Globular Adiponectin Inhibits Lipopolysaccharide-Primed Inflammasomes Activation in Macrophages via Autophagy Induction: The Critical Role of AMPK Signaling

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
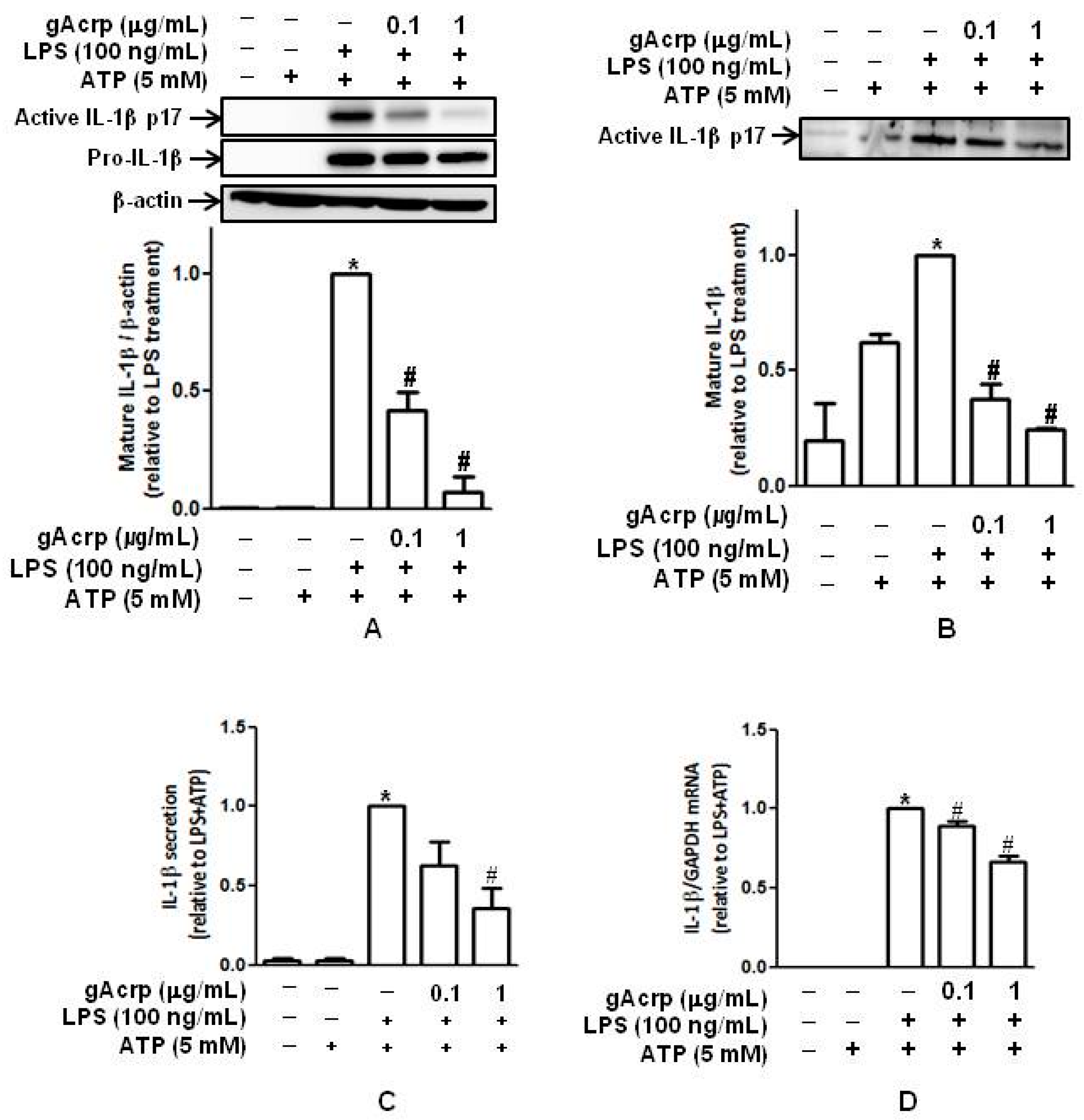
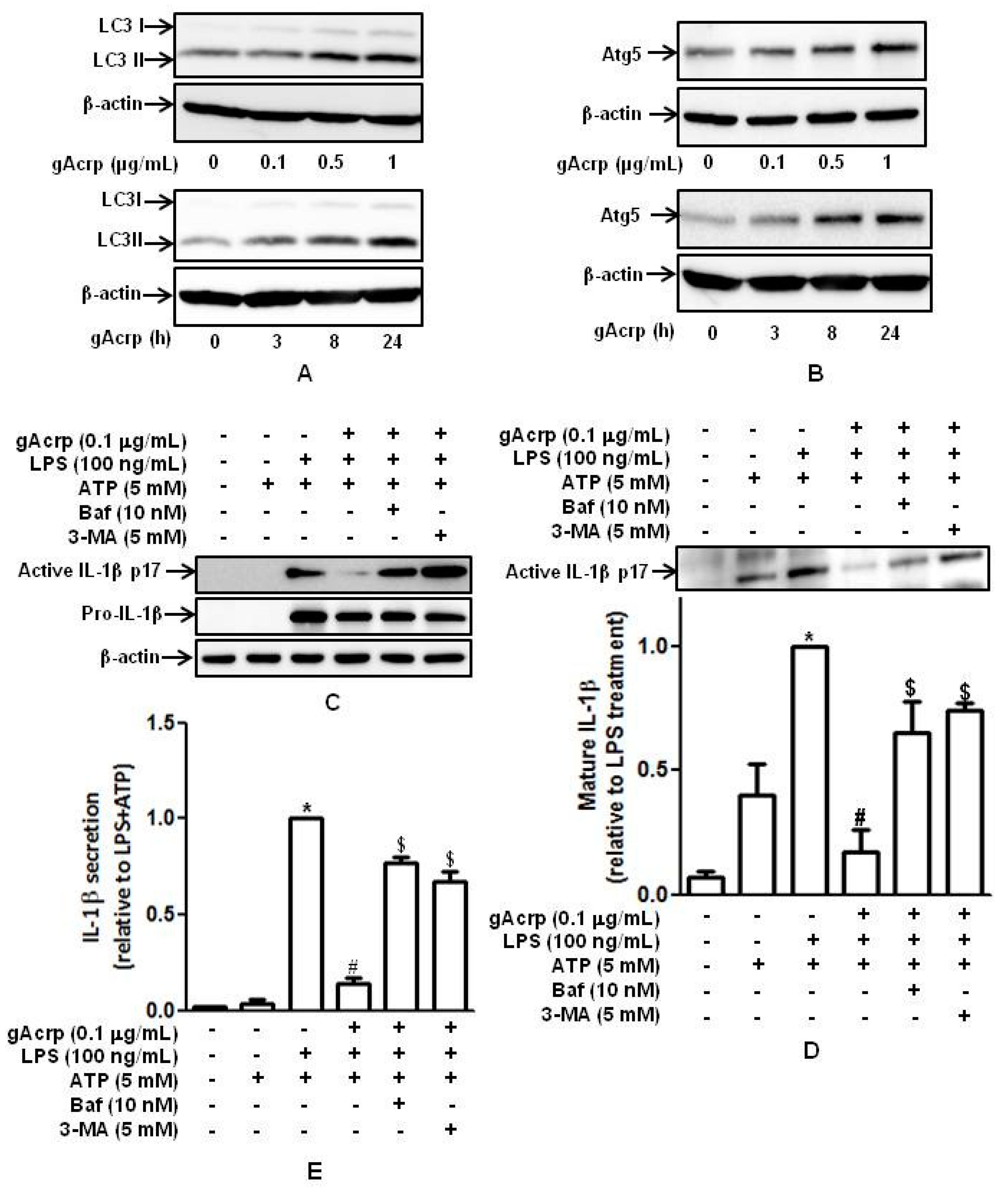
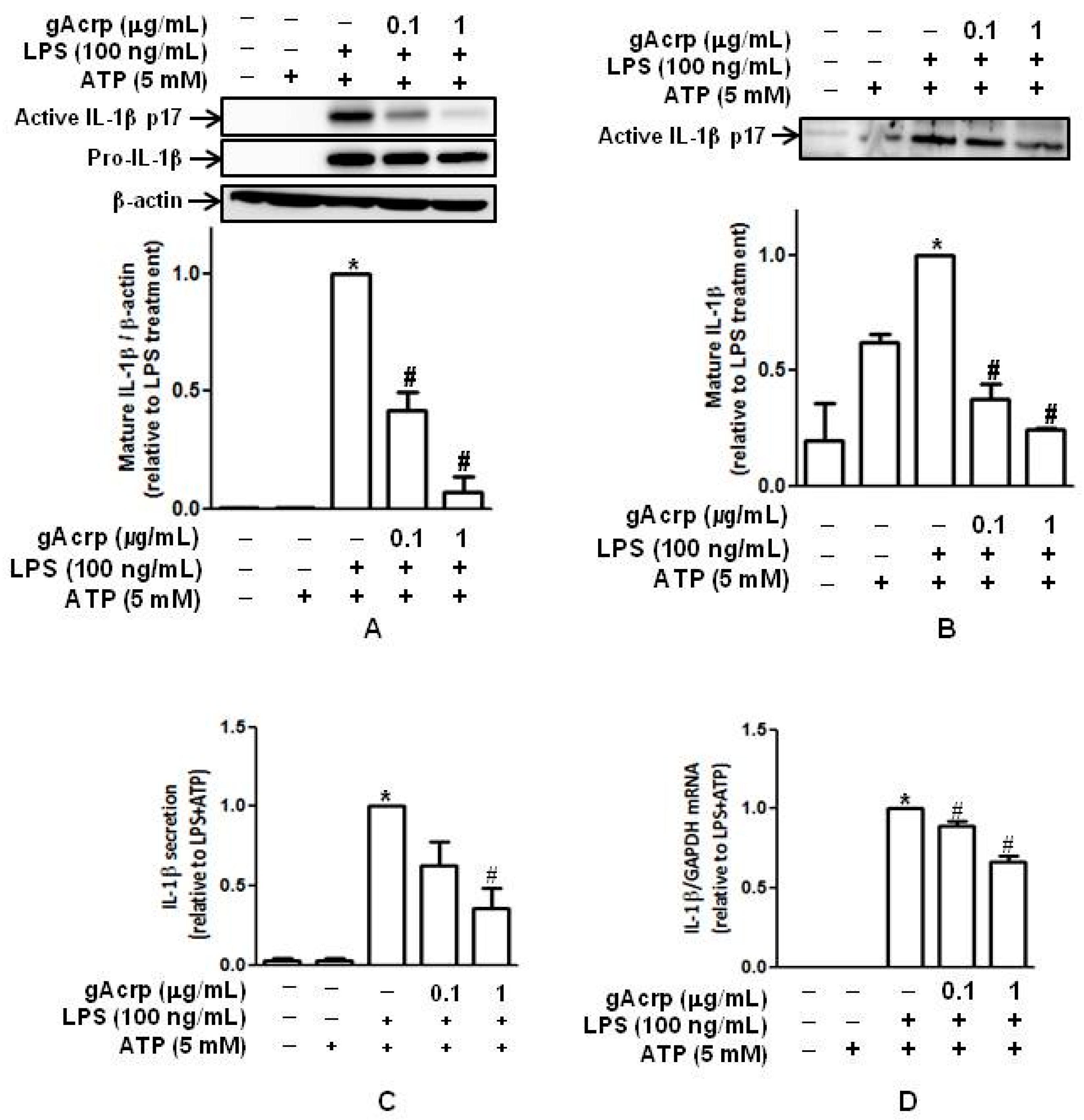
2.1. Globular Adiponectin Inhibits LPS-Stimulated IL-1β Maturation and Secretion by Murine Peritoneal Macrophages
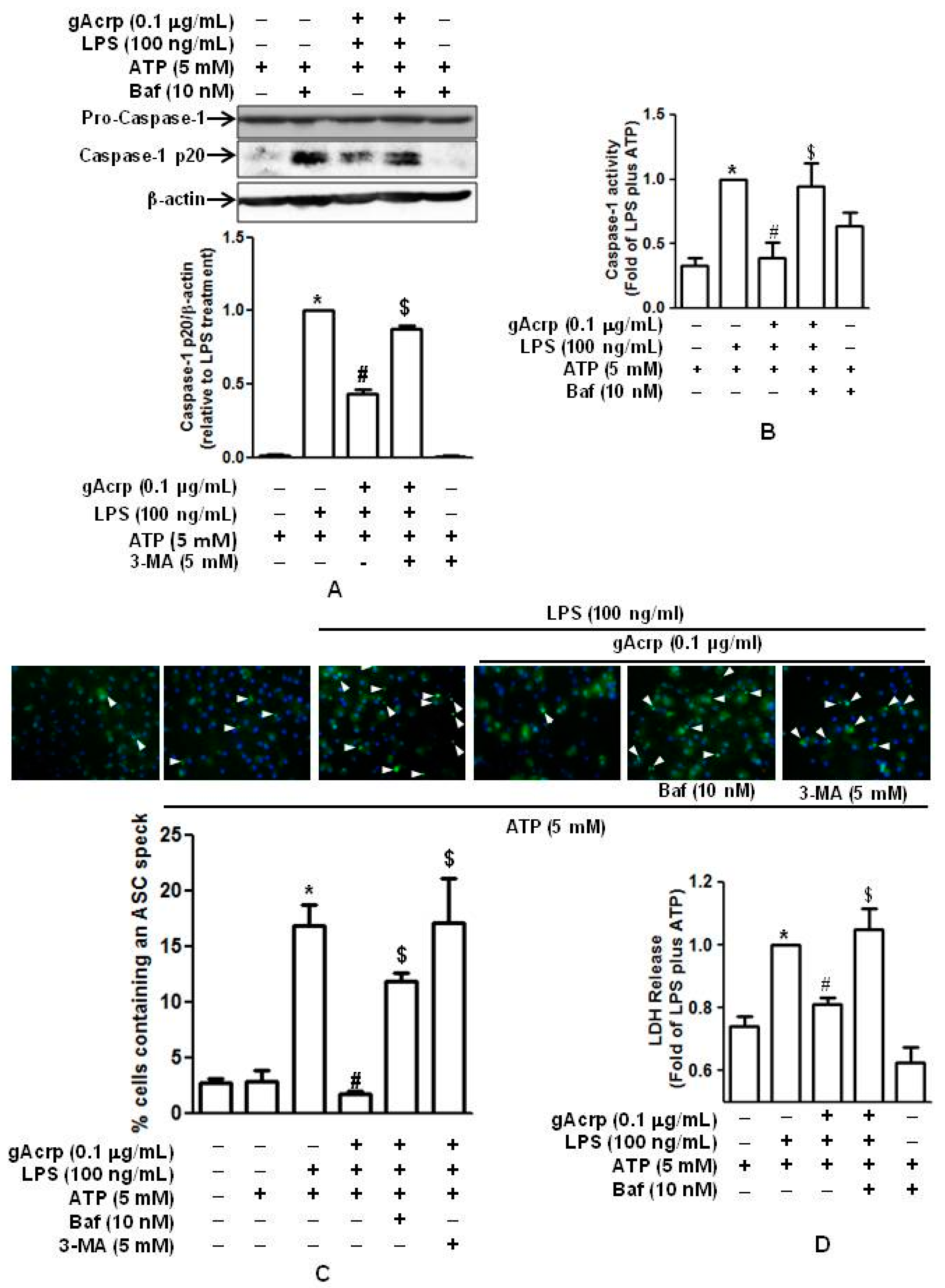
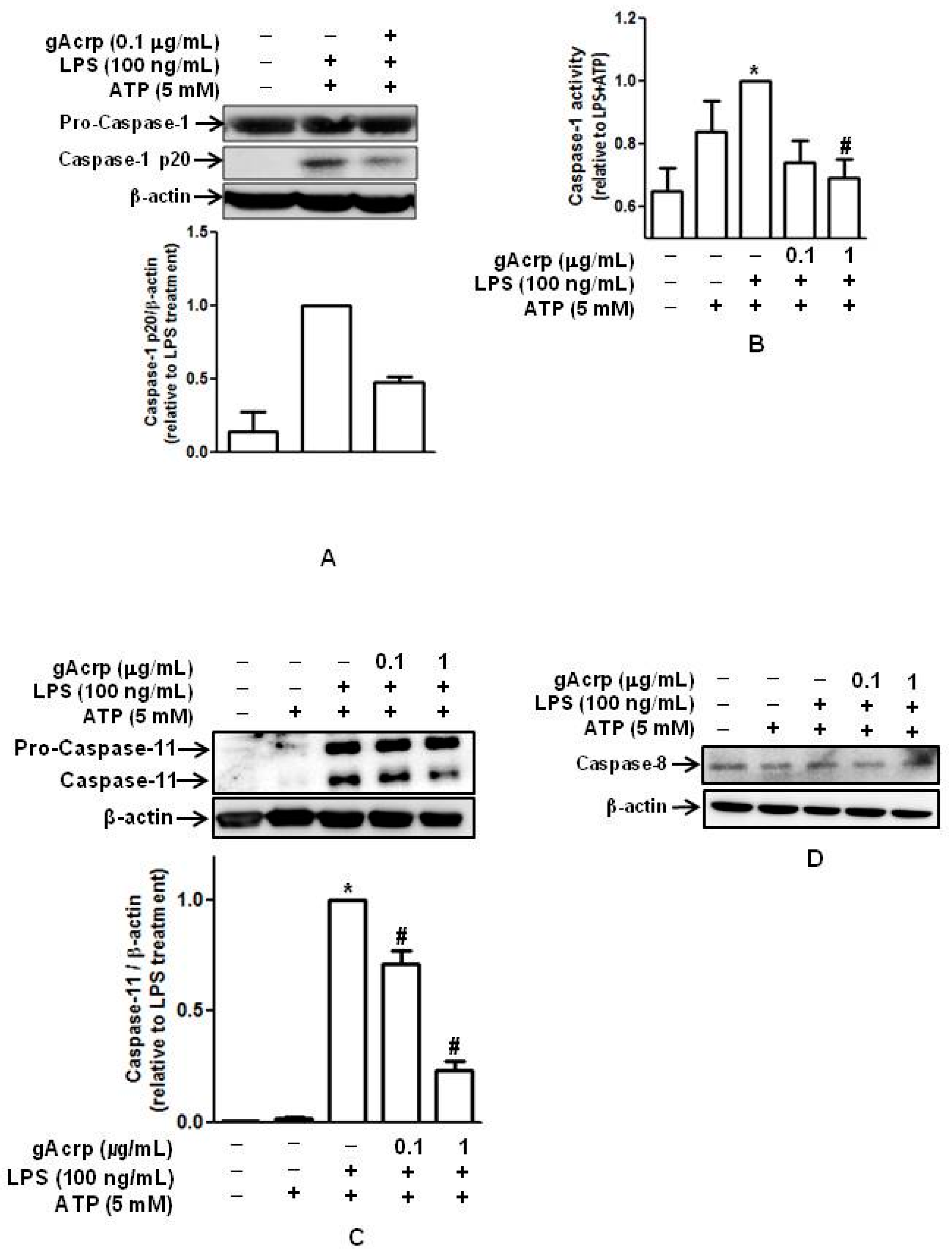
2.2. Globular Adiponectin Suppresses LPS-Primed Activation of Caspase in Peritoneal Macrophages
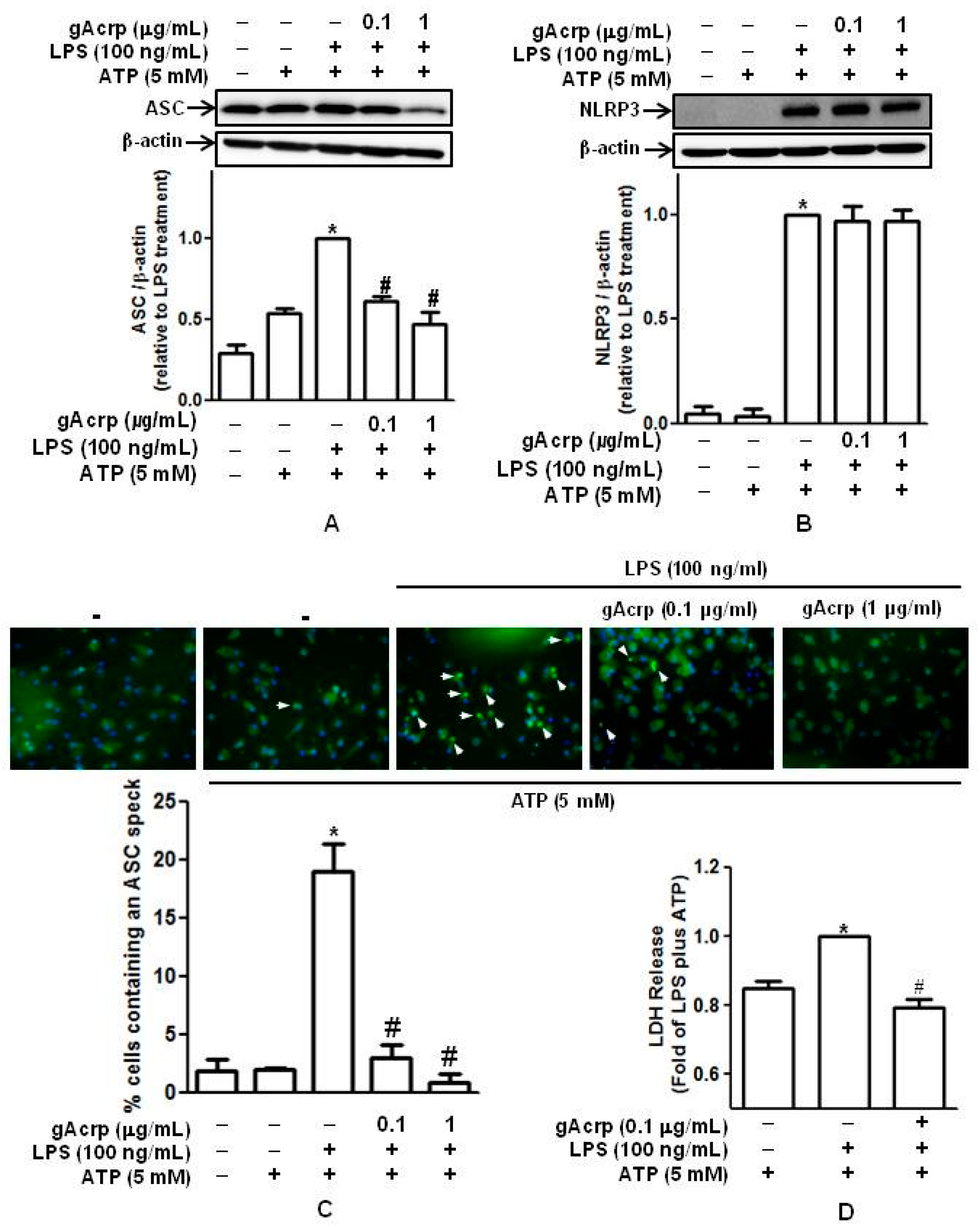
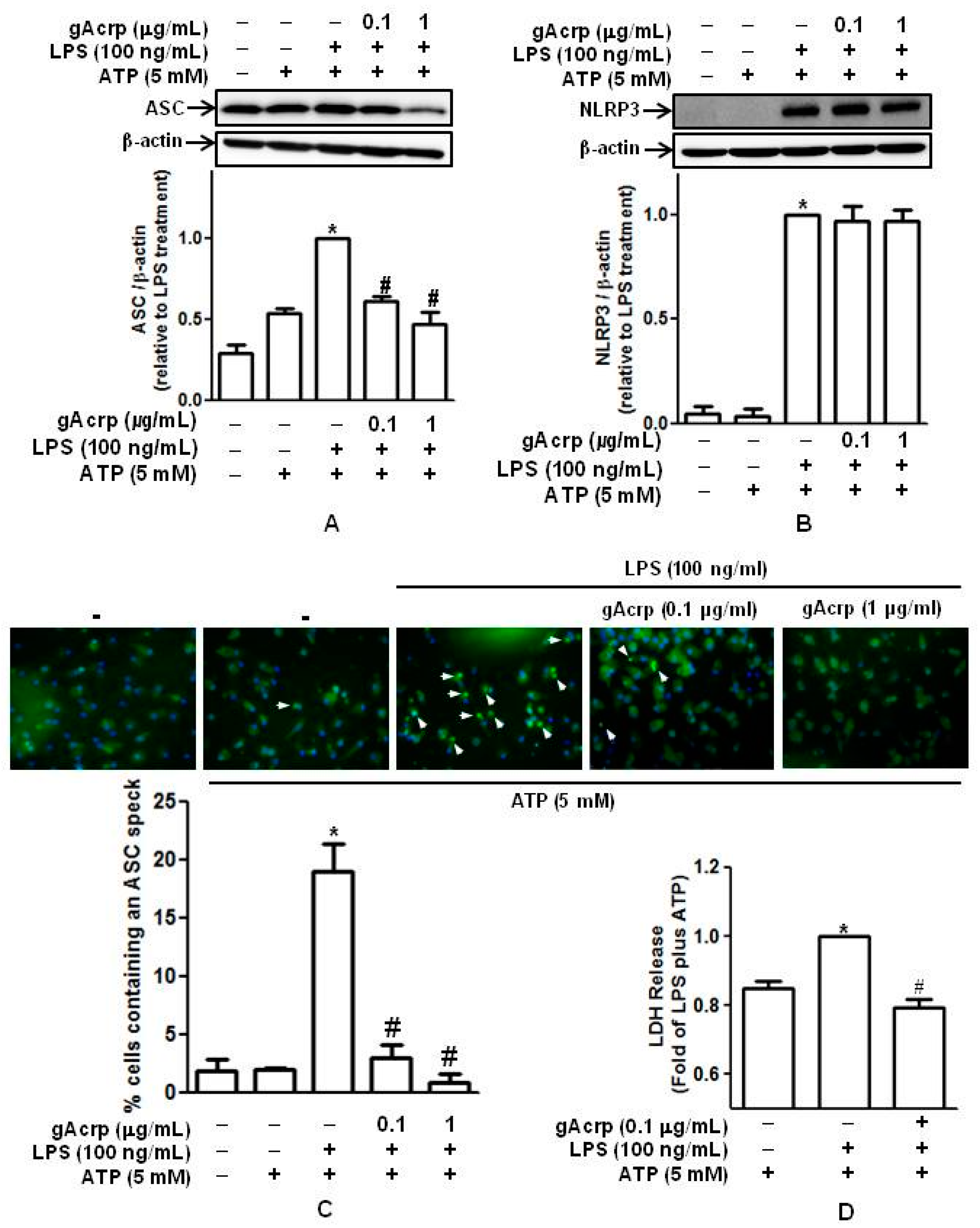
2.3. Globular Adiponectin Suppresses Inflammasomes Activation and Pyroptosis in LPS-Primed Peritoneal Macrophages
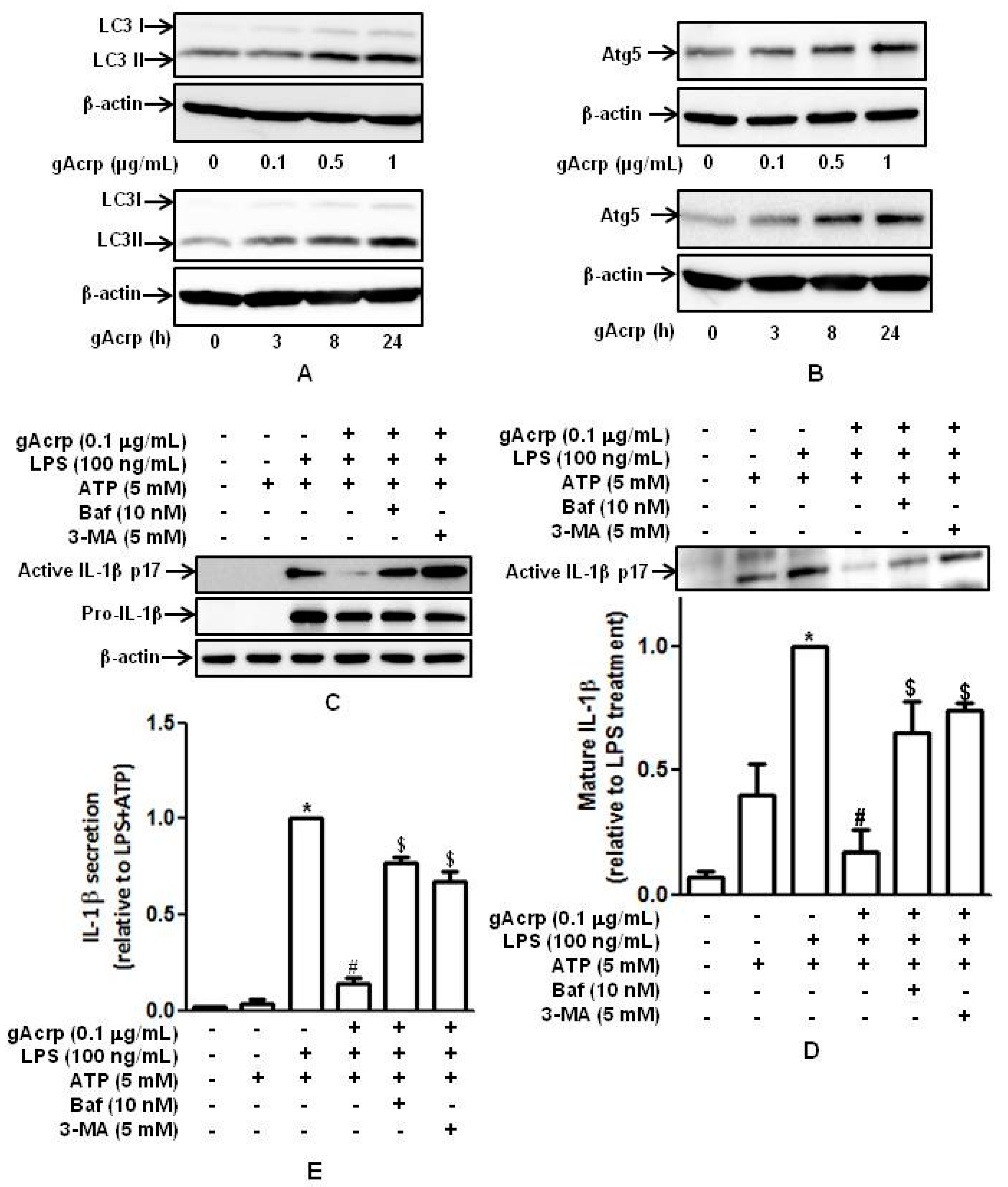
2.4. Autophagy Induction Plays A Critical Role in The Suppression of IL-1β Production by Globular Adiponectin in Murine Peritoneal Macrophages
2.5. Autophagy Induction Contributes to The Suppression of Caspase-1 Activation, ASC Speck Formation, and Pyroptosis by Globular Adiponectin
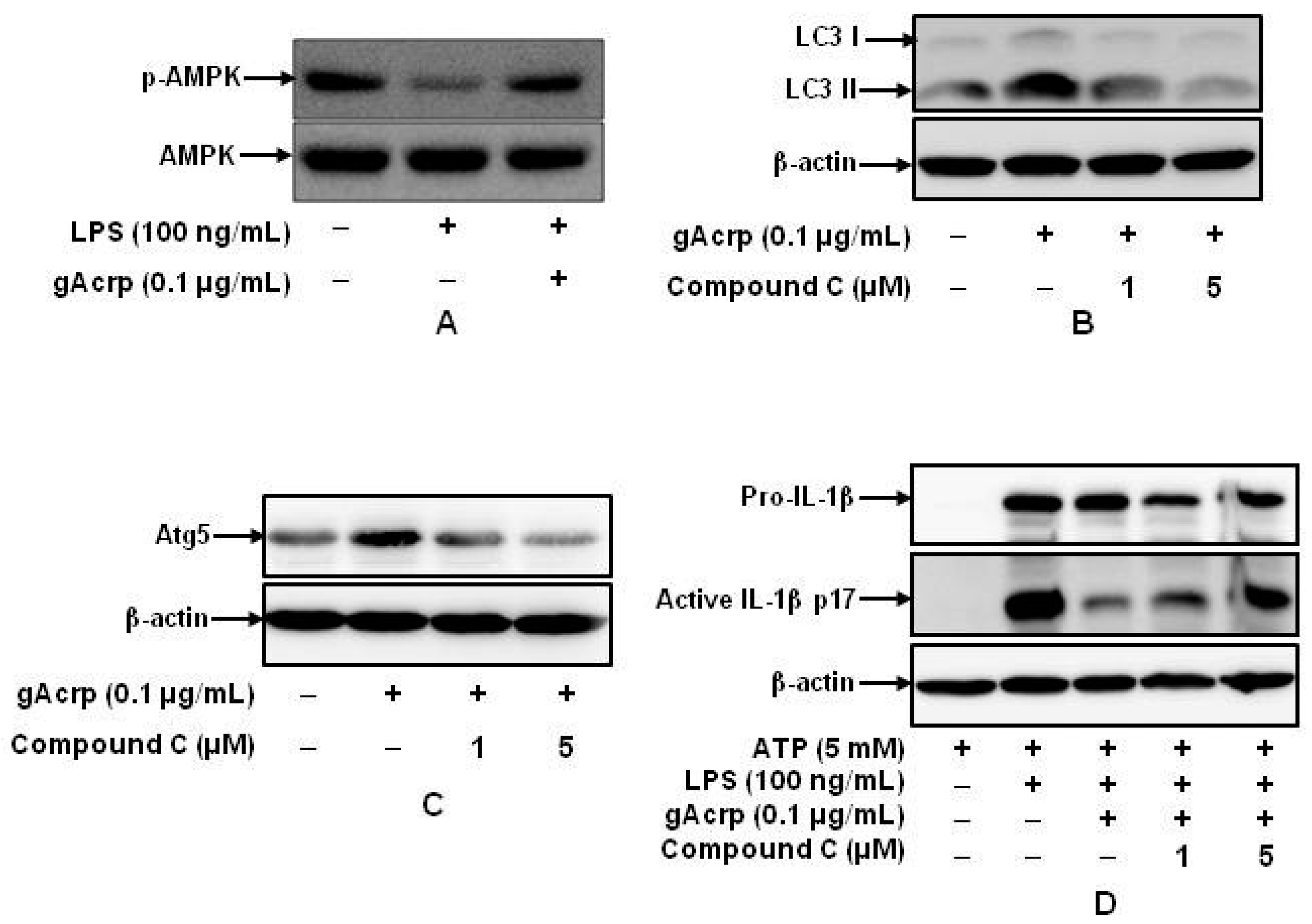
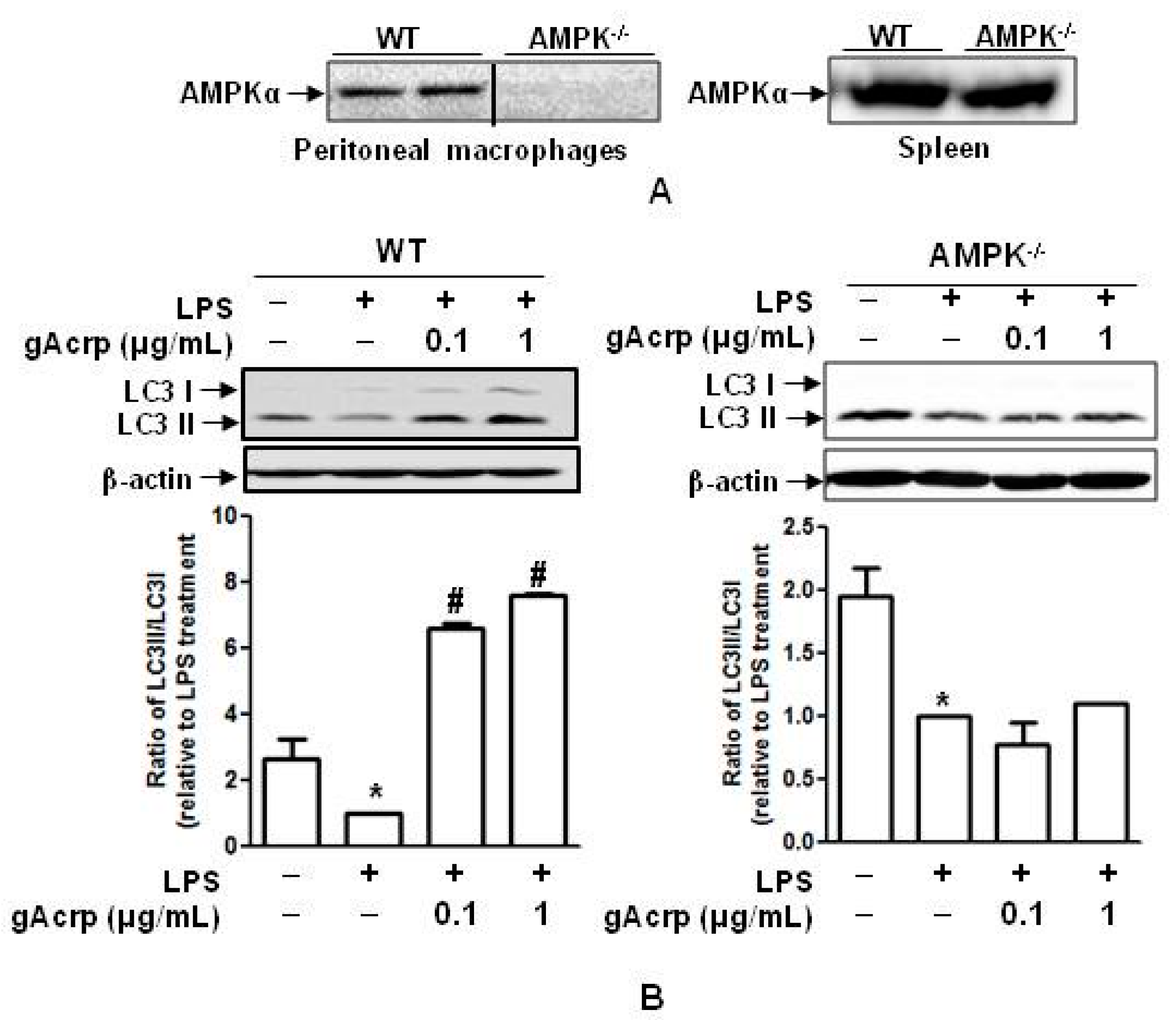
2.6. AMPK Signaling Plays A Crucial Role in Autophagy Induction and supprEssion of IL-1β Production by gAcrp in Peritoneal Macrophages
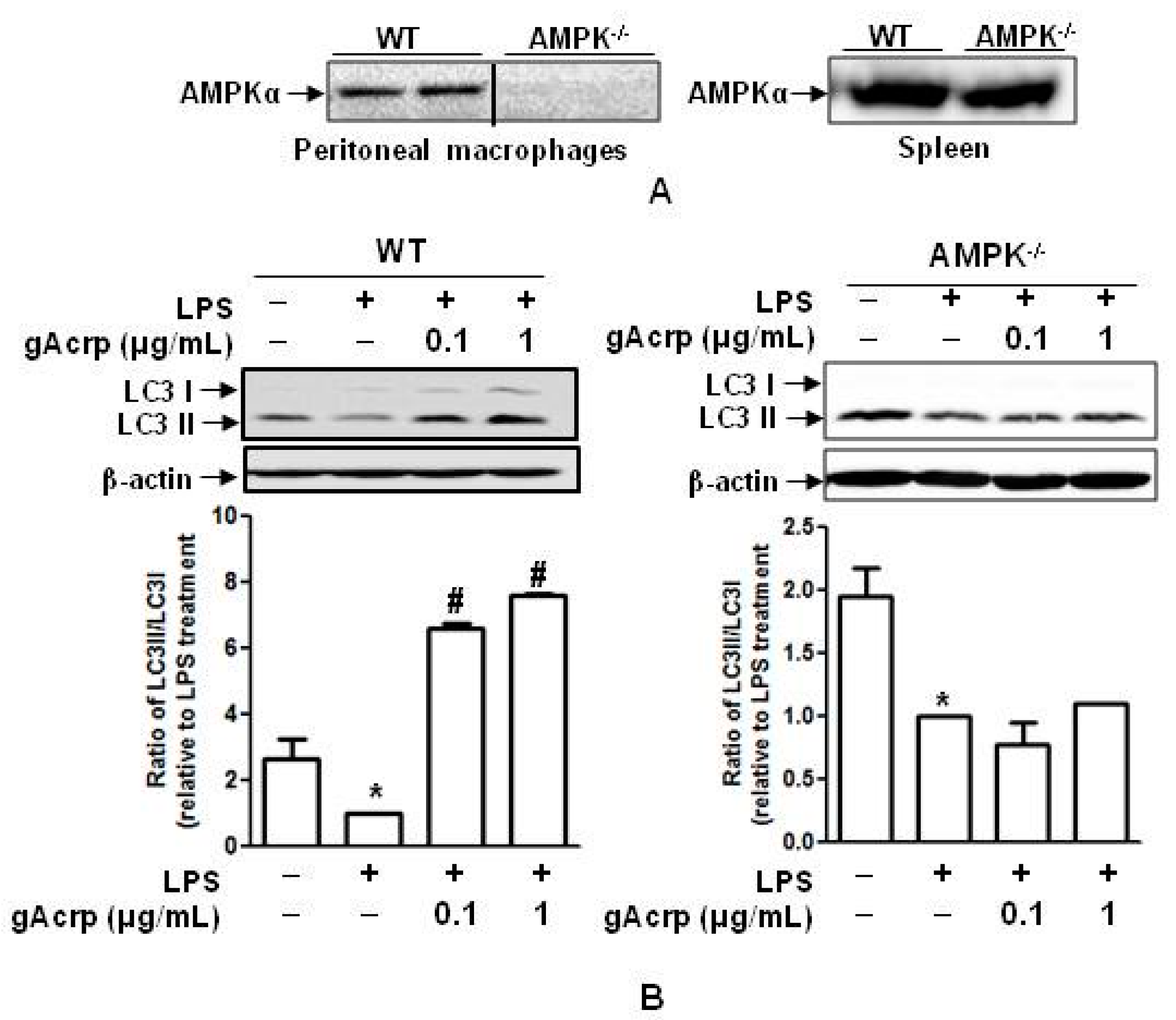
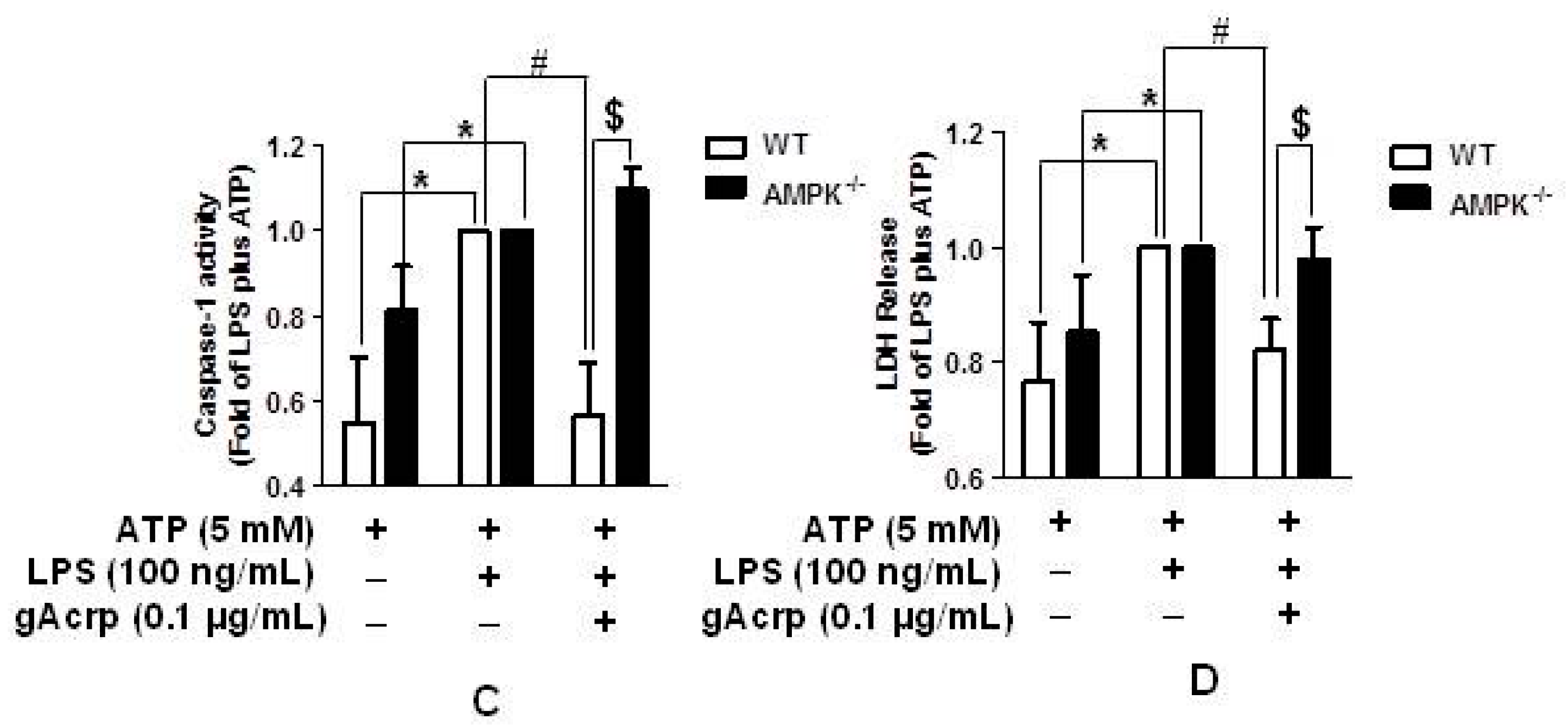
