
Involvement of Astrocytes in Mediating the Central Effects of Ghrelin


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Ghrelin and Its Receptor
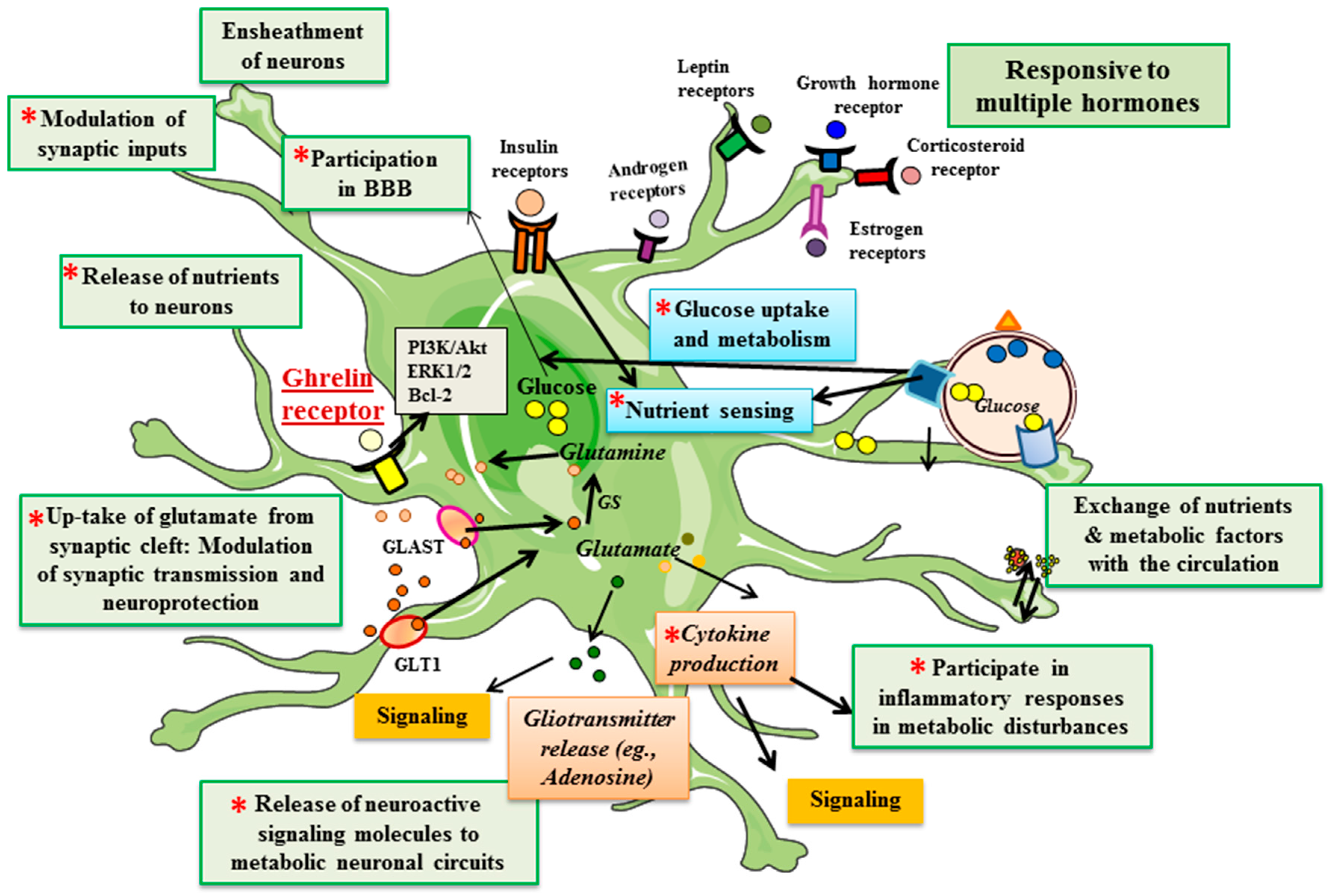
3. Astrocytes
4. Ghrelin Receptors in Astrocytes
5. Ghrelin and Astrocytes in Metabolic Control
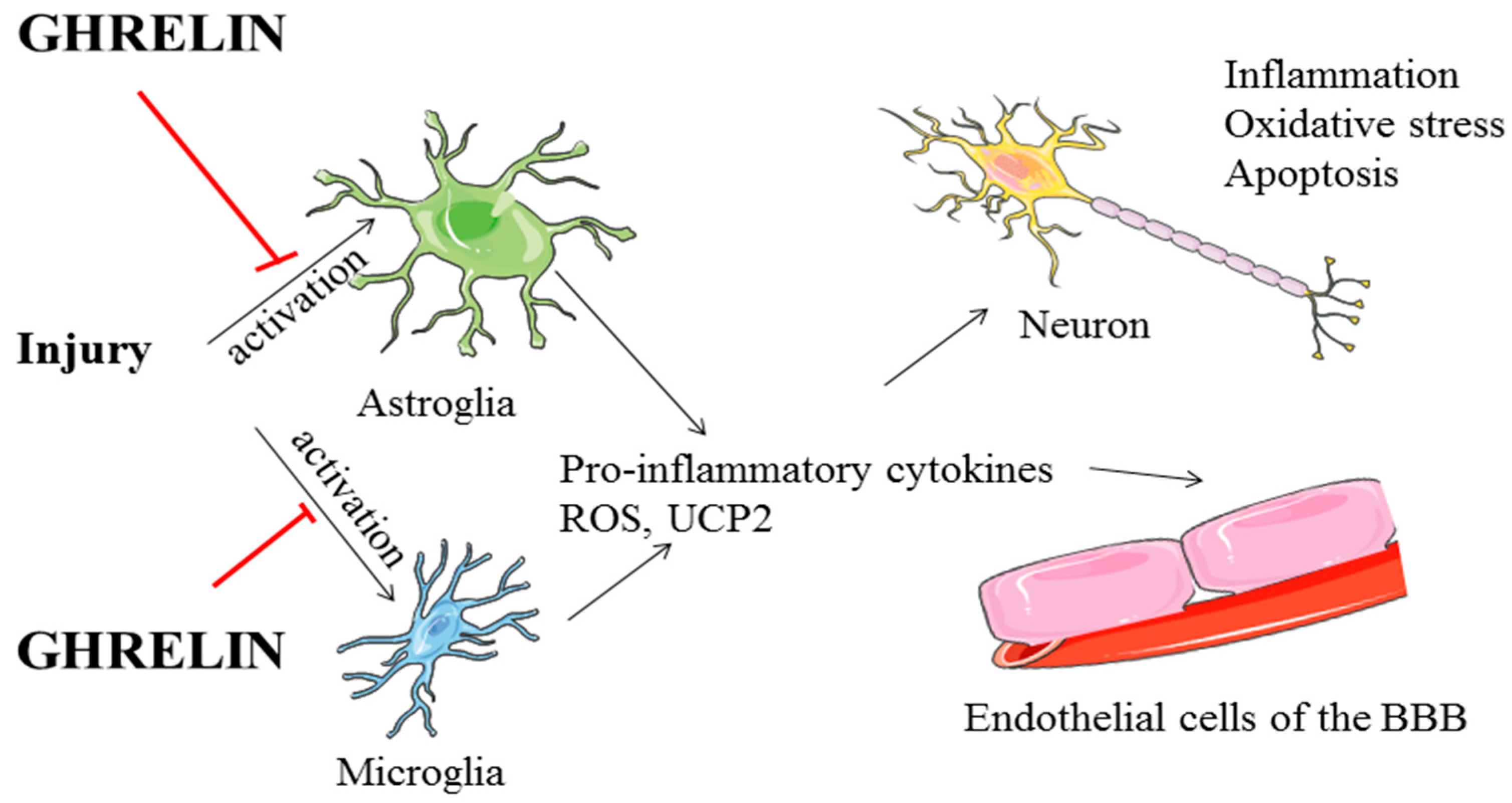
6. Ghrelin and Astrocytes in Neuroprotection
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Cowley, M.A.; Smith, R.G.; Diano, S.; Tschöp, M.; Pronchuk, N.; Grove, K.L.; Strasburger, C.J.; Bidlingmaier, M.; Esterman, M.; Heiman, M.L. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 2003, 37, 649–661. [Google Scholar] [CrossRef]
- Müller, T.D.; Nogueiras, R.; Andermann, M.L.; Andrews, Z.B.; Anker, S.D.; Argente, J.; Batterham, R.L.; Benoit, S.C.; Bowers, C.Y.; Broglio, F.; et al. Ghrelin. Mol. Metab. 2015, 4, 437–460. [Google Scholar] [CrossRef] [PubMed]
- Nakazato, M.; Murakami, N.; Date, Y.; Kojima, M.; Matsuo, H.; Kangawa, K.; Matsukura, S. A role for ghrelin in the central regulation of feeding. Nature 2001, 409, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Schmid, S.M.; Hallschmid, M.; Jauch-Chara, K.; Born, J.; Schultes, B. A single night of sleep deprivation increases ghrelin levels and feelings of hunger in normal-weight healthy men. J. Sleep Res. 2008, 17, 331–334. [Google Scholar] [CrossRef] [PubMed]
- Dickson, S.L.; Luckman, S.M. Induction of c-fos messenger ribonucleic acid in neuropeptide y and growth hormone (GH)-releasing factor neurons in the rat arcuate nucleus following systemic injection of the GH secretagogue, GH-releasing peptide-6. Endocrinology 1997, 138, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Jerlhag, E.; Egecioglu, E.; Landgren, S.; Salomé, N.; Heilig, M.; Moechars, D.; Datta, R.; Perrissoud, D.; Dickson, S.L.; Engel, J.A. Requirement of central ghrelin signaling for alcohol reward. Proc. Natl. Acad. Sci. USA 2009, 106, 11318–11323. [Google Scholar] [CrossRef] [PubMed]
- Ahima, R.S.; Antwi, D.A. Brain regulation of appetite and satiety. Endocrinol. Metab. Clin. N. Am. 2008, 37, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Dickson, S.L.; Egecioglu, E.; Landgren, S.; Skibicka, K.P.; Engel, J.A.; Jerlhag, E. The role of the central ghrelin system in reward from food and chemical drugs. Mol. Cell. Endocrinol. 2011, 340, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Skibicka, K.P.; Hansson, C.; Egecioglu, E.; Dickson, S.L. Role of ghrelin in food reward: Impact of ghrelin on sucrose self-administration and mesolimbic dopamine and acetylcholine receptor gene expression. Addict. Biol. 2012, 17, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Diano, S.; Farr, S.A.; Benoit, S.C.; McNay, E.C.; da Silva, I.; Horvath, B.; Gaskin, F.S.; Nonaka, N.; Jaeger, L.B.; Banks, W.A. Ghrelin controls hippocampal spine synapse density and memory performance. Nat. Neurosci. 2006, 9, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Kanoski, S.E.; Fortin, S.M.; Ricks, K.M.; Grill, H.J. Ghrelin signaling in the ventral hippocampus stimulates learned and motivational aspects of feeding via PI3K-Akt signaling. Biol. Psychiatry 2013, 73, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Andrews, Z.B.; Erion, D.; Beiler, R.; Liu, Z.-W.; Abizaid, A.; Zigman, J.; Elsworth, J.D.; Savitt, J.M.; DiMarchi, R.; Tschoep, M. Ghrelin promotes and protects nigrostriatal dopamine function via a UCP2-dependent mitochondrial mechanism. J. Neurosci. 2009, 29, 14057–14065. [Google Scholar] [CrossRef] [PubMed]
