
Cellular Repair of DNA–DNA Cross-Links Induced by 1,2,3,4-Diepoxybutane

Abstract
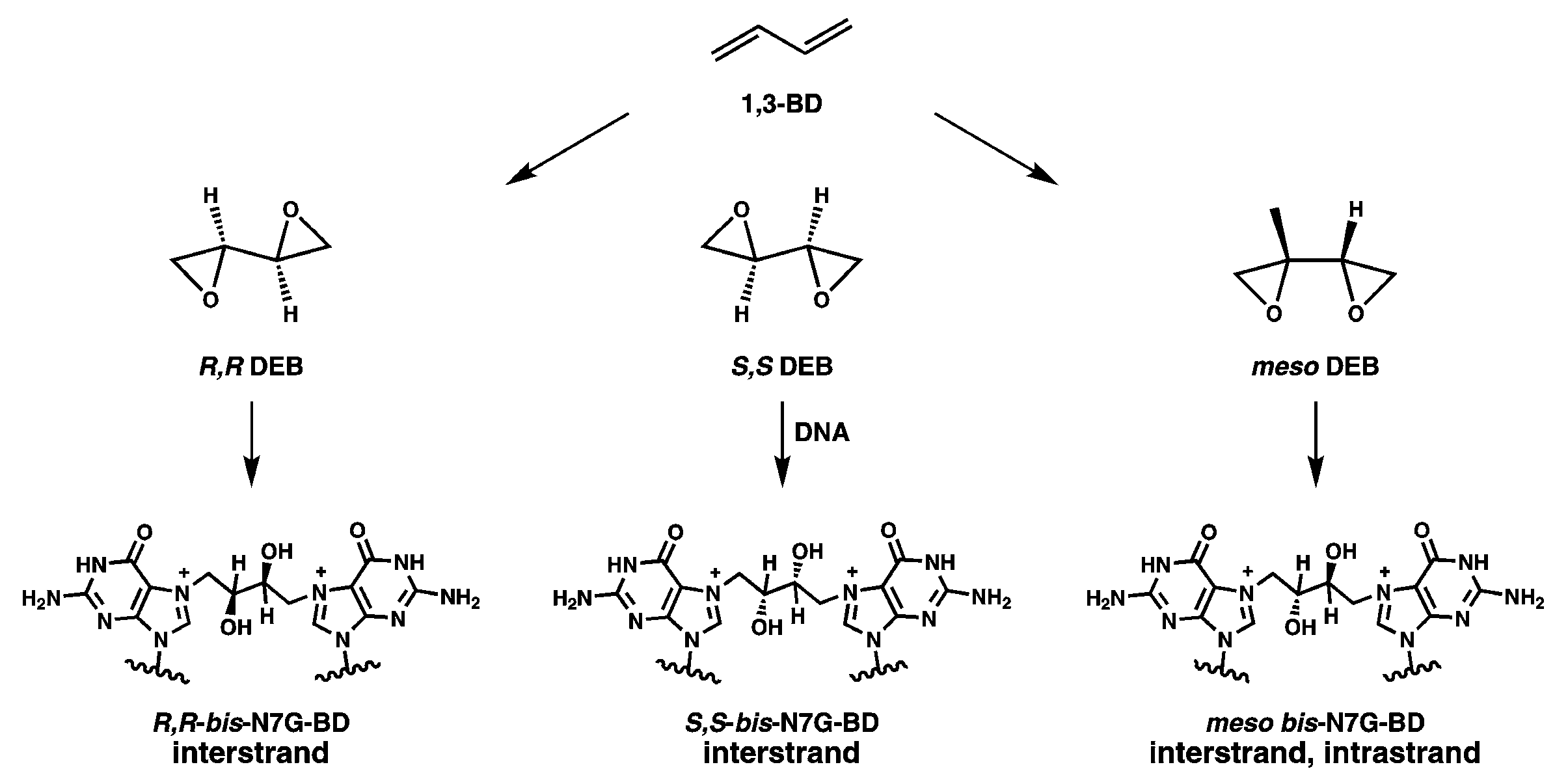
:1. Introduction
2. Results
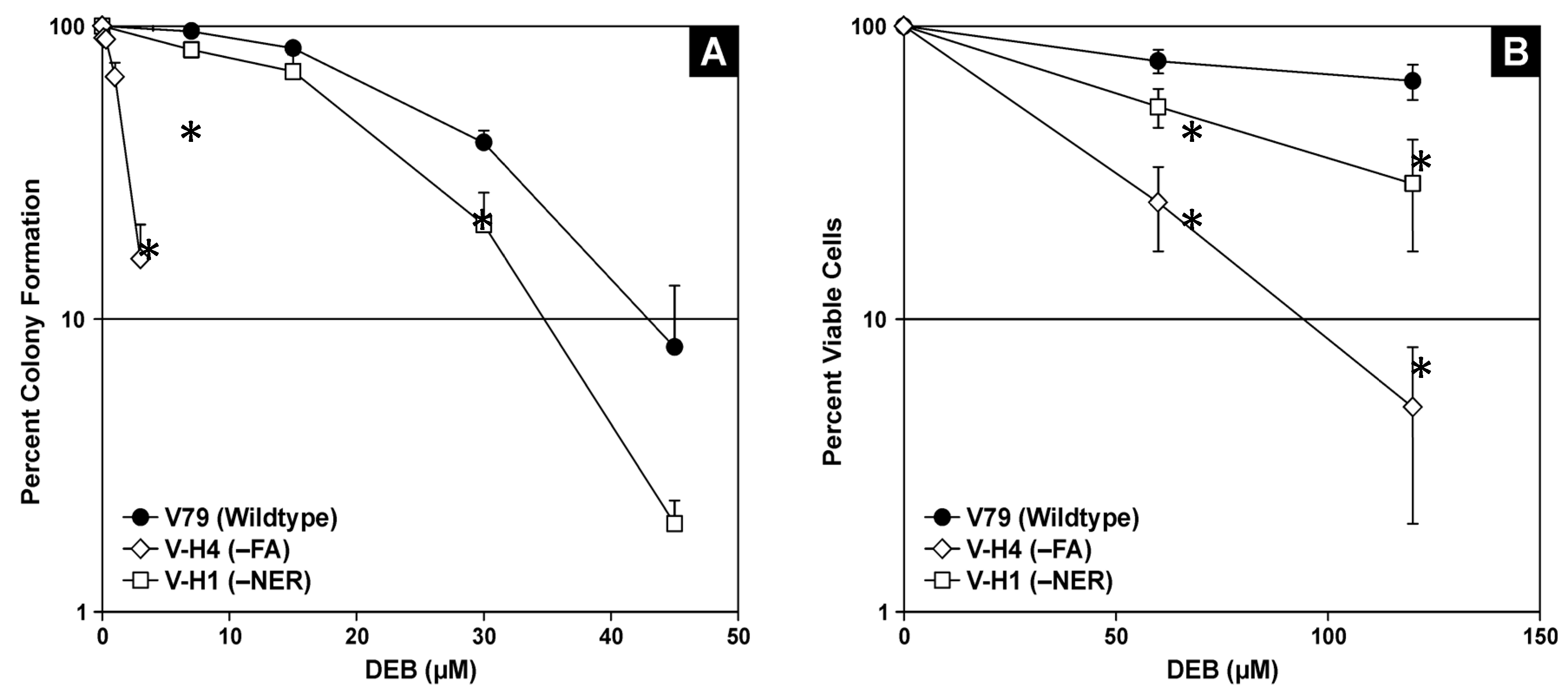
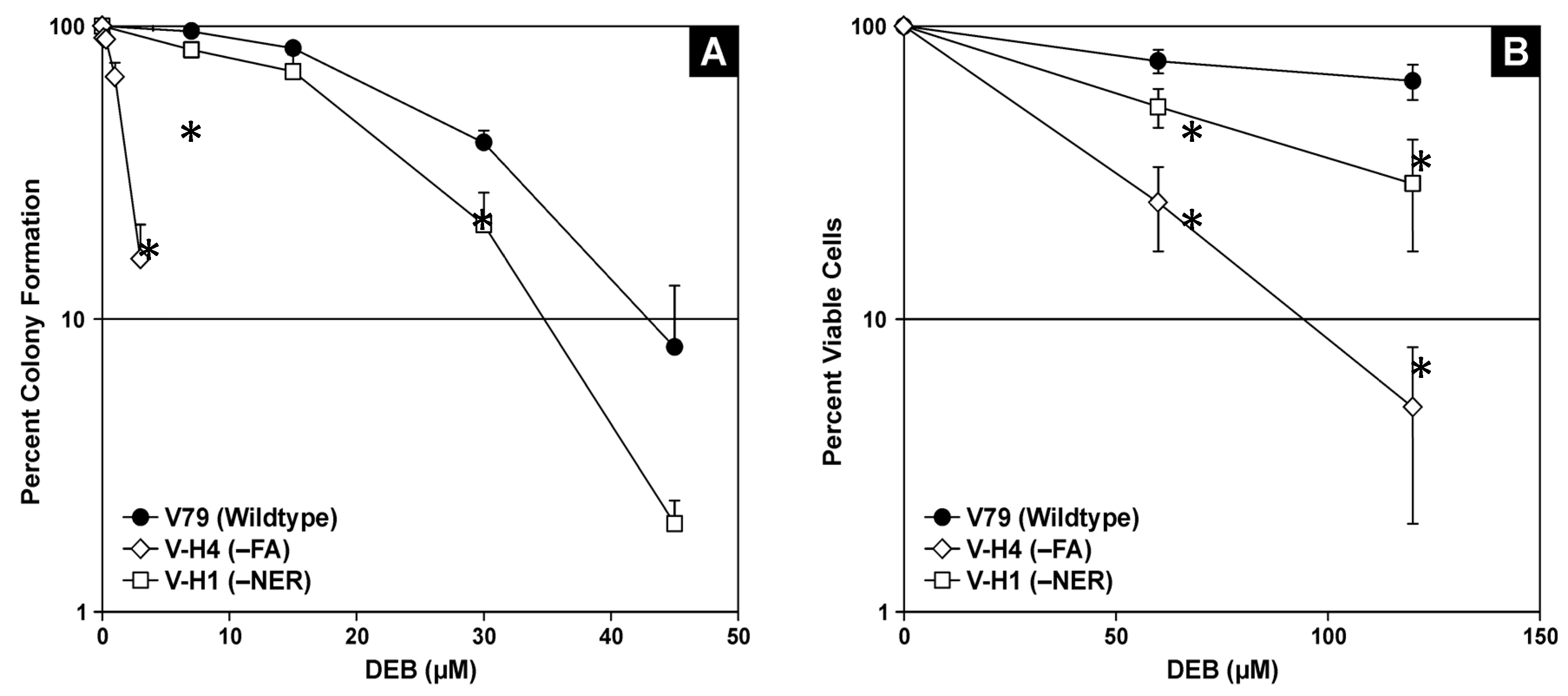
2.1. Cell Viability in the Presence of 1,2,3,4-Diepoxybutane (DEB)
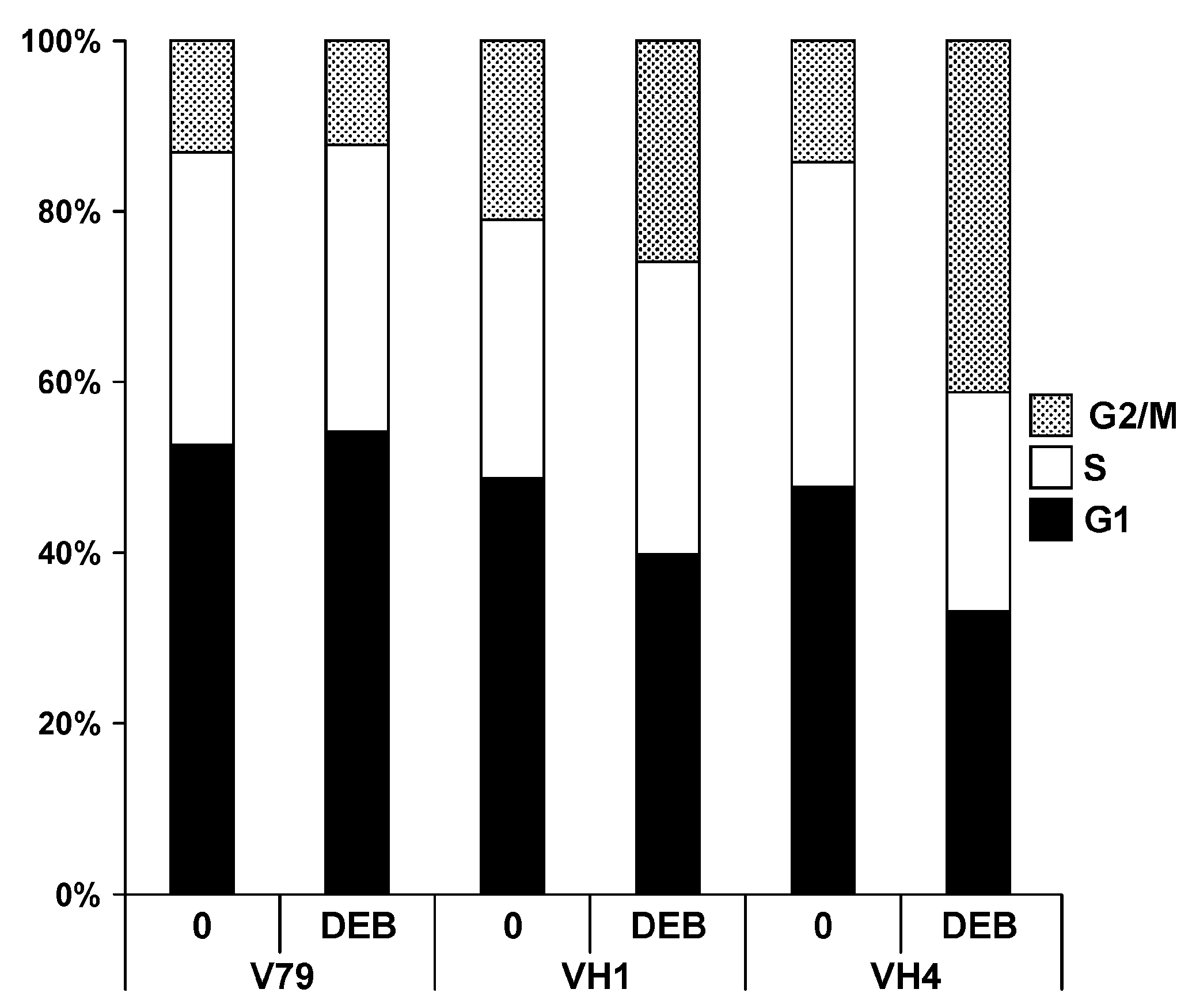
2.2. Effects of DEB Exposure on Cell Cycle Dynamics
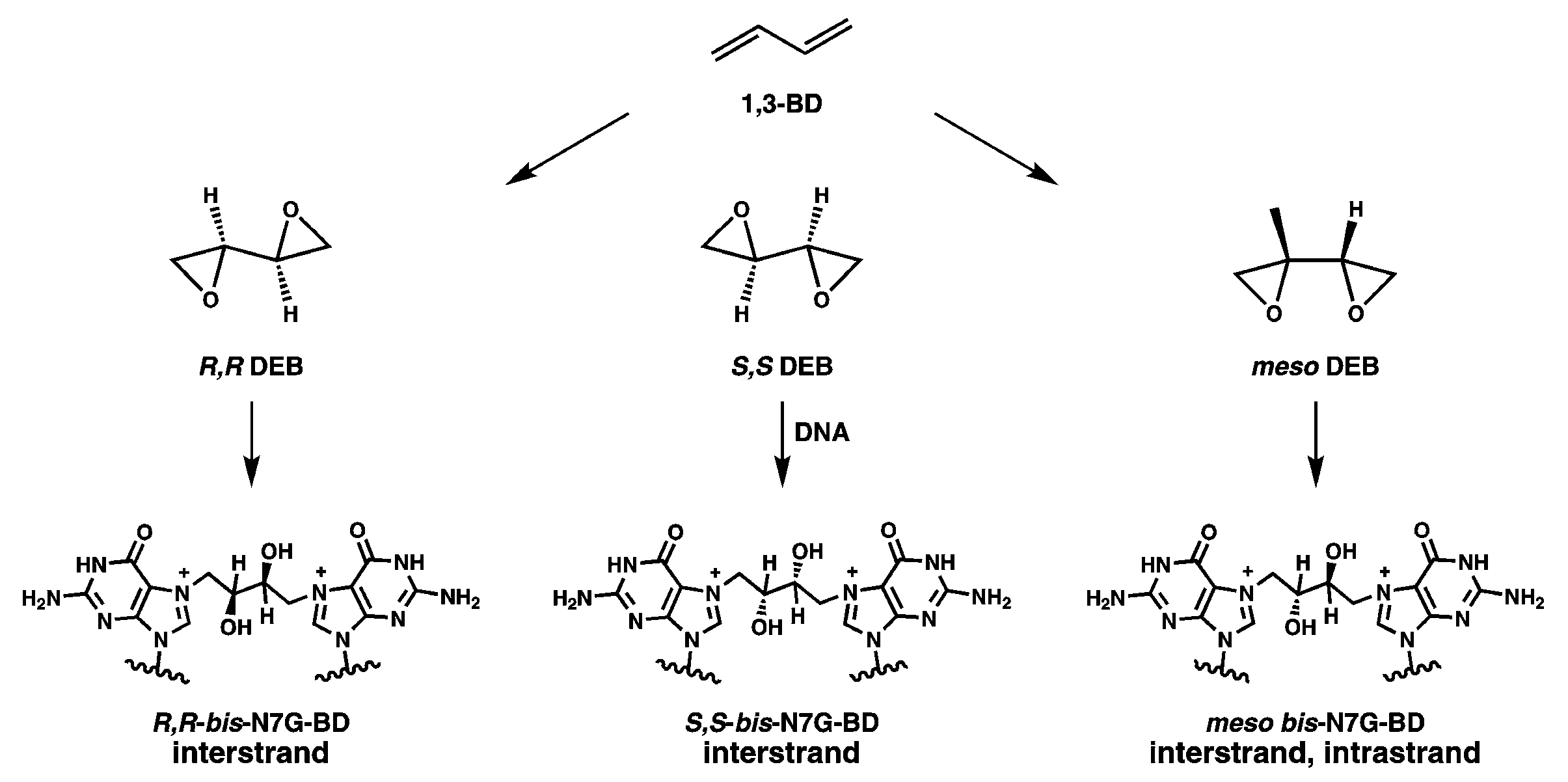
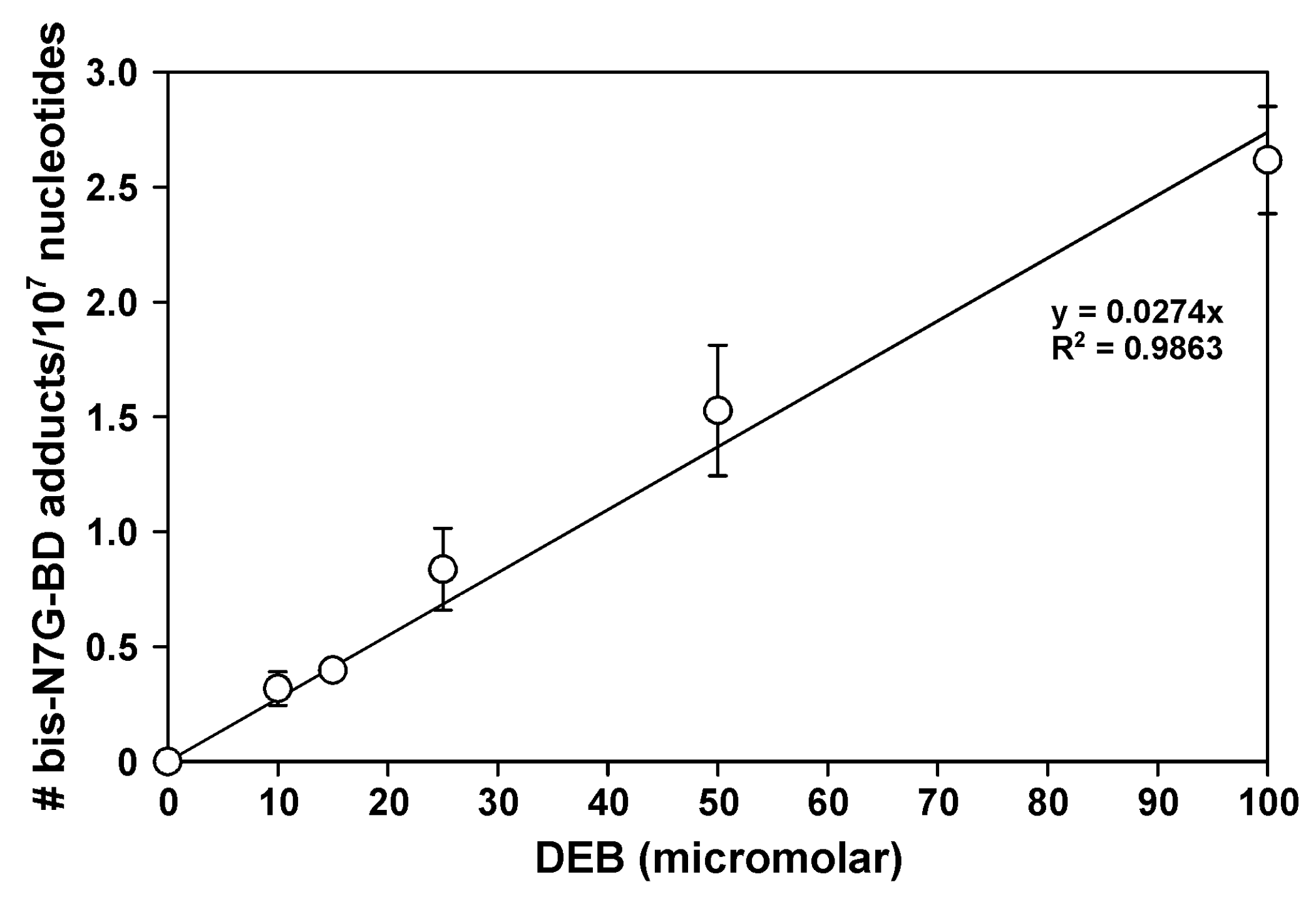
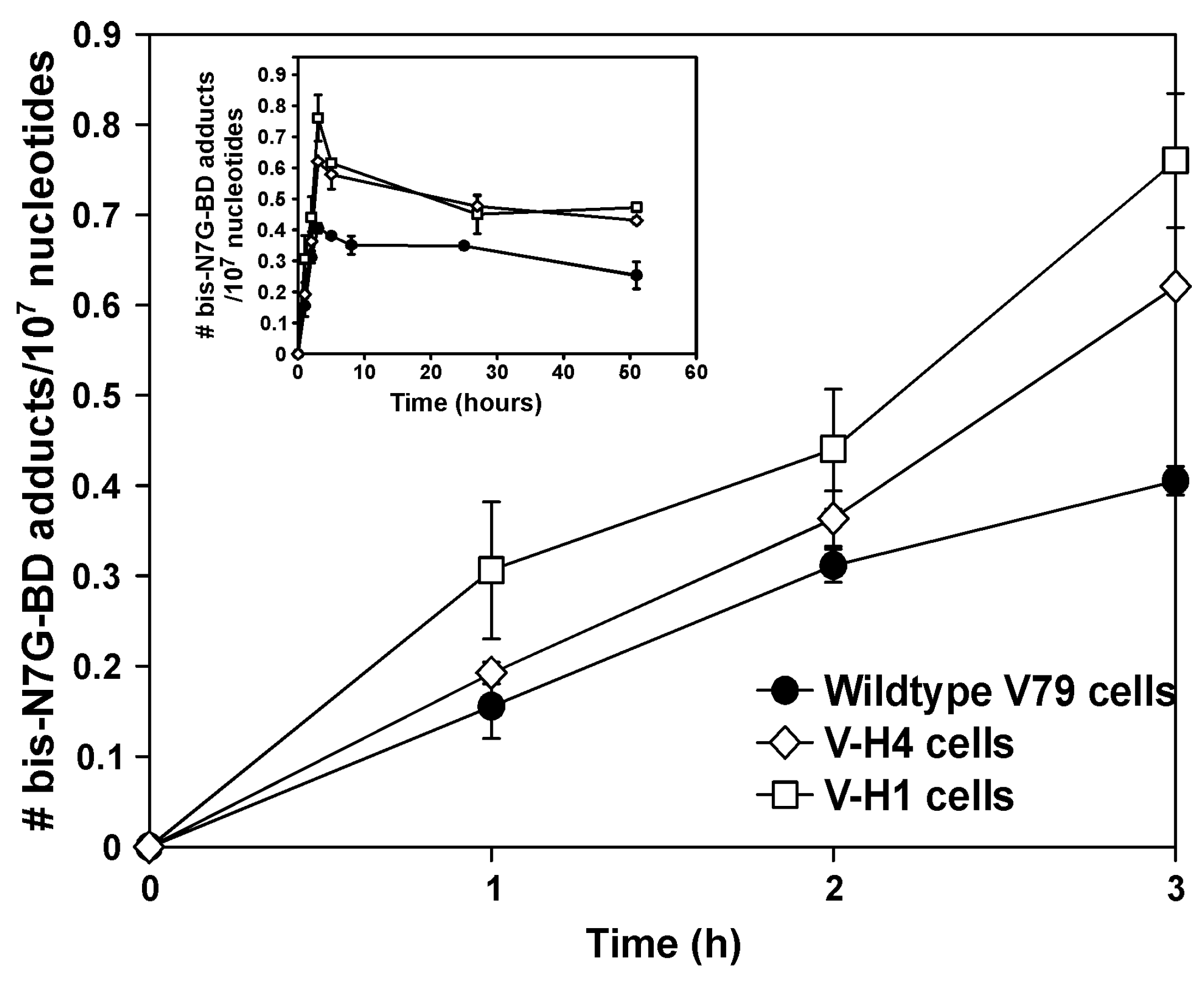
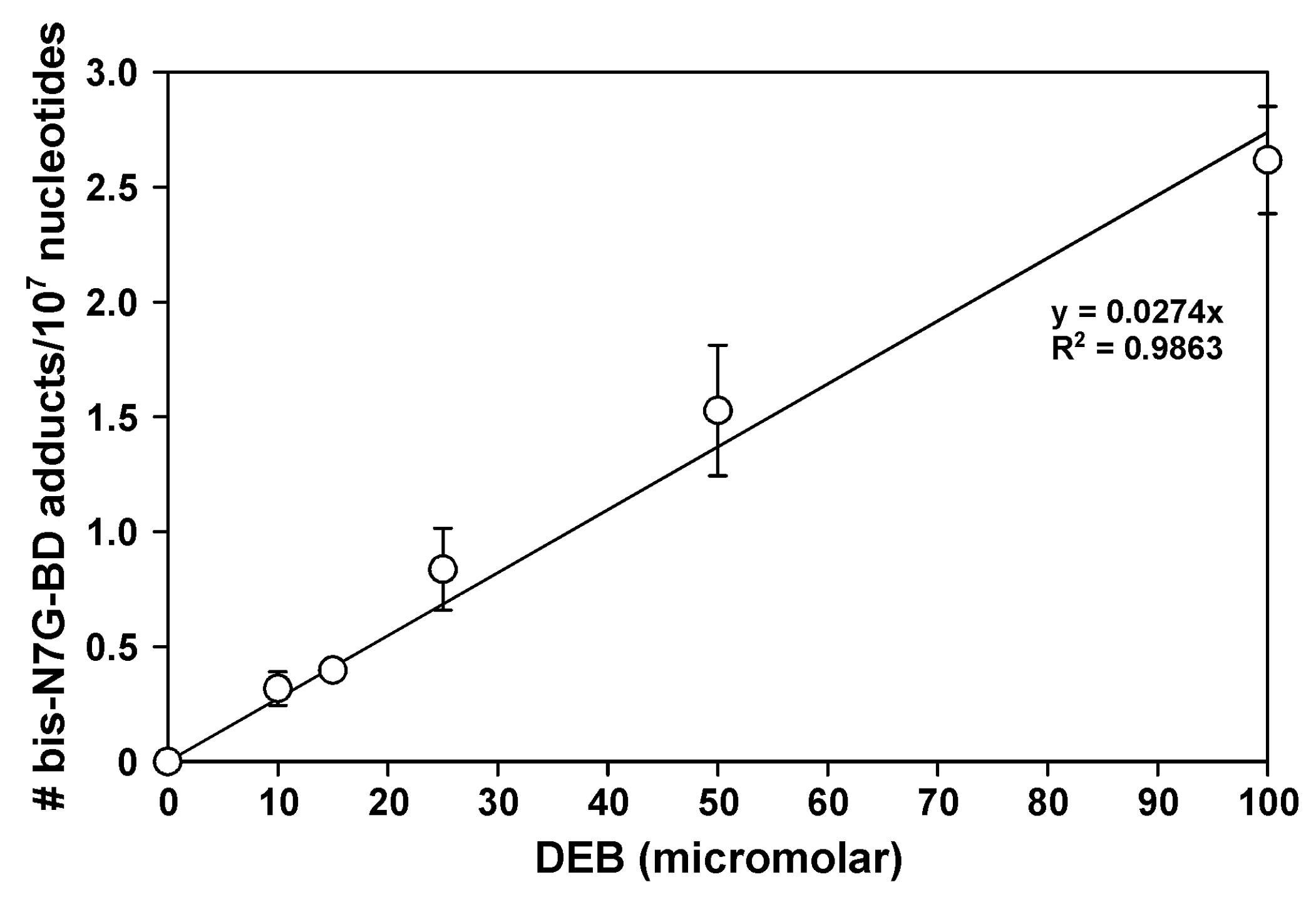
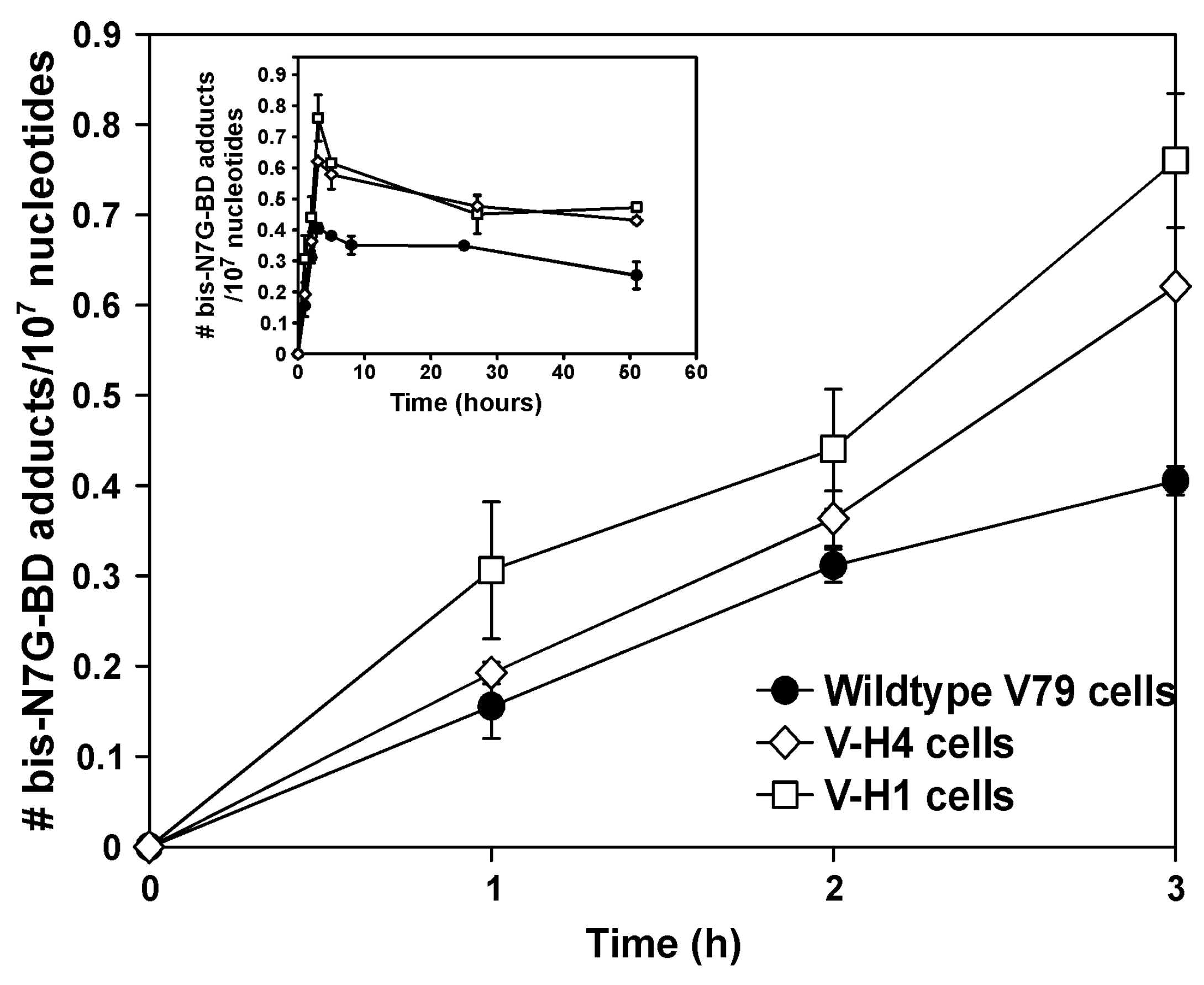
2.3. Bis-N7G-BD Adduct Formation in DEB-Treated Hamster Cells
2.4. Bis-N7G-BD Adduct Formation in DEB-Treated Human Cells
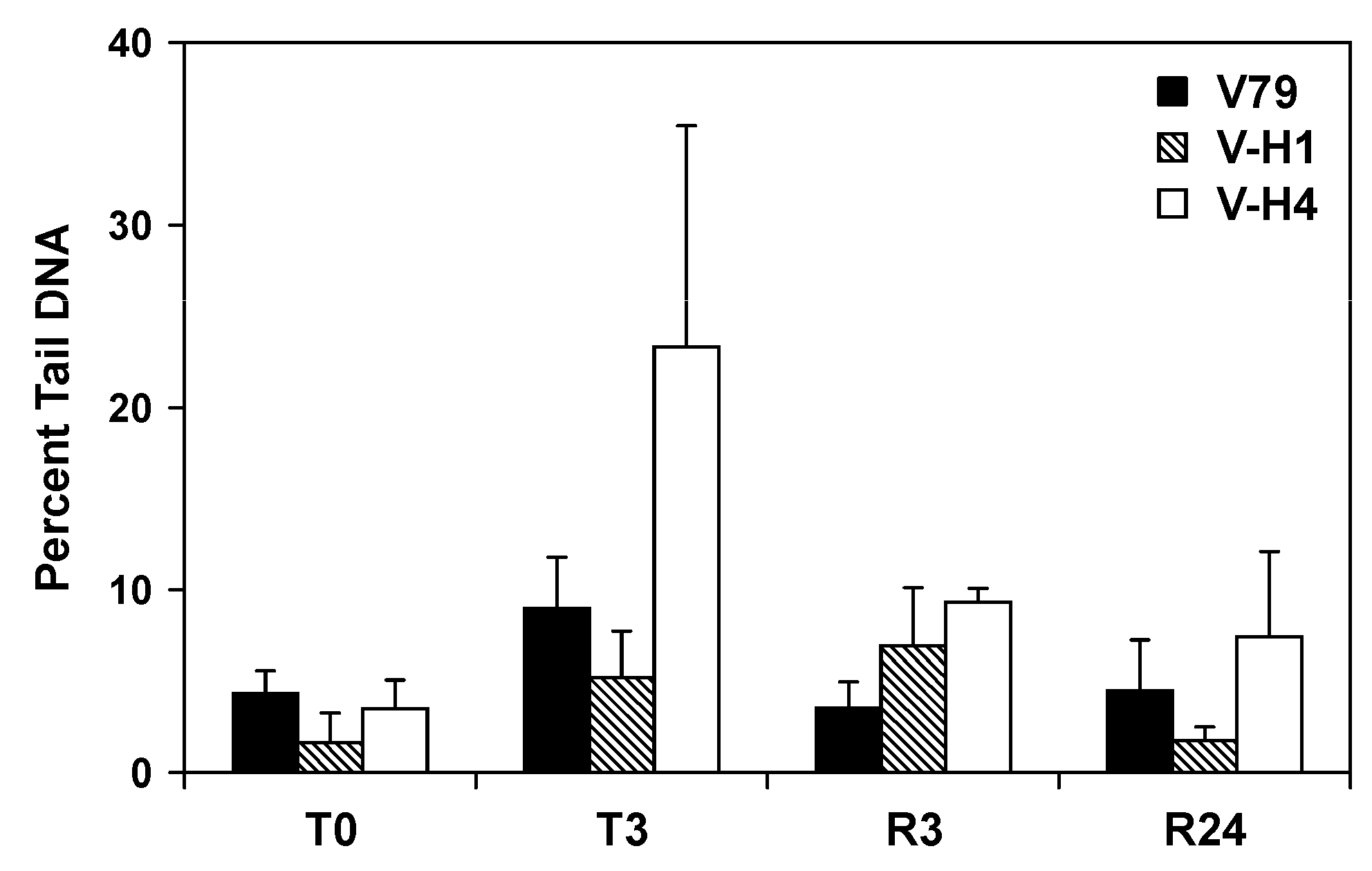
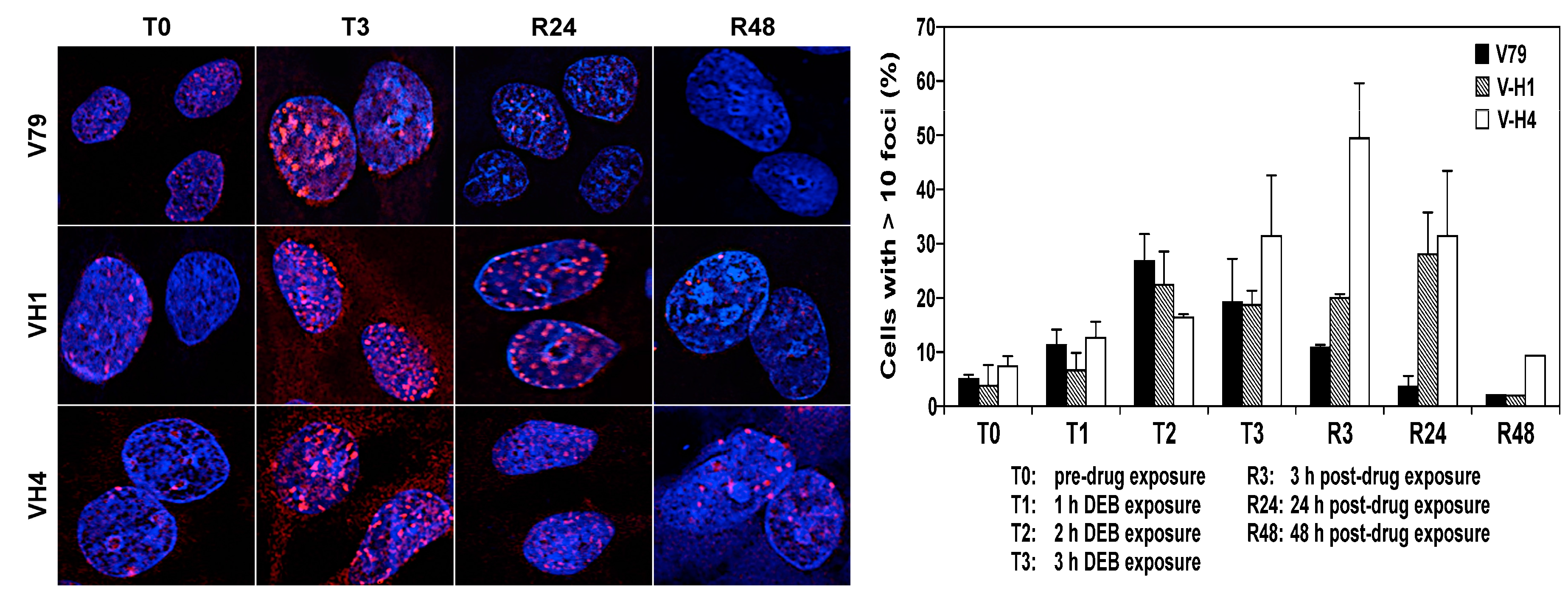
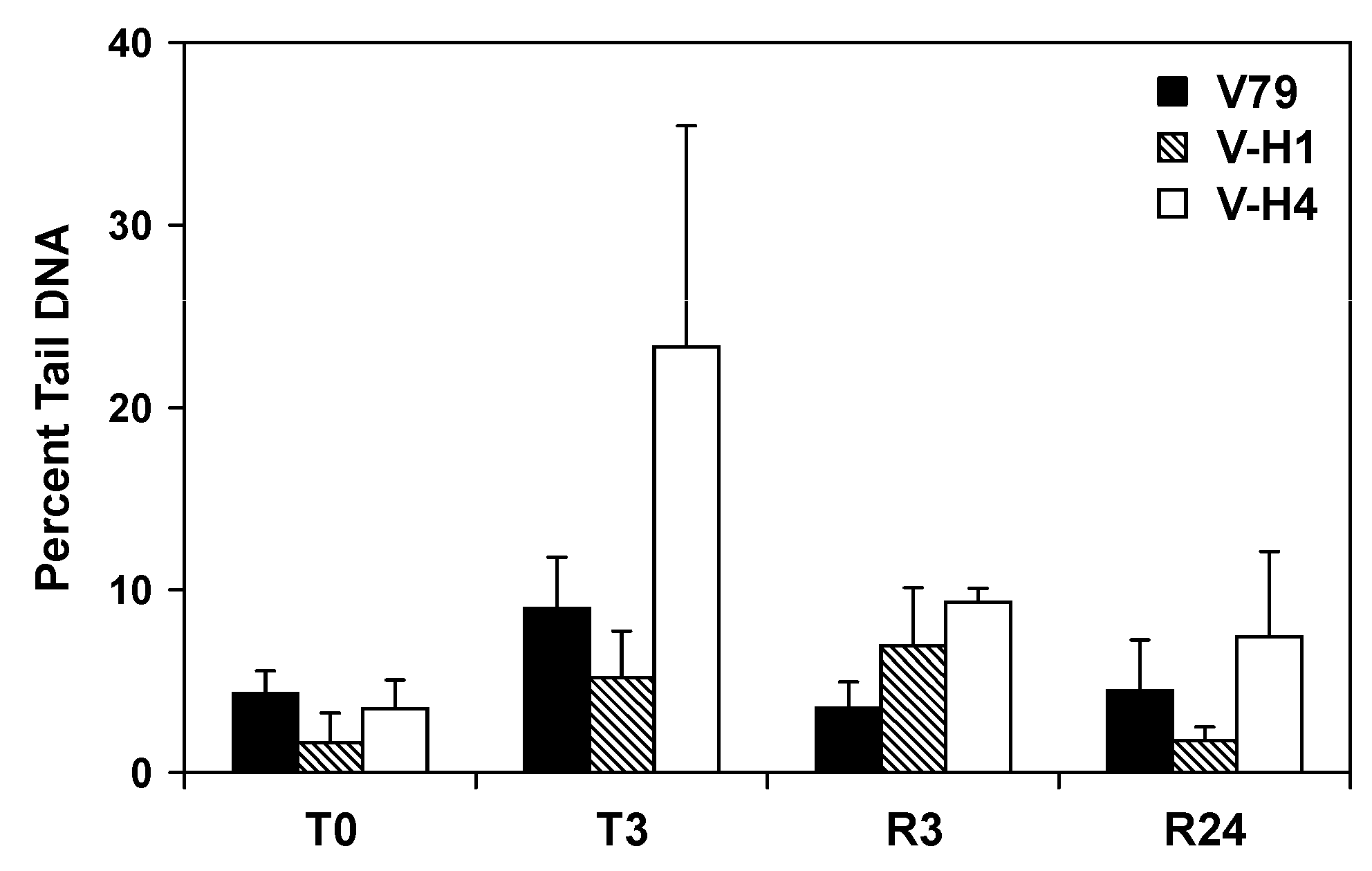
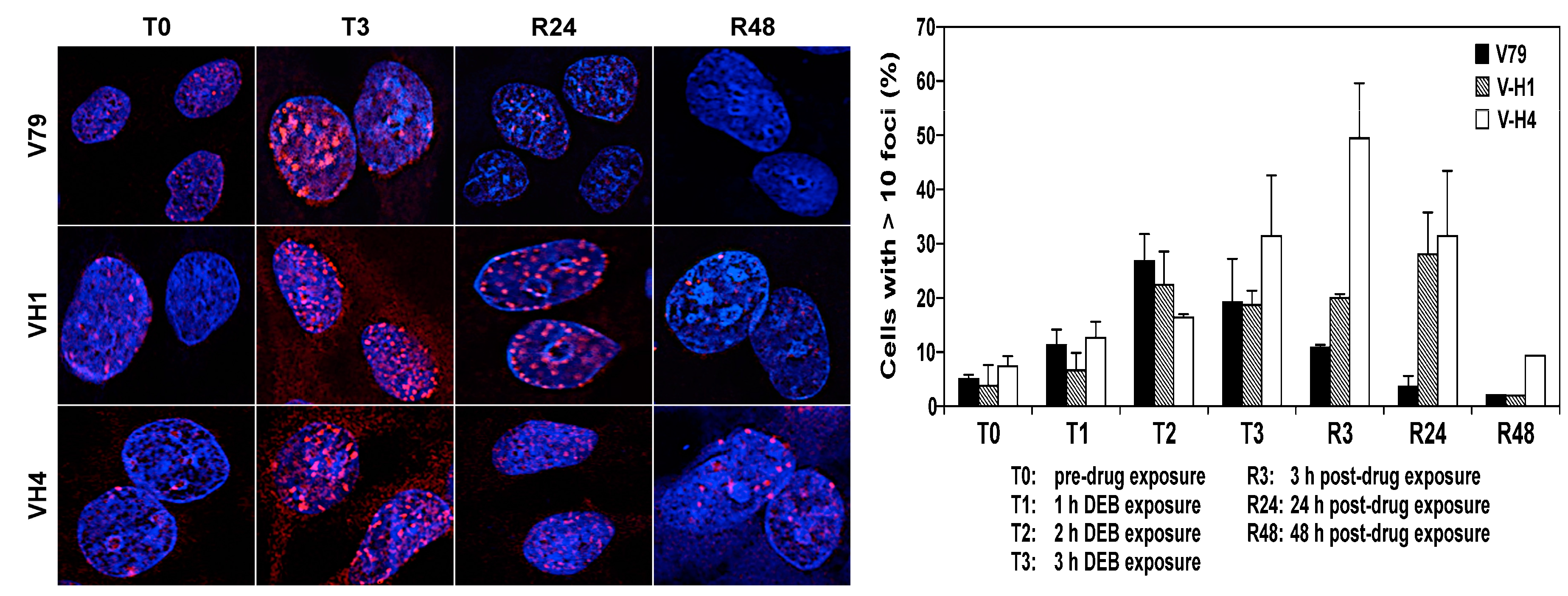
2.5. Formation and Repair of DEB-Induced DNA Double Strand Breaks
2.6. Effect of a Non-Homologous End-Joining (NHEJ) Inhibitor on Cellular Sensitivity of V79 and V-H4 Cells to DEB
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cytotoxicity Assays
4.3. Effects of DEB Exposure on Cell Cycle Distribution
4.4. Bis-N7G-BD Adduct Detection in DEB-Treated Cells
4.5. Native Comet Assay
4.6. Immunocytochemistry for Visualization of γH2AX Foci and Cyclin B1
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ICL | Interstrand DNA–DNA cross-links |
| DSB | DNA double strand break |
| bis-N7G-BD | DNA double strand break |
| DEB | 1,2,3,4-diepoxybutane |
| NER | Nucleotide excision repair |
| HR | Homologous recombinational repair |
| FA | Fanconi anemia |
| NHEJ | Non-homologous DNA end-joining |
| DNA-PK | DNA-dependent protein kinase |
| PBS | Phosphate buffered saline |
| DAPI | 4′,6-diamidino-2phenylindole dihydrochloride dihydrate |
References
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Kozekov, I.D.; Nechev, L.V.; Moseley, M.S.; Harris, C.M.; Rizzo, C.J.; Stone, M.P.; Harris, T.M. DNA interchain cross-links formed by acrolein and crotonaldehyde. J. Am. Chem. Soc. 2003, 125, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Stone, M.P.; Cho, Y.J.; Huang, H.; Kim, H.Y.; Kozekov, I.D.; Kozekova, A.; Wang, H.; Minko, I.G.; Lloyd, R.S.; Harris, T.M.; et al. Interstrand DNA cross-links induced by alpha, beta-unsaturated aldehydes derived from lipid peroxidation and environmental sources. Acc. Chem Res. 2008, 41, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Tretyakova, N. Structural characterization of the major DNA-DNA cross-link of 1,2,3,4-diepoxybutane. Chem. Res. Toxicol. 