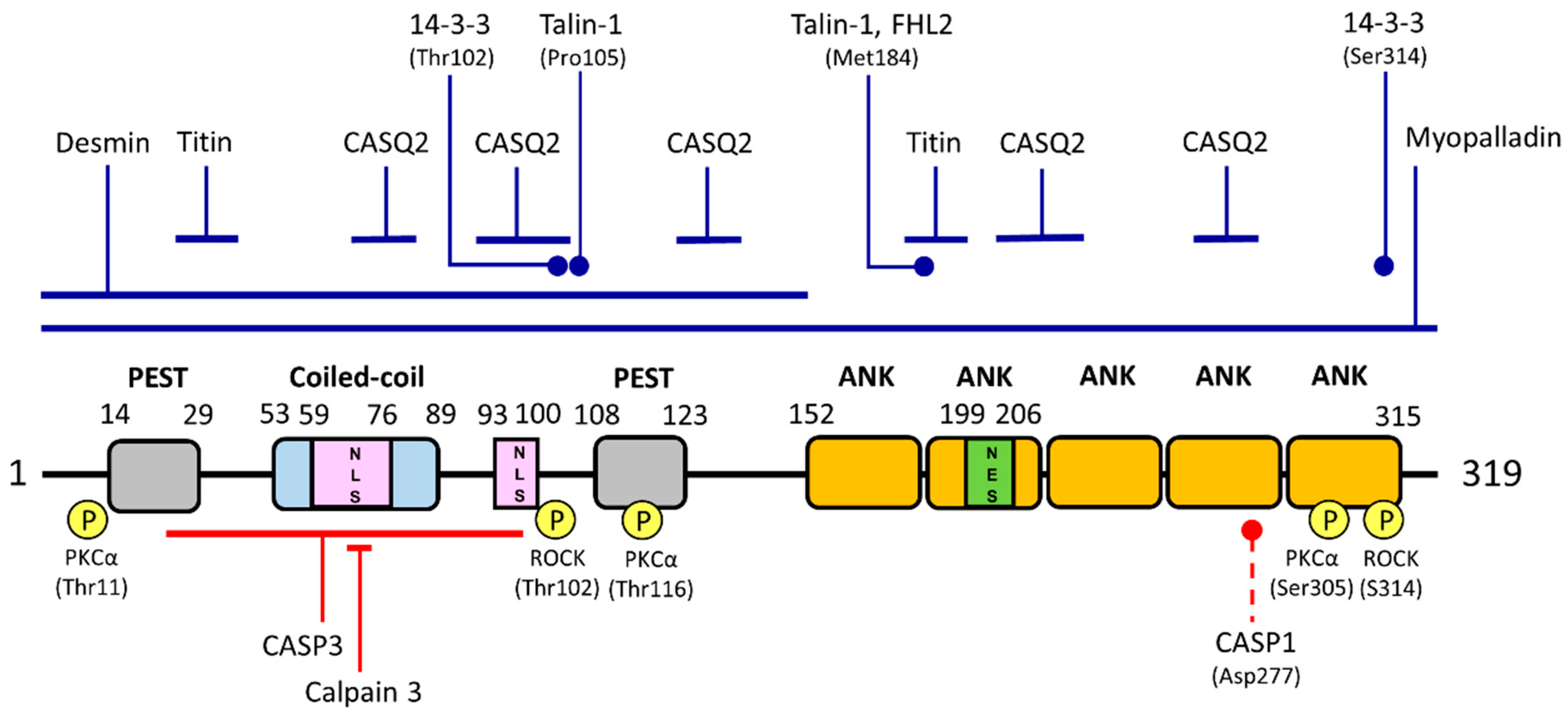

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interacting Partner | Functional Effects | Verified by | Association with Disease | Ref. |
|---|---|---|---|---|
| Transcription factors | ||||
| YB-1 | negative transcriptional co-factor of YB-1; cardiomyogenesis | Y2H, Co-IP GST-pulldown | - | [18] |
| NF-κB | negative transcriptional co-factor of NF-κB; anti-inflammation | Co-IP | - | [27] |
| Nucleolin | co-repression of MMP13 gene transcription; wound healing | Y2H, Co-IP | - | [28] |
| p53 | positive transcriptional co-activator of p53; regulation during development and stress response | Co-IP, GST-pulldown | - | [29] |
| GATA-4 | positive transcriptional co-activator of GATA-4; anti-apoptosis | Co-IP | - | [30] |
| Structural components | ||||
| Myopalladin | maintaining sarcomere structural integrity | Y2H, GST-pulldown | Pro52Ala, Thr123Met and Ile280Val mutations in ANKRD1 increase its binding to myopalladin and are associated with hypertrophic cardiomyopathy | [24,31] |
| Titin | Mechano-sensing, regulation of gene expression | Y2H, Blot overlay, Fluorescence spectroscopy | Pro52Ala, Thr123Met and Ile280Val mutations in ANKRD1 increase its binding to titin and are associated with hypertrophic cardiomyopathy | [1,31] |
| Desmin | Unknown | Y2H | - | [13] |
| Talin-1 | Mechano-sensing, regulation of gene expression | Y2H | Met184Ile and Pro105Ser mutations in ANKRD1 decrease its binding to talin-1 and are associated with dilated cardiomyopathy | [8] |
| Signaling molecules | ||||
| FHL2 | Mechano-sensing, regulation of gene expression | Y2H | Met184Ile mutation in ANKRD1 decreases its binding to FHL2 and is associated with dilated cardiomyopathy | [8] |
| CASQ2 | Sequestration of CASQ2, resulting in lower Ca2+ concentration to regulate various signaling pathways | FLAG-pulldown, Blot overlay, Co-IP | - | [32] |
| 14-3-3 proteins | Cytoplasmic retention of ANKRD1 and thus inhibiting its nuclear functions | GST-pulldown | - | [21] |
| PKCα | Sequestration of PKCα at intercalated discs | GST-pulldown, Co-IP | Sequestration of PKCα at the intercalated discs results in chronic PKCα stress signaling and is associated with heart failure | [33] |
| MicroRNA | Relative Expression in HF vs. Control | Sample Type | Ref. | TargetScan | miRanda | ||
|---|---|---|---|---|---|---|---|
