AA-NAT, MT1 and MT2 Correlates with Cancer Stem-Like Cell Markers in Colorectal Cancer: Study of the Influence of Stage and p53 Status of Tumors

 ,
,
Abstract
:
1. Introduction
2. Results
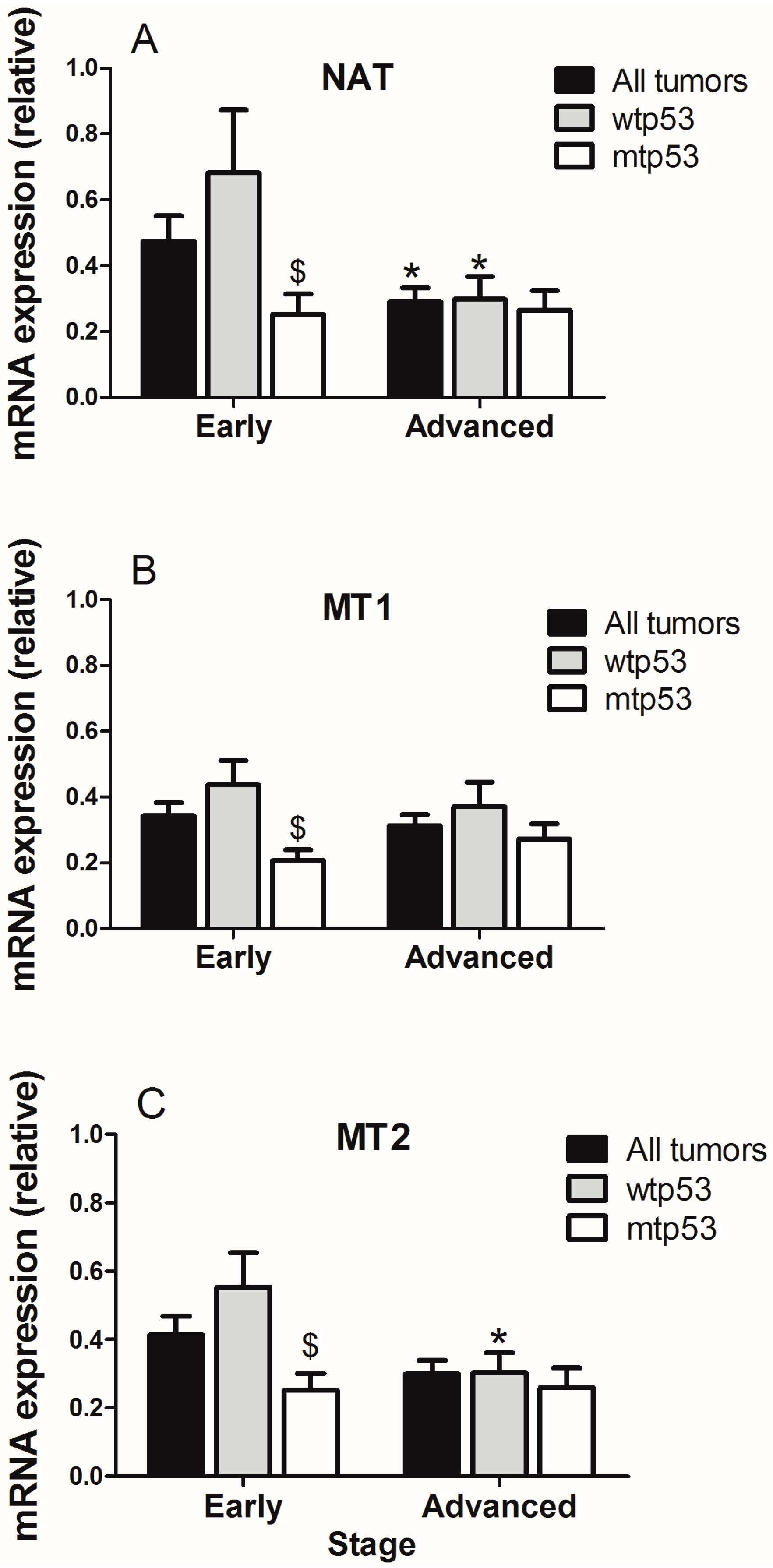
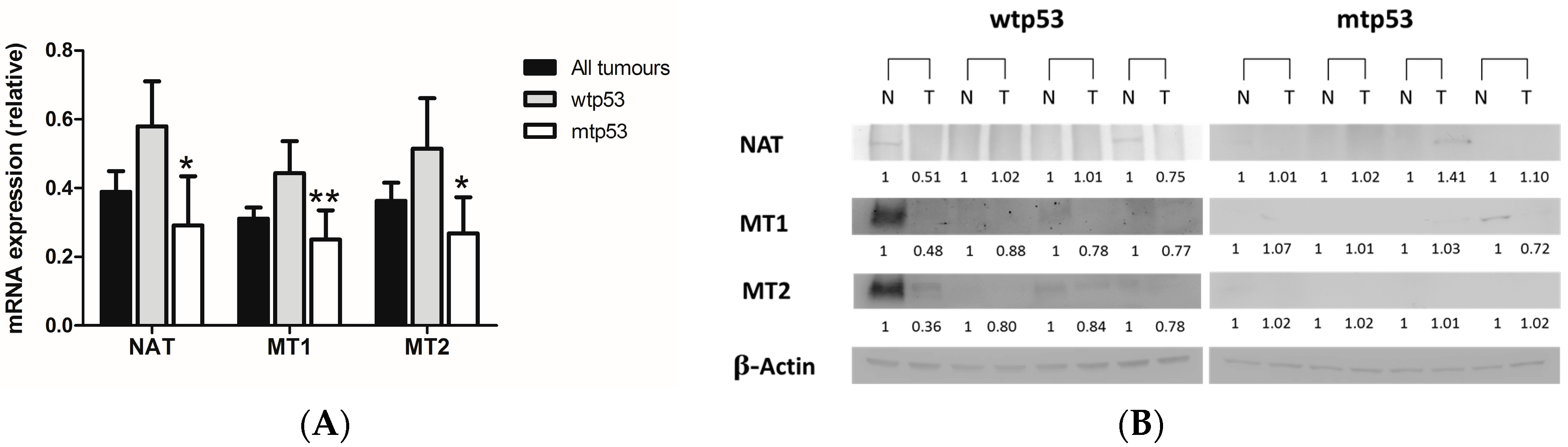
2.1. Evaluation of Arylalkylamine N-acetyltransferase (AA-NAT), MT1, and MT2 Expression in Samples from Human colorectal cancer(CRC) with Different Status of p53
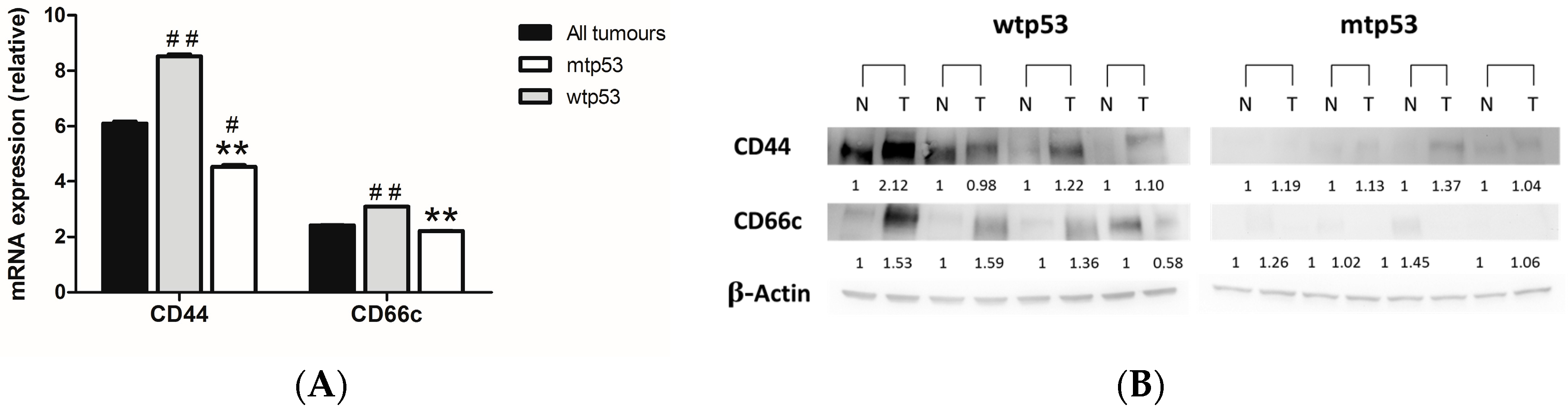
2.2. Evaluation of the Expression of Cancer Stem Cells (CSC) Markers in Samples from Human CRC with Different p53 Status
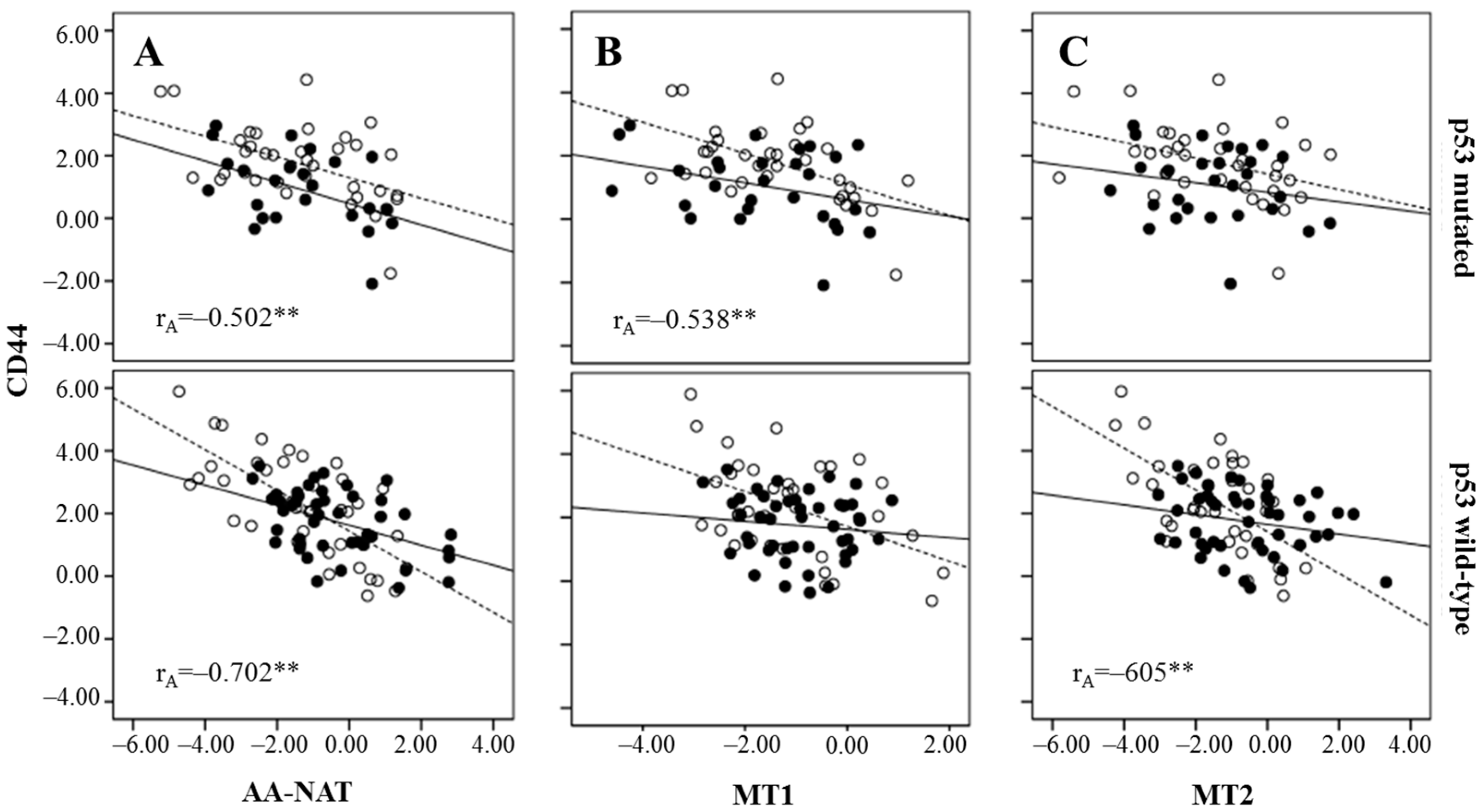
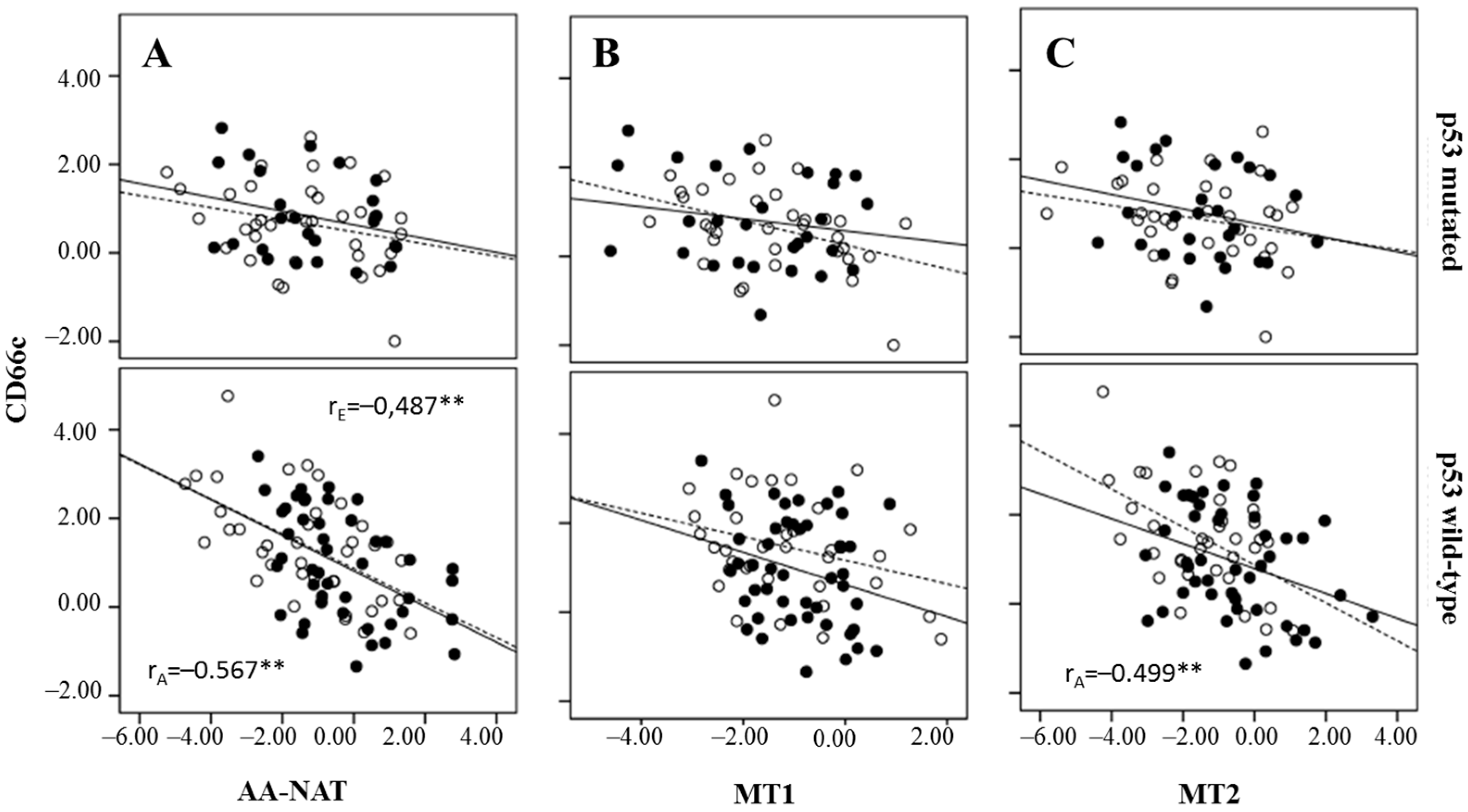
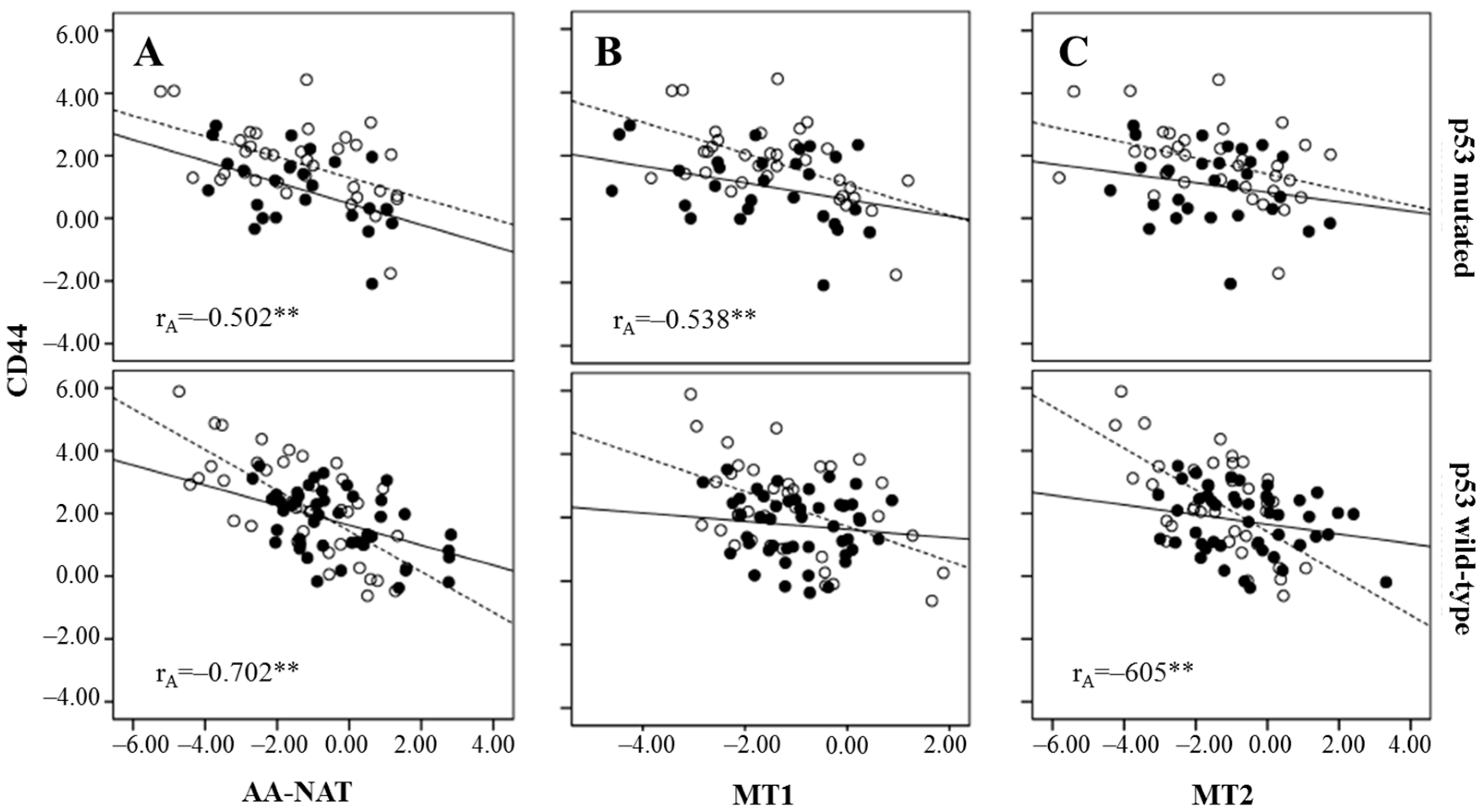
2.3. Correlation Between Markers of Cancer Stem Cells and Synthesis and Signaling Melatonin-Related Genes in Colorectal Cancer
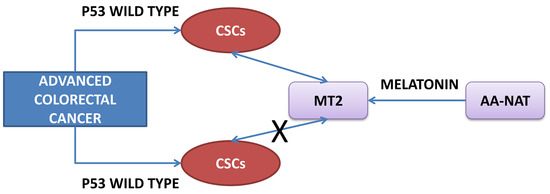
3. Discussion
4. Materials and Methods
4.1. CRC Human Samples
4.2. RNA Isolation and cDNA Synthesis
4.3. Real-Time PCR (RT-PCR)
4.4. DNA Extraction
4.5. Sequencing of p53 Mutations
4.6. Western Blotting
4.7. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ALDH1 | Aldehyde dehydrogenase-1 |
| AA-NAT | Arylalkylamine N-acetyltransferase |
| ASMT | N-Acetylserotonin O-methyltransferase |
| CRC | Colorectal cancer |
| CSCs | Cancer stem cells |
| HGVS | Human Gene Variation Society |
| Lgr5 | Leucine-rich-repeat containing G protein-coupled receptor 5 |
| MSCs | Mesenchymal stem cells |
| Mtp53 | Mutated p53 |
| Wtp53 | Wild-type p53 |
References
- Haggar, F.A.; Boushey, R.P. Colorectal cancer epidemiology: Incidence, mortality, survival, and risk factors. Clin. Colon Rectal Surg. 2009, 22, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Patil, H.; Saxena, S.G.; Barrow, C.J.; Kanwar, J.R.; Kapat, A.; Kanwar, R.K. Chasing the personalized medicine dream through biomarker validation in colorectal cancer. Drug Discov. Today 2017, 22, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Botchkina, G. Colon cancer stem cells—From basic to clinical application. Cancer Lett. 2013, 338, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Fanali, C.; Lucchetti, D.; Farina, M.; Corbi, M.; Cufino, V.; Cittadini, A.; Sgambato, A. Cancer stem cells in colorectal cancer from pathogenesis to therapy: Controversies and perspectives. World J. Gastroenterol. 2014, 20, 923–942. [Google Scholar] [CrossRef] [PubMed]
- Todaro, M.; Francipane, M.G.; Medema, J.P.; Stassi, G. Colon cancer stem cells: Promise of targeted therapy. Gastroenterology 2010, 138, 2151–2162. [Google Scholar] [CrossRef] [PubMed]