2004, 17, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Vare, D.; Groth, P.; Carlsson, R.; Johansson, F.; Erixon, K.; Jenssen, D. DNA interstrand crosslinks induce a potent replication block followed by formation and repair of double strand breaks in intact mammalian cells. DNA Repair 2012, 11, 976–985. [Google Scholar] [CrossRef] [PubMed]
- Eastman, A. Reevaluation of interaction of cis-dichloro(ethylenediamine)platinum(II) with DNA. Biochemistry 1986, 25, 3912–3915. [Google Scholar] [CrossRef] [PubMed]
- Gargiulo, D.; Kumar, G.S.; Musser, S.S.; Tomasz, M. Structural and function modification of DNA by mitomycin C. Mechanism of the DNA sequence specificity of mitomycins. Nucl. Acids Symp. Ser. 1995, 169–170. [Google Scholar]
- Vare, D.; Johansson, F.; Persson, J.O.; Erixon, K.; Jenssen, D. Quantification and repair of psoralen-induced interstrand crosslinks in human cells. Toxicol. Lett. 2014, 226, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Fleer, R.; Brendel, M. Toxicity, interstrand cross-links and DNA fragmentation induced by “activated” cyclophosphamide in yeast: Comparative studies on 4-hydroperoxy-cyclophosphamide, its monofunctional analogon, acrolein, phosphoramide mustard, and nor-nitrogen mustard. Chem. Boil. Interact. 1982, 39, 1–15. [Google Scholar] [CrossRef]
- Cole, R.S. Repair of DNA containing interstrand crosslinks in Escherichia coli: Sequential excision and recombination. Proc. Natl. Acad. Sci. USA 1973, 70, 1064–1068. [Google Scholar] [CrossRef] [PubMed]
- Dronkert, M.L.; Kanaar, R. Repair of DNA interstrand cross-links. Mutat. Res. 2001, 486, 217–247. [Google Scholar] [CrossRef]
- Lehoczky, P.; McHugh, P.J.; Chovanec, M. DNA interstrand cross-link repair in Saccharomyces cerevisiae. FEMS Microb. Rev. 2007, 31, 109–133. [Google Scholar] [CrossRef] [PubMed]