2.7. Autophagy Induction and Suppression of The Inflammasome Activation by Globular Adiponectin Is Abrogated in AMPK Deficient Macrophages
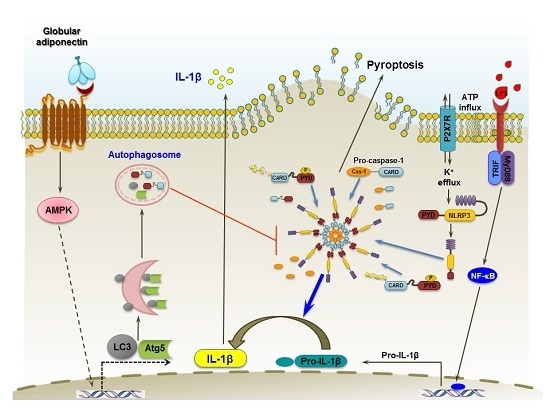
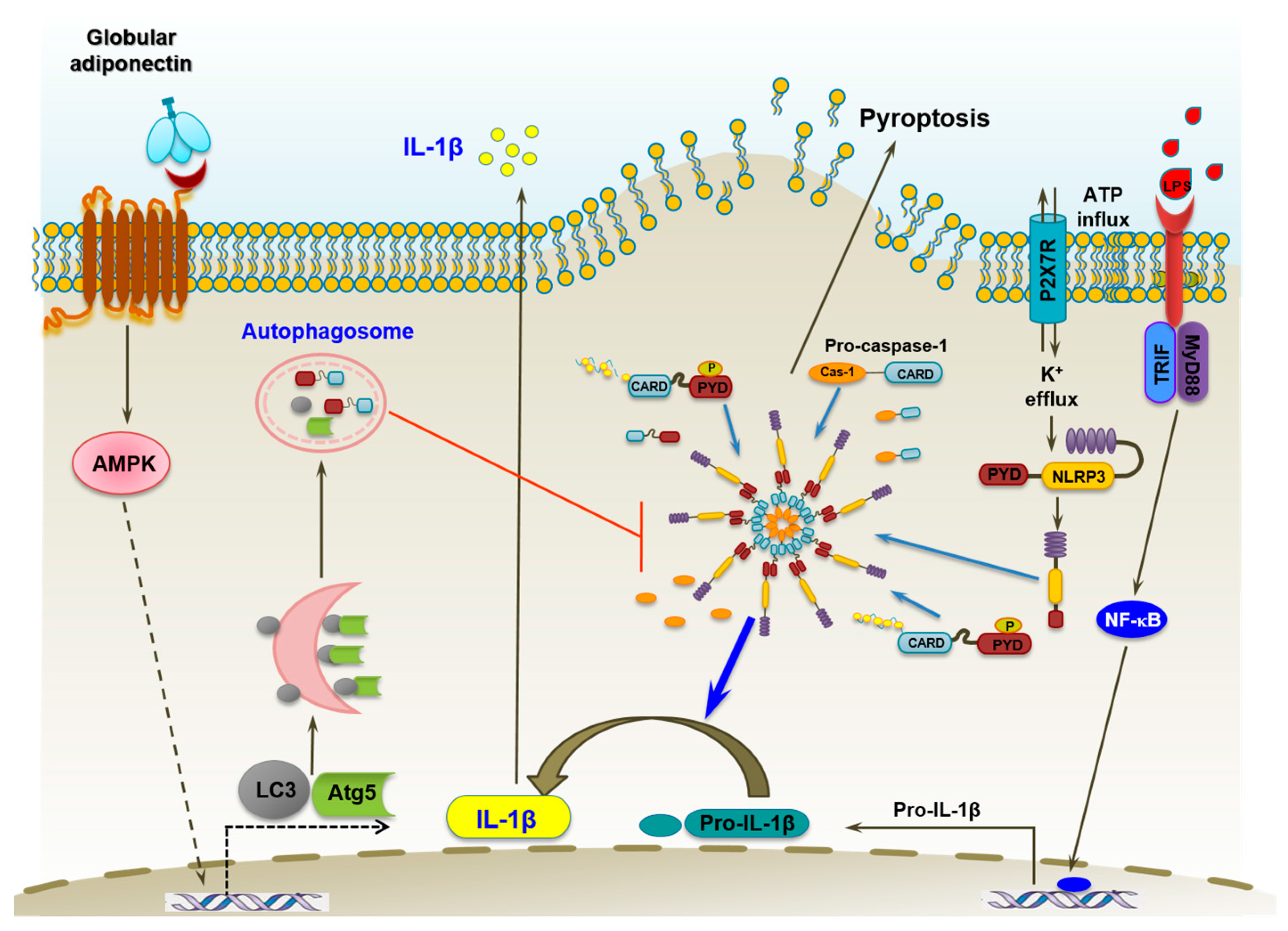
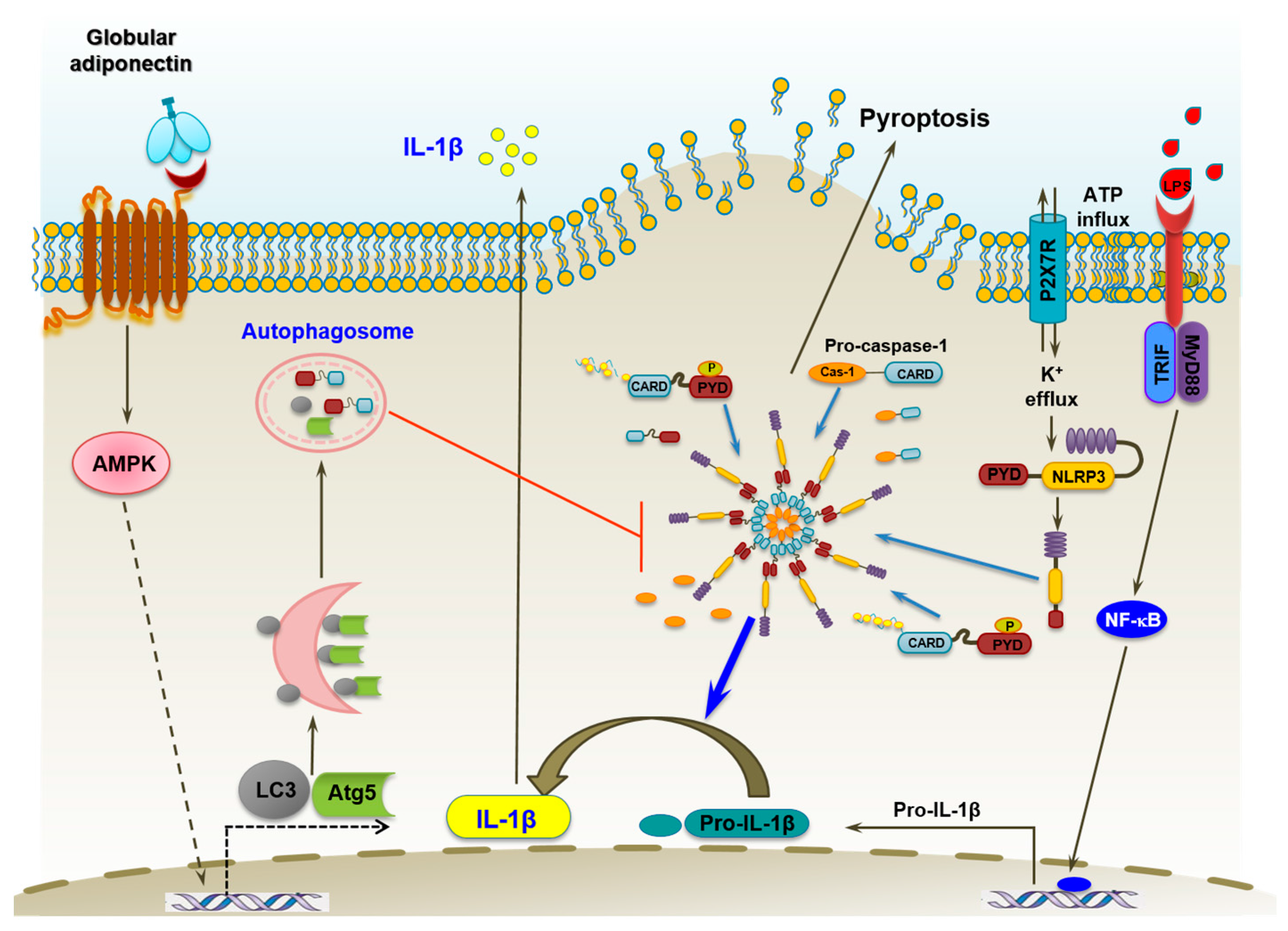
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Generation of Macrophage-Specific AMPK Knockout Mice
4.3. Isolation and Culture of Murine Peritoneal Macrophages
4.4. Preparation of Cellular Extracts and Western Blot Analysis
4.5. Immunocytochemistry
4.6. Enzyme-Linked Immunosorbent Assay (ELISA)
4.7. Caspase-1 Enzyme Activity Assay
4.8. Measurement of Lactate Dehydrogenase (LDH) Release
4.9. RNA Isolation, Reverse Transcription (RT), and Quantitative Real-Time PCR (qPCR)
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stehlik, C.; Dorfleutner, A. COPs and POPs: Modulators of inflammasome activity. J. Immunol. 2007, 179, 7993–7998. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Miao, E.A.; Rajan, J.V.; Aderem, A. Caspase-1-induced pyroptotic cell death. Immunol. Rev. 2011, 243, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef] [PubMed]
- So, A.; de Smedt, T.; Revaz, S.; Tschopp, J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res. Ther. 2007, 9, R28. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.K.; Wen, H.; Ting, J.P.Y. The Inflammasome NLRs in Immunity, Inflammation, and Associated Diseases. Annu. Rev. Immunol. 2011, 29, 707–735. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef]