- Gemei, M.; Mirabelli, P.; di Noto, R.; Corbo, C.; Iaccarino, A.; Zamboli, A.; Troncone, G.; Galizia, G.; Lieto, E.; del Vecchio, L.; et al. CD66c is a novel marker for colorectal cancer stem cell isolation, and its silencing halts tumor growth in vivo. Cancer 2013, 119, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Weinberg, R.A. How does multistep tumorigenesis really proceed? Cancer Discov. 2015, 5, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Li, X.L.; Zhou, J.; Chen, Z.R.; Chng, W.J. p53 mutations in colorectal cancer-molecular pathogenesis and pharmacological reactivation. World J. Gastroenterol. 2015, 21, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Shetzer, Y.; Solomon, H.; Koifman, G.; Molchadsky, A.; Horesh, S.; Rotter, V. The paradigm of mutant p53-expressing cancer stem cells and drug resistance. Carcinogenesis 2014, 35, 1196–1208. [Google Scholar] [CrossRef] [PubMed]
- Aloni-Grinstein, R.; Shetzer, Y.; Kaufman, T.; Rotter, V. p53: The barrier to cancer stem cell formation. FEBS Lett. 2014, 588, 2580–2589. [Google Scholar] [CrossRef] [PubMed]
- Jung, B.; Ahmad, N. Melatonin in cancer management: Progress and promise. Cancer Res. 2006, 66, 9789–9793. [Google Scholar] [CrossRef] [PubMed]
- Mayo, J.C.; Sainz, R.M.; Gonzalez Menendez, P.; Cepas, V.; Tan, D.X.; Reiter, R.J. Melatonin and sirtuins: A “not-so unexpected” relationship. J. Pineal Res. 2017, 62. [Google Scholar] [CrossRef] [PubMed]
- Reppert, S.M.; Weaver, D.R.; Ebisawa, T. Cloning and characterization of a mammalian melatonin receptor that mediates reproductive and circadian responses. Neuron 1994, 13, 1177–1185. [Google Scholar] [CrossRef]
- Reppert, S.M.; Godson, C.; Mahle, C.D.; Weaver, D.R.; Slaugenhaupt, S.A.; Gusella, J.F. Molecular characterization of a second melatonin receptor expressed in human retina and brain: The Mel1b melatonin receptor. Proc. Natl. Acad. Sci. USA 1995, 92, 8734–8738. [Google Scholar] [CrossRef] [PubMed]
- Becker-Andre, M.; Wiesenberg, I.; Schaeren-Wiemers, N.; Andre, E.; Missbach, M.; Saurat, J.H.; Carlberg, C. Pineal gland hormone melatonin binds and activates an orphan of the nuclear receptor superfamily. J. Biol. Chem. 1994, 269, 28531–28534. [Google Scholar] [CrossRef] [PubMed]
- Manchester, L.C.; Coto-Montes, A.; Boga, J.A.; Andersen, L.P.; Zhou, Z.; Galano, A.; Vriend, J.; Tan, D.X.; Reiter, R.J. Melatonin: An ancient molecule that makes oxygen metabolically tolerable. J. Pineal Res. 2015, 59, 403–419. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Manchester, L.C.; Esteban-Zubero, E.; Zhou, Z.; Reiter, R.J. Melatonin as a potent and inducible endogenous antioxidant: Synthesis and metabolism. Molecules 2015, 20, 18886–18906. [Google Scholar] [CrossRef] [PubMed]