| Conserved | Poorly Conserved | Good mirSVR Score and Conserved | Non-Good mirSVR Score and Conserved | ||||
| miR-101 | Up | Serum | [91] | 0 | 0 | 0 | 1 |
| miR-129-5p | Up | Plasma | [92] | 0 | 1 | 0 | 1 |
| miR-139-5p | Down | PBMC | [93] | 0 | 0 | 0 | 1 |
| miR-17 | Up | Serum | [91] | 0 | 0 | 0 | 1 |
| miR-199a-5p | Up | Cardiac biopsy | [85] | 0 | 0 | 0 | 1 |
| miR-211-5p | Down | Whole blood | [94] | 0 | 1 | 0 | 0 |
| miR-34a-5p | Up | Serum | [95] | 0 | 1 | 1 | 2 |
| miR-545-5p | Up | Whole blood | [94] | 0 | 1 | 0 | 0 |
| MicroRNA Seed Family | Seed Abundance | Seed Sequence |
|---|---|---|
| miR-1ab/206/613 | 0.083384693 | GGAATGT |
| miR-133abc | 0.031080626 | TTGGTCC |
| miR-29abcd | 0.023655364 | AGCACCA |
| miR-199ab-5p | 0.003286278 | CCAGTGT |
| miR-28-5p/708/1407/1653/3139 | 0.003077991 | AGGAGCT |
| miR-34ac/34bc-5p/449abc/449c-5p | 0.001174053 | GGCAGTG |
| miR-574-5p | 0.000356476 | GAGTGTG |
| miR-193a-5p | 0.00024827 | GGGTCTT |
| miR-9/9ab | 0.000246405 | CTTTGGT |
| miR-132/212/212-3p | 0.000215371 | AACAGTC |
| Residue | Amino Acid Sequence P5–P5′ | Prediction Score |
|---|---|---|
| 304 | KAIFD-SLREN | 0.913 |
| 150 | PDVCD-EYKRT | 0.875 |
| 30 | EDFRD-GEYEA | 0.853 |
| 258 | TPLHD-AVRLN | 0.788 |
| 120 | TEPVD-VPTFL | 0.782 |
| 183 | IEFRD-MLEST | 0.697 |
| 244 | ACEAD-LNAKD | 0.682 |
| 200 | GGNLD-VLKLL | 0.664 |
| 290 | KTPMD-LVLHW | 0.642 |
| 216 | ISARD-KLLST | 0.624 |
| 253 | DREGD-TPLHD | 0.573 |
| 277 | MYGAD-LNIKN | 0.561 |
| 142 | KFLSD-KNNPD | 0.544 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ling, S.S.M.; Chen, Y.-T.; Wang, J.; Richards, A.M.; Liew, O.W. Ankyrin Repeat Domain 1 Protein: A Functionally Pleiotropic Protein with Cardiac Biomarker Potential. Int. J. Mol. Sci. 2017, 18, 1362. https://doi.org/10.3390/ijms18071362
Ling SSM, Chen Y-T, Wang J, Richards AM, Liew OW. Ankyrin Repeat Domain 1 Protein: A Functionally Pleiotropic Protein with Cardiac Biomarker Potential. International Journal of Molecular Sciences. 2017; 18(7):1362. https://doi.org/10.3390/ijms18071362
Chicago/Turabian StyleLing, Samantha S. M., Yei-Tsung Chen, Juan Wang, Arthur M. Richards, and Oi Wah Liew. 2017. "Ankyrin Repeat Domain 1 Protein: A Functionally Pleiotropic Protein with Cardiac Biomarker Potential" International Journal of Molecular Sciences 18, no. 7: 1362. https://doi.org/10.3390/ijms18071362