
Combining Low Temperature Fluorescence DNA-Hybridization, Immunostaining, and Super-Resolution Localization Microscopy for Nano-Structure Analysis of ALU Elements and Their Influence on Chromatin Structure



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
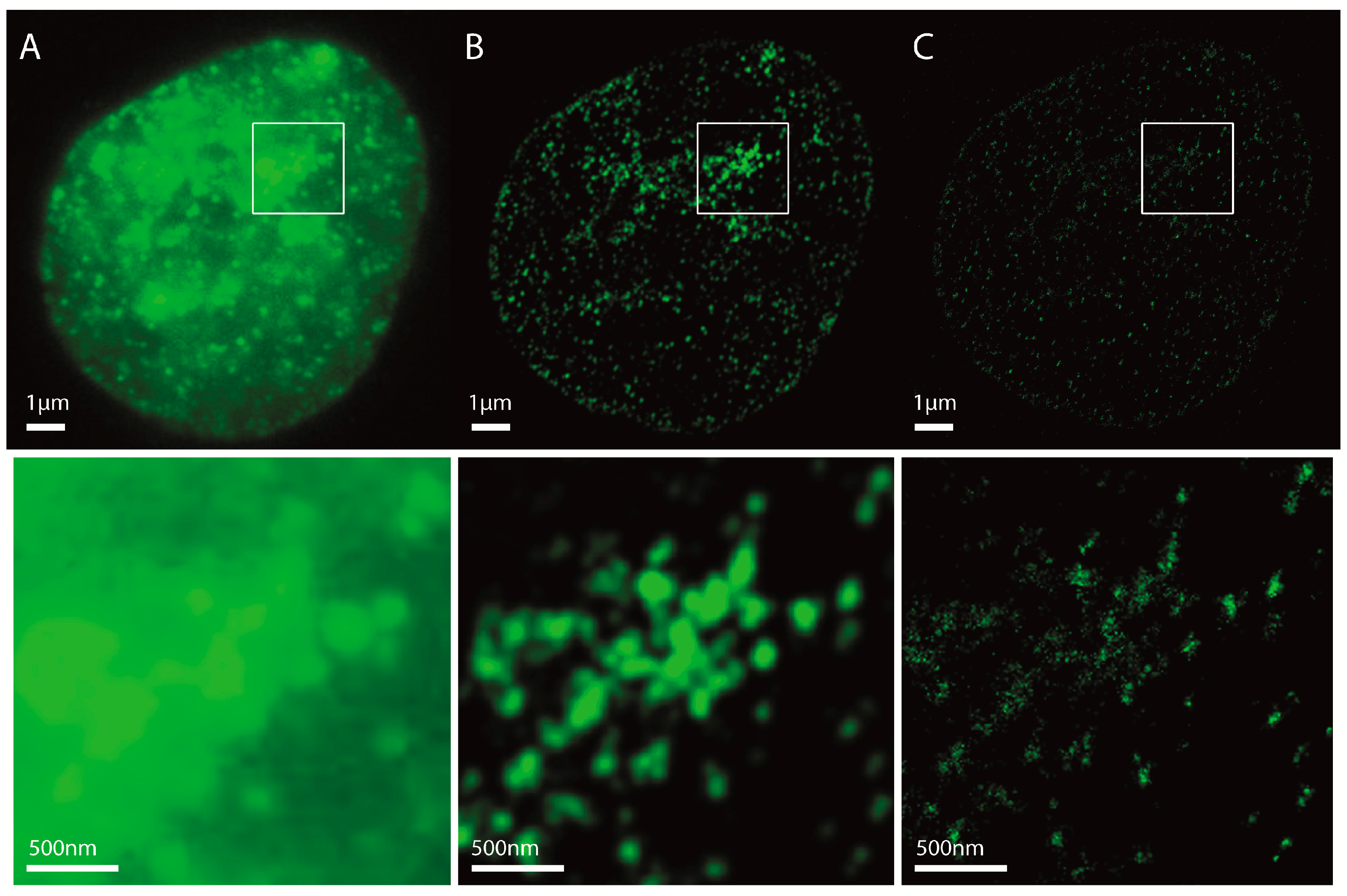
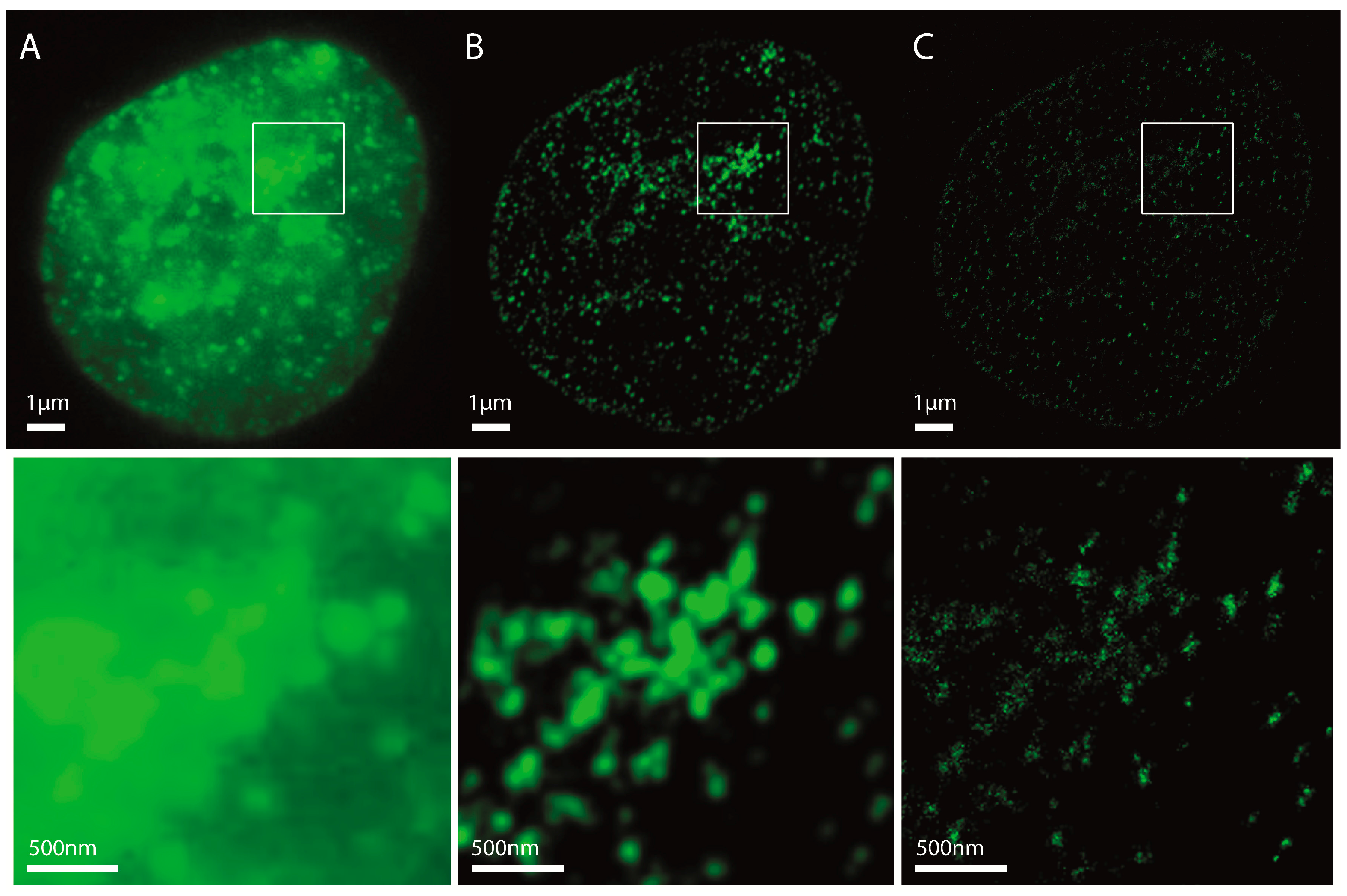
2.1. Localization Microscopy after Immunohistochemistry Labelling
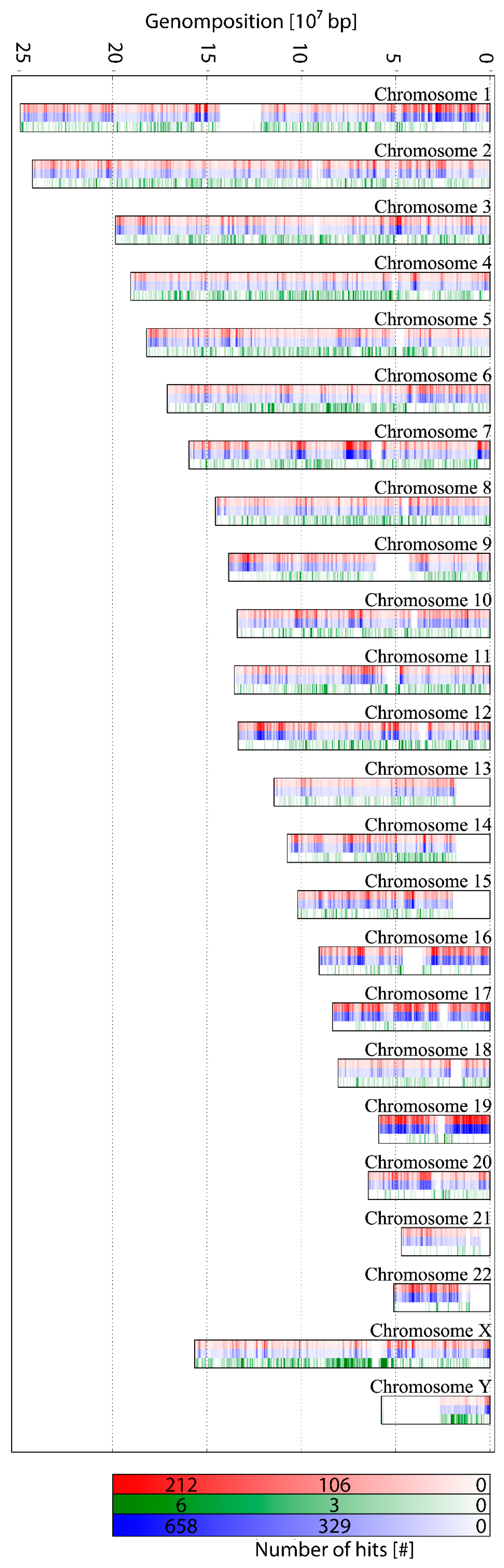
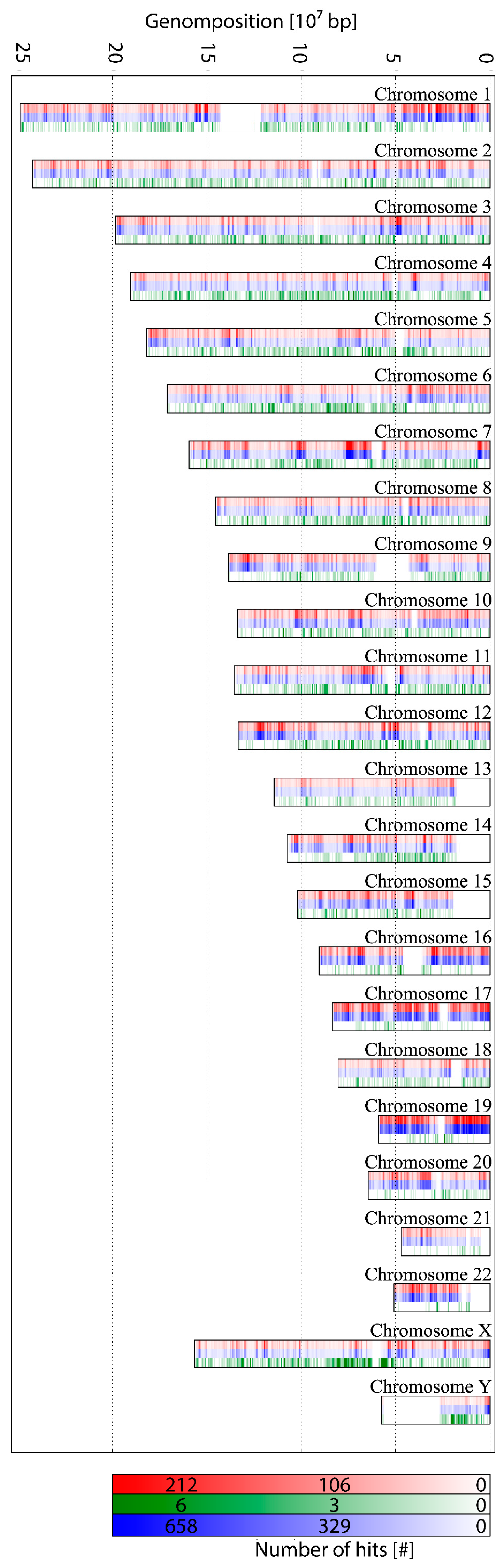
2.2. Probe Design for ALU-Combinatorial Oligonucleotide (COMBO)-Fluorescence In Situ Hybridization (FISH)-Staining
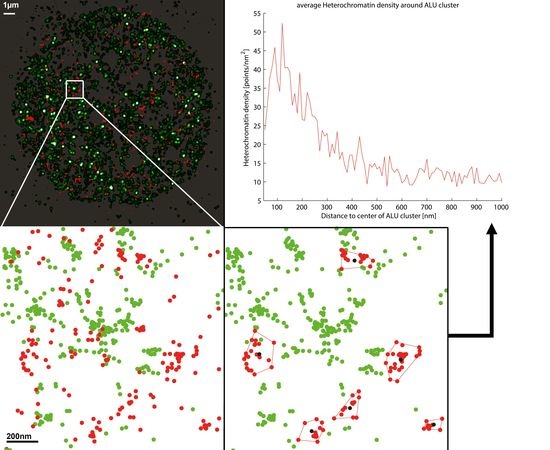
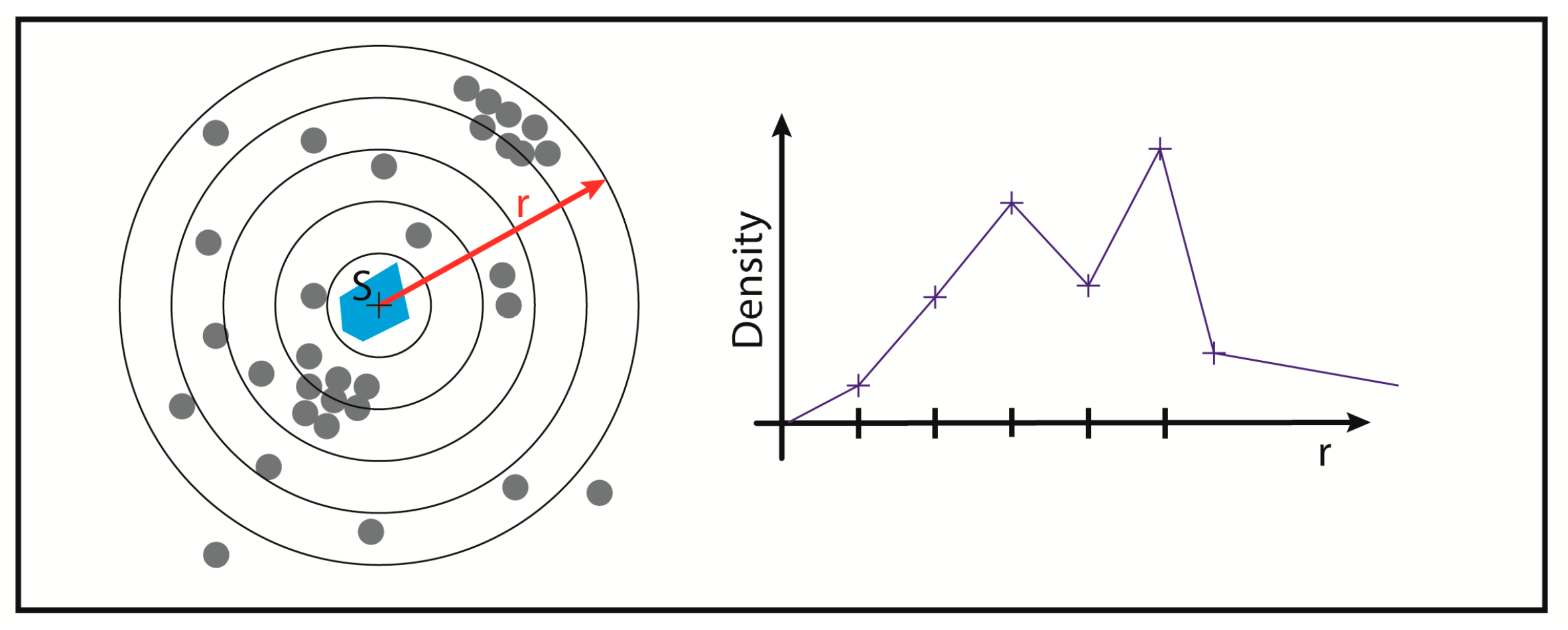
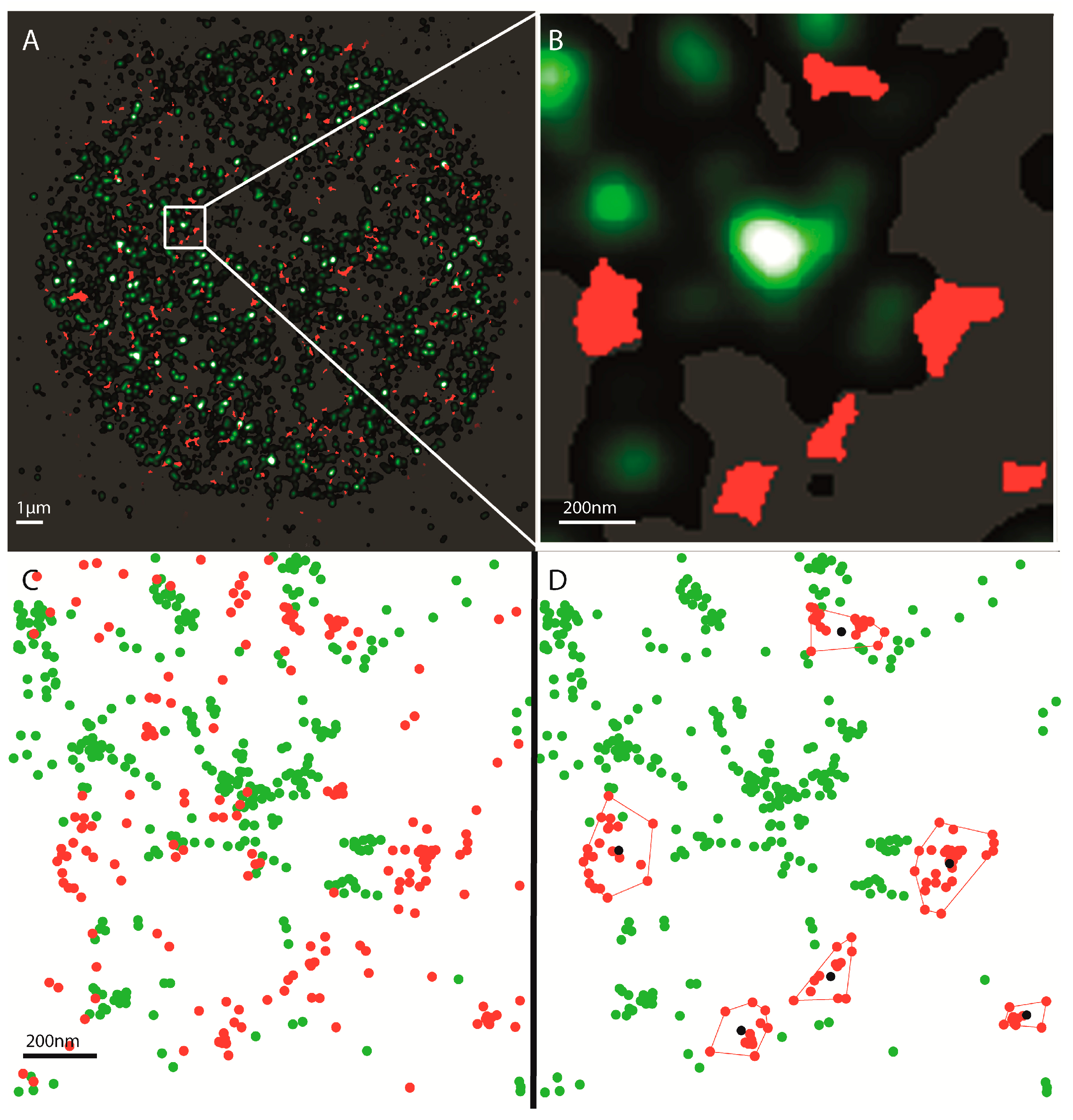
2.3. Two-Color Localization Microscopy and ALU Clustering
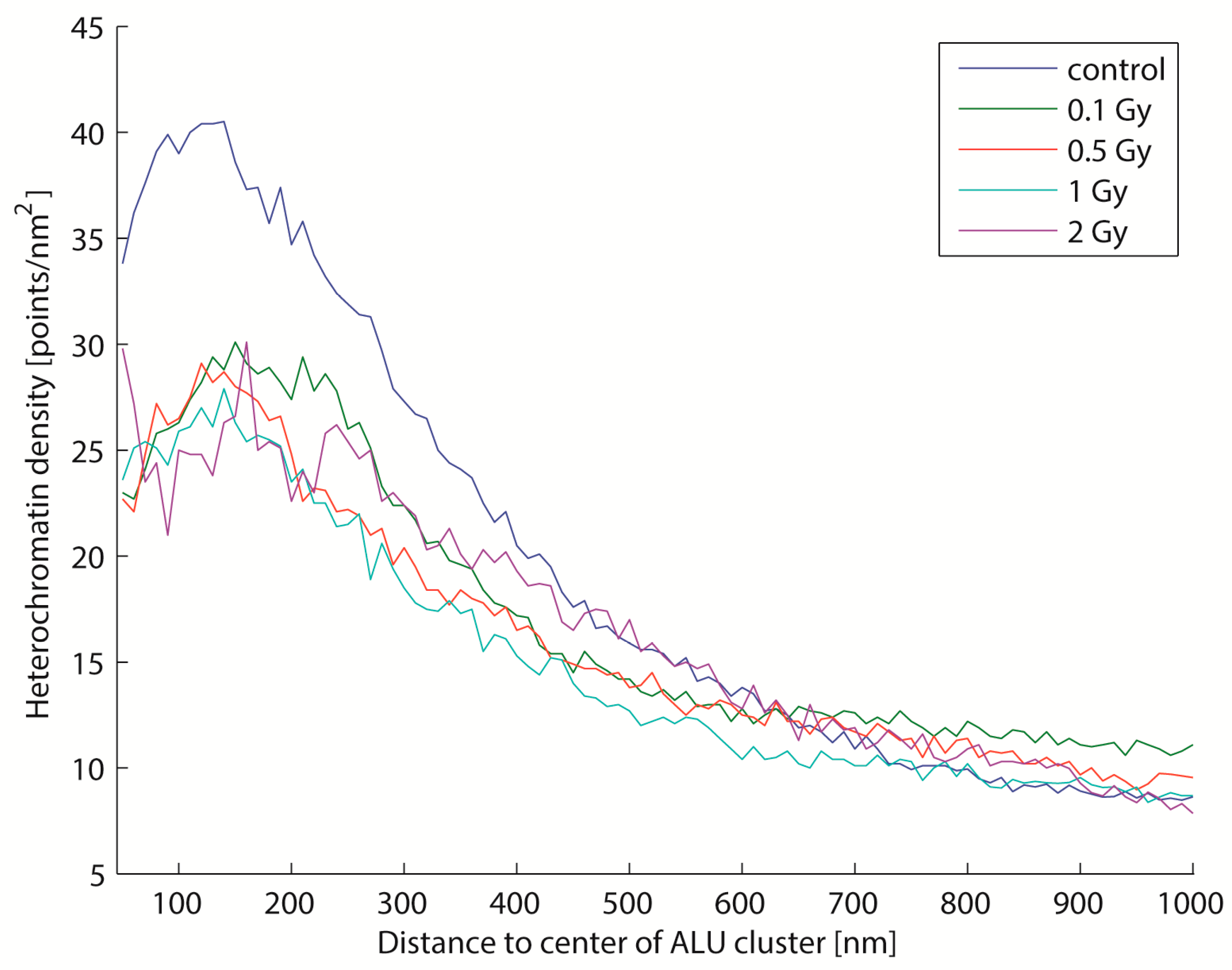
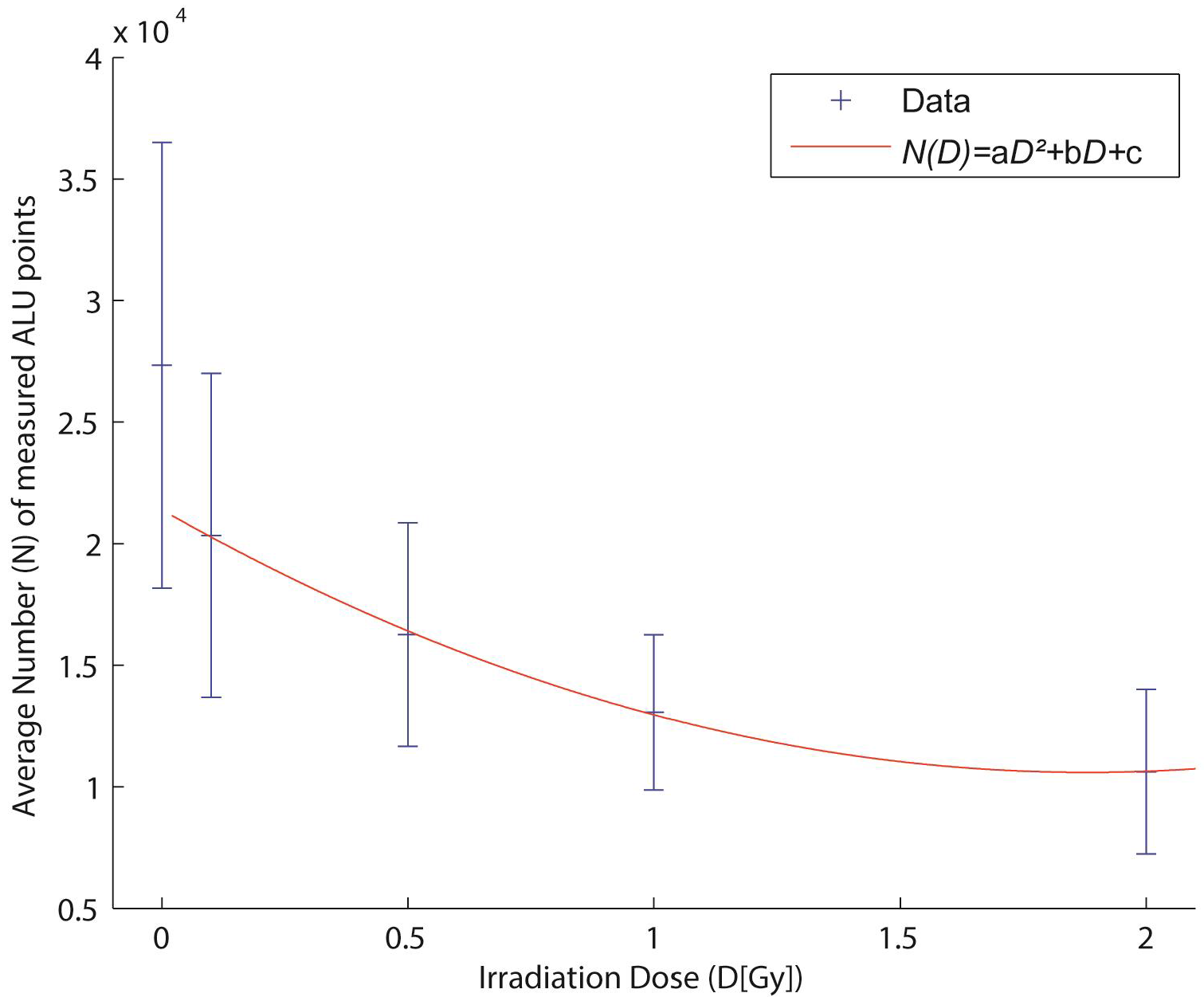
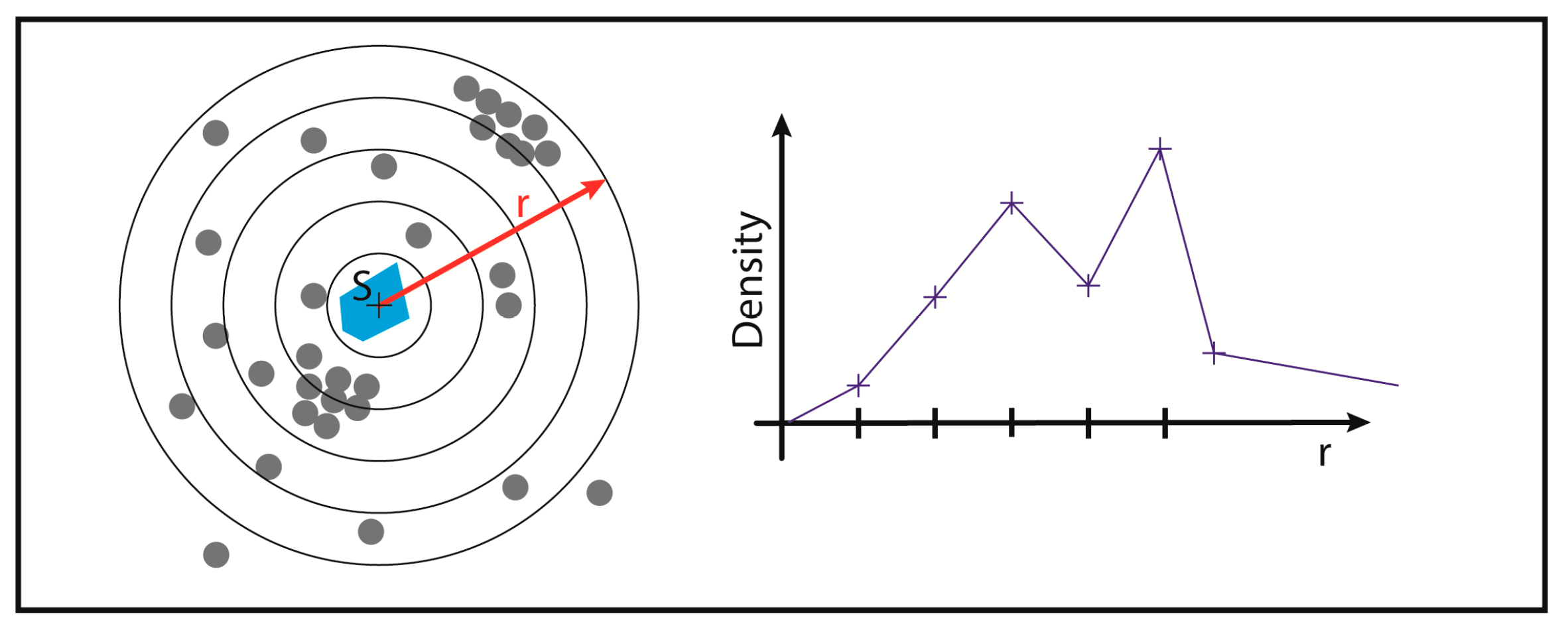
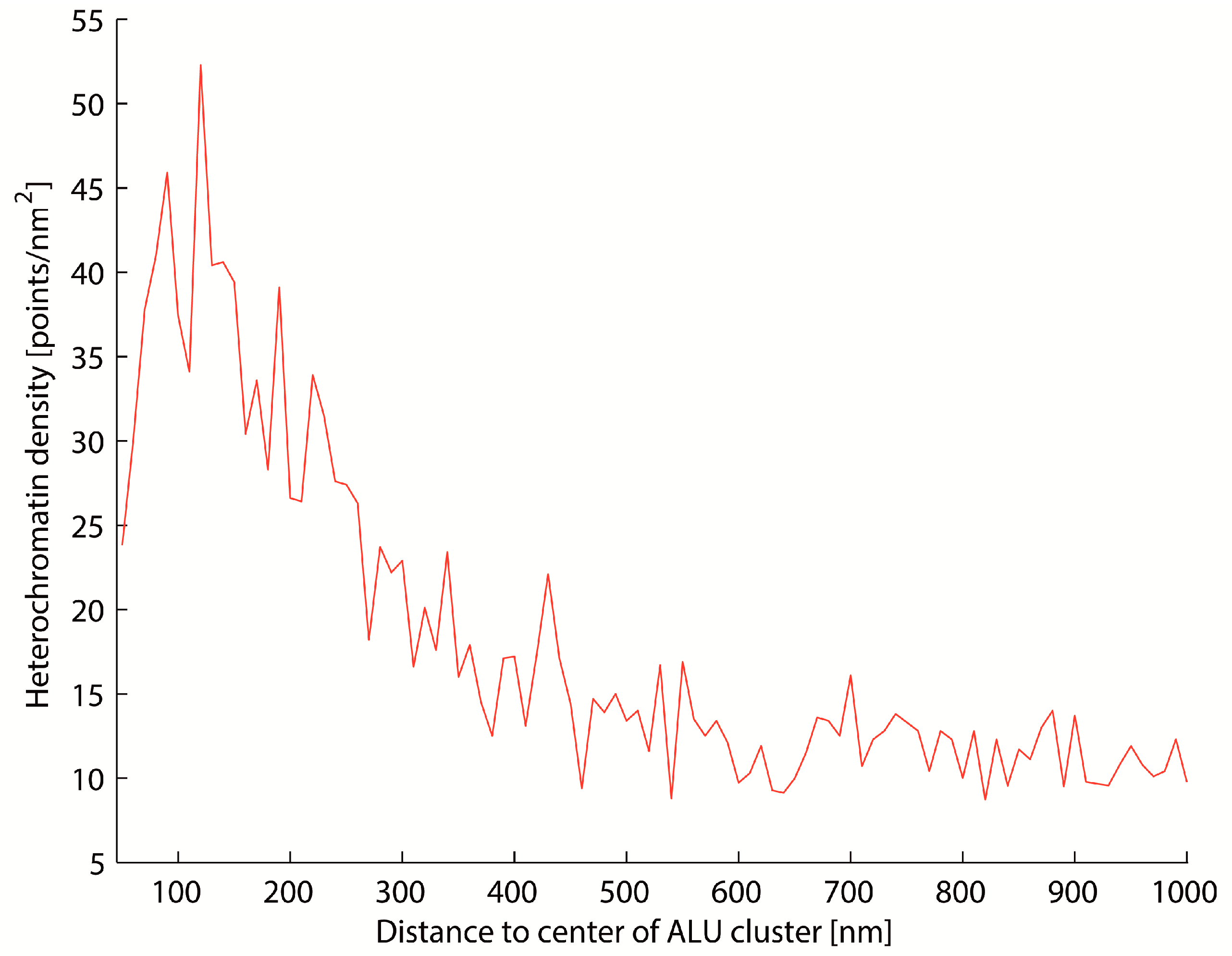
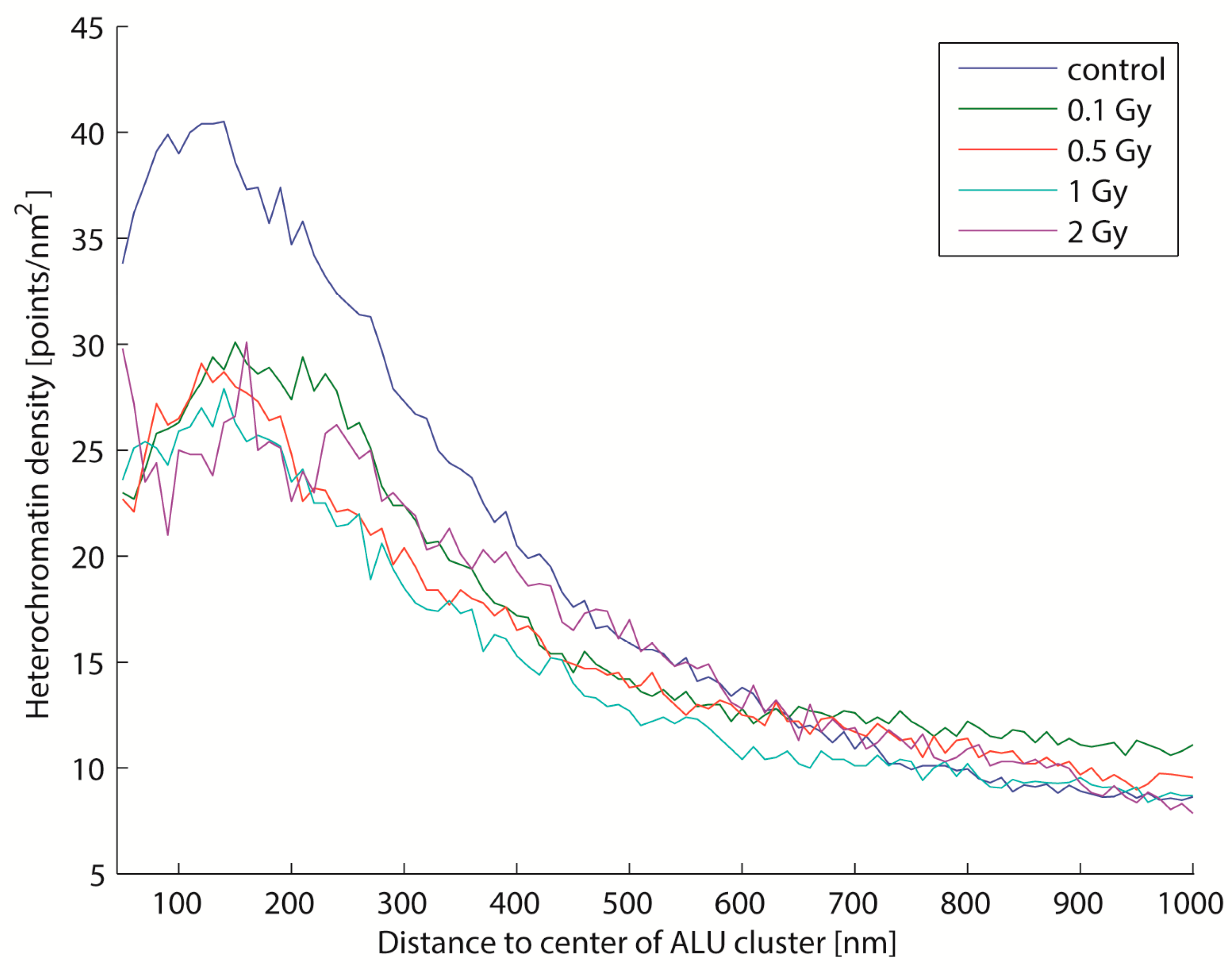
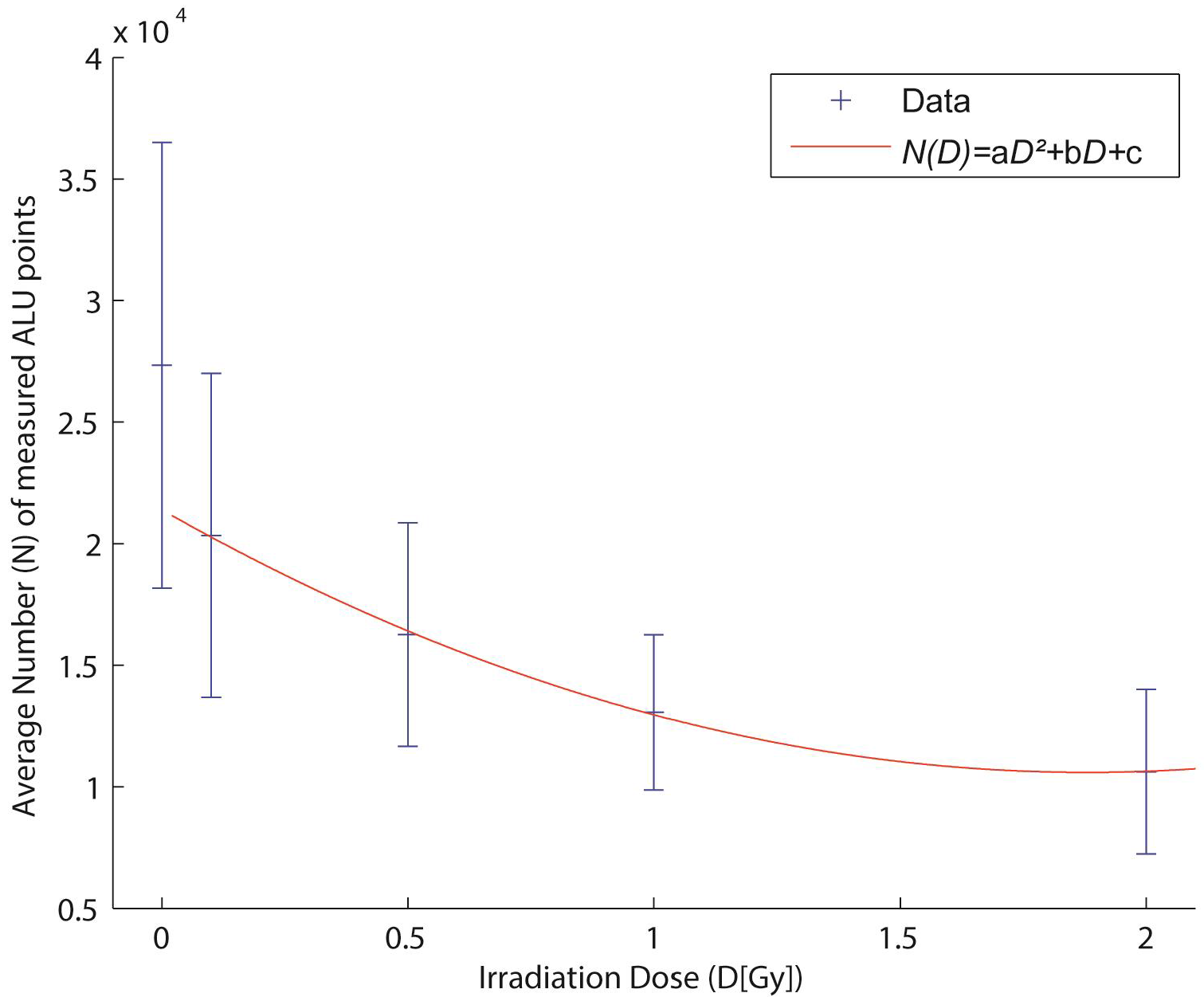
2.4. Biological Dosimetry by ALU Point Counting
3. Discussion
4. Materials and Methods
4.1. Staining Materials and Concentrations (Immunostaining and COMBO-FISH)
4.2. Handling Steps
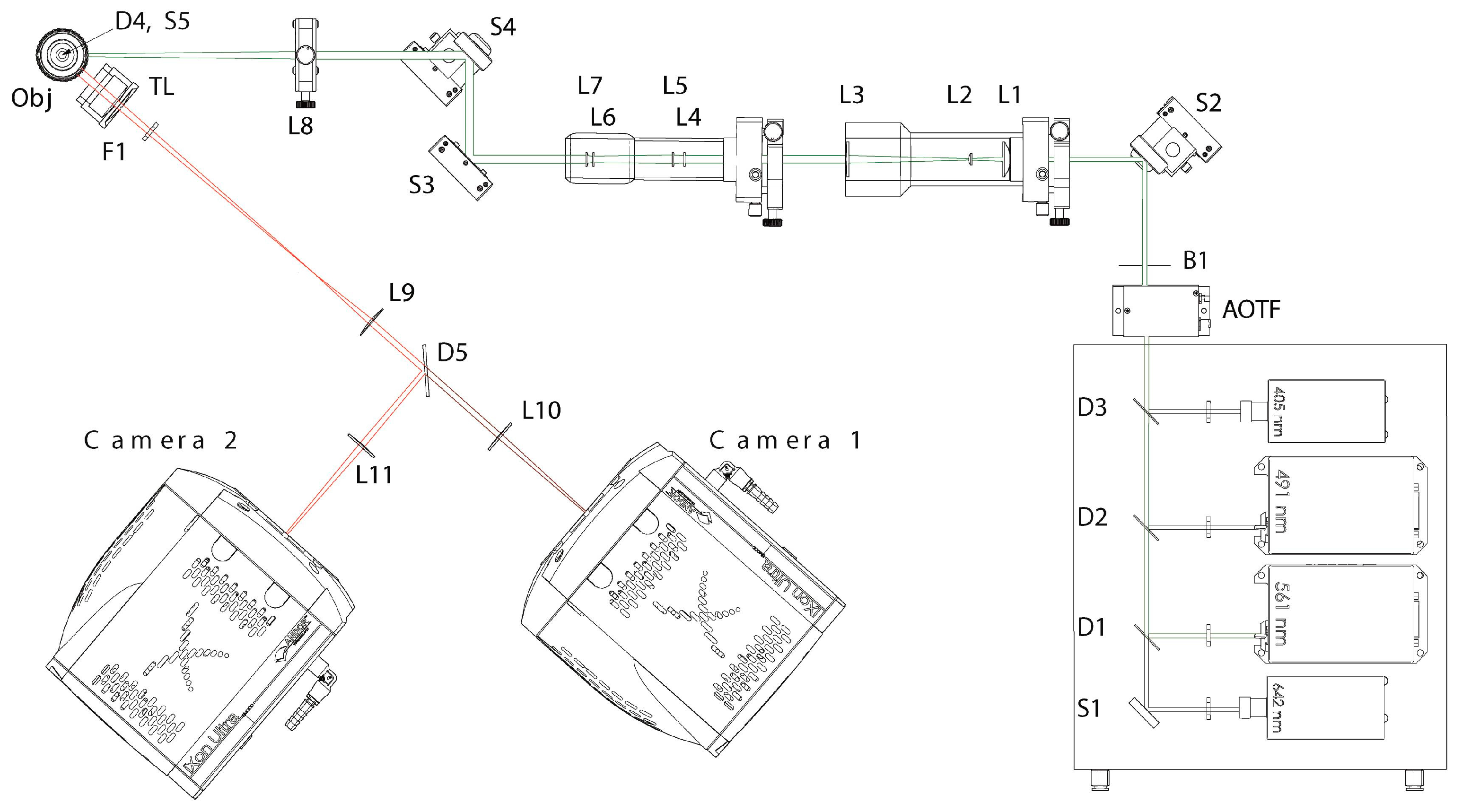
4.3. Microscopy Setup
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hausmann, M.; Cremer, C. Standardisation of FISH-procedures: Summary of the first discussion workshop. Anal. Cell. Pathol. 2003, 25, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Schermelleh, L.; Heintzmann, R.; Leonhardt, H. A guide to super-resolution fluorescence microscopy. J. Cell Biol. 2010, 190, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Cremer, C.; Masters, B.R. Resolution enhancement techniques in microscopy. Eur. Phys. J. H 2013, 38, 281–344. [Google Scholar] [CrossRef]
- Markaki, Y.; Smeets, D.; Fiedler, S.; Schmid, V.J.; Schermelleh, L.; Cremer, T.; Cremer, M. The potential of 3D-FISH and super-resolution structured illumination microscopy for studies of 3D nuclear architecture. Bioessays 2012, 34, 412–426. [Google Scholar] [CrossRef] [PubMed]