- Luchetti, F.; Canonico, B.; Bartolini, D.; Arcangeletti, M.; Ciffolilli, S.; Murdolo, G.; Piroddi, M.; Papa, S.; Reiter, R.J.; Galli, F. Melatonin regulates mesenchymal stem cell differentiation: A review. J. Pineal Res. 2014, 56, 3823–3897. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Chen, S.; Li, Y.; Liu, Y. Melatonin as a promising agent of regulating stem cell biology and its application in disease therapy. Pharmacol. Res. 2017, 117, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Navarro, A.; Gonzalez-Puga, C.; Escames, G.; Lopez, L.C.; Lopez, A.; Lopez-Cantarero, M.; Camacho, E.; Espinosa, A.; Gallo, M.A.; Acuna-Castroviejo, D. Cellular mechanisms involved in the melatonin inhibition of HT-29 human colon cancer cell proliferation in culture. J. Pineal Res. 2007, 43, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Winczyk, K.; Pawlikowski, M.; Lawnicka, H.; Kunert-Radek, J.; Spadoni, G.; Tarzia, G.; Karasek, M. Effects of melatonin and melatonin receptors ligand N-[(4-methoxy-1H-indol-2-yl)methyl]propanamide on murine colon 38 cancer growth in vitro and in vivo. Neuro Endocrinol. Lett. 2002, 23, S50–S54. [Google Scholar]
- Farriol, M.; Venereo, Y.; Orta, X.; Castellanos, J.M.; Segovia-Silvestre, T. In vitro effects of melatonin on cell proliferation in a colon adenocarcinoma line. J. Appl. Toxicol. 2000, 20, 21–24. [Google Scholar] [CrossRef]
- Kannen, V.; Marini, T.; Zanette, D.L.; Frajacomo, F.T.; Silva, G.E.; Silva, W.A., Jr.; Garcia, S.B. The melatonin action on stromal stem cells within pericryptal area in colon cancer model under constant light. Biochem. Biophys. Res. Commun. 2011, 405, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Santoro, R.; Mori, F.; Marani, M.; Grasso, G.; Cambria, M.A.; Blandino, G.; Muti, P.; Strano, S. Blockage of melatonin receptors impairs p53-mediated prevention of DNA damage accumulation. Carcinogenesis 2013, 34, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Santoro, R.; Marani, M.; Blandino, G.; Muti, P.; Strano, S. Melatonin triggers p53 ser phosphorylation and prevents DNA damage accumulation. Oncogene 2012, 31, 2931–2942. [Google Scholar] [CrossRef] [PubMed]
- Leon, J.; Casado, J.; Carazo, A.; Sanjuan, L.; Mate, A.; Munoz de Rueda, P.; de la Cueva, P.; Quiles, R.; Ruiz, S.; Ruiz-Extremera, A.; et al. Gender-related invasion differences associated with mRNA expression levels of melatonin membrane receptors in colorectal cancer. Mol. Carcinog. 2012, 51, 608–618. [Google Scholar] [CrossRef] [PubMed]
