
Perturbations in the Replication Program Contribute to Genomic Instability in Cancer

1
Department of Microbiology and Molecular Genetics, IMRIC, Faculty of Medicine, Hebrew University of Jerusalem, Jerusalem 91120, Israel
2
Pharmacology and Experimental Therapeutics Unit, The Institute for Drug Research, School of Pharmacy, Faculty of Medicine, The Hebrew University of Jerusalem, Jerusalem 91120, Israel
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2017, 18(6), 1138; https://doi.org/10.3390/ijms18061138
Submission received: 6 April 2017
/
Revised: 8 May 2017
/
Accepted: 21 May 2017
/
Published: 25 May 2017
(This article belongs to the Special Issue Mechanisms Leading to Genomic Instability)
Abstract
:Cancer and genomic instability are highly impacted by the deoxyribonucleic acid (DNA) replication program. Inaccuracies in DNA replication lead to the increased acquisition of mutations and structural variations. These inaccuracies mainly stem from loss of DNA fidelity due to replication stress or due to aberrations in the temporal organization of the replication process. Here we review the mechanisms and impact of these major sources of error to the replication program.
1. Introduction
Malignant transformation is characterized by uncontrolled cell growth, which is often caused by an accumulation of mutations due to increased mutation rates and genome instability. Accurate deoxyribonucleic acid (DNA) replication is crucial for preventing such genomic instability [1,2] since during replication there is great potential for the introduction of mutations. Three main mechanisms contribute to mutations during DNA replication: (1) Mismatches inserted during replication due to the inherent error rates in DNA polymerase [3] that escaped mismatch repair [4]; (2) DNA damage that occurred prior to replication initiation that was not repaired in time; and (3) DNA damage that occurred during replication itself [5].
In this review we will discuss two major types of changes in replication that may lead to genome instability. The first potential alteration of the replication program is an increase in replication stress, which is defined as replication fork slowing, stalling, or collapse. Heightened replication stress frequently leads to higher levels of DNA damage and genomic instability [6]. This phenomenon plays an important role in the development of cancer as proposed by the oncogene-induced replication stress model. According to this model, cancerous transformation is driven by increased replication stress, which creates an environment of genomic instability [7]. This model suggests that the increased mutation rate essential for cancer development stems from replication stress rather than from random mutations in genes involved in genome surveillance [8]. The oncogene-induced replication stress model is supported by recent high-throughput sequencing studies that discovered a paucity of mutations in DNA repair genes in primary cancer [7].
The second replication-related change linked to genomic instability involves changes in the temporal organization of the replication process, also known as replication timing. DNA replication is a strictly regulated process, in which origins of replication fire at distinct times during synthesis (S) phase creating distinct early and late replication zones [9]. Aberrant order of replication initiation has been implicated in cancer and in the generation of genomic instability [10,11], though the frequency and significance of such changes during cancer transformation is not yet clear.
The relationship between replication stress and replication timing is highly complex and interdependent. Nonetheless, replication stress and replication timing remain distinct. Replication stress refers to the fidelity of the replication fork, whereas replication timing refers to the relative time at which different regions are replicated. Changes in replication timing may potentially result in discoordination of the replication process and lead to stress. Conversely, replication stress may affect the replication timing program as many stalled or collapsed forks in a given region may delay the time at which it is replicated. However, in yeast it was shown that induction of replication stress through depletion of the nucleotide pool or through mutations in certain cell cycle proteins, can alter the duration of the replication but does not affect the general replication timing profile [12,13]. Experiments validating this in mammals remain to be performed.
2. Replication Stress
2.1. Mechanisms of Replication Stress
During S phase of the cell cycle, the cell must accurately replicate its entire DNA content. Eukaryotic replication is initiated from multiple licensed origins and expands via bidirectional replication forks in order to fully replicate the entire genome [14]. Replication stress occurring during this process results in slow or stalled DNA replication forks [15] and can prevent accurate replication of DNA. These stalled or collapsed forks expose single stranded DNA (ssDNA) as the helicase continues to unwind the adjacent DNA, leaving the region susceptible to double strand breaks (DSB) [6,16,17]. Normally, to prevent such damage, replication protein A (RPA) coats the ssDNA and activates ataxia telangiectasia and Rad3-related protein (ATR) initiating a cascade of events to resolve the stalled forks or bypass collapsed forks [18]. Failure in this DNA repair pathway will lead to cell cycle arrest, whereas partial recovery from stalled forks may lead to genomic instability. Thus, high levels of replication stress frequently result in the accumulation of mutations as well as larger chromosomal aberrations such as copy number variations and translocations [6,19].
There are a number of potential sources of replication stress (reviewed in [18,20]). First, replication stress can be caused by direct barriers to the replication fork. This includes lesions in the DNA induced by UV light or chemical mutagens [21], which physically block replication fork progression. Production of reactive oxygen species (ROS) can result in oxidized or abasic sites, which will impede fork progression [22]. Repetitive regions or unusual DNA structures such as the G-quadruplex are also challenging to fork progression [23]. Additionally, accidental incorporation of ribonucleotides in the DNA can serve as a barrier to fork progression [24].
Another major source of replication stress is a lack of resources. The DNA replication program requires many different factors and fork progression can be limited by their deficiency. Adequate levels of deoxynucleotide triphosphates (dNTPs) are crucial for the progression of the replication fork [25,26]. Histone deficiency can also induce slowing of the replication fork although this appears to occur only after a long time, since transient histone depletion does not induce stress [27,28]. RPA consumption due to a large number of stalled forks can create an additional level of stress as the remaining stalled forks will collapse due to lack of RPA [29]. In addition, it was shown in yeast that reduced expression of replisome proteins can lead to replication stress and DNA damage [30]. Depletion of resources can occur as a result of extensive use due to over replication or due to a lack of coordination between pathways responsible for supplying such factors [31]. For example, early entry into S phase may cause the cell to begin replication before it has acquired the necessary factors [31].
Transcription interference can also induce replication stress. The replication timing program maintains coordination between replication and transcription in order to minimize interference between the two. Should this system be perturbed, collisions of the DNA polymerase and RNA polymerase can occur. During collision, the excess of the transcription and replication machineries creates topological constraint and supercoiling of the DNA, which can lead to knotting and prevent fork progression [32]. Transcription also results in the formation of R loops, which are ribonucleic acid (RNA):DNA hybrids that occur during transcription when the newly synthesized RNA anneals to the DNA leaving a displaced ssDNA strand. In the event of an R loop formation, the fork can be blocked by the RNA polymerase, the trinucleotide structure, or a lesion created in the ssDNA [33]. Transcriptional interference can be resolved by topoisomerase, helicase or ribonuclease H (RNase H), which prevents formation of R loops [34,35,36] and reduces topological stress.
Extent of origin usage is another critical factor, which can contribute to replication stress (reviewed in [37]). Intact origin usage has been implicated in a number of diseases such as Meier–Gorlin syndrome [38]. Re-replication of origins can occur when licensed origins initiate replication for a second time before mitosis, ultimately resulting in replication stress [39]. Over activation of origins can lead to stress and instability through depletion of nucleotides [40], RPA [29], or other replisome machinery or through disruption of the coordination between transcription and replication resulting in an increase in collisions [41], as described above. Conversely, under usage of origins has also been implicated in the accumulation of replication stress. Experiments in yeast [42,43], mice [44] and human cells [45] exhibiting licensing deficiency have demonstrated the link between origin sparsity and genomic instability. It has been shown that sufficient licensing of origins is needed to deal with replication stress [46,47], because the activation of dormant origins adjacent to stalled forks is a crucial mechanism for overcoming stress [48]. Thus, insufficient licensing may result in a lack of dormant origins and facilitate entry to mitosis before replication is completed [42,43,49].
The oncogene-induced replication stress model proposes that oncogenes facilitate replication stress [7]. Various well-established oncogenes have been shown to be involved in all the above described causes of replication stress (Table 1). Oncogene overexpression impairs normal cell cycle regulation and stimulates increased growth, abnormal replication initiation and abnormal transcription, all of which result in sustained proliferation and increased stress via the mechanisms described above. Increased cell metabolism associated with high levels of cell growth can contribute to ROS levels. Hyper-replication associated with cancer can lead to depletion of resources as well as early entry into S phase. Abnormal regulation of cell cycle processes can result in discoordination of transcription and replication resulting in increased collisions. Further, cancer has been associated with deregulation of origin usage through impairment of licensing mechanisms or due to general cell cycle deregulation. The mechanisms through which oncogenes induce replication stress are not exclusive as different oncogenes can induce stress through similar mechanisms. Additionally, a given oncogene can lead to stress through a number of different mechanisms occurring simultaneously. For example, a recent review of the contribution of c-myc to replication stress [50] suggested two coexisting mechanisms for fork stalling induction: over usage of origins resulting in depletion of resources and increased interference between transcription and replication.
2.2. Resolution of Replication Stress
There are a number of cellular mechanisms which are involved in the response to and resolution of replication stress. The DNA damage response (DDR) pathways respond to stress and DNA damage through either repair or cell cycle arrest. The main DDR pathway in the response to replication stress is the ATR pathway. RPA coats the ssDNA near a stalled fork and recruits and activates ATR. ATR localizes to and stabilizes the stalled forks [68,69] until either the source of stress is removed [20], or the region is replicated due to activation of nearby dormant origins [48,70,71] or through translesion synthesis [72]. The ATR pathway inhibits cell cycle progression by the activation of the intra-S checkpoint by activation of the checkpoint kinase 1 (Chk1). Phosphorylated Chk1 also inhibits further initiation of origins of replication and thus ensures more resources are available to deal with the current stress [29,73,74]. If the damage is insurmountable, cells will not return to the cell cycle but will rather enter senescence or apoptosis [75,76,77]. In this way, the DNA repair proteins together with the cell cycle arrest proteins protect the cell from reaching a state of genomic instability [75,78].
While the ATR pathway is activated by ssDNA, the ataxia-telangiectasia mutated (ATM) pathway is activated in response to DSBs and therefore also plays a role in the response to stress, particularly at collapsed replication forks associated with DSBs [79]. The Mre11-Rad50-Nbs1 (MRN) complex senses DSBs and recruits and activates ATM. ATM and the MRN complex can then facilitate homologous recombination to repair the DSB [80]. Further, localized ATM phosphorylates a number of substrates including checkpoint kinase 2 (Chk2) and H2A Histone Family Member X (H2AX). Phosphorylated H2AX, γH2AX, helps to recruit more ATM to the site and potentially opens the chromatin to allow for the recruitment of more proteins to fix the DSB [80]. The phosphorylated Chk2 acts as a checkpoint leading to cell cycle arrest. ATM also aids in fork restart through its interaction with DNA helicases involved in this process [81]. If the damage is not repaired, the ATM pathway will result in high levels of phosphorylated p53 which can lead to senescence or apoptosis [82].
The DNA damage tolerance (DDT) pathway, also known as post-replicative repair, is another pathway involved in the repair of stalled forks (reviewed in [5]). The DDT pathway is activated by PCNA ubiquitination which is induced by ssDNA at stalled forks [83]. DDT entails activation of specialized low fidelity translesion DNA polymerases which can bypass lesions in the DNA [72,84]. There are a range of different translesion polymerases, suitable for different types of damage and with different levels of fidelity (reviewed in [85]). In addition to translesion polymerases, DDT also employs template switching which is generally error-free and uses the already copied sister chromatid as a template to repair damage [86].