- Mihalache, C.C.; Simon, H.U. Autophagy regulation in macrophages and neutrophils. Exp. Cell Res. 2012, 318, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein ATG16L1 enhances endotoxin-induced IL-1β production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.; Hartman, M.; Roche, C.; Zeng, S.G.; O'Shea, A.; Sharp, F.A.; Lambe, E.M.; Creagh, E.M.; Golenbock, D.T.; Tschopp, J.; et al. Autophagy controls IL-1β secretion by targeting pro-IL-1β for degradation. J. Biol. Chem. 2011, 286, 9587–9597. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Nagy, L.E.; Park, P.H. Globular adiponectin inhibits ethanol-induced reactive oxygen species production through modulation of NADPH oxidase in macrophages: Involvement of liver kinase B1/AMP-activated protein kinase pathway. Mol. Pharmacol. 2014, 86, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Pun, N.T.; Subedi, A.; Kim, M.J.; Park, P.H. Globular Adiponectin Causes Tolerance to LPS-Induced TNF-α Expression via Autophagy Induction in RAW 264.7 Macrophages: Involvement of SIRT1/FoxO3A Axis. PLoS ONE 2015, 10, e0124636. [Google Scholar]
- Nepal, S.; Kim, M.J.; Subedi, A.; Lee, E.S.; Yong, C.S.; Kim, J.A.; Kang, W.; Kwak, M.K.; Arya, D.S.; Park, P.H. Globular adiponectin inhibits ethanol-induced apoptosis in HepG2 cells through heme oxygenase-1 induction. Biochem. Pharmacol. 2012, 84, 974–983. [Google Scholar] [CrossRef]