- Muniandy, P.A.; Thapa, D.; Thazhathveetil, A.K.; Liu, S.T.; Seidman, M.M. Repair of laser-localized DNA interstrand cross-links in G1 phase mammalian cells. J. Biol. Chem. 2009, 284, 27908–27917. [Google Scholar] [CrossRef] [PubMed]
- Enoiu, M.; Jiricny, J.; Scharer, O.D. Repair of cisplatin-induced DNA interstrand crosslinks by a replication-independent pathway involving transcription-coupled repair and translesion synthesis. Nucleic Acids Res. 2012, 40, 8953–8964. [Google Scholar] [CrossRef] [PubMed]
- Dardalhon, M.; Averbeck, D. Pulsed-field gel electrophoresis analysis of the repair of psoralen plus UVA induced DNA photoadducts in Saccharomyces cerevisiae. Mutat. Res. 1995, 336, 49–60. [Google Scholar] [CrossRef]
- Magana-Schwencke, N.; Henriques, J.A.; Chanet, R.; Moustacchi, E. The fate of 8-methoxypsoralen photoinduced crosslinks in nuclear and mitochondrial yeast DNA: Comparison of wild-type and repair-deficient strains. Proc. Natl. Acad. Sci. USA 1982, 79, 1722–1726. [Google Scholar] [CrossRef] [PubMed]
- Saffran, W.A.; Ahmed, S.; Bellevue, S.; Pereira, G.; Patrick, T.; Sanchez, W.; Thomas, S.; Alberti, M.; Hearst, J.E. DNA repair defects channel interstrand DNA cross-links into alternate recombinational and error-prone repair pathways. J. Biol. Chem. 2004, 279, 36462–36469. [Google Scholar] [CrossRef] [PubMed]
- De Silva, I.U.; McHugh, P.J.; Clingen, P.H.; Hartley, J.A. Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol. Cell. Biol. 2000, 20, 7980–7990. [Google Scholar] [CrossRef] [PubMed]
- Niedernhofer, L.J.; Odijk, H.; Budzowska, M.; van Drunen, E.; Maas, A.; Theil, A.F.; de Wit, J.; Jaspers, N.G.; Beverloo, H.B.; Hoeijmakers, J.H.J.; et al. The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol. Cell. Biol. 2004, 24, 5776–5787. [Google Scholar] [CrossRef] [PubMed]
- Rothfuss, A.; Grompe, M. Repair kinetics of genomic interstrand DNA cross-links: Evidence for DNA double-strand break-dependent activation of the Fanconi anemia/BRCA pathway. Mol. Cell. Biol. 2004, 24, 123–134. [Google Scholar] [CrossRef] [PubMed]
- McHugh, P.J.; Sones, W.R.; Hartley, J.A. Repair of intermediate structures produced at DNA interstrand cross-links in Saccharomyces cerevisiae. Mol. Cell. Biol. 2000, 20, 3425–3433. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Walter, J.C. , Mechanism and regulation of incisions during DNA interstrand cross-link repair. DNA Repair 2014, 19, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.V.; Scharer, O.D. Translesion DNA synthesis polymerases in DNA interstrand crosslink repair. Environ. Mol. Mutagen. 2010, 51, 552–566. [Google Scholar] [CrossRef] [PubMed]