- Moon, M.; Kim, H.G.; Hwang, L.; Seo, J.-H.; Kim, S.; Hwang, S.; Kim, S.; Lee, D.; Chung, H.; Oh, M.S. Neuroprotective effect of ghrelin in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of parkinson’s disease by blocking microglial activation. Neurotox Res. 2009, 15, 332–347. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.V.; Krenick, R.; Ullian, E.; Tsai, H.-H.; Deneen, B.; Richardson, W.D.; Barres, B.A.; Rowitch, D.H. Astrocytes and disease: A neurodevelopmental perspective. Genes Dev. 2012, 26, 891–907. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, M.; Magistretti, P.J. The role of astroglia in neuroprotection. Dialogues Clin. Neurosci. 2009, 11, 281–295. [Google Scholar] [PubMed]
- Fuente-Martin, E.; Garcia-Caceres, C.; Morselli, E.; Clegg, D.; Chowen, J.; Finan, B.; Brinton, R.; Tschöp, M. Estrogen, astrocytes and the neuroendocrine control of metabolism. Rev. Endocr. Metab. Disord. 2013, 14, 331–338. [Google Scholar] [CrossRef] [PubMed]
- García-Cáceres, C.; Fuente-Martín, E.; Argente, J.; Chowen, J.A. Emerging role of glial cells in the control of body weight. Mol. Metab. 2012, 1, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Baquedano, E.; Chowen, J.A.; Argente, J.; Frago, L.M. Differential effects of GH and GH-releasing peptide-6 on astrocytes. J. Endocrinol. 2013, 218, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Fuente-Martín, E.; García-Cáceres, C.; Argente-Arizón, P.; Díaz, F.; Granado, M.; Freire-Regatillo, A.; Castro-González, D.; Ceballos, M.L.; Frago, L.M.; Dickson, S.L.; et al. Ghrelin regulates glucose and glutamate transporters in hypothalamic astrocytes. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Caceres, C.; Fuente-Martin, E.; Diaz, F.; Granado, M.; Argente-Arizon, P.; Frago, L.M.; Freire-Regatillo, A.; Barrios, V.; Argente, J.; Chowen, J.A. The opposing effects of ghrelin on hypothalamic and systemic inflammatory processes are modulated by its acylation status and food intake in male rats. Endocrinology 2014, 155, 2868–2880. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Inui, A. Ghrelin: An orexigenic and somatotrophic signal from the stomach. Nat. Rev. Neurosci. 2001, 2, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Asakawa, A.; Inui, A.; Kaga, O.; Yuzuriha, H.; Nagata, T.; Ueno, N.; Makino, S.; Fujimiya, M.; Niijima, A.; Fujino, M.A.; et al. Ghrelin is an appetite-stimulatory signal from stomach with structural resemblance to motilin. Gastroenterology 2001, 120, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Tschöp, M.; Smiley, D.L.; Heiman, M.L. Ghrelin induces adiposity in rodents. Nature 2000, 407, 908–913. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.-M.; Yu, H.; Palyha, O.C.; McKee, K.K.; Feighner, S.D.; Sirinathsinghji, D.J.; Smith, R.G.; Van der Ploeg, L.H.; Howard, A.D. Distribution of mRNA encoding the growth hormone secretagogue receptor in brain and peripheral tissues. Mol. Brain Res. 1997, 48, 23–29. [Google Scholar] [CrossRef]