- Park, P.H.; Huang, H.; McMullen, M.R.; Mandal, P.; Sun, L.; Nagy, L.E. Suppression of lipopolysaccharide-stimulated tumor necrosis factor-α production by adiponectin is mediated by transcriptional and post-transcriptional mechanisms. J. Biol. Chem. 2008, 283, 26850–26858. [Google Scholar] [CrossRef]
- Fruebis, J.; Tsao, T.S.; Javorschi, S.; Ebbets-Reed, D.; Erickson, M.R.; Yen, F.T.; Bihain, B.E.; Lodish, H.F. Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc. Natl. Acad. Sci. USA 2001, 98, 2005–2010. [Google Scholar] [CrossRef] [PubMed]
- Qi, G.M.; Jia, L.X.; Li, Y.L.; Li, H.H.; Du, J. Adiponectin suppresses angiotensin II-induced inflammation and cardiac fibrosis through activation of macrophage autophagy. Endocrinology 2014, 155, 2254–2265. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.T.; Kola, B.; Korbonits, M. AMPK as a mediator of hormonal signalling. J. Mol. Endocrinol. 2010, 44, 87–97. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Puissant, A.; Robert, G.; Fenouille, N.; Luciano, F.; Cassuto, J.P.; Raynaud, S.; Auberger, P. Resveratrol promotes autophagic cell death in chronic myelogenous leukemia cells via JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer Res. 2010, 70, 1042–1252. [Google Scholar] [CrossRef]
- Nepal, S.; Park, P.H. Activation of autophagy by globular adiponectin attenuates ethanol-induced apoptosis in HepG2 cells: Involvement of AMPK/FoxO3A axis. Biochim. Biophys. Acta 2013, 1833, 2111–2125. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickey, W.J.; Ting, J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef]