- Abaza, H.M.; Youssef, S.R.; Saad, A.A.; Kamal, G.M.; Hegazy, M.G.; Ibrahim, R.I.; Annaka, L.M. Detection of 14q32 rearrangements in multiple myeloma, using simultaneous FISH analysis combined with immunofluorescence. Hematol. Oncol. Stem Cell Ther. 2015, 8, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Mongelard, F.; Vourch, C.; Robert-Nicoud, M.; Usson, Y. Quantitative assessment of the alteration of chromatin during the course of FISH procedures. Cytometry 1999, 36, 96–101. [Google Scholar] [CrossRef]
- Winkler, R.; Perner, B.; Rapp, A.; Durm, M.; Cremer, C.; Greulich, K.O.; Hausmann, M. Labelling quality and chromosome morphology after low temperature FISH analysed by scanning far-field and near-field optical microscopy. J. Microsc. 2003, 209, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.I.; Röhse, H.; Rizzoli, S.O.; Opazo, F. Fluorescent in situ hybridization of synaptic proteins imaged with super-resolution STED microscopy. Microsc. Res. Tech. 2014, 77, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Raj, A.; van den Bogaard, P.; Rifkin, S.A.; van Oudenaarden, A.; Tyagi, S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods 2008, 5, 877. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.; Fiederling, F.; Bastmeyer, M. Practical limitations of superresolution imaging due to conventional sample preparation revealed by a direct comparison of CLSM, SIM and dSTORM. J. Microsc. 2015, 262, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Fornasiero, E.F.; Opazo, F. Super-resolution imaging for cell biologists. Bioessays 2015, 37, 436–451. [Google Scholar] [CrossRef] [PubMed]
- Anguiano, A. Fluorescence in situ hybridization (FISH): Overview and medical applications. J. Clin. Ligand Assay 2000, 23, 33–42. [Google Scholar]
- Wolf, D.; Rauch, J.; Hausmann, M.; Cremer, C. Comparison of the thermal denaturation behaviour of DNA-solutions and metaphase chromosome preparations in suspension. Biophys. Chem. 1999, 81, 207–221. [Google Scholar] [CrossRef]