2.3. Replication Stress Leads to Genomic Instability, Common Fragile Sites Breakage, and Cancer
Failure to resolve replication stress can result in point mutations, loss of heterozygosity (LOH), insertions, deletions, translocations and aneuploidy [6,19,87]. Point mutations may stem from usage of translesion synthesis polymerases [84] whereas copy number variations commonly stem from aberrant restoration of stalled forks either by non-homologous end joining or through inaccurate fork restarting caused by error-free post-replication repair [88]. Mitotic catastrophe can occur in the event that the stalled forks are not resolved and the cell enters mitosis with only partially replicated DNA [6,21]. This can result in nondisjunction, anaphase bridges and lagging chromosomes leading to large-scale chromosomal damage [89,90,91,92].
While replication stress results in global instability, large genomic regions spanning a few 100 kilobases, termed common fragile sites (CFSs), are particularly vulnerable to replication stress and are more susceptible to double strand breaks (reviewed in [93]). Though these regions are typically stable under normal conditions, they frequently break under stress. The predisposition of CFSs to DNA damage has made fragile sites hotspots for translocations and deletions [94,95]. Many CFS properties contribute to their predisposition to DNA damage including increased transcriptional interference, late replication, origin shortage, AT-rich repeats and unique chromatin modifications [93]. Though CFSs are often associated with late replication, recently a new class of CFSs have been discovered, which are associated with early replication [96]. These early replication fragile sites (ERFSs) differ from CFSs (reviewed in [97]) and likely involve a different mechanism leading to their fragility. Late replicating CFSs are likely unable to complete replication before mitosis in replication stress conditions due to a combination of the aforementioned reasons [98,99], whereas ERFSs are probably mainly impacted by replication–transcription collisions [96].
On a broader scale, the oncogene induced replication stress model proposes that replication stress plays an important role in cancer progression. Studies have found increased levels of replication stress particularly in the early stages of cancers [58,60,100] suggesting that replication stress and the resulting genomic instability are primary hallmarks of cancer and not just downstream events [7]. Indeed, oncogene induced replication stress was shown to induce allelic imbalance at CFSs at the early stages of cancer even before the DDR was compromised [62,100]. Genome-wide studies have confirmed these findings and demonstrated that stress induced instability at CFSs is an early phenomenon in cancer development [101].
As cancer is associated with increased levels of replication stress [7], in pre-cancerous and cancerous cells CFSs are associated with deletions, translocations and amplifications [95,102]. The fragile site landscape in cancer is dependent on the unique combination of oncogenes in the cell [61]. Further support for the involvement of replication stress in generating genome instability in early stage of cancer, comes from mutation pattern analyses [31]. In cancer there are two main classes of mutations: driver mutations, referring to mutations which are selected for due to their conferring an advantage, and passenger mutations, referring to the many other mutations in the genome which seem to provide no benefit to the cancer cell. As passenger mutations do not supply any advantage to the cell and are therefore not influenced by selection, their pattern tells of the underlying mechanism of their generation during cancer development. Analyses of the pattern of the most common passenger copy number variations (CNVs) in many types of cancer revealed that they are enriched in late replicating regions or in very large genes. Similarly, it was demonstrated that the most common passenger point mutations are CG to TG located in large genes. While there are many mechanisms that can lead to CNVs in late replicating regions (see below), the enrichment of both CNVs and point mutations in large genes suggests that replication stress is a leading cause of these mutations and an early driver of cancer transformation.
2.4. The DDR in Cancer Progression and Therapy
Genomic instability induced by replication stress is not sufficient for generating complete cancer transformation. Under normal conditions, the cellular DDR drives defective cells into senescence or apoptosis and prevents transformation. Thus, cancer will develop only if oncogene induced replication stress is followed by evasion of the DDR. Accordingly, in preneoplastic cells, hyper-replication [60] and ROS [103] induced by oncogenic stress activate the DDR and ultimately lead to senescence, whereas the malfunctioning DDR of cancerous cells enables circumvention of cell death and ultimately results in complete transformation.
There is selective pressure for the mutation or elimination of tumor suppressors and other agents of the DDR pathway, such as downstream targets of ATR including p53, in order to allow the cell to proliferate malignantly. Studies have found that tissues in the early stages of cancer showed signs of functional apoptosis or senescence while only more advanced cancerous tissues were lacking in p53 and other DNA repair genes [7,58,100]. Similarly, Ras induction results in tumors only after evasion of the senescence checkpoints. Though high levels of Ras generally induce senescence, elimination of p53-dependent senescence checkpoints results in cancer transformation [104].
Though impairment of the DDR facilitates the progression of cancer, by conferring the ability to proliferate despite sustained damage, the DDR may also act as a powerful potential therapeutic target. Extreme DNA damage or impairment of DDR activity can ultimately result in mitotic catastrophe and cell death. This is due to the delicate balance of DDR and genomic instability in cancer cells. Mild levels of replication stress foster genomic instability and tumorigenesis, but high levels of stress will induce mitotic catastrophe [105]. Radiation and chemotherapies work according to this principle, as they induce general high levels of DNA damage which the cell cannot tolerate due to its defective DDR.
More recently, specific therapies have relied on increasing replication stress by directly targeting DDR pathways involved in the resolution of stress. Inhibitors of the DDR are useful targets in cancer therapy as their inhibition will speed up mitotic catastrophe (reviewed in [106,107]). Several Chk1 and ATR inhibitors are in clinical trials for anticancer therapy. Targeted therapies also aim to manipulate cell cycle regulator proteins such as WEE1 in order to speed up entry into mitosis before the DNA is fully replicated. Another major class of cancer drugs includes poly(ADP-ribose) polymerase 1 (PARP1) inhibitors that inhibit PARP1 proteins, which are crucial to repairing single strand breaks in DNA. Inhibition of PARP1 leads to accumulation of double strand breaks and ultimately cell death. In normal cells, fully functioning DDR as well as lower levels of cell growth, result in higher tolerance to the loss of these mechanisms and therefore these therapies are highly specific to cancer cells. Another potential target is gain of function p53 (GOF p53), a mutant p53 with oncogenic qualities. GOF p53 increases expression of necessary replication proteins and has recently been shown to stabilize replication forks preventing unmanageable levels of stress [108].
3. Replication Timing
3.1. Background
The cellular replication program is highly regulated and temporally coordinated according to a strict program [109]. During S phase of the cell cycle, each genomic region is replicated at a distinct time through the activation of an origin of replication. The time each region is replicated is a function of its distance from an active origin and of the time the origin was activated. Adjacent origins are usually activated simultaneously and give rise to large chromosomal regions that are replicated synchronously, called constant timing regions (CTRs). These regions are activated at various time points along S, giving rise to early, middle and late time zones. Between early and late zones there are large regions, in which the replication timing changes gradually, termed timing transition regions (TTRs) [9,110] (Figure 1a).
Replication timing has proven to be a measurable and stable epigenetic feature. In both humans and mice, it has been demonstrated that a large portion of the genome has a relatively constant replication timing, which does not vary between different developmental states and tissue types. However, the rest of the genome has been termed developmentally switching [112,113] and differs between specific tissue types.
The replication timing of a region, seems to reflect higher order genomic organization, since it correlates with basic genetic features such as the regional GC content, Giemsa banding, and gene density, as well as other epigenetic features such as chromatin state, and the three-dimensional (3D) structure of the genome [9,114]. Detailed analysis of the replication timing of individual genes has revealed a striking correlation between transcription and early replication. Expressed genes, such as constitutively transcribed housekeeping genes, replicate at early stages of S phase, whereas repressed tissue specific genes may replicate in most tissues at late stages of S phase and become early-replicating only in the expressing tissue [115].
There are two main methods used to determine replication timing. The first is based on copy number [111] and relies on the fact that in unsynchronized culture, the percentage of cells in S phase in which a locus has replicated is indicative of its replication timing. The second method is based on bromodeoxyuridine (BrdU) incorporation [116]. Cells are exposed to BrdU and sorted to early and late fractions in S phase. BrdU labeled DNA is immunoprecipitated from the different S fractions and measured in order to determine the replication timing.
3.2. Effects of Replication Timing on Mutation Rates and Structural Variations
Replication timing is known to correlate with the rate of many types of mutations and chromosomal aberrations (Table 2 and Figure 1b) (reviewed in [117]).
Point mutation rates are associated with replication timing in a range of organisms. Studies of divergence between chimpanzee and human [118,119], rat and mouse [119,122], and within different Drosophila species [123], have demonstrated increased mutation prevalence in late replicating regions. Analyses of single nucleotide polymorphism (SNP) data in both human [118] and mouse [119] have confirmed this bias. This bias has even been demonstrated in yeast, where the URA3 gene, encoding orotidine-5′-phosphate decarboxylase, was inserted in different replication timing zones and displayed corresponding changes in mutation rate [131]. In addition to these studies in germline mutation rates, studies using multiple cancer types [124,125] as well as studies of individual cancers such as liver [126] and lung cancer [129], have also exhibited a bias for higher point mutation rates in late replicating zones. Despite the general association of mutation rate and replication timing, mutation signature analyses revealed that the correlation appears to be extremely nuanced and different mutation signatures correlate differently to replication timing [120,130,146].
Larger structural changes in DNA such as amplifications and deletions have also been linked to replication timing patterns. Studies in flies discovered enrichment of duplications in late regions and deletions in early regions [136,137]. However, studies in human cancers have found the opposite trend demonstrating that amplifications are more likely to be in early regions whereas deletions tend to be in late regions [133]. It is unclear whether this difference is due to the difference in species or due to the cancerous effects. As noted earlier, most common fragile sites are enriched in late replicating regions. The fact that replication timing varies between tissues has been used to explain the variation in CFS instability between different tissue types, supporting the strong connection between replication timing and fragility [142]. Certain late replicating regions do not manage to complete replication by the end of S phase leading to fragility [99,143,144]. Other CFSs, which generally replicate in early/mid S may be delayed to late S replication due to stress and are thereby made fragile [147,148,149].
Several studies in cancer cells found that rearrangements or translocations occur preferentially in early replicating regions [139,140]. However, one study found a subset of rearrangements in specific cancers, which occur in particularly late regions [138], suggesting that this phenomenon may be cancer type specific. Interestingly, translocation partners tend to be from the same timing regions [141], probably reflecting the spatial proximity of regions replicating at the same time [150,151].
TTRs also exhibit enrichment of specific mutation types. As TTRs are regions located between early and late zones, which contain few or no origins [109,152,153,154], each replication fork replicates a large genomic region and is therefore more prone to fork stalling and collapse [155]. Indeed TTRs exhibit higher levels of SNP density [121] as well as amplifications [134].
The mechanism, by which replication timing may affect mutation rates, appears to be multifaceted and a number of theories have been proposed. It has been shown that the increased mutation rate characteristic of late replicating regions impacts different mutation types similarly, suggesting that the bias stems from the general accumulation of ssDNA in late regions [118]. However, other more recent studies, have found that the mutational bias differentially favors specific mutation types [120,146,156] and suggests that factors unique to the different stages of S phase may be contributing to the differential rates.