- Leon, J.; Casado, J.; Jimenez Ruiz, S.M.; Zurita, M.S.; Gonzalez-Puga, C.; Rejon, J.D.; Gila, A.; Munoz de Rueda, P.; Pavon, E.J.; et al. Melatonin reduces endothelin-1 expression and secretion in colon cancer cells through the inactivation of FoxO-1 and NF-κB. J. Pineal Res. 2014, 56, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, C.; Humpeler, S.; Kallay, E.; Mesteri, I.; Svoboda, M.; Rogelsperger, O.; Klammer, N.; Thalhammer, T.; Ekmekcioglu, C. Decreased expression of the melatonin receptor 1 in human colorectal adenocarcinomas. J. Biol. Regul. Homeost Agents 2011, 25, 531–542. [Google Scholar] [PubMed]
- Wielenga, V.J.; Heider, K.H.; Offerhaus, G.J.; Adolf, G.R.; van den Berg, F.M.; Ponta, H.; Herrlich, P.; Pals, S.T. Expression of CD44 variant proteins in human colorectal cancer is related to tumor progression. Cancer Res. 1993, 53, 4754–4756. [Google Scholar] [PubMed]
- Zeilstra, J.; Joosten, S.P.; Vermeulen, L.; Koster, J.; Medema, J.P.; Versteeg, R.; Spaargaren, M.; Pals, S.T. CD44 expression in intestinal epithelium and colorectal cancer is independent of p53 status. PLoS ONE 2013, 8, e72849. [Google Scholar] [CrossRef] [PubMed]
- Zeuner, A.; Todaro, M.; Stassi, G.; de Maria, R. Colorectal cancer stem cells: From the crypt to the clinic. Cell Stem Cell 2014, 15, 692–705. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Sanchez-Sanchez, A.M.; Puente-Moncada, N.; Gomez-Lobo, M.; Alvarez-Vega, M.A.; Antolin, I.; Rodriguez, C. Involvement of autophagy in melatonin-induced cytotoxicity in glioma-initiating cells. J. Pineal Res. 2014, 57, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Sanchez-Sanchez, A.M.; Herrera, F.; Gomez-Manzano, C.; Fueyo, J.; Alvarez-Vega, M.A.; Antolin, I.; Rodriguez, C. Melatonin-induced methylation of the ABCG2/BCRP promoter as a novel mechanism to overcome multidrug resistance in brain tumour stem cells. Br. J. Cancer 2013, 108, 2005–2012. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hao, A.; Li, X.; Du, Z.; Li, H.; Wang, H.; Yang, H.; Fang, Z. Melatonin inhibits tumorigenicity of glioblastoma stem-like cells via the AKT-EZH2-STAT3 signaling axis. J. Pineal Res. 2016, 61, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Goncalves Ndo, N.; Colombo, J.; Lopes, J.R.; Gelaleti, G.B.; Moschetta, M.G.; Sonehara, N.M.; Hellmen, E.; Zanon Cde, F.; Oliani, S.M.; Zuccari, D.A. Effect of melatonin in epithelial mesenchymal transition markers and invasive properties of breast cancer stem cells of canine and human cell lines. PLoS ONE 2016, 11, e0150407. [Google Scholar] [CrossRef]