- Rauch, J.; Wolf, D.; Hausmann, M.; Cremer, C. The influence of formamide on thermal denaturation profiles of DNA and metaphase chromosomes in suspension. Z. Naturforsch. C 2000, 55, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Hausmann, M.; Winkler, R.; Hildenbrand, G.; Finsterle, J.; Weisel, A.; Rapp, A.; Schmitt, E.; Janz, S.; Cremer, C. COMBO-FISH: Specific labeling of nondenatured chromatin targets by computer-selected DNA oligonucleotide probe combinations. Biotechniques 2003, 35, 564–577. [Google Scholar] [PubMed]
- Stuhlmüller, M.; Hausmann, M. Selection of COMBO-FISH probes for multi-purpose applications. J. Theor. Comput. Sci. 2015, 2, 131–132. [Google Scholar] [CrossRef]
- Schmitt, E.; Schwarz-Finsterle, J.; Stein, S.; Boxler, C.; Müller, P.; Mokhir, A.; Krämer, R.; Cremer, C.; Hausmann, M. COMBinatorial Oligo FISH: Directed labeling of specific genome domains in differentially fixed cell material and live cells. In Fluorescence In Situ Hybridization (FISH), Protocols and Applications; Bridger, J.M., Volpi, E.V., Eds.; Humana Press: New York, NY, USA, 2010; pp. 185–202. [Google Scholar]
- Schmitt, E.; Wagner, J.; Hausmann, M. Combinatorial selection of short triplex forming oligonucleotides for fluorescence in situ hybridisation COMBO-FISH. J. Comput. Sci. 2012, 3, 328–334. [Google Scholar] [CrossRef]
- Lemmer, P.; Gunkel, M.; Baddeley, D.; Kaufmann, R.; Urich, A.; Weiland, Y.; Reymann, J.; Müller, P.; Hausmann, M.; Cremer, C. SPDM: Light microscopy with single-molecule resolution at the nanoscale. Appl. Phys. B 2008, 93, 1–12. [Google Scholar] [CrossRef]
- Cremer, C.; Kaufmann, R.; Gunkel, M.; Pres, S.; Weiland, Y.; Müller, P.; Ruckelshausen, T.; Lemmer, P.; Geiger, F.; Gegenhard, S.; et al. Superresolution imaging of biological nanostructures by spectral precision distance microscopy. Biotechnol. J. 2011, 6, 1037–1051. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.; Schmitt, E.; Jacob, A.; Hoheisel, J.; Kaufmann, R.; Cremer, C.; Hausmann, M. COMBO-FISH enables high precision localization microscopy as a prerequisite for nanostructure analysis of genome loci. Int. J. Mol. Sci. 2010, 11, 4094–4105. [Google Scholar] [CrossRef] [PubMed]