- Walden, H.; Deans, A.J. The Fanconi anemia DNA repair pathway: Structural and functional insights into a complex disorder. Annu. Rev. Biophys. 2014, 43, 257–278. [Google Scholar] [CrossRef] [PubMed]
- Bogliolo, M.; Schuster, B.; Stoepker, C.; Derkunt, B.; Su, Y.; Raams, A.; Trujillo, J.P.; Minguillon, J.; Ramirez, M.J.; Pujol, R.; et al. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am. J. hum. Genet. 2013, 92, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Kashiyama, K.; Nakazawa, Y.; Pilz, D.T.; Guo, C.; Shimada, M.; Sasaki, K.; Fawcett, H.; Wing, J.F.; Lewin, S.O.; Carr, L.; et al. Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes Cockayne syndrome, xeroderma pigmentosum, and Fanconi anemia. Am. J. Hum. Genet. 2013, 92, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Scharer, O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Boil. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Hoy, C.A.; Thompson, L.H.; Mooney, C.L.; Salazar, E.P. Defective DNA cross-link removal in Chinese hamster cell mutants hypersensitive to bifunctional alkylating agents. Cancer Res. 1985, 45, 1737–1743. [Google Scholar] [PubMed]
- Wood, R.D. Mammalian nucleotide excision repair proteins and interstrand crosslink repair. Environ. Mol. Mutagen. 2010, 51, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Mouw, K.W.; D’Andrea, A.D. Crosstalk between the nucleotide excision repair and Fanconi anemia/BRCA pathways. DNA Repair 2014, 19, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Crossan, G.P.; Patel, K.J. The Fanconi anaemia pathway orchestrates incisions at sites of crosslinked DNA. J. Pathol. 2012, 226, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, J.E.; Skopek, T.R. Mutagenicity of butadiene and its epoxide metabolites: II. Mutational spectra of butadiene, 1,2-epoxybutene and diepoxybutane at the hprt locus in splenic T cells from exposed B6C3F1 mice. Carcinogenesis 1994, 15, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Tretyakova, N.; Chiang, S.Y.; Walker, V.E.; Swenberg, J.A. Quantitative analysis of 1,3-butadiene-induced DNA adducts in vivo and in vitro using liquid chromatography electrospray ionization tandem mass spectrometry. J. Mass Spectrum. JMS 1998, 33, 363–376. [Google Scholar] [CrossRef]