- Denes, A.; Lopez-Castejon, G.; Brough, D. Caspase-1: Is IL-1 just the tip of the ICEberg? Cell Death Dis. 2012, 3, e338. [Google Scholar] [CrossRef] [PubMed]
- Maelfait, J.; Vercammen, E.; Janssens, S.; Schotte, P.; Haegman, M.; Magez, S.; Beyaert, R. Stimulation of Toll-like receptor 3 and 4 induces interleukin-1β maturation by caspase-8. J. Exp. Med. 2008, 205, 1967–1973. [Google Scholar] [CrossRef]
- Gurung, P.; Anand, P.K.; Malireddi, R.K.; Vande Walle, L.; Van Opdenbosch, N.; Dillon, C.P.; Weinlich, R.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.D. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 2014, 192, 1835–1846. [Google Scholar] [CrossRef] [PubMed]
- Fink, S.L.; Cookson, B.T. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell. Microbiol. 2006, 8, 1812–1825. [Google Scholar] [CrossRef] [PubMed]
- Brennan, M.A.; Cookson, B.T. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol. Microbiol. 2000, 38, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Alnemri, T.; Wu, J.; Yu, J.W.; Datta, P.; Miller, B.; Jankowski, W.; Rosenberg, S.; Zhang, J.; Alnemri, E.S. The pyroptosome: A supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007, 14, 1590–1604. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef]