- Stuhlmüller, M.; Schwarz-Finsterle, J.; Fey, E.; Lux, J.; Bach, M.; Cremer, C.; Hinderhofer, K.; Hausmann, M.; Hildenbrand, G. In situ optical sequencing and structure analysis of a trinucleotide repeat genome region by localization microscopy after specific COMBO-FISH nano-probing. Nanoscale 2015, 7, 17938–17946. [Google Scholar] [CrossRef] [PubMed]
- Müller, P.; Rößler, J.; Schwarz-Finsterle, J.; Schmitt, E.; Hausmann, M. PNA-COMBO-FISH: From combinatorial probe design in silico to vitality compatible, specific labelling of gene targets in cell nuclei. Exp. Cell Res. 2016, 345, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Deininger, P. ALU elements: Know the SINEs. Genome Biol. 2011, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Batzer, M.A.; Deininger, P.L. ALU repeats and human genomic diversity. Nat. Rev. Genet. 2002, 3, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ara, T.; Gautheret, D. Using ALU elements as polyadenylation sites: A case of retroposon exaptation. Mol. Biol. Evol. 2009, 26, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Morales, M.E.; White, T.B.; Streva, V.A.; DeFreece, C.B.; Hedges, D.J.; Deininger, P.L. The contribution of ALU elements to mutagenic DNA double-strand break repair. PLoS Genet. 2015, 11, e1005016. [Google Scholar] [CrossRef] [PubMed]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Jin, K.; Crabbe, M.J.C.; Zhang, Y.; Liu, X.; Huang, Y.; Hua, M.; Nan, P.; Zhang, Z.; Zhong, Y. Enrichment analysis of ALU elements with different spatial chromatin proximity in the human genome. Protein Cell 2016, 7, 250–266. [Google Scholar] [CrossRef] [PubMed]
- Tsirigos, A.; Rigoutsos, I. ALU and b1 repeats have been selectively retained in the upstream and intronic regions of genes of specific functional classes. PLOS Comput. Biol. 2009, 5, e1000610. [Google Scholar] [CrossRef] [PubMed]
- Welsch, P.L.; King, M.C. BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum. Mol. Genet. 2001, 10, 705–713. [Google Scholar]