It has been suggested that the differential mutation rates along S phase are formed due to different replication and repair machinery present at different stages of S. For example, a study in yeast demonstrated that elimination of the rev1 translesion polymerase, which is more active in late S, removed the bias of point mutations in late regions [131]. Studies in humans have also pinned mutational correlation on differential DNA machinery, for example by showing that mutations are not enriched in late regions in mismatch repair deficient cells [127]. Similarly, the replication timing bias of CNVs can be explained by their method of formation. It has been shown that homologous recombination based CNVs are enriched in early replicating regions whereas CNVs generated by other mechanisms such as non-homologous end-joining (NHEJ) and microhomology-mediated break-induced replication occur more frequently in late replicating regions [120]. Another potential mechanism contributing to the replication timing mutational bias is the temporal variation in the composition of the dNTP pool, which favors different nucleotides at different times in S phase [156]. Alternatively, the connection between replication timing and mutational frequency may be impacted by other factors, which correlate closely with replication timing such as open versus closed chromatin state which may affect the accessibility of the DNA to different DNA repair mechanisms [157].
3.3. Replication Timing Changes and Cancer
A number of replication timing changes have been linked to cancerous transformation (Table 3). In a genome-wide study of replication timing in cancer [10], 17 pediatric leukemia tumors were analyzed to determine the changes in replication timing after cancer transformation. The cancerous replication timing profiles varied in comparison to normal B and T cell replication timing. Additionally, replication timing heterogeneity was found within the different tumor specimens. Interestingly, replication timing changes common to all cancer specimens included the region next to the Runt-related transcription factor 1 (RUNX1) locus, a common leukemic translocation site though only some specimens displayed the actual translocation itself. This suggests the possibility that the replication timing change is an early step predisposing the cancer cell to the translocation. Another study in tumor-derived cell lines [158] also found a connection between translocations and replication timing, however, suggesting that rearrangements can result in chromosome-wide replication delays.
Studies in different cancer cells have reported changes in replication timing of specific cancer-related genes. Among these are tumor suppressor genes like tumor protein P53 (p53), retinoblastoma 1 (RB1); DNA repair related genes like ATM; and classical oncogenes like c-myc, HER-2/neu [11,159,160]. These replication timing changes can be specific to one allele resulting in asynchronous replication of the two alleles [11,159,160,162], or biallelic, causing a uniform change of the replication timing of the gene [11]. The B-cell lymphoma 2 (Bcl2) gene, which is frequently translocated in human follicular lymphoma is associated with a change from middle to early replication in the translocated gene in correlation with its overexpression [163].
3.4. Causes of Replication Timing Changes in Cancer
It is unclear what exactly leads to the changes in replication timing associated with cancer. It has been shown in yeast that most global changes in replication timing stem from local changes in replication fork speed and origin activation [164]. Thus, oncogene-induced replication stress, which slows down fork progression and inhibits global initiation of origins of replication, may play a role in changing the replication timing landscape. Indeed, specific oncogenes have been shown to modulate the replication program. For example, the c-myc proto-oncogene is involved in replication initiation and its deregulation can result in premature origin firing [53] potentially bringing about global changes in the replication timing program.
Misregulation of other replication-related genes can also effect changes in the replication timing program and have been implicated in the development of cancer [165]. In yeast, forkhead box proteins [166] as well as the replication timing regulatory factor 1 (Rif1) protein [167] play important roles in the maintenance of accurate replication timing [166]. The role of Rif1 in replication timing has been demonstrated in mice [168,169] and humans [170] as well. Changes in the expression of proteins involved in the minichromosome maintenance (MCM) complex, which is involved in initiation of origins of replication, may also affect replication timing since the amount of MCM complexes at an origin influences its timing [171]. Interestingly, polymerase theta also plays an important role in determination of replication timing, and its depletion or overexpression leads to large scale replication timing changes [172].
In addition to changes in the expression of specific genes associated with DNA replication, replication timing may be indirectly affected by alterations to closely related epigenomic features. For example, changes in histone modifying proteins can lead to changes in the chromatin state, which may then alter the replication timing program [173,174].
Translocations can affect replication timing locally since the edges of the translocated gene may propagate its timing to its new neighbors [141]. Alternatively, translocated genes may adopt the timing of their new region [163]. Translocations may also influence replication timing on a more global level by delaying replication of the entire chromosome [158].
3.5. Effects of Changes in Replication Timing in Cancer
Replication timing changes can result in genomic instability and cancer development in a number of ways. First, replication timing changes may potentially influence the progression of cancer through their impact on the gene expression profile. The causal relationship between replication timing and expression is quite complicated. Though replication timing is associated with gene expression [110,175] this correlation does not hold true for all genes [112]. Further, it is unclear if replication timing changes are sufficient to induce changes in expression [176]. Nonetheless, changes in the replication timing of specific genes in cancer cells have been associated with their overexpression [11], possibly due to their repackaging as open chromatin [177]. It is possible that genes which are switched to earlier replication may exhibit higher expression levels, though further research is required to elucidate this point.
Second, replication timing can affect cancer development through its mutational impact. As established earlier, replication timing plays an important role in determining the mutational landscape of the genome. In fact, it has been shown that cell type specific replication timing can best explain the mutational landscape of different cancer tissues [127,178]. This suggests that replication timing may be a contributing factor in cancer progression as changes in replication timing of important oncogenes or tumor suppressor genes may lead to their further mutagenesis. Indeed, it has been suggested that changes in the replication timing of the RUNX1 locus, precede chromosomal breakage and translocations common in leukemia [10].
Third, particularly aberrant changes in the replication timing program can induce stress and thus propagate genomic instability. Unscheduled origin licensing and discoordination of replication forks can impede on fork progression and overload the system resulting in a lack of resources in the form of nucleotides or other factors [6]. A change in replication timing may also interfere with the proper coordination of replication and transcription resulting in increased collisions [41]. Delayed replication induced by cancer-related chromosomal rearrangements has also been associated with overall instability and increased acquisition of translocations [158].
4. Conclusions
Faithfulness of the DNA replication program is crucial to cell viability and its impairment can ultimately lead to the development of cancer. Both management and prevention of replication stress as well as maintenance of ordered replication timing are crucial to cell viability and the prevention of genomic instability. Further studies are required in order to comprehensively address the relationship between replication timing and replication stress. Insight into their interplay may shed light on the development of genomic instability during aberrant DNA replication.
Increased replication stress is a hallmark of cancer progression and can be particularly useful as both a biomarker and as a therapeutic target. Currently, biomarkers of stress rely mainly on the appearance of γH2AX. Discovery of more biomarkers, specifically those also involved in the ATR pathway, may be particularly effective as a diagnostic method for detecting levels of replication stress in early stages of cancer development. Further understanding of replication stress mechanisms and DDR pathways could aid in the discovery of additional therapeutic targets.
Despite many years of research, there are limited new findings highlighting the association between replication timing and cancer. As sequencing costs decrease and experimental methods improve we expect to gain new insights into replication timing and its connection to genomic instability. Replication timing data from cancer tissues themselves will not only improve our grasp of the impact of specific replication timing changes, but may also serve as an important step in the establishment of replication timing as a biomarker in cancer diagnostics [10]. Furthermore, time course studies of replication timing changes in cancer progression will advance our understanding of the specific causes and effects of such changes.
The limiting factor in such research is the availability of large amounts of dividing cancer cells. Recently, new methods have been developed which will aid in the study of replication timing in human cancers. For example, it was demonstrated that mouse xenografts retains the replication timing of the original human transformed cells in acute lymphoblastic leukemia (ALL) cancers [179]. An alternative approach would require measuring replication timing from few thousands of cells. This advancement would open the field for in vivo studies and will allow genome-wide study of the replication timing in cancer cells.
Recent studies [180] have expanded analyses of the replication program to include data on processes differentially affecting the leading and lagging strands. These studies remain to be expanded and will likely shed light on more mutational processes involved in replication, which contribute to genomic instability.
Acknowledgments
We would like to thank Sharon Eden (Hebrew University of Jerusalem, Israel) for fruitful discussion and critical reading of the manuscript. Work in the Itamar Simon group was supported by the Israel Science Foundation grants No. 567/10, 2555/16 and 184/16; the European Research Council Starting Grant #281306 and the Binational Science Foundation grant # 2015272.