- Tretyakova, N.; Lin, Y.; Sangaiah, R.; Upton, P.B.; Swenberg, J.A. Identification and quantitation of DNA adducts from calf thymus DNA exposed to 3,4-epoxy-1-butene. Carcinogenesis 1997, 18, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Lawley, P.D.; Brookes, P. Interstrand cross-linking of DNA by difunctional alkylating agents. J. Mol. Boil. 1967, 25, 143–160. [Google Scholar] [CrossRef]
- Park, S.; Anderson, C.; Loeber, R.; Seetharaman, M.; Jones, R.; Tretyakova, N. Interstrand and intrastrand DNA–DNA cross-linking by 1,2,3,4-diepoxybutane: Role of stereochemistry. J. Am. Chem. Soc. 2005, 127, 14355–14365. [Google Scholar] [CrossRef] [PubMed]
- Goggin, M.; Sangaraju, D.; Walker, V.E.; Wickliffe, J.; Swenberg, J.A.; Tretyakova, N. Persistence and repair of bifunctional DNA adducts in tissues of laboratory animals exposed to 1,3-butadiene by inhalation. Chem. Res. Toxicol. 2011, 24, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Zdzienicka, M.Z.; Arwert, F.; Neuteboom, I.; Rooimans, M.; Simons, J.W. The Chinese hamster V79 cell mutant V-H4 is phenotypically like Fanconi anemia cells. Somat. Cell Mol. Genet. 1990, 16, 575–581. [Google Scholar] [CrossRef]
- Kang, S.G.; Chung, H.; Yoo, Y.D.; Lee, J.G.; Choi, Y.I.; Yu, Y.S. Mechanism of growth inhibitory effect of Mitomycin-C on cultured human retinal pigment epithelial cells: Apoptosis and cell cycle arrest. Curr. Eye Res. 2001, 22, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Seyschab, H.; Friedl, R.; Sun, Y.; Schindler, D.; Hoehn, H.; Hentze, S.; Schroeder-Kurth, T. Comparative evaluation of diepoxybutane sensitivity and cell cycle blockage in the diagnosis of Fanconi anemia. Blood 1995, 85, 2233–2237. [Google Scholar] [PubMed]