- Willesen, M.G.; Kristensen, P.; Rømer, J. Co-localization of growth hormone secretagogue receptor and NPY mRNA in the arcuate nucleus of the rat. Neuroendocrinology 1999, 70, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Schellekens, H.; Finger, B.C.; Dinan, T.G.; Cryan, J.F. Ghrelin signalling and obesity: At the interface of stress, mood and food reward. Pharmacol. Ther. 2012, 135, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Kenny, P.J. Reward mechanisms in obesity: New insights and future directions. Neuron 2011, 69, 664–679. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Q.; Du, X.; Li, Y.; Gong, B.; Shi, L.; Tang, T.; Jiang, H. The neurological effects of ghrelin in brain diseases: Beyond metabolic functions. Neurosci. Biobehav. Rev. 2017, 73, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Du, X.; Jiang, H.; Xie, J. Ghrelin and neurodegenerative disorders—A review. Mol. Neurobiol. 2016, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Stoyanova, I.I.; le Feber, J. Ghrelin accelerates synapse formation and activity development in cultured cortical networks. BMC Neurosci. 2014, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, J.A.; Solenberg, P.J.; Perkins, D.R.; Willency, J.A.; Knierman, M.D.; Jin, Z.; Witcher, D.R.; Luo, S.; Onyia, J.E.; Hale, J.E. Ghrelin octanoylation mediated by an orphan lipid transferase. Proc. Natl. Acad. Sci. USA 2008, 105, 6320–6325. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Brown, M.S.; Liang, G.; Grishin, N.V.; Goldstein, J.L. Identification of the acyltransferase that octanoylates ghrelin, an appetite-stimulating peptide hormone. Cell 2008, 132, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Hosoda, H.; Kojima, M.; Mizushima, T.; Shimizu, S.; Kangawa, K. Structural divergence of human ghrelin identification of multiple ghrelin-derived molecules produced by post-translational processing. J. Biol. Chem. 2003, 278, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Verhagen, L.A.W.; Egecioglu, E.; Luijendijk, M.C.M.; Hillebrand, J.J.G.; Adan, R.A.H.; Dickson, S.L. Acute and chronic suppression of the central ghrelin signaling system reveals a role in food anticipatory activity. Eur. Neuropsychopharmacol. 2011, 21, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Howard, A.D.; Feighner, S.D.; Cully, D.F.; Arena, J.P.; Liberator, P.A.; Rosenblum, C.I.; Hamelin, M.; Hreniuk, D.L.; Palyha, O.C.; Anderson, J.; et al. A receptor in pituitary and hypothalamus that functions in growth hormone release. Science 1996, 273, 974–977. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Johno, Y.; Hayashi, K.; Yakabi, K.; Tanaka, T.; Ro, S. Expression of c-fos protein in the brain after intravenous injection of ghrelin in rats. Neurosci. Lett. 2007, 417, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Tschöp, M.; Robinson, S.M.; Heiman, M.L. Extent and direction of ghrelin transport across the blood–brain barrier is determined by its unique primary structure. J. Pharmacol. Exp. Ther. 2002, 302, 822–827. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, M.; Langlet, F.; Lafont, C.; Molino, F.; Hodson, D.J.; Roux, T.; Lamarque, L.; Verdié, P.; Bourrier, E.; Dehouck, B. Rapid sensing of circulating ghrelin by hypothalamic appetite-modifying neurons. Proc. Natl. Acad. Sci. USA 2013, 110, 1512–1517. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S. Physiology and neurobiology of stress and adaptation: Central role of the brain. Physiol. Rev. 2007, 87, 873–904. [Google Scholar] [CrossRef] [PubMed]