Author Contributions
Britny Blumenfeld designed and drafted the manuscript; Itamar Simon and Micha Ben-Zimra discussed and revised the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| DNA | Deoxyribonucleic acid |
| S phase | Synthesis phase |
| ssDNA | Single stranded DNA |
| DSB | Double strand breaks |
| RPA | Replication protein A |
| ATR | Ataxia telangiectasia and Rad3-related protein |
| ROS | Reactive oxygen species |
| dNTP | Deoxynucleotide triphosphate |
| RNA | Ribonucleic acid |
| RNase H | Ribonuclease H |
| DDR | DNA damage response |
| Chk1 | Checkpoint kinase 1 |
| MRN | Mre11-Rad50-Nbs1 |
| Chk2 | Checkpoint kinase 2 |
| H2AX | H2A histone family member X |
| DDT | DNA damage tolerance |
| LOH | Loss of heterozygosity |
| CFS | Common fragile site |
| ERFS | Early replication fragile site |
| CNV | Copy number variations |
| PARP1 | Poly(ADP-ribose) polymerase 1 |
| GOF p53 | Gain of function p53 |
| CTR | Constant timing regions |
| TTR | Timing transition regions |
| MEF | Mouse embryonic fibroblasts |
| BrdU | Bromodeoxyuridine |
| SNP | Single nucleotide polymorphism |
| NHEJ | Non-homologous end-joining |
| RUNX1 | Runt-related transcription factor 1 |
| p53 | Tumor protein P53 |
| RB1 | Retinoblastoma 1 |
| Bcl2 | B-cell lymphoma 2 |
| Rif1 | Replication timing regulatory factor 1 |
| MCM | Minichromosome maintenance |
| ALL | Acute lymphoblastic leukemia |
References
- Tomasetti, C.; Li, L.; Vogelstein, B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 2017, 355, 1330–1334. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Vogelstein, B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, T.A. Evolving views of DNA replication (in)fidelity. Cold Spring Harb. Symp. Quant. Biol. 2009, 74, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Li, G.-M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Mutter-Rottmayer, E.; Zlatanou, A.; Vaziri, C.; Yang, Y. Mechanisms of post-replication DNA repair. Genes 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Farkash-Amar, S.; Simon, I. Genome-wide analysis of the replication program in mammals. Chromosome Res. 2010, 18, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Ryba, T.; Battaglia, D.; Chang, B.H.; Shirley, J.W.; Buckley, Q.; Pope, B.D.; Devidas, M.; Druker, B.J.; Gilbert, D.M. Abnormal developmental control of replication-timing domains in pediatric acute lymphoblastic leukemia. Genome Res. 2012, 22, 1833–1844. [Google Scholar] [CrossRef] [PubMed]
- Fritz, A.; Sinha, S.; Marella, N.; Berezney, R. Alterations in replication timing of cancer-related genes in malignant human breast cancer cells. J. Cell. Biochem. 2013, 114, 1074–1083. [Google Scholar] [CrossRef] [PubMed]
- Alvino, G.M.; Collingwood, D.; Murphy, J.M.; Delrow, J.; Brewer, B.J.; Raghuraman, M.K. Replication in Hydroxyurea: It is a Matter of Time. Mol. Cell. Biol. 2007, 27, 6396–6406. [Google Scholar] [CrossRef] [PubMed]
- Koren, A.; Soifer, I.; Barkai, N. MRC1-dependent scaling of the budding yeast DNA replication timing program. Genome Res. 2010, 20, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Heintz, N.H.; Dailey, L.; Held, P.; Heintz, N. Eukaryotic replication origins as promoters of bidirectional DNA synthesis. Trends Genet. TIG 1992, 8, 376–381. [Google Scholar] [CrossRef]
- Burhans, W.C.; Weinberger, M. DNA replication stress, genome instability and aging. Nucleic Acids Res. 2007, 35, 7545–7556. [Google Scholar] [CrossRef] [PubMed]
- Van, C.; Yan, S.; Michael, W.M.; Waga, S.; Cimprich, K.A. Continued primer synthesis at stalled replication forks contributes to checkpoint activation. J. Cell Biol. 2010, 189, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Byun, T.S.; Pacek, M.; Yee, M.; Walter, J.C.; Cimprich, K.A. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005, 19, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 2010, 11, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.-Q.; Zhang, K.; Wu, X.-C.; Mieczkowski, P.A.; Petes, T.D. Global analysis of genomic instability caused by DNA replication stress in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2016, 113, E8114–E8121. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A.; Lindsey-Boltz, L.A.; Ünsal-Kaçmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, S.M. DNA replication: Driving past four-stranded snags. Nature 2013, 497, 449–450. [Google Scholar] [CrossRef] [PubMed]
- McElhinny, S.A.N.; Kumar, D.; Clark, A.B.; Watt, D.L.; Watts, B.E.; Lundström, E.-B.; Johansson, E.; Chabes, A.; Kunkel, T.A. Genome instability due to ribonucleotide incorporation into DNA. Nat. Chem. Biol. 2010, 6, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Beck, H.; Nähse-Kumpf, V.; Larsen, M.S.Y.; O’Hanlon, K.A.; Patzke, S.; Holmberg, C.; Mejlvang, J.; Groth, A.; Nielsen, O.; Syljuåsen, R.G.; et al. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol. Cell. Biol. 2012, 32, 4226–4236. [Google Scholar] [CrossRef] [PubMed]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Mejlvang, J.; Feng, Y.; Alabert, C.; Neelsen, K.J.; Jasencakova, Z.; Zhao, X.; Lees, M.; Sandelin, A.; Pasero, P.; Lopes, M.; et al. New histone supply regulates replication fork speed and PCNA unloading. J. Cell Biol. 2014, 204, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Groth, A.; Corpet, A.; Cook, A.J.L.; Roche, D.; Bartek, J.; Lukas, J.; Almouzni, G. Regulation of replication fork progression through histone supply and demand. Science 2007, 318, 1928–1931. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Altmeyer, M.; Rask, M.-B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Nakato, R.; Katou, Y.; Shirahige, K.; Araki, H. Origin Association of Sld3, Sld7, and Cdc45 Proteins Is a Key Step for Determination of Origin-Firing Timing. Curr. Biol. 2011, 21, 2055–2063. [Google Scholar] [CrossRef] [PubMed]
- Macheret, M.; Halazonetis, T.D. DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. 2015, 10, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Olavarrieta, L.; Hernández, P.; Krimer, D.B.; Schvartzman, J.B. DNA knotting caused by head-on collision of transcription and replication. J. Mol. Biol. 2002, 322, 1–6. [Google Scholar] [CrossRef]
- Aguilera, A.; García-Muse, T. R loops: From transcription byproducts to threats to genome stability. Mol. Cell 2012, 46, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Tuduri, S.; Crabbé, L.; Conti, C.; Tourrière, H.; Holtgreve-Grez, H.; Jauch, A.; Pantesco, V.; De Vos, J.; Thomas, A.; Theillet, C.; et al. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 2009, 11, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, R.; Capra, T.; Gonzalez-Huici, V.; Fachinetti, D.; Cocito, A.; Natoli, G.; Katou, Y.; Mori, H.; Kurokawa, K.; Shirahige, K.; et al. Genome-organizing factors Top2 and Hmo1 prevent chromosome fragility at sites of S phase transcription. Cell 2009, 138, 870–884. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Shatalin, K.; Epshtein, V.; Gottesman, M.E.; Nudler, E. Linking RNA polymerase backtracking to genome instability in E. coli. Cell 2011, 146, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Hills, S.A.; Diffley, J.F.X. DNA replication and oncogene-induced replicative stress. Curr. Biol. 2014, 24, R435–R444. [Google Scholar] [CrossRef] [PubMed]
- Stiff, T.; Alagoz, M.; Alcantara, D.; Outwin, E.; Brunner, H.G.; Bongers, E.M.H.F.; O’Driscoll, M.; Jeggo, P.A. Deficiency in origin licensing proteins impairs cilia formation: Implications for the aetiology of Meier-Gorlin syndrome. PLoS Genet. 2013, 9, e1003360. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.L.; Orr-Weaver, T.L. Replication fork instability and the consequences of fork collisions from rereplication. Genes Dev. 2016, 30, 2241–2252. [Google Scholar] [CrossRef] [PubMed]
- Mantiero, D.; Mackenzie, A.; Donaldson, A.; Zegerman, P. Limiting replication initiation factors execute the temporal programme of origin firing in budding yeast. EMBO J. 2011, 30, 4805–4814. [Google Scholar] [CrossRef] [PubMed]
- Helmrich, A.; Ballarino, M.; Nudler, E.; Tora, L. Transcription-replication encounters, consequences and genomic instability. Nat. Struct. Mol. Biol. 2013, 20, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Diffley, J.F.X. Deregulated G1-cyclin expression induces genomic instability by preventing efficient pre-RC formation. Genes Dev. 2002, 16, 2639–2649. [Google Scholar] [CrossRef] [PubMed]
- Lengronne, A.; Schwob, E. The yeast CDK inhibitor Sic1 prevents genomic instability by promoting replication origin licensing in late G1. Mol. Cell 2002, 9, 1067–1078. [Google Scholar] [CrossRef]
- Kunnev, D.; Rusiniak, M.E.; Kudla, A.; Freeland, A.; Cady, G.K.; Pruitt, S.C. DNA damage response and tumorigenesis in Mcm2-deficient mice. Oncogene 2010, 29, 3630–3638. [Google Scholar] [CrossRef] [PubMed]