- Nieto, A.; Cabrera, C.M.; Catalina, P.; Cobo, F.; Barnie, A.; Cortes, J.L.; Barroso del Jesus, A.; Montes, R.; Concha, A. Effect of mitomycin-C on human foreskin fibroblasts used as feeders in human embryonic stem cells: Immunocytochemistry MIB1 score and DNA ploidy and apoptosis evaluated by flow cytometry. Cell Biol. Int. 2007, 31, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Goggin, M.; Loeber, R.; Park, S.; Walker, V.; Wickliffe, J.; Tretyakova, N. HPLC-ESI+-MS/MS analysis of N7-guanine-N7-guanine DNA cross-links in tissues of mice exposed to 1,3-butadiene. Chem. Res. Toxicol. 2007, 20, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Clingen, P.H.; Wu, J.Y.; Miller, J.; Mistry, N.; Chin, F.; Wynne, P.; Prise, K.M.; Hartley, J.A. Histone H2AX phosphorylation as a molecular pharmacological marker for DNA interstrand crosslink cancer chemotherapy. Biochem. Pharmacol. 2008, 76, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Pace, P.; Mosedale, G.; Hodskinson, M.R.; Rosado, I.V.; Sivasubramaniam, M.; Patel, K.J. Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science 2010, 329, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Adamo, A.; Collis, S.J.; Adelman, C.A.; Silva, N.; Horejsi, Z.; Ward, J.D.; Martinez-Perez, E.; Boulton, S.J.; La Volpe, A. Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol. Cell 2010, 39, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Chesner, L.N. University of Minnesota, Minneapolis, MN, USA. Unpublished work. 2017. [Google Scholar]
- Lai, C.; Cao, H.; Hearst, J.E.; Corash, L.; Luo, H.; Wang, Y. Quantitative analysis of DNA interstrand cross-links and monoadducts formed in human cells induced by psoralens and UVA irradiation. Anal. Chem. 2008, 80, 8790–8798. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Hearst, J.E.; Corash, L.; Wang, Y. LC-MS/MS for the detection of DNA interstrand cross-links formed by 8-methoxypsoralen and UVA irradiation in human cells. Anal. Chem. 2008, 80, 2932–2938. [Google Scholar] [CrossRef] [PubMed]
- Paz, M.M.; Ladwa, S.; Champeil, E.; Liu, Y.; Rockwell, S.; Boamah, E.K.; Bargonetti, J.; Callahan, J.; Roach, J.; Tomasz, M. Mapping DNA adducts of mitomycin C and decarbamoyl mitomycin C in cell lines using liquid chromatography/ electrospray tandem mass spectrometry. Chem. Res. Toxicol. 2008, 21, 2370–2378. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, Y. A quantitative mass spectrometry-based approach for assessing the repair of 8-methoxypsoralen-induced DNA interstrand cross-links and monoadducts in mammalian cells. Anal. Chem. 2013, 85, 6732–6739. [Google Scholar] [CrossRef] [PubMed]