- Ferrini, F.; Salio, C.; Lossi, L.; Merighi, A. Ghrelin in central neurons. Curr. Neuropharmacol. 2009, 7, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Bednarek, M.A.; Feighner, S.D.; Pong, S.-S.; McKee, K.K.; Hreniuk, D.L.; Silva, M.V.; Warren, V.A.; Howard, A.D.; van der Ploeg, L.H.Y.; Heck, J.V. Structure-function studies on the new growth hormone-releasing peptide, ghrelin: Minimal sequence of ghrelin necessary for activation of growth hormone secretagogue receptor 1a. J. Med. Chem. 2000, 43, 4370–4376. [Google Scholar] [CrossRef] [PubMed]
- Gauna, C.; van de Zande, B.; van Kerkwijk, A.; Themmen, A.P.N.; van der Lely, A.-J.; Delhanty, P.J.D. Unacylated ghrelin is not a functional antagonist but a full agonist of the type 1A growth hormone secretagogue receptor (GHS-R). Mol. Cell. Endocrinol. 2007, 274, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Heppner, K.M.; Piechowski, C.L.; Müller, A.; Ottaway, N.; Sisley, S.; Smiley, D.L.; Habegger, K.M.; Pfluger, P.T.; DiMarchi, R.; Biebermann, H. Both acyl and des-acyl ghrelin regulate adiposity and glucose metabolism via central nervous system ghrelin receptors. Diabetes 2014, 63, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Ariyasu, H.; Takaya, K.; Iwakura, H.; Hosoda, H.; Akamizu, T.; Arai, Y.; Kangawa, K.; Nakao, K. Transgenic mice overexpressing des-acyl ghrelin show small phenotype. Endocrinology 2005, 146, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Chow, K.B.; Sun, J.; Chu, K.M.; Cheung, W.T.; Cheng, C.H.; Wise, H. The truncated ghrelin receptor polypeptide (GHS-R1b) is localized in the endoplasmic reticulum where it forms heterodimers with ghrelin receptors (GHS-R1a) to attenuate their cell surface expression. Mol. Cell. Endocrinol. 2012, 348, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Navarro, G.; Aguinaga, D.; Angelats, E.; Medrano, M.; Moreno, E.; Mallol, J.; Cortes, A.; Canela, E.I.; Casado, V.; McCormick, P.J.; et al. A significant role of the truncated ghrelin receptor GHS-R1b in ghrelin-induced signaling in neurons. J. Biol. Chem. 2016, 291, 13048–13062. [Google Scholar] [CrossRef] [PubMed]
- Freeman, M.R. Specification and morphogenesis of astrocytes. Science 2010, 330, 774–778. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Reactive astrocytes in neural repair and protection. Neuroscientist 2005, 11, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Haim, L.B.; Rowitch, D.H. Functional diversity of astrocytes in neural circuit regulation. Nat. Rev. Neurosci 2017, 18, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Mong, J.; Blutstein, T. Estradiol modulation of astrocytic form and function: Implications for hormonal control of synaptic communication. Neuroscience 2006, 138, 967–975. [Google Scholar] [CrossRef] [PubMed]
- García-Cáceres, C.; Quarta, C.; Varela, L.; Gao, Y.; Gruber, T.; Legutko, B.; Jastroch, M.; Johansson, P.; Ninkovic, J.; Yi, C.X.; et al. Astrocytic insulin signaling couples brain glucose uptake with nutrient availability. Cell 2016, 166, 867–880. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Garzón, E.; Fernandez, A.M.; Perez-Alvarez, A.; Genis, L.; Bascuñana, P.; Fernandez de la Rosa, R.; Delgado, M.; Angel Pozo, M.; Moreno, E.; McCormick, P.J.; et al. The insulin-like growth factor I receptor regulates glucose transport by astrocytes. Glia 2016, 64, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Hosli, L.; Hosli, E. Receptors for dopamine and serotonin on astrocytes of cultured rat central nervous system. J. Physiol. 1987, 82, 191–195. [Google Scholar]