- Shreeram, S.; Sparks, A.; Lane, D.P.; Blow, J.J. Cell type-specific responses of human cells to inhibition of replication licensing. Oncogene 2002, 21, 6624–6632. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Q.; Jackson, D.A.; Blow, J.J. Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev. 2007, 21, 3331–3341. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, A.; Schwob, E.; Méndez, J. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc. Natl. Acad. Sci. USA 2008, 105, 8956–8961. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, D.; Blow, J.J. Dormant origins, the licensing checkpoint, and the response to replicative stresses. Cold Spring Harb. Perspect. Biol. 2012, 4, a012955. [Google Scholar] [CrossRef] [PubMed]
- Shima, N.; Alcaraz, A.; Liachko, I.; Buske, T.R.; Andrews, C.A.; Munroe, R.J.; Hartford, S.A.; Tye, B.K.; Schimenti, J.C. A viable allele of Mcm4 causes chromosome instability and mammary adenocarcinomas in mice. Nat. Genet. 2007, 39, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Rohban, S.; Campaner, S. Myc induced replicative stress response: How to cope with it and exploit it. Biochim. Biophys. Acta 2015, 1849, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Neiman, P.E.; Kimmel, R.; Icreverzi, A.; Elsaesser, K.; Bowers, S.-J.; Burnside, J.; Delrow, J. Genomic instability during Myc-induced lymphomagenesis in the bursa of Fabricius. Oncogene 2006, 25, 6325–6335. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.; Asawachaicharn, N.; Galloway, D.A.; Grandori, C. c-Myc accelerates S-phase and requires WRN to avoid replication stress. PLoS ONE 2009, 4, e5951. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.V.; Dominguez-Sola, D.; Wang, L.C.; Hyrien, O.; Gautier, J. Cdc45 is a critical effector of myc-dependent DNA replication stress. Cell Rep. 2013, 3, 1629–1639. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Sola, D.; Ying, C.Y.; Grandori, C.; Ruggiero, L.; Chen, B.; Li, M.; Galloway, D.A.; Gu, W.; Gautier, J.; Dalla-Favera, R. Non-transcriptional control of DNA replication by c-Myc. Nature 2007, 448, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Maya-Mendoza, A.; Ostrakova, J.; Kosar, M.; Hall, A.; Duskova, P.; Mistrik, M.; Merchut-Maya, J.M.; Hodny, Z.; Bartkova, J.; Christensen, C.; et al. Myc and Ras oncogenes engage different energy metabolism programs and evoke distinct patterns of oxidative and DNA replication stress. Mol. Oncol. 2015, 9, 601–616. [Google Scholar] [CrossRef] [PubMed]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Lindström, M.S.; Wiman, K.G. Myc and E2F1 induce p53 through p14ARF-independent mechanisms in human fibroblasts. Oncogene 2003, 22, 4993–5005. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.-V.F.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Davidson, I.F.; Li, A.; Blow, J.J. Deregulated replication licensing causes DNA fragmentation consistent with head-to-tail fork collision. Mol. Cell 2006, 24, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre’, M.; Nuciforo, P.G.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Miron, K.; Golan-Lev, T.; Dvir, R.; Ben-David, E.; Kerem, B. Oncogenes create a unique landscape of fragile sites. Nat. Commun. 2015, 6, 7094. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Horejsí, Z.; Koed, K.; Krämer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Matsumura, I.; Ezoe, S.; Satoh, Y.; Sakamaki, T.; Albanese, C.; Machii, T.; Pestell, R.G.; Kanakura, Y. E2F1 and c-Myc potentiate apoptosis through inhibition of NF-κB activity that facilitates MnSOD-mediated ROS elimination. Mol. Cell 2002, 9, 1017–1029. [Google Scholar] [CrossRef]
- Costantino, L.; Sotiriou, S.K.; Rantala, J.K.; Magin, S.; Mladenov, E.; Helleday, T.; Haber, J.E.; Iliakis, G.; Kallioniemi, O.P.; Halazonetis, T.D. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 2014, 343, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Ekholm-Reed, S.; Méndez, J.; Tedesco, D.; Zetterberg, A.; Stillman, B.; Reed, S.I. Deregulation of cyclin E in human cells interferes with prereplication complex assembly. J. Cell Biol. 2004, 165, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Mortusewicz, O.; Afzal, I.; Lorvellec, M.; García, P.; Helleday, T.; Petermann, E. Increased replication initiation and conflicts with transcription underlie Cyclin E-induced replication stress. Oncogene 2013, 32, 3744–3753. [Google Scholar] [CrossRef] [PubMed]
- Ray, D.; Terao, Y.; Fuhrken, P.G.; Ma, Z.-Q.; DeMayo, F.J.; Christov, K.; Heerema, N.A.; Franks, R.; Tsai, S.Y.; Papoutsakis, E.T.; et al. Deregulated CDC25A expression promotes mammary tumorigenesis with genomic instability. Cancer Res. 2007, 67, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Dungrawala, H.; Rose, K.L.; Bhat, K.P.; Mohni, K.N.; Glick, G.G.; Couch, F.B.; Cortez, D. The replication checkpoint prevents two types of fork collapse without regulating replisome stability. Mol. Cell 2015, 59, 998–1010. [Google Scholar] [CrossRef] [PubMed]
- Couch, F.B.; Bansbach, C.E.; Driscoll, R.; Luzwick, J.W.; Glick, G.G.; Bétous, R.; Carroll, C.M.; Jung, S.Y.; Qin, J.; Cimprich, K.A.; et al. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 2013, 27, 1610–1623. [Google Scholar] [CrossRef] [PubMed]
- Blow, J.J.; Ge, X.Q.; Jackson, D.A. How dormant origins promote complete genome replication. Trends Biochem. Sci. 2011, 36, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, T.; Luebben, S.W.; Yamaguchi, S.; Ilves, I.; Matise, I.; Buske, T.; Botchan, M.R.; Shima, N. Stalled fork rescue via dormant replication origins in unchallenged S phase promotes proper chromosome segregation and tumor suppression. Mol. Cell 2011, 41, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Washington, M.T. Translesion synthesis: Insights into the selection and switching of DNA polymerases. Genes 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Q.; Blow, J.J. Chk1 inhibits replication factory activation but allows dormant origin firing in existing factories. J. Cell Biol. 2010, 191, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. Interplay of replication checkpoints and repair proteins at stalled replication forks. DNA Repair 2007, 6, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Murga, M.; Gutierrez-Martinez, P.; Soria, R.; Fernandez-Capetillo, O. ATR signaling can drive cells into senescence in the absence of DNA breaks. Genes Dev. 2008, 22, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, C.; Lukas, J. Checking on DNA damage in S phase. Nat. Rev. Mol. Cell Biol. 2004, 5, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Lomonosov, M.; Anand, S.; Sangrithi, M.; Davies, R.; Venkitaraman, A.R. Stabilization of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Genes Dev. 2003, 17, 3017–3022. [Google Scholar] [CrossRef] [PubMed]
- Mazouzi, A.; Velimezi, G.; Loizou, J.I. DNA replication stress: Causes, resolution and disease. Exp. Cell Res. 2014, 329, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Zou, L. DNA damage sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef] [PubMed]
- Ammazzalorso, F.; Pirzio, L.M.; Bignami, M.; Franchitto, A.; Pichierri, P. ATR and ATM differently regulate WRN to prevent DSBs at stalled replication forks and promote replication fork recovery. EMBO J. 2010, 29, 3156–3169. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Kannouche, P.L.; Lehmann, A.R. Ubiquitination of PCNA and the polymerase switch in human cells. Cell Cycle Georget. Tex. 2004, 3, 1011–1013. [Google Scholar] [CrossRef]
- Sale, J.E. Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012708. [Google Scholar] [CrossRef] [PubMed]
- Waters, L.S.; Minesinger, B.K.; Wiltrout, M.E.; D’Souza, S.; Woodruff, R.V.; Walker, G.C. Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol. Mol. Biol. Rev. 2009, 73, 134–154. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D. Ubiquitin family modifications and template switching. FEBS Lett. 2011, 585, 2810–2817. [Google Scholar] [CrossRef] [PubMed]
- Arlt, M.F.; Mulle, J.G.; Schaibley, V.M.; Ragland, R.L.; Durkin, S.G.; Warren, S.T.; Glover, T.W. Replication stress induces genome-wide copy number changes in human cells that resemble polymorphic and pathogenic variants. Am. J. Hum. Genet. 2009, 84, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Arlt, M.F.; Wilson, T.E.; Glover, T.W. Replication stress and mechanisms of CNV formation. Curr. Opin. Genet. Dev. 2012, 22, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, T.; Magdalou, I.; Barascu, A.; Técher, H.; Debatisse, M.; Lopez, B.S. Spontaneous slow replication fork progression elicits mitosis alterations in homologous recombination-deficient mammalian cells. Proc. Natl. Acad. Sci. USA 2014, 111, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.; van der Burg, M.; Szuhai, K.; Kops, G.J.P.L.; Medema, R.H. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 2011, 333, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Neelsen, K.J.; Zanini, I.M.Y.; Herrador, R.; Lopes, M. Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. J. Cell Biol. 2013, 200, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.-C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Ozeri-Galai, E.; Tur-Sinai, M.; Bester, A.C.; Kerem, B. Interplay between genetic and epigenetic factors governs common fragile site instability in cancer. Cell. Mol. Life Sci. 2014, 71, 4495–4506. [Google Scholar] [CrossRef] [PubMed]