- Bunting, S.F.; Callen, E.; Kozak, M.L.; Kim, J.M.; Wong, N.; Lopez-Contreras, A.J.; Ludwig, T.; Baer, R.; Faryabi, R.B.; Malhowski, A.; et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol. Cell 2012, 46, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Howard, S.M.; Yanez, D.A.; Stark, J.M. DNA damage response factors from diverse pathways, including DNA crosslink repair, mediate alternative end joining. PLoS Genet. 2015, 11, e1004943. [Google Scholar] [CrossRef] [PubMed]
- Larminat, F.; Germanier, M.; Papouli, E.; Defais, M. Impairment of homologous recombination control in a Fanconi anemia-like Chinese hamster cell mutant. Biol. Cell 2004, 96, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Studzian, K.; Telleman, P.; van der Schans, G.P.; Zdzienicka, M.Z. Mutagenic response and repair of cis-DDP-induced DNA cross-links in the Chinese hamster V79 cell mutant V-H4 which is homologous to Fanconi anemia (group A). Mutat. Res. 1994, 314, 115–120. [Google Scholar] [CrossRef]
- Telleman, P.; Overkamp, W.J.; van Wessel, N.; Studzian, K.; Wetselaar, L.; Natarajan, A.T.; Zdzienicka, M.Z. A new complementation group of mitomycin C-hypersensitive Chinese hamster cell mutants that closely resembles the phenotype of fanconi anemia cells. Cancer Res. 1995, 55, 3412–3416. [Google Scholar] [PubMed]
- Arwert, F.; Rooimans, M.A.; Westerveld, A.; Simons, J.W.; Zdzienicka, M.Z. The Chinese hamster cell mutant V-H4 is homologous to Fanconi anemia (complementation group A). Cytogenet. Cell Genet. 1991, 56, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Kadkhodayan, S.; Salazar, E.P.; Ramsey, M.J.; Takayama, K.; Zdzienicka, M.Z.; Tucker, J.D.; Weber, C.A. Molecular analysis of ERCC2 mutations in the repair deficient hamster mutants UVL-1 and V-H1. Mutat. Res. 1997, 385, 47–57. [Google Scholar] [CrossRef]
- Jakobs, P.M.; Fiddler-Odell, E.; Reifsteck, C.; Olson, S.; Moses, R.E.; Grompe, M. Complementation group assignments in Fanconi anemia fibroblast cell lines from North America. Somat. Cell Mol. Genet. 1997, 23, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Timmers, C.; Taniguchi, T.; Hejna, J.; Reifsteck, C.; Lucas, L.; Bruun, D.; Thayer, M.; Cox, B.; Olson, S.; D’Andrea, A.D.; et al. Positional cloning of a novel Fanconi anemia gene, FANCD2. Mol. Cell 2001, 7, 241–248. [Google Scholar] [CrossRef]