- Li, H.; Willingham, S.B.; Ting, J.P.; Re, F. Cutting edge: Inflammasome activation by alum and alum’s adjuvant effect are mediated by NLRP3. J. Immunol. 2008, 181, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Ehsan, M.; Singh, K.K.; Lovren, F.; Pan, Y.; Quan, A.; Mantella, L.E.; Sandhu, P.; Teoh, H.; Al-Omran, M.; Verma, S. Adiponectin limits monocytic microparticle-induced endothelial activation by modulation of the AMPK, Akt and NFκB signaling pathways. Atherosclerosis 2016, 245, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bryan, N.B.; Dorfleutner, A.; Rojanasakul, Y.; Stehlik, C. Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J. Immunol. 2009, 182, 3173–3182. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Yaginuma, K.; Tsutsui, H.; Sagara, J.; Guan, X.; Seki, E.; Yasuda, K.; Yamamoto, M.; Akira, S.; Nakanishi, K.; et al. ASC is essential for LPS-induced activation of procaspase-1 independently of TLR-associated signal adaptor molecules. Genes Cells 2004, 9, 1055–1067. [Google Scholar] [CrossRef] [PubMed]
- Compan, V.; Martin-Sanchez, F.; Baroja-Mazo, A.; Lopez-Castejon, G.; Gomez, A.I.; Verkhratsky, A.; Brough, D.; Pelegrin, P. Apoptosis-associated speck-like protein containing a CARD forms specks but does not activate caspase-1 in the absence of NLRP3 during macrophage swelling. J. Immunol. 2015, 194, 1261–1273. [Google Scholar] [CrossRef] [PubMed]