- Admire, A.; Shanks, L.; Danzl, N.; Wang, M.; Weier, U.; Stevens, W.; Hunt, E.; Weinert, T. Cycles of chromosome instability are associated with a fragile site and are increased by defects in DNA replication and checkpoint controls in yeast. Genes Dev. 2006, 20, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Arlt, M.F.; Durkin, S.G.; Ragland, R.L.; Glover, T.W. Common fragile sites as targets for chromosome rearrangements. DNA Repair 2006, 5, 1126–1135. [Google Scholar] [CrossRef] [PubMed]
- Barlow, J.H.; Faryabi, R.B.; Callén, E.; Wong, N.; Malhowski, A.; Chen, H.T.; Gutierrez-Cruz, G.; Sun, H.-W.; McKinnon, P.; Wright, G.; et al. Identification of early replicating fragile sites that contribute to genome instability. Cell 2013, 152, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Mortusewicz, O.; Herr, P.; Helleday, T. Early replication fragile sites: Where replication-transcription collisions cause genetic instability. EMBO J. 2013, 32, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Le Beau, M.M.; Rassool, F.V.; Neilly, M.E.; Espinosa, R.; Glover, T.W.; Smith, D.I.; McKeithan, T.W. Replication of a common fragile site, FRA3B, occurs late in S phase and is delayed further upon induction: Implications for the mechanism of fragile site induction. Hum. Mol. Genet. 1998, 7, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.G.; Vassiliou, L.-V.F.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Tsantoulis, P.K.; Kotsinas, A.; Sfikakis, P.P.; Evangelou, K.; Sideridou, M.; Levy, B.; Mo, L.; Kittas, C.; Wu, X.-R.; Papavassiliou, A.G.; et al. Oncogene-induced replication stress preferentially targets common fragile sites in preneoplastic lesions. A genome-wide study. Oncogene 2008, 27, 3256–3264. [Google Scholar] [CrossRef] [PubMed]
- Burrow, A.A.; Williams, L.E.; Pierce, L.C.T.; Wang, Y.-H. Over half of breakpoints in gene pairs involved in cancer-specific recurrent translocations are mapped to human chromosomal fragile sites. BMC Genom. 2009, 10, 59. [Google Scholar] [CrossRef] [PubMed]
- Courtois-Cox, S.; Jones, S.L.; Cichowski, K. Many roads lead to oncogene-induced senescence. Oncogene 2008, 27, 2801–2809. [Google Scholar] [CrossRef] [PubMed]
- Sarkisian, C.J.; Keister, B.A.; Stairs, D.B.; Boxer, R.B.; Moody, S.E.; Chodosh, L.A. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat. Cell Biol. 2007, 9, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Berti, M.; Vindigni, A. Replication stress: Getting back on track. Nat. Struct. Mol. Biol. 2016, 23, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.-J.; Peng, G. Cellular responses to replication stress: Implications in cancer biology and therapy. DNA Repair 2017, 49, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Dai, Q.; Park, D.; Deng, X. Targeting DNA replication stress for cancer therapy. Genes 2016, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Klusmann, I.; Rodewald, S.; Müller, L.; Friedrich, M.; Wienken, M.; Li, Y.; Schulz-Heddergott, R.; Dobbelstein, M. p53 Activity results in DNA replication fork processivity. Cell Rep. 2016, 17, 1845–1857. [Google Scholar] [CrossRef] [PubMed]
- Rhind, N.; Gilbert, D.M. DNA replication timing. Cold Spring Harb. Perspect. Biol. 2013, 5, a010132. [Google Scholar] [CrossRef] [PubMed]
- Hiratani, I.; Ryba, T.; Itoh, M.; Yokochi, T.; Schwaiger, M.; Chang, C.-W.; Lyou, Y.; Townes, T.M.; Schübeler, D.; Gilbert, D.M. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 2008, 6, e245. [Google Scholar] [CrossRef] [PubMed]
- Yehuda, Y.; Blumenfeld, B.; Lehmann, D.; Simon, I. Genome-wide determination of mammalian replication timing by DNA content measurement. J. Vis. Exp. JoVE 2017. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Mulia, J.C.; Buckley, Q.; Sasaki, T.; Zimmerman, J.; Didier, R.A.; Nazor, K.; Loring, J.F.; Lian, Z.; Weissman, S.; Robins, A.J.; et al. Dynamic changes in replication timing and gene expression during lineage specification of human pluripotent stem cells. Genome Res. 2015, 25, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Hiratani, I.; Ryba, T.; Itoh, M.; Rathjen, J.; Kulik, M.; Papp, B.; Fussner, E.; Bazett-Jones, D.P.; Plath, K.; Dalton, S.; et al. Genome-wide dynamics of replication timing revealed by in vitro models of mouse embryogenesis. Genome Res. 2010, 20, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Dileep, V.; Rivera-Mulia, J.C.; Sima, J.; Gilbert, D.M. Large-Scale Chromatin Structure-Function Relationships during the cell cycle and development: Insights from replication timing. Cold Spring Harb. Symp. Quant. Biol. 2015, 80, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Hiratani, I.; Takebayashi, S.; Lu, J.; Gilbert, D.M. Replication timing and transcriptional control: Beyond cause and effect—Part II. Curr. Opin. Genet. Dev. 2009, 19, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Ryba, T.; Battaglia, D.; Pope, B.D.; Hiratani, I.; Gilbert, D.M. Genome-scale analysis of replication timing: From bench to bioinformatics. Nat. Protoc. 2011, 6, 870–895. [Google Scholar] [CrossRef] [PubMed]
- Sima, J.; Gilbert, D.M. Complex correlations: Replication timing and mutational landscapes during cancer and genome evolution. Curr. Opin. Genet. Dev. 2014, 25, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Stamatoyannopoulos, J.A.; Adzhubei, I.; Thurman, R.E.; Kryukov, G.V.; Mirkin, S.M.; Sunyaev, S.R. Human mutation rate associated with DNA replication timing. Nat. Genet. 2009, 41, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-L.; Rappailles, A.; Duquenne, L.; Huvet, M.; Guilbaud, G.; Farinelli, L.; Audit, B.; d’Aubenton-Carafa, Y.; Arneodo, A.; Hyrien, O.; et al. Impact of replication timing on non-CpG and CpG substitution rates in mammalian genomes. Genome Res. 2010, 20, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Koren, A.; Polak, P.; Nemesh, J.; Michaelson, J.J.; Sebat, J.; Sunyaev, S.R.; McCarroll, S.A. Differential relationship of DNA replication timing to different forms of human mutation and variation. Am. J. Hum. Genet. 2012, 91, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Fujiyama, A.; Ichiba, Y.; Hattori, M.; Yada, T.; Sakaki, Y.; Ikemura, T. Chromosome-wide assessment of replication timing for human chromosomes 11q and 21q: Disease-related genes in timing-switch regions. Hum. Mol. Genet. 2002, 11, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Pink, C.J.; Hurst, L.D. Timing of replication is a determinant of neutral substitution rates but does not explain slow Y chromosome evolution in rodents. Mol. Biol. Evol. 2010, 27, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.C.; Pink, C.J.; Hurst, L.D. Late-replicating domains have higher divergence and diversity in Drosophila melanogaster. Mol. Biol. Evol. 2012, 29, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.H.; Li, W.-H. DNA replication timing and selection shape the landscape of nucleotide variation in cancer genomes. Nat. Commun. 2012, 3, 1004. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, A.; Furuta, M.; Totoki, Y.; Tsunoda, T.; Kato, M.; Shiraishi, Y.; Tanaka, H.; Taniguchi, H.; Kawakami, Y.; Ueno, M.; et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet. 2016, 48, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Lehner, B. Differential DNA mismatch repair underlies mutation rate variation across the human genome. Nature 2015, 521, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; De, S.; Michor, F. DNA replication timing and higher-order nuclear organization determine single-nucleotide substitution patterns in cancer genomes. Nat. Commun. 2013, 4, 1502. [Google Scholar] [CrossRef] [PubMed]