- Michaelson-Richie, E.D.; Ming, X.; Codreanu, S.G.; Loeber, R.L.; Liebler, D.C.; Campbell, C.; Tretyakova, N.Y. Mechlorethamine-induced DNA-protein cross-linking in human fibrosarcoma (HT1080) cells. J. Proteome Res. 2011, 10, 2785–2796. [Google Scholar] [CrossRef] [PubMed]
- Sangaraju, D.; Goggin, M.; Walker, V.; Swenberg, J.; Tretyakova, N. NanoHPLC-nanoESI+-MS/MS quantitation of bis-N7-guanine DNA-DNA cross-links in tissues of B6C3F1 mice exposed to subppm levels of 1,3-butadiene. Anal. Chem. 2012, 84, 1732–1739. [Google Scholar] [CrossRef] [PubMed]
- Fairbairn, D.W.; Olive, P.L.; O’Neill, K.L. The comet assay: A comprehensive review. Mutat. Res. 1995, 339, 37–59. [Google Scholar] [CrossRef]
- Kumaravel, T.S.; Vilhar, B.; Faux, S.P.; Jha, A.N. Comet Assay measurements: A perspective. Cell Boil. Toxicol. 2009, 25, 53–64. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | PDcorr (WT) | PD20 (-FANCD2) | V79 (WT) | V-H4 (-FANCA) | ||||
|---|---|---|---|---|---|---|---|---|
| Nu7026 | - | + | - | + | - | + | - | + |
| Percent cell death (± SEM) | 21 (± 1.4) | 20 (± 7.8) | 40 (± 2.8) | 42 (± 8) | 23 (± 1.4) | 22 (± 1.4) | 42 (± 2.1) | 42 (± 3.5) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chesner, L.N.; Degner, A.; Sangaraju, D.; Yomtoubian, S.; Wickramaratne, S.; Malayappan, B.; Tretyakova, N.; Campbell, C. Cellular Repair of DNA–DNA Cross-Links Induced by 1,2,3,4-Diepoxybutane. Int. J. Mol. Sci. 2017, 18, 1086. https://doi.org/10.3390/ijms18051086
Chesner LN, Degner A, Sangaraju D, Yomtoubian S, Wickramaratne S, Malayappan B, Tretyakova N, Campbell C. Cellular Repair of DNA–DNA Cross-Links Induced by 1,2,3,4-Diepoxybutane. International Journal of Molecular Sciences. 2017; 18(5):1086. https://doi.org/10.3390/ijms18051086
Chicago/Turabian StyleChesner, Lisa N., Amanda Degner, Dewakar Sangaraju, Shira Yomtoubian, Susith Wickramaratne, Bhaskar Malayappan, Natalia Tretyakova, and Colin Campbell. 2017. "Cellular Repair of DNA–DNA Cross-Links Induced by 1,2,3,4-Diepoxybutane" International Journal of Molecular Sciences 18, no. 5: 1086. https://doi.org/10.3390/ijms18051086