- Krausova, M.; Korinek, V. Wnt signaling in adult intestinal stem cells and cancer. Cell. Signal. 2014, 26, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Ashley, N. Regulation of intestinal cancer stem cells. Cancer Lett. 2013, 338, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Park, K.H.; Kang, J.W.; Lee, E.M.; Kim, J.S.; Rhee, Y.H.; Kim, M.; Jeong, S.J.; Park, Y.G.; Kim, S.H. Melatonin promotes osteoblastic differentiation through the BMP/ERK/Wnt signaling pathways. J. Pineal Res. 2011, 51, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Margheri, M.; Pacini, N.; Tani, A.; Nosi, D.; Squecco, R.; Dama, A.; Masala, E.; Francini, F.; Zecchi-Orlandini, S.; Formigli, L. Combined effects of melatonin and all-trans retinoic acid and somatostatin on breast cancer cell proliferation and death: Molecular basis for the anticancer effect of these molecules. Eur. J. Pharmacol. 2012, 681, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.E.; Hart, L.S.; Dicker, D.T.; Wang, W.; El-Deiry, W.S. Visualization and enrichment of live putative cancer stem cell populations following p53 inactivation or Bax deletion using non-toxic fluorescent dyes. Cancer Biol. Ther. 2009, 8, 2194–2205. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Zhang, X.M.; Tavaluc, R.T.; Hart, L.S.; Dicker, D.T.; Wang, W.; El-Deiry, W.S. The combination of 5-fluorouracil plus p53 pathway restoration is associated with depletion of p53-deficient or mutant p53-expressing putative colon cancer stem cells. Cancer Biol. Ther. 2009, 8, 2186–2193. [Google Scholar] [CrossRef] [PubMed]
- Puca, F.; Colamaio, M.; Federico, A.; Gemei, M.; Tosti, N.; Bastos, A.U.; del Vecchio, L.; Pece, S.; Battista, S.; Fusco, A. HMGA1 silencing restores normal stem cell characteristics in colon cancer stem cells by increasing p53 levels. Oncotarget 2014, 5, 3234–3245. [Google Scholar] [CrossRef] [PubMed]
- Godar, S.; Ince, T.A.; Bell, G.W.; Feldser, D.; Donaher, J.L.; Bergh, J.; Liu, A.; Miu, K.; Watnick, R.S.; Reinhardt, F.; et al. Growth-inhibitory and tumor-suppressive functions of p53 depend on its repression of CD44 expression. Cell 2008, 134, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Mediavilla, M.D.; Cos, S.; Sanchez-Barcelo, E.J. Melatonin increases p53 and p21WAF1 expression in MCF-7 human breast cancer cells in vitro. Life Sci. 1999, 65, 415–420. [Google Scholar] [CrossRef]
- Kim, C.H.; Yoo, Y.M. Melatonin induces apoptotic cell death via p53 in LNCaP cells. Korean J. Physiol. Pharmacol. 2010, 14, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Bubenik, G.A. Thirty four years since the discovery of gastrointestinal melatonin. J. Physiol. Pharmacol. 2008, 59 (Suppl. S2), 33–51. [Google Scholar] [PubMed]