- Jia, P.; Jin, H.; Meador, C.B.; Xia, J.; Ohashi, K.; Liu, L.; Pirazzoli, V.; Dahlman, K.B.; Politi, K.; Michor, F.; et al. Next-generation sequencing of paired tyrosine kinase inhibitor-sensitive and -resistant EGFR mutant lung cancer cell lines identifies spectrum of DNA changes associated with drug resistance. Genome Res. 2013, 23, 1434–1445. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Kucab, J.E.; Morganella, S.; Glodzik, D.; Alexandrov, L.B.; Arlt, V.M.; Weninger, A.; Hollstein, M.; Stratton, M.R.; Phillips, D.H. The genome as a record of environmental exposure. Mutagenesis 2015, 30, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Lang, G.I.; Murray, A.W. Mutation rates across budding yeast chromosome VI are correlated with replication timing. Genome Biol. Evol. 2011, 3, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Agier, N.; Fischer, G. The mutational profile of the yeast genome is shaped by replication. Mol. Biol. Evol. 2012, 29, 905–913. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Michor, F. DNA replication timing and long-range DNA interactions predict mutational landscapes of cancer genomes. Nat. Biotechnol. 2011, 29, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Ikemura, T.; Sugimura, H. Amplicons on human chromosome 11q are located in the early/late-switch regions of replication timing. Genomics 2004, 84, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Li, H.; Hu, M.; Sasaki, T.; Baccei, A.; Gilbert, D.M.; Liu, J.S.; Collins, J.J.; Lerou, P.H. The distribution of genomic variations in human iPSCs is related to replication-timing reorganization during reprogramming. Cell Rep. 2014, 7, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Cardoso-Moreira, M.M.; Long, M. Mutational bias shaping fly copy number variation: Implications for genome evolution. Trends Genet. TIG 2010, 26, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Cardoso-Moreira, M.; Emerson, J.J.; Clark, A.G.; Long, M. Drosophila duplication hotspots are associated with late-replicating regions of the genome. PLoS Genet. 2011, 7, e1002340. [Google Scholar] [CrossRef] [PubMed]
- Drier, Y.; Lawrence, M.S.; Carter, S.L.; Stewart, C.; Gabriel, S.B.; Lander, E.S.; Meyerson, M.; Beroukhim, R.; Getz, G. Somatic rearrangements across cancer reveal classes of samples with distinct patterns of DNA breakage and rearrangement-induced hypermutability. Genome Res. 2013, 23, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Janoueix-Lerosey, I.; Hupé, P.; Maciorowski, Z.; La Rosa, P.; Schleiermacher, G.; Pierron, G.; Liva, S.; Barillot, E.; Delattre, O. Preferential occurrence of chromosome breakpoints within early replicating regions in neuroblastoma. Cell Cycle Georget. Tex. 2005, 4, 1842–1846. [Google Scholar] [CrossRef] [PubMed]
- Shugay, M.; Ortiz de Mendíbil, I.; Vizmanos, J.L.; Novo, F.J. Genomic hallmarks of genes involved in chromosomal translocations in hematological cancer. PLoS Comput. Biol. 2012, 8, e1002797. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, E.; Farkash-Amar, S.; Polten, A.; Yakhini, Z.; Tanay, A.; Simon, I. Comparative analysis of DNA replication timing reveals conserved large-scale chromosomal architecture. PLoS Genet. 2010, 6, e1001011. [Google Scholar] [CrossRef] [PubMed]
- Letessier, A.; Millot, G.A.; Koundrioukoff, S.; Lachagès, A.-M.; Vogt, N.; Hansen, R.S.; Malfoy, B.; Brison, O.; Debatisse, M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 2011, 470, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Palakodeti, A.; Han, Y.; Jiang, Y.; Le Beau, M.M. The role of late/slow replication of the FRA16D in common fragile site induction. Genes Chromosomes Cancer 2004, 39, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Darling, J.; Zhang, J.S.; Huang, H.; Liu, W.; Smith, D.I. Allele-specific late replication and fragility of the most active common fragile site, FRA3B. Hum. Mol. Genet. 1999, 8, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.S.; De, S. Loss of heterozygosity preferentially occurs in early replicating regions in cancer genomes. Nucleic Acids Res. 2013, 41, 7615–7624. [Google Scholar] [CrossRef] [PubMed]
- Morganella, S.; Alexandrov, L.B.; Glodzik, D.; Zou, X.; Davies, H.; Staaf, J.; Sieuwerts, A.M.; Brinkman, A.B.; Martin, S.; Ramakrishna, M.; et al. The topography of mutational processes in breast cancer genomes. Nat. Commun. 2016, 7, 11383. [Google Scholar] [CrossRef] [PubMed]
- Hellman, A.; Rahat, A.; Scherer, S.W.; Darvasi, A.; Tsui, L.C.; Kerem, B. Replication delay along FRA7H, a common fragile site on human chromosome 7, leads to chromosomal instability. Mol. Cell. Biol. 2000, 20, 4420–4427. [Google Scholar] [CrossRef] [PubMed]
- Hellman, A.; Zlotorynski, E.; Scherer, S.W.; Cheung, J.; Vincent, J.B.; Smith, D.I.; Trakhtenbrot, L.; Kerem, B. A role for common fragile site induction in amplification of human oncogenes. Cancer Cell 2002, 1, 89–97. [Google Scholar] [CrossRef]
- Pelliccia, F.; Bosco, N.; Curatolo, A.; Rocchi, A. Replication timing of two human common fragile sites: FRA1H and FRA2G. Cytogenet. Genome Res. 2008, 121, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Engreitz, J.M.; Agarwala, V.; Mirny, L.A. Three-dimensional genome architecture influences partner selection for chromosomal translocations in human disease. PLoS ONE 2012, 7, e44196. [Google Scholar] [CrossRef] [PubMed]
- Pope, B.D.; Ryba, T.; Dileep, V.; Yue, F.; Wu, W.; Denas, O.; Vera, D.L.; Wang, Y.; Hansen, R.S.; Canfield, T.K.; et al. Topologically associating domains are stable units of replication-timing regulation. Nature 2014, 515, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Farkash-Amar, S.; David, Y.; Polten, A.; Hezroni, H.; Eldar, Y.C.; Meshorer, E.; Yakhini, Z.; Simon, I. Systematic determination of replication activity type highlights interconnections between replication, chromatin structure and nuclear localization. PLoS ONE 2012, 7, e48986. [Google Scholar] [CrossRef] [PubMed]
- Ryba, T.; Hiratani, I.; Lu, J.; Itoh, M.; Kulik, M.; Zhang, J.; Schulz, T.C.; Robins, A.J.; Dalton, S.; Gilbert, D.M. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res. 2010, 20, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Guilbaud, G.; Rappailles, A.; Baker, A.; Chen, C.-L.; Arneodo, A.; Goldar, A.; d’Aubenton-Carafa, Y.; Thermes, C.; Audit, B.; Hyrien, O. Evidence for sequential and increasing activation of replication origins along replication timing gradients in the human genome. PLoS Comput. Biol. 2011, 7, e1002322. [Google Scholar] [CrossRef] [PubMed]
- Donley, N.; Thayer, M.J. DNA replication timing, genome stability and cancer: Late and/or delayed DNA replication timing is associated with increased genomic instability. Semin. Cancer Biol. 2013, 23, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Kenigsberg, E.; Yehuda, Y.; Marjavaara, L.; Keszthelyi, A.; Chabes, A.; Tanay, A.; Simon, I. The mutation spectrum in genomic late replication domains shapes mammalian GC content. Nucleic Acids Res. 2016, 44, 4222–4232. [Google Scholar] [CrossRef] [PubMed]
- Murga, M.; Jaco, I.; Fan, Y.; Soria, R.; Martinez-Pastor, B.; Cuadrado, M.; Yang, S.-M.; Blasco, M.A.; Skoultchi, A.I.; Fernandez-Capetillo, O. Global chromatin compaction limits the strength of the DNA damage response. J. Cell Biol. 2007, 178, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.; Plug, A.; Thayer, M. Delayed replication timing leads to delayed mitotic chromosome condensation and chromosomal instability of chromosome translocations. Proc. Natl. Acad. Sci. USA 2001, 98, 13300–13305. [Google Scholar] [CrossRef] [PubMed]
- Reish, O.; Orlovski, A.; Mashevitz, M.; Sher, C.; Libman, V.; Rosenblat, M.; Avivi, L. Modified allelic replication in lymphocytes of patients with neurofibromatosis type 1. Cancer Genet. Cytogenet. 2003, 143, 133–139. [Google Scholar] [CrossRef]
- Grinberg-Rashi, H.; Cytron, S.; Gelman-Kohan, Z.; Litmanovitch, T.; Avivi, L. Replication timing aberrations and aneuploidy in peripheral blood lymphocytes of breast cancer patients. Neoplasia N. Y. 2010, 12, 668–674. [Google Scholar] [CrossRef]
- Korenstein-Ilan, A.; Amiel, A.; Lalezari, S.; Lishner, M.; Avivi, L. Allele-specific replication associated with aneuploidy in blood cells of patients with hematologic malignancies. Cancer Genet. Cytogenet. 2002, 139, 97–103. [Google Scholar] [CrossRef]
- Dotan, Z.A.; Dotan, A.; Litmanovitch, T.; Ravia, Y.; Oniashvili, N.; Leibovitch, I.; Ramon, J.; Avivi, L. Modification in the inherent mode of allelic replication in lymphocytes of patients suffering from renal cell carcinoma: A novel genetic alteration associated with malignancy. Genes Chromosomes Cancer 2000, 27, 270–277. [Google Scholar] [CrossRef]
- Sun, Y.; Wyatt, R.T.; Bigley, A.; Krontiris, T.G. Expression and replication timing patterns of wildtype and translocated BCL2 genes. Genomics 2001, 73, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Gispan, A.; Carmi, M.; Barkai, N. Model-based analysis of DNA replication profiles: Predicting replication fork velocity and initiation rate by profiling free-cycling cells. Genome Res. 2017, 27, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Lau, E.; Tsuji, T.; Guo, L.; Lu, S.-H.; Jiang, W. The role of pre-replicative complex (pre-RC) components in oncogenesis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2007, 21, 3786–3794. [Google Scholar] [CrossRef] [PubMed]