- Laliberte, R.E.; Perregaux, D.G.; Hoth, L.R.; Rosner, P.J.; Jordan, C.K.; Peese, K.M.; Eggler, J.F.; Dombroski, M.A.; Geoghegan, K.F.; Gabel, C.A. Glutathione s-transferase ω1–1 is a target of cytokine release inhibitory drugs and may be responsible for their effect on interleukin-1β posttranslational processing. J. Biol. Chem. 2003, 278, 16567–16578. [Google Scholar] [CrossRef] [PubMed]
- Coll, R.C.; Robertson, A.; Butler, M.; Cooper, M.; O’Neill, L.A. The cytokine release inhibitory drug CRID3 targets ASC oligomerisation in the NLRP3 and AIM2 inflammasomes. PLoS ONE 2011, 6, e29539. [Google Scholar] [CrossRef] [PubMed]
- Dowling, J.K.; O′Neill, L.A. Biochemical regulation of the inflammasome. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 424–443. [Google Scholar] [CrossRef]
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007, 462, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Jagannath, C.; Liu, X.D.; Sharafkhaneh, A.; Kolodziejska, K.E.; Eissa, N.T. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 2007, 27, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Delgado Mó, A.; Elmaoued, R.A.; Davis, A.S.; Kyei, G.; Deretic, V. Toll-like receptors control autophagy. EMBO J. 2008, 27, 1110–1121. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell. Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, N.; Walsh, K. Adiponectin as an anti-inflammatory factor. Clin. Chim. Acta 2007, 380, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.M.; Kim, J.J.; Kim, H.J.; Shong, M.; Ku, B.J.; Jo, E.K. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 2013, 62, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Li, J.; Hou, F.; Wang, X.; Liu, B. Mangiferin inhibits endoplasmic reticulum stress-associated thioredoxin-interacting protein/NLRP3 inflammasome activation with regulation of AMPK in endothelial cells. Metabolism 2015, 64, 428–437. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.J.; Kim, E.H.; Pun, N.T.; Chang, J.-H.; Kim, J.-A.; Jeong, J.-H.; Choi, D.Y.; Kim, S.-H.; Park, P.-H. Globular Adiponectin Inhibits Lipopolysaccharide-Primed Inflammasomes Activation in Macrophages via Autophagy Induction: The Critical Role of AMPK Signaling. Int. J. Mol. Sci. 2017, 18, 1275. https://doi.org/10.3390/ijms18061275
Kim MJ, Kim EH, Pun NT, Chang J-H, Kim J-A, Jeong J-H, Choi DY, Kim S-H, Park P-H. Globular Adiponectin Inhibits Lipopolysaccharide-Primed Inflammasomes Activation in Macrophages via Autophagy Induction: The Critical Role of AMPK Signaling. International Journal of Molecular Sciences. 2017; 18(6):1275. https://doi.org/10.3390/ijms18061275
Chicago/Turabian StyleKim, Mi Jin, Eun Hye Kim, Nirmala TiliJa Pun, Jae-Hoon Chang, Jung-Ae Kim, Jee-Heon Jeong, Dong Young Choi, Sang-Hyun Kim, and Pil-Hoon Park. 2017. "Globular Adiponectin Inhibits Lipopolysaccharide-Primed Inflammasomes Activation in Macrophages via Autophagy Induction: The Critical Role of AMPK Signaling" International Journal of Molecular Sciences 18, no. 6: 1275. https://doi.org/10.3390/ijms18061275