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical causes of cancer. Nat. Rev. Cancer 2003, 3, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Ziolko, E.; Kokot, T.; Skubis, A.; Sikora, B.; Szota-Czyz, J.; Kruszniewska-Rajs, C.; Wierzgon, J.; Mazurek, U.; Grochowska-Niedworok, E.; Muc-Wierzgon, M. The profile of melatonin receptors gene expression and genes associated with their activity in colorectal cancer: A preliminary report. J. Biol. Regul. Homeost Agents 2015, 29, 823–828. [Google Scholar] [PubMed]
- Dalerba, P.; Kalisky, T.; Sahoo, D.; Rajendran, P.S.; Rothenberg, M.E.; Leyrat, A.A.; Sim, S.; Okamoto, J.; Johnston, D.M.; Qian, D.; et al. Single-cell dissection of transcriptional heterogeneity in human colon tumors. Nat. Biotechnol. 2011, 29, 1120–1127. [Google Scholar] [CrossRef] [PubMed]
- Punt, C.J.; Koopman, M.; Vermeulen, L. From tumour heterogeneity to advances in precision treatment of colorectal cancer. Nat. Rev. Clin. Oncol. 2017, 14, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Bunz, F.; Hwang, P.M.; Torrance, C.; Waldman, T.; Zhang, Y.; Dillehay, L.; Williams, J.; Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J. Clin. Investig. 1999, 104, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Pilat, N.; Grunberger, T.; Langle, F.; Mittlbock, M.; Perisanidis, B.; Kappel, S.; Wolf, B.; Starlinger, P.; Kuhrer, I.; Muhlbacher, F.; et al. Assessing the TP53 marker type in patients treated with or without neoadjuvant chemotherapy for resectable colorectal liver metastases: A p53 research group study. Eur. J. Surg. Oncol. 2015, 41, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.; Atkin, W.; Lenz, H.J.; Lynch, H.T.; Minsky, B.; Nordlinger, B.; Starling, N. Colorectal cancer. Lancet 2010, 375, 1030–1047. [Google Scholar] [CrossRef]
- Gao, Y.; Xiao, X.; Zhang, C.; Yu, W.; Guo, W.; Zhang, Z.; Li, Z.; Feng, X.; Hao, J.; Zhang, K.; et al. Melatonin synergizes the chemotherapeutic effect of 5-fluorouracil in colon cancer by suppressing PI3K/AKT and NF-κB/iNOS signaling pathways. J. Pineal Res. 2017, 62. [Google Scholar] [CrossRef] [PubMed]