- Chénais, B.; Caruso, A.; Hiard, S.; Casse, N. The impact of transposable elements on eukaryotic genomes: From genome size increase to genetic adaptation to stressful environments. Gene 2012, 509, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Huang, S. Nuclear function of ALUs. Nucleus 2014, 5, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [PubMed]
- Hess, S.T.; Girirajan, T.P.; Mason, M.D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 2006, 91, 4258–4272. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Bates, M.; Zhuang, X. Super-resolution fluorescence microscopy. Annu. Rev. Biochem. 2009, 78, 993–1016. [Google Scholar] [CrossRef] [PubMed]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Heilemann, M.; van de Linde, S.; Schüttpelz, M.; Kaspar, R.; Seefeldt, B.; Mukherjee, A.; Tinnefeld, P.; Sauer, M. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew. Chem. Int. Ed. 2008, 47, 6172–6176. [Google Scholar] [CrossRef] [PubMed]
- Fölling, J.; Bossi, M.; Bock, H.; Medda, R.; Wurm, C.A.; Hein, B.; Jakobs, S.; Eggeling, C.; Hell, S.W. Fluorescence nanoscopy by ground-state depletion and single-molecule return. Nat. Methods 2008, 5, 943–945. [Google Scholar] [CrossRef] [PubMed]
- Dertinger, T.; Colyer, R.; Iyer, G.; Weiss, S.; Enderlein, J. Fast, background-free, 3D super-resolution optical fluctuation imaging (SOFI). Proc. Natl. Acad. Sci. USA 2009, 106, 22287–22292. [Google Scholar] [CrossRef] [PubMed]
- Baddeley, D.; Crossman, D.; Rossberger, S.; Cheyne, J.E.; Montgomery, J.M.; Jayasinghe, I.D.; Cremer, C.; Cannell, M.B.; Soeller, C. 4D super-resolution microscopy with conventional fluorophores and single wavelength excitation in optically thick cells and tissues. PLoS ONE 2011, 6, e20645. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, R.; Lemmer, P.; Gunkel, M.; Weiland, Y.; Müller, P.; Hausmann, M.; Baddeley, D.; Amberger, R.; Cremer, C. SPDM: Single Molecule Superresolution of Cellular Nanostructures. In Single Molecule Spectroscopy and Imaging II, Proceedings of SPIE; SPIE BiOS: Biomedical Optics; International Society for Optics and Photonics: San Jose, CA, USA, 2009. [Google Scholar]