- Kruijver, F.P.; Balesar, R.; Espila, A.M.; Unmehopa, U.A.; Swaab, D.F. Estrogen-receptor-β distribution in the human hypothalamus: Similarities and differences with ERα distribution. J. Comp. Neurol. 2003, 466, 251–277. [Google Scholar] [CrossRef] [PubMed]
- Langub, M.C., Jr.; Watson, R., Jr. Estrogen receptor-immunoreactive glia, endothelia, and ependyma in guinea pig preoptic area and median eminence: Electron microscopy. Endocrinology 1992, 130, 364–372. [Google Scholar] [PubMed]
- Lorenz, B.; Garcia-Segura, L.M.; DonCarlos, L.L. Cellular phenotype of androgen receptor-immunoreactive nuclei in the developing and adult rat brain. J. Comp. Neurol. 2005, 492, 456–468. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.M.; Swanson, R.A. Astrocyte glutamate transport: Review of properties, regulation, and physiological functions. Glia 2000, 32, 1–14. [Google Scholar] [CrossRef]
- Diano, S.; Naftolin, F.; Horvath, T.L. Kainate glutamate receptors (GluR5–7) in the rat arcuate nucleus: Relationship to tanycytes, astrocytes, neurons and gonadal steroid receptors. J. Neuroendocrinol. 1998, 10, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Schousboe, A. Role of astrocytes in the maintenance and modulation of glutamatergic and gabaergic neurotransmission. Neurochem. Res. 2003, 28, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Steinhäuser, C. Ion channels in glial cells. Brain Res. Brain Res. Rev. 2000, 32, 380–412. [Google Scholar] [CrossRef]
- Sharma, G.; Vijayaraghavan, S. Nicotinic cholinergic signaling in hippocampal astrocytes involves calcium-induced calcium release from intracellular stores. Proc. Natl. Acad. Sci. USA 2001, 98, 4148–4153. [Google Scholar] [CrossRef] [PubMed]
- Cheunsuang, O.; Morris, R. Astrocytes in the arcuate nucleus and median eminence that take up a fluorescent dye from the circulation express leptin receptors and neuropeptide Y Y1 receptors. Glia 2005, 52, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.; Barres, B. Snapshot: Astrocytes in health and disease. Cell 2015, 162, 1170. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.L.; Magistretti, P.J.; Allaman, I. Regulation of neurotrophic factors and energy metabolism by antidepressants in astrocytes. Curr. Drug Targets 2013, 14, 1308–1321. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.-S.; Allen, N.J.; Eroglu, C. Astrocytes control synapse formation, function, and elimination. Cold Spring Harb. Perspect. Biol. 2015, 7, a020370. [Google Scholar] [CrossRef] [PubMed]
- Cabezas, R.; Avila, M.; Gonzalez, J.; El-Bacha, R.S.; Baez, E.; Garcia-Segura, L.M.; Jurado Coronel, J.C.; Capani, F.; Cardona-Gomez, G.P.; Barreto, G.E. Astrocytic modulation of blood–brain barrier: Perspectives on Parkinson’s disease. Front. Cell. Neurosci. 2014, 8, 211. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.A.; Burda, J.E.; Ren, Y.; Ao, Y.; O’Shea, T.M.; Kawaguchi, R.; Coppola, G.; Khakh, B.S.; Deming, T.J.; Sofroniew, M.V. Astrocyte scar formation aids central nervous system axon regeneration. Nature 2016, 532, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, J.R.; Herrmann, J.E.; Woo, M.J.; Tansey, K.E.; Doan, N.B.; Sofroniew, M.V. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J. Neurosci. 2004, 24, 2143–2155. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A. Astrocytes going live: Advances and challenges. J. Physiol. 2009, 587, 1639–1647. [Google Scholar] [CrossRef] [PubMed]