- Ostrow, A.Z.; Kalhor, R.; Gan, Y.; Villwock, S.K.; Linke, C.; Barberis, M.; Chen, L.; Aparicio, O.M. Conserved forkhead dimerization motif controls DNA replication timing and spatial organization of chromosomes in S. cerevisiae. Proc. Natl. Acad. Sci. USA 2017, 114, E2411–E2419. [Google Scholar] [CrossRef] [PubMed]
- Mattarocci, S.; Shyian, M.; Lemmens, L.; Damay, P.; Altintas, D.M.; Shi, T.; Bartholomew, C.R.; Thomä, N.H.; Hardy, C.F.J.; Shore, D. Rif1 controls DNA replication timing in yeast through the PP1 phosphatase Glc7. Cell Rep. 2014, 7, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Cornacchia, D.; Dileep, V.; Quivy, J.-P.; Foti, R.; Tili, F.; Santarella-Mellwig, R.; Antony, C.; Almouzni, G.; Gilbert, D.M.; Buonomo, S.B.C. Mouse Rif1 is a key regulator of the replication-timing programme in mammalian cells. EMBO J. 2012, 31, 3678–3690. [Google Scholar] [CrossRef] [PubMed]
- Foti, R.; Gnan, S.; Cornacchia, D.; Dileep, V.; Bulut-Karslioglu, A.; Diehl, S.; Buness, A.; Klein, F.A.; Huber, W.; Johnstone, E.; et al. Nuclear architecture organized by Rif1 underpins the replication-timing program. Mol. Cell 2016, 61, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Ishii, A.; Kanoh, Y.; Oda, M.; Nishito, Y.; Masai, H. Rif1 regulates the replication timing domains on the human genome. EMBO J. 2012, 31, 3667–3677. [Google Scholar] [CrossRef] [PubMed]
- Das, S.P.; Borrman, T.; Liu, V.W.T.; Yang, S.C.-H.; Bechhoefer, J.; Rhind, N. Replication timing is regulated by the number of MCMs loaded at origins. Genome Res. 2015, 25, 1886–1892. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Vidal, A.; Guitton-Sert, L.; Cadoret, J.-C.; Drac, M.; Schwob, E.; Baldacci, G.; Cazaux, C.; Hoffmann, J.-S. A role for DNA polymerase θ in the timing of DNA replication. Nat. Commun. 2014, 5, 4285. [Google Scholar] [CrossRef] [PubMed]
- Casas-Delucchi, C.S.; van Bemmel, J.G.; Haase, S.; Herce, H.D.; Nowak, D.; Meilinger, D.; Stear, J.H.; Leonhardt, H.; Cardoso, M.C. Histone hypoacetylation is required to maintain late replication timing of constitutive heterochromatin. Nucleic Acids Res. 2012, 40, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Goren, A.; Tabib, A.; Hecht, M.; Cedar, H. DNA replication timing of the human β-globin domain is controlled by histone modification at the origin. Genes Dev. 2008, 22, 1319–1324. [Google Scholar] [CrossRef] [PubMed]
- Farkash-Amar, S.; Lipson, D.; Polten, A.; Goren, A.; Helmstetter, C.; Yakhini, Z.; Simon, I. Global organization of replication time zones of the mouse genome. Genome Res. 2008, 18, 1562–1570. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Mulia, J.C.; Gilbert, D.M. Replication timing and transcriptional control: Beyond cause and effect-part III. Curr. Opin. Cell Biol. 2016, 40, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xu, F.; Hashimshony, T.; Keshet, I.; Cedar, H. Establishment of transcriptional competence in early and late S phase. Nature 2002, 420, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Polak, P.; Karlić, R.; Koren, A.; Thurman, R.; Sandstrom, R.; Lawrence, M.S.; Reynolds, A.; Rynes, E.; Vlahoviček, K.; Stamatoyannopoulos, J.A.; et al. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature 2015, 518, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Rivera-Mulia, J.C.; Vera, D.; Zimmerman, J.; Das, S.; Padget, M.; Nakamichi, N.; Chang, B.H.; Tyner, J.; Druker, B.J.; et al. Stability of patient-specific features of altered DNA replication timing in xenografts of primary human acute lymphoblastic leukemia. Exp. Hematol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Haradhvala, N.J.; Polak, P.; Stojanov, P.; Covington, K.R.; Shinbrot, E.; Hess, J.M.; Rheinbay, E.; Kim, J.; Maruvka, Y.E.; Braunstein, L.Z.; et al. Mutational strand asymmetries in cancer genomes reveal mechanisms of DNA damage and repair. Cell 2016, 164, 538–549. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Representative replication timing profiles. (a) An example of a replication timing map of mouse embryonic fibroblasts (MEF) [111] for a 2 Mb-sized region labeled by region type: TTR (timing transition region) or CTR (constant timing region); (b) The same replication timing map with annotation of the regions prone to various types of mutations.
Figure 1.
Representative replication timing profiles. (a) An example of a replication timing map of mouse embryonic fibroblasts (MEF) [111] for a 2 Mb-sized region labeled by region type: TTR (timing transition region) or CTR (constant timing region); (b) The same replication timing map with annotation of the regions prone to various types of mutations.
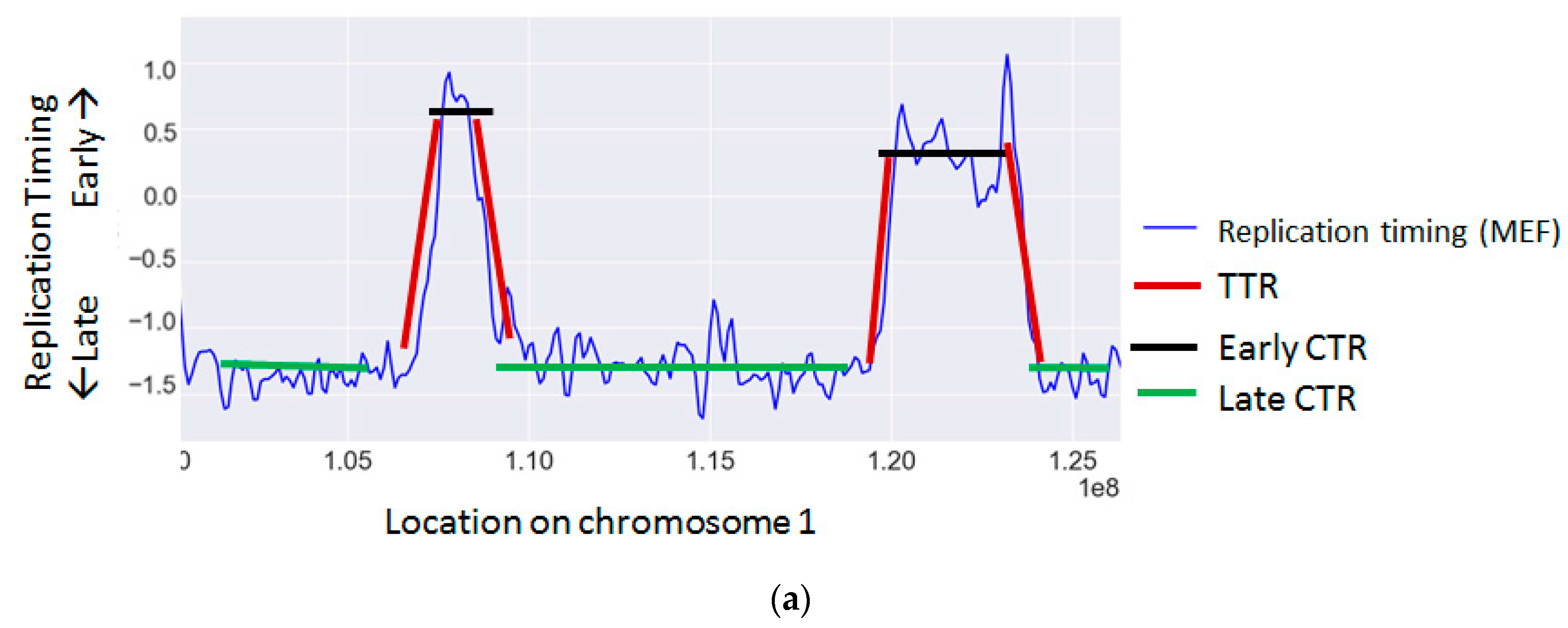
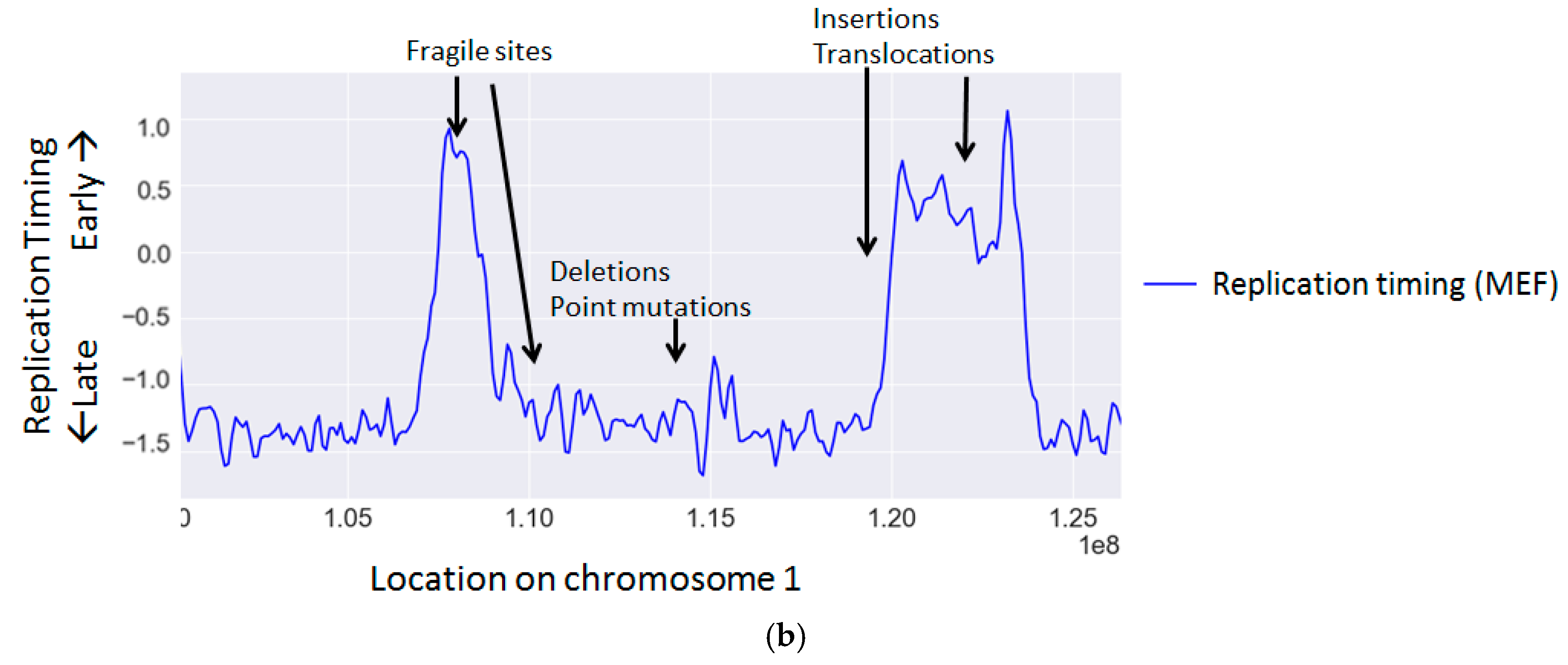

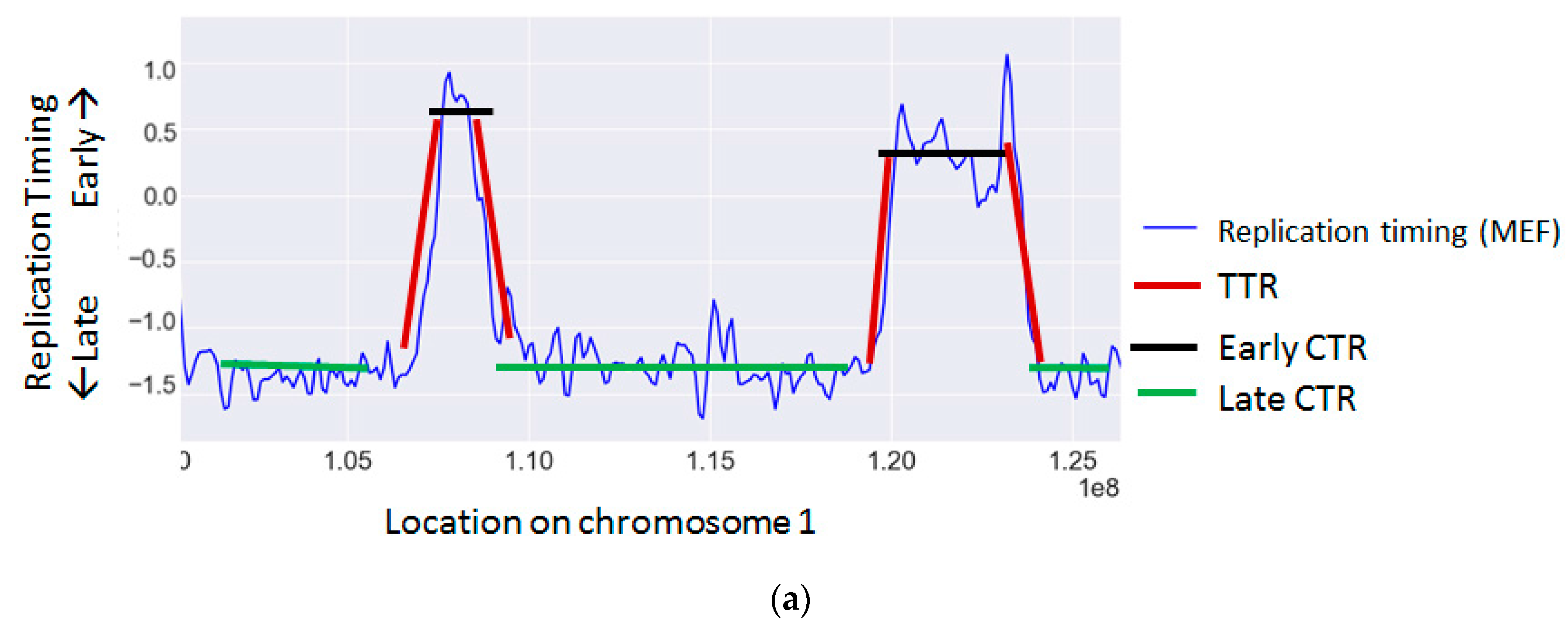{kind=link}
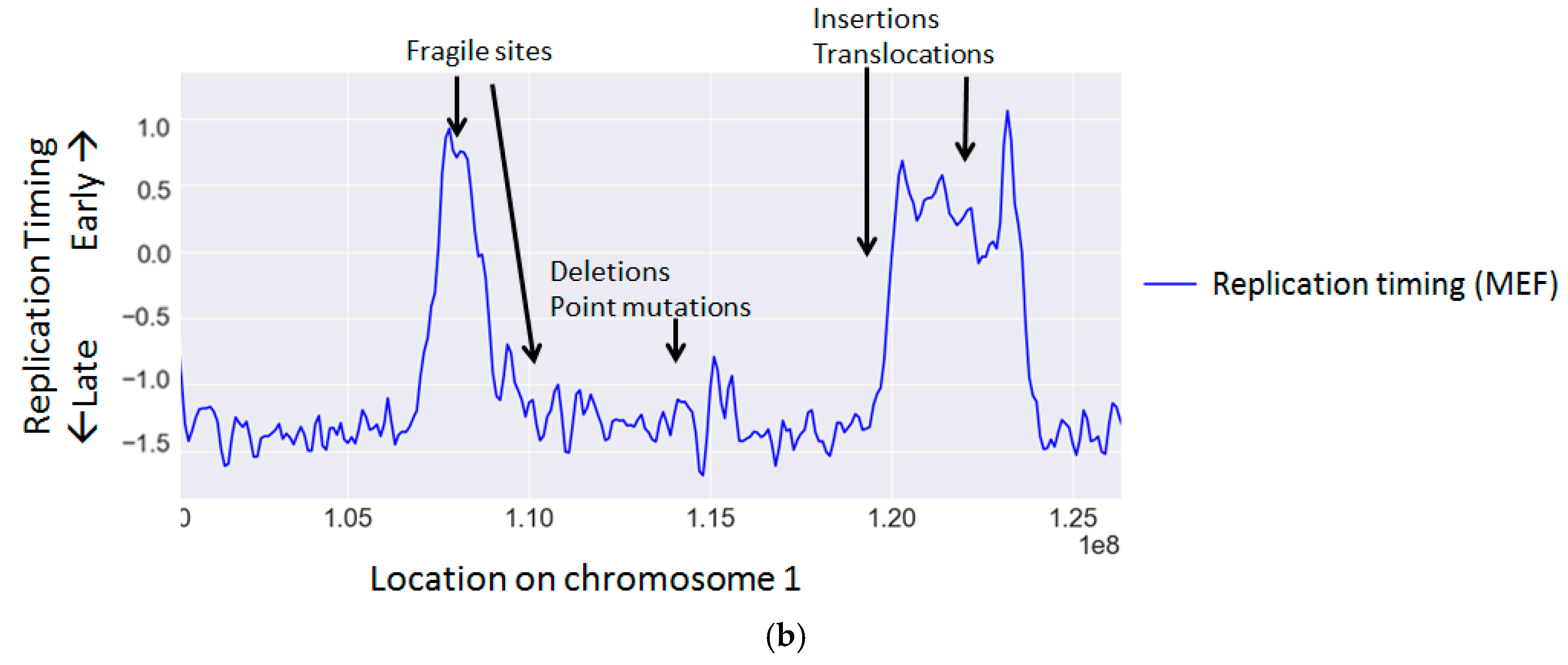{kind=link}
Table 1.
Oncogenes involved in replication stress.
| Gene | Mechanisms | References |
|---|---|---|
| c-myc | Accelerated S phase, increased origin activity, ROS, transcriptional interference | [51,52,53,54,55,56,57,58] |
| Cdt1 | Re-replication | [59] |
| H-RasV12 | Oxidative stress, hyperreplication | [55,58,60,61] |
| E2F1 | Deregulated replication , Oxidative stress | [57,62,63] |
| MOS | Unknown | [58] |
| Cdc6 | Unknown | [58] |
| Cyclin E | Deregulated replication, deficient licensing, transcriptional interference, origin over usage, nucleotide depletion | [26,58,61,62,64,65,66] |
| Cdc25A | Deregulated replication | [62,67] |
| HPV-16 E6/E7 | Nucleotide depletion | [26] |
c-myc: MYC proto-oncogene; Cdt1: Chromatin Licensing And DNA Replication Factor 1; H-RasV12: HRas proto-oncogene mutant (constitutively active); E2F1: E2F Transcription Factor 1; MOS: MOS Proto-Oncogene, Serine/Threonine Kinase; Cdc6: cell division cycle 6; Cdc25A: Cell Division Cycle 25A; HPV-16 E6/E7: Human papillomavirus 16 E6/E7 ; ROS: Reactive oxygen species.
Table 2.
Studies linking replication timing to mutations.
| Mutation type | Measurement | Higher in | References |
|---|---|---|---|
| Germline point mutations | Human SNP | Late and TTR | [118,119,120,121] |
| Mouse SNP | Late | [119] | |
| Mouse–rat divergence | Late | [119,122] | |
| Human–chimp divergence | Late | [118,119] | |
| Drosophila divergence | Late | [123] | |
| Somatic point mutations | Human cancer | Late | [124,125,126,127,128,129,130] |
| Yeast point mutations | Yeast URA3 gene | Late | [131,132] |
| Insertions | Human cancer | Early and TTR | [126,133,134] |
| Human iPSC | Early | [135] | |
| Fly | Late | [136,137] | |
| Translocations | Human cancer | Early (and late in [138]) | [126,133,138,139,140] |
| Mammalian divergence | TTR/early | [134,141] | |
| Deletions | Human cancer | Late | [126] |
| Human iPSC | Late | [135] | |
| Fly | Early | [136] | |
| Fragile sites | Cancer | Early or late | [96,99,142,143,144] |
| LOH | Cancer | Early | [145] |
SNP: single nucleotide polymorphism; TTR: timing transition regions; URA3: yeast gene encoding orotidine-5′-phosphate decarboxylase; iPSC: induced pluripotent stem cells; LOH: loss of heterozygosity.
Table 3.
Studies linking replication timing changes to cancer.
| Replication Timing Change | Genes | Cancer Type | Reference |
|---|---|---|---|
| Global replication timing changes | Global changes | Bone marrow from ALL patients | [10] |
| Asynchronous replication | HER-2, AML1, RB1, c-myc, p53 | Peripheral blood lymphocyte of cancer patients, MCF10CA1a | [11,159,160,161,162] |
| Replication timing changes of cancer-related genes | p53, ATM, c-myc, RAD51, PTEN, translocated Bcl2 | MCF10CA1a, SU-DHL-6, Jurkat | [11,163] |
HER2: human epidermal growth factor receptor 2; AML1: acute myeloid leukemia 1 protein; RB1: Retinoblastoma 1; c-myc: MYC proto-oncogene; ATM: ataxia-telangiectasia mutated; Bcl2: B-cell lymphoma 2; ALL: Acute lymphoblastic leukemia; MCF10CA1a: malignant human breast cell line; SU-DHL-6: Stanford University-Diffuse Histiocytic Lymphoma-6 cell line.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Blumenfeld, B.; Ben-Zimra, M.; Simon, I. Perturbations in the Replication Program Contribute to Genomic Instability in Cancer. Int. J. Mol. Sci. 2017, 18, 1138. https://doi.org/10.3390/ijms18061138
AMA Style
Blumenfeld B, Ben-Zimra M, Simon I. Perturbations in the Replication Program Contribute to Genomic Instability in Cancer. International Journal of Molecular Sciences. 2017; 18(6):1138. https://doi.org/10.3390/ijms18061138
Chicago/Turabian StyleBlumenfeld, Britny, Micha Ben-Zimra, and Itamar Simon. 2017. "Perturbations in the Replication Program Contribute to Genomic Instability in Cancer" International Journal of Molecular Sciences 18, no. 6: 1138. https://doi.org/10.3390/ijms18061138
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.