- Grull, F.; Kirchgessner, M.; Kaufmann, R.; Hausmann, M.; Kebschull, U. Accelerating Image Analysis for Localization Microscopy with FPGAs. In Proceedings of the 21st International Conference on Field Programmable Logic and Applications (FPL), Chania, Greece, 5–7 September 2011. [Google Scholar]
- Lemmer, P.; Gunkel, M.; Weiland, Y.; Müller, P.; Baddeley, D.; Kaufmann, R.; Urich, A.; Eipel, H.; Amberger, R.; Hausmann, M.; et al. Using conventional fluorescent markers for far-field fluorescence localization nanoscopy allows resolution in the 10-nm range. J. Microsc. 2009, 235, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Máté, G.; Müller, P.; Hillebrandt, S.; Krufczik, M.; Bach, M.; Kaufmann, R.; Hausmann, M.; Heermann, D.W. Radiation induced chromatin conformation changes analysed by fluorescent localization microscopy, statistical physics, and graph theory. PLoS ONE 2015, 10, e0128555. [Google Scholar] [CrossRef] [PubMed]
- Boyd, P.S.; Struve, N.; Bach, M.; Eberle, J.P.; Gote, M.; Schock, F.; Cremer, C.; Kriegs, M.; Hausmann, M. Clustered localization of EGFRvIII in glioblastoma cells as detected by high precision localization microscopy. Nanoscale 2016, 8, 20037–20047. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, R.; Müller, P.; Hildenbrand, G.; Hausmann, M.; Cremer, C. Analysis of Her2/neu membrane protein clusters in different types of breast cancer cells using localization microscopy. J. Microsc. 2011, 242, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.S.; Nicetto, D.; Zaret, K.S. H3K9me3-dependent heterochromatin: Barrier to cell fate changes. Trends Genet. 2016, 32, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Bolzer, A.; Kreth, G.; Solovei, I.; Koehler, D.; Saracoglu, K.; Fauth, C.; Muller, S.; Eils, R.; Cremer, C.; Speicher, M.R.; et al. Three-dimensional maps of all chromosomes in human male fibroblast nuclei and prometaphase rosettes. PLoS Biol. 2005, 3, e157. [Google Scholar] [CrossRef] [PubMed]