- Theodosis, D.T.; Poulain, D.A.; Oliet, S.H. Activity-dependent structural and functional plasticity of astrocyte-neuron interactions. Physiol. Rev. 2008, 88, 983–1008. [Google Scholar] [CrossRef] [PubMed]
- Raichle, M.E.; Mintun, M.A. Brain work and brain imaging. Annu. Rev. Neurosci. 2006, 29, 449–476. [Google Scholar] [CrossRef] [PubMed]
- Halassa, M.M.; Fellin, T.; Haydon, P.G. Tripartite synapses: Roles for astrocytic purines in the control of synaptic physiology and behavior. Neuropharmacology 2009, 57, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Petzold, G.C.; Albeanu, D.F.; Sato, T.F.; Murthy, V.N. Coupling of neural activity to blood flow in olfactory glomeruli is mediated by astrocytic pathways. Neuron 2008, 58, 897–910. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lou, N.; Xu, Q.; Tian, G.-F.; Peng, W.G.; Han, X.; Kang, J.; Takano, T.; Nedergaard, M. Astrocytic Ca2+ signaling evoked by sensory stimulation in vivo. Nat. Neurosci. 2006, 9, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Lushnikova, I.; Skibo, G.; Muller, D.; Nikonenko, I. Synaptic potentiation induces increased glial coverage of excitatory synapses in CA1 hippocampus. Hippocampus 2009, 19, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, J.; Lammert, G.; Meyer, U.; Krug, M. The influence of long-term potentiation on the spatial relationship between astrocyte processes and potentiated synapses in the dentate gyrus neuropil of rat brain. Brain Res. 1991, 560, 122–131. [Google Scholar] [CrossRef]
- Halassa, M.M.; Haydon, P.G. Integrated brain circuits: Astrocytic networks modulate neuronal activity and behavior. Annu. Rev. Physiol. 2010, 72, 335. [Google Scholar] [CrossRef] [PubMed]
- Ridet, J.; Privat, A.; Malhotra, S.; Gage, F. Reactive astrocytes: Cellular and molecular cues to biological function. Trends Neurosci. 1997, 20, 570–577. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed]
- Silver, J.; Miller, J.H. Regeneration beyond the glial scar. Nat. Rev. Neurosci. 2004, 5, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Pekny, M.; Nilsson, M. Astrocyte activation and reactive gliosis. Glia 2005, 50, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Robel, S.; Berninger, B.; Götz, M. The stem cell potential of glia: Lessons from reactive gliosis. Nat. Rev. Neurosci. 2011, 12, 88–104. [Google Scholar] [CrossRef] [PubMed]
- Halassa, M.M.; Fellin, T.; Haydon, P.G. The tripartite synapse: Roles for gliotransmission in health and disease. Trends Mol. Med. 2007, 13, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Volterra, A. Astrocytic dysfunction: Insights on the role in neurodegeneration. Brain Res. Bull. 2009, 80, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Oberheim, N.A.; Goldman, S.A.; Nedergaard, M. Heterogeneity of astrocytic form and function. Methods Mol. Biol. 2012, 814, 23–45. [Google Scholar] [PubMed]