- Cerea, G.; Vaghi, M.; Ardizzoia, A.; Villa, S.; Bucovec, R.; Mengo, S.; Gardani, G.; Tancini, G.; Lissoni, P. Biomodulation of cancer chemotherapy for metastatic colorectal cancer: A randomized study of weekly low-dose irinotecan alone versus irinotecan plus the oncostatic pineal hormone melatonin in metastatic colorectal cancer patients progressing on 5-fluorouracil-containing combinations. Anticancer Res. 2003, 23, 1951–1954. [Google Scholar] [PubMed]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; de Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Kemper, K.; Grandela, C.; Medema, J.P. Molecular identification and targeting of colorectal cancer stem cells. Oncotarget 2010, 1, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Kemper, K.; Sprick, M.R.; de Bree, M.; Scopelliti, A.; Vermeulen, L.; Hoek, M.; Zeilstra, J.; Pals, S.T.; Mehmet, H.; Stassi, G.; et al. The AC133 epitope, but not the CD133 protein, is lost upon cancer stem cell differentiation. Cancer Res. 2010, 70, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Shmelkov, S.V.; Butler, J.M.; Hooper, A.T.; Hormigo, A.; Kushner, J.; Milde, T.; St Clair, R.; Baljevic, M.; White, I.; Jin, D.K.; et al. CD133 expression is not restricted to stem cells, and both CD133+ and CD133− metastatic colon cancer cells initiate tumors. J. Clin. Investig. 2008, 118, 2111–2120. [Google Scholar] [CrossRef] [PubMed]
- Horst, D.; Kriegl, L.; Engel, J.; Kirchner, T.; Jung, A. CD133 expression is an independent prognostic marker for low survival in colorectal cancer. Br. J. Cancer 2008, 99, 1285–1289. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Ishii, G.; Atsumi, N.; Fujii, S.; Saito, N.; Ochiai, A. Immunohistochemical detection of CD133 expression in colorectal cancer: A clinicopathological study. Cancer Sci. 2008, 99, 1578–1583. [Google Scholar] [CrossRef] [PubMed]
- Leroy, B.; Fournier, J.L.; Ishioka, C.; Monti, P.; Inga, A.; Fronza, G.; Soussi, T. The TP53 website: An integrative resource centre for the TP53 mutation database and TP53 mutant analysis. Nucleic Acids Res. 2013, 41, D962–D969. [Google Scholar] [CrossRef] [PubMed]
- Pietrantonio, F.; Biondani, P.; Perrone, F.; di Bartolomeo, M.; Pacifici, M.; Milione, M.; Melotti, F.; Maggi, C.; Montemurro, G.; Bossi, I.; et al. TP53 mutations in advanced colorectal cancer: The dark side of the moon. Oncology 2014, 86, 289–294. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Covariates | TP53 a Wild-Type % (n) | TP53 Mutant % (n) | pb |
|---|---|---|---|
| Gender | |||
| Male | 62.0 (66) | 63.2 (48) | 0.893 |
| Female | 38.0 (41) | 36.8 (28) | |
| Differentiation Grade | |||
| Well differentiated | 16.8 (18) | 35.5 (27) | 0.095 |
| Moderately differentiated | 69.1 (74) | 51.3 (39) | |
| Poorly differentiated | 14 (15) | 13.2 (10) | |
| Stage | |||
| Stage I + II (Early) | 59.8 (64) | 33.3 (32) | 0.025 |
| Stage III + IV (Advanced) | 40.2 (43) | 57.9 (44) | |
| Location | |||