- RepeatMasker. Available online: http://www.repeatmasker.org (accessed on 7 August 2015).
- Su, M.; Han, D.; Boyd-Kirkup, J.; Yu, X.; Han, J.J. Evolution of ALU elements toward enhancers. Cell Rep. 2014, 7, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.C.; Karpen, G.H. Epigenetic regulation of heterochromatic DNA stability. Curr. Opin. Genet. Dev. 2008, 18, 204. [Google Scholar] [CrossRef] [PubMed]
- Sander, J.; Ester, M.; Kriegel, H.P.; Xu, X. Density-based clustering in spatial databases: The algorithm gdbscan and its applications. Data Min. Knowl. Discov. 1998, 2, 169–194. [Google Scholar] [CrossRef]
- Braden, B. The surveyor’s area formula. Coll. Math. J. 1986, 17, 326–337. [Google Scholar] [CrossRef]
- Chen, C.; Gentles, A.J.; Jurka, J.; Karlin, S. Genes, pseudogenes, and ALU sequence organization across human chromosomes 21 and 22. Proc. Natl. Acad. Sci. USA 2002, 99, 2930–2935. [Google Scholar] [CrossRef] [PubMed]
- Lorat, Y.; Schanz, S.; Schuler, N.; Wennemuth, G.; Rübe, C.; Rübe, C.E. Beyond repair foci: DNA double-strand break repair in euchromatic and heterochromatic compartments analyzed by transmission electron microscopy. PLoS ONE 2012, 7, e38165. [Google Scholar] [CrossRef] [PubMed]
- Schwarz-Finsterle, J.; Stein, S.; Großmann, C.; Schmitt, E.; Trakhtenbrot, L.; Rechavi, G.; Amariglio, N.; Cremer, C.; Hausmann, M. Comparison of triple helical COMBO-FISH and standard FISH by means of quantitative microscopic image analysis of abl/bcr positions in cell nuclei. J. Biochem. Biophys. Methods 2007, 70, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Weiland, Y.; Lemmer, P.; Cremer, C. Combining FISH with localisation microscopy: Super-resolution imaging of nuclear genome nanostructures. Chromosome Res. 2011, 19, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.; Hausmann, M.; Lukášová, E.; Biswas, A.; Hildenbrand, G.; Davídková, M.; Krasavin, E.; Kleibl, Z.; Falkova, I.; Jezkova, L.; et al. Giving OMICS spatiotemporal dimensions by challenging microscopy: From functional networks to structural organization of cell nuclei elucidating mechanisms of complex radiation damage response and chromatin repair—PART A (Radiomics). Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.; Hausmann, M.; Lukášová, E.; Biswas, A.; Hildenbrand, G.; Davídková, M.; Krasavin, E.; Kleibl, Z.; Falkova, I.; Jezkova, L.; et al. Giving OMICS spatiotemporal dimensions by challenging microscopy: From functional networks to structural organization of cell nuclei elucidating mechanisms of complex radiation damage response and chromatin repair—PART B (Structuromics). Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 225–247. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, C.; Schiff, R. HER2: Biology, detection, and clinical implications. Arch. Pathol. Lab. Med. 2011, 135, 55–62. [Google Scholar] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krufczik, M.; Sievers, A.; Hausmann, A.; Lee, J.-H.; Hildenbrand, G.; Schaufler, W.; Hausmann, M. Combining Low Temperature Fluorescence DNA-Hybridization, Immunostaining, and Super-Resolution Localization Microscopy for Nano-Structure Analysis of ALU Elements and Their Influence on Chromatin Structure. Int. J. Mol. Sci. 2017, 18, 1005. https://doi.org/10.3390/ijms18051005
Krufczik M, Sievers A, Hausmann A, Lee J-H, Hildenbrand G, Schaufler W, Hausmann M. Combining Low Temperature Fluorescence DNA-Hybridization, Immunostaining, and Super-Resolution Localization Microscopy for Nano-Structure Analysis of ALU Elements and Their Influence on Chromatin Structure. International Journal of Molecular Sciences. 2017; 18(5):1005. https://doi.org/10.3390/ijms18051005
Chicago/Turabian StyleKrufczik, Matthias, Aaron Sievers, Annkathrin Hausmann, Jin-Ho Lee, Georg Hildenbrand, Wladimir Schaufler, and Michael Hausmann. 2017. "Combining Low Temperature Fluorescence DNA-Hybridization, Immunostaining, and Super-Resolution Localization Microscopy for Nano-Structure Analysis of ALU Elements and Their Influence on Chromatin Structure" International Journal of Molecular Sciences 18, no. 5: 1005. https://doi.org/10.3390/ijms18051005