- Haydon, P.G. Glia: Listening and talking to the synapse. Nat. Rev. Neurosci. 2001, 2, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Segura, L.M.; McCarthy, M.M. Minireview: Role of glia in neuroendocrine function. Endocrinology 2004, 145, 1082–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zigman, J.M.; Jones, J.E.; Lee, C.E.; Saper, C.B.; Elmquist, J.K. Expression of ghrelin receptor mRNA in the rat and the mouse brain. J. Comp. Neurol. 2006, 494, 528–548. [Google Scholar] [CrossRef] [PubMed]
- Gnanapavan, S.; Kola, B.; Bustin, S.A.; Morris, D.G.; McGee, P.; Fairclough, P.; Bhattacharya, S.; Carpenter, R.; Grossman, A.B.; Korbonits, M. The tissue distribution of the mRNA of ghrelin and subtypes of its receptor, GHS-R, in humans. J. Clin. Endocrinol. Metab. 2002, 87, 2988. [Google Scholar] [CrossRef]
- Kojima, M.; Kangawa, K. Ghrelin: Structure and function. Physiol. Rev. 2005, 85, 495–522. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Huang, S.M.; Chen, C.C.; Tsai, C.F.; Yeh, W.L.; Chou, S.J.; Hsieh, W.T.; Lu, D.Y. Ghrelin induces cell migration through GHS-R, CaMkII, AMPK, and NF-κB signaling pathway in glioma cells. J. Cell. Biochem. 2011, 112, 2931–2941. [Google Scholar] [CrossRef] [PubMed]
- Dixit, V.D.; Weeraratna, A.T.; Yang, H.; Bertak, D.; Cooper-Jenkins, A.; Riggins, G.J.; Eberhart, C.G.; Taub, D.D. Ghrelin and the growth hormone secretagogue receptor constitute a novel autocrine pathway in astrocytoma motility. J. Biol. Chem. 2006, 281, 16681–16690. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Enriori, P.J. Ghrelin: A journey from GH secretagogue to regulator of metabolism. Trans. Gastrointest Cancer 2014, 4, 14–27. [Google Scholar]
- Argente-Arizon, P.; Guerra-Cantera, S.; Garcia-Segura, L.M.; Argente, J.; Chowen, J.A. Glial cells and energy balance. J. Mol. Endocrinol. 2017, 58, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Chowen, J.A.; Argente-Arizon, P.; Freire-Regatillo, A.; Frago, L.M.; Horvath, T.L.; Argente, J. The role of astrocytes in the hypothalamic response and adaptation to metabolic signals. Prog. Neurobiol. 2016, 144, 68–87. [Google Scholar] [CrossRef] [PubMed]
- Levin, B.E.; Magnan, C.; Dunn-Meynell, A.; Le Foll, C. Metabolic sensing and the brain: Who, what, where, and how? Endocrinology 2011, 152, 2552–2557. [Google Scholar] [CrossRef] [PubMed]
- Pénicaud, L.; Leloup, C.; Lorsignol, A.; Alquier, T.; Guillod, E. Brain glucose sensing mechanism and glucose homeostasis. Curr. Opin. Clin. Nutr. Metab. Care 2002, 5, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Bak, L.K.; Schousboe, A.; Waagepetersen, H.S. The glutamate/GABA-glutamine cycle: Aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 2006, 98, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Prebil, M.; Vardjan, N.; Jensen, J.; Zorec, R.; Kreft, M. Dynamic monitoring of cytosolic glucose in single astrocytes. Glia 2011, 59, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Mangia, S.; Simpson, I.A.; Vannucci, S.J.; Carruthers, A. The in vivo neuron-to-astrocyte lactate shuttle in human brain: Evidence from modeling of measured lactate levels during visual stimulation. J. Neurochem. 2009, 109, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.K.T.; Gutierrez-Juarez, R.; Pocai, A.; Rossetti, L. Regulation of blood glucose by hypothalamic pyruvate metabolism. Science 2005, 309, 943–947. [Google Scholar] [CrossRef] [PubMed]