| Proximal | 47.7 (51) | 38.2 (29) | 0.437 |
| Distal | 47.7 (51) | 56.6 (43) | |
| Rectal | 4.6 (5) | 5.2 (4) |
| p53 Wild-Type | ||||||
| Gene | Early Stages | Advanced Stages | ||||
| CD44lowCD66clow n (%) | CD44highCD66chigh n (%) | p a | CD44lowCD66clow n (%) | CD44highCD66chigh n (%) | p a | |
| NAT b | ns | 0.045 | ||||
| Low | 7 (63.6) | 17 (89.5) | 4 (40.0) | 16 (80.0) | ||
| High | 4 (36.4) | 2 (10.5) | 6 (60.0) | 4(20.0) | ||
| MT1 | ns | ns | ||||
| Low | 11 (91.7) | 19 (95.0) | 7 (77.8) | 18 (90.0) | ||
| High | 1 (8.3) | 1 (5.0) | 2 (22.2) | 2 (10) | ||
| MT2 b | ns | |||||
| Low | 8 (66.7) | 17 (85.0) | 4 (44.4) | 20 (100.0) | ||
| High | 4 (33.3) | 3 (15.0) | 5 (55.6) | 0 (0.0) | ||
| p53 Mutated | ||||||
| Gene | Early Stages | Advanced Stages | ||||
| CD44lowCD66clow n (%) | CD44highCD66chigh n (%) | pa | CD44lowCD66clow n (%) | CD44highCD66chigh n (%) | pa | |
| NAT b | ns | ns | ||||
| Low | 6 (75.0) | 4 (80.0) | 4 (40.0) | 10 (83.3) | ||
| High | 2 (25.0) | 1 (20.0) | 6 (60.0) | 2 (16.7) | ||
| MT1 | ns | ns | ||||
| Low | 9 (100.0) | 5 (71.4) | 7 (77.8) | 11 (100.0) | ||
| High | 0 (0.0) | 2 (28.6) | 2 (22.2) | 0 (0.0) | ||
| MT2 b | ns | ns | ||||
| Low | 7 (77.8) | 6 (85.7) | 6 (66.7) | 9 (81.8) | ||
| High | 2 (22.2) | 1 (14.3) | 3 (33.3) | 2 (18.2) | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casado, J.; Iñigo-Chaves, A.; Jiménez-Ruiz, S.M.; Ríos-Arrabal, S.; Carazo-Gallego, Á.; González-Puga, C.; Núñez, M.I.; Ruíz-Extremera, Á.; Salmerón, J.; León, J. AA-NAT, MT1 and MT2 Correlates with Cancer Stem-Like Cell Markers in Colorectal Cancer: Study of the Influence of Stage and p53 Status of Tumors. Int. J. Mol. Sci. 2017, 18, 1251. https://doi.org/10.3390/ijms18061251
Casado J, Iñigo-Chaves A, Jiménez-Ruiz SM, Ríos-Arrabal S, Carazo-Gallego Á, González-Puga C, Núñez MI, Ruíz-Extremera Á, Salmerón J, León J. AA-NAT, MT1 and MT2 Correlates with Cancer Stem-Like Cell Markers in Colorectal Cancer: Study of the Influence of Stage and p53 Status of Tumors. International Journal of Molecular Sciences. 2017; 18(6):1251. https://doi.org/10.3390/ijms18061251
Chicago/Turabian StyleCasado, Jorge, Almudena Iñigo-Chaves, Sergio M. Jiménez-Ruiz, Sandra Ríos-Arrabal, Ángel Carazo-Gallego, Cristina González-Puga, María Isabel Núñez, Ángeles Ruíz-Extremera, Javier Salmerón, and Josefa León. 2017. "AA-NAT, MT1 and MT2 Correlates with Cancer Stem-Like Cell Markers in Colorectal Cancer: Study of the Influence of Stage and p53 Status of Tumors" International Journal of Molecular Sciences 18, no. 6: 1251. https://doi.org/10.3390/ijms18061251