- Fuente-Martin, E.; Garcia-Caceres, C.; Granado, M.; de Ceballos, M.L.; Sanchez-Garrido, M.A.; Sarman, B.; Liu, Z.W.; Dietrich, M.O.; Tena-Sempere, M.; Argente-Arizon, P.; et al. Leptin regulates glutamate and glucose transporters in hypothalamic astrocytes. J. Clin. Investig. 2012, 122, 3900–3913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.G.; Suyama, S.; Koch, M.; Jin, S.; Argente-Arizon, P.; Argente, J.; Liu, Z.-W.; Zimmer, M.R.; Jeong, J.K.; Szigeti-Buck, K.; et al. Leptin signaling in astrocytes regulates hypothalamic neuronal circuits and feeding. Nat. Neurosci. 2014, 17, 908–910. [Google Scholar] [CrossRef] [PubMed]
- Burdakov, D.; Luckman, S.M.; Verkhratsky, A. Glucose-sensing neurons of the hypothalamus. Philos. Trans. R. Soc. B Biol. Sci. 2005, 360, 2227–2235. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L.; Bouzier-Sore, A.K.; Aubert, A.; Serres, S.; Merle, M.; Costalat, R.; Magistretti, P.J. Activity-dependent regulation of energy metabolism by astrocytes: An update. Glia 2007, 55, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L.; Halestrap, A.P.; Pierre, K. Cellular and subcellular distribution of monocarboxylate transporters in cultured brain cells and in the adult brain. J. Neurosci. Res. 2005, 79, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Pierre, K.; Pellerin, L.; Debernardi, R.; Riederer, B.M.; Magistretti, P.J. Cell-specific localization of monocarboxylate transporters, MCT1 and MCT2, in the adult mouse brain revealed by double immunohistochemical labeling and confocal microscopy. Neuroscience 2000, 100, 617–627. [Google Scholar] [CrossRef]
- Dringen, R.; Gebhardt, R.; Hamprecht, B. Glycogen in astrocytes: Possible function as lactate supply for neighboring cells. Brain Res. 1993, 623, 208–214. [Google Scholar] [CrossRef]
- Sobrino Crespo, C.; Perianes Cachero, A.; Puebla Jiménez, L.; Barrios, V.; Arilla Ferreiro, E. Peptides and food intake. Front. Endocrinol. 2014, 5, 58. [Google Scholar] [CrossRef] [PubMed]
- Pinto, S.; Roseberry, A.G.; Liu, H.; Diano, S.; Shanabrough, M.; Cai, X.; Friedman, J.M.; Horvath, T.L. Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science 2004, 304, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Horvath, T.L.; Sarman, B.; García-Cáceres, C.; Enriori, P.J.; Sotonyi, P.; Shanabrough, M.; Borok, E.; Argente, J.; Chowen, J.A.; Perez-Tilve, D.; et al. Synaptic input organization of the melanocortin system predicts diet-induced hypothalamic reactive gliosis and obesity. Proc. Natl. Acad. Sci. USA 2010, 107, 14875–14880. [Google Scholar] [CrossRef] [PubMed]
- Argente-Arizón, P.; Freire-Regatillo, A.; Argente, J.; Chowen, J.A. Role of non-neuronal cells in body weight and appetite control. Front. Endocrinol. 2015, 6, 42. [Google Scholar]
- Belgardt, B.F.; Okamura, T.; Brüning, J.C. Hormone and glucose signalling in POMC and AGRP neurons. J. Physiol. 2009, 587, 5305–5314. [Google Scholar] [CrossRef] [PubMed]
- García-Cáceres, C.; Fuente-Martín, E.; Burgos-Ramos, E.; Granado, M.; Frago, L.M.; Barrios, V.; Horvath, T.; Argente, J.; Chowen, J.A. Differential acute and chronic effects of leptin on hypothalamic astrocyte morphology and synaptic protein levels. Endocrinology 2011, 152, 1809–1818. [Google Scholar] [CrossRef] [PubMed]
- Fuente-Martin, E.; Garcia-Caceres, C.; Granado, M.; Sanchez-Garrido, M.A.; Tena-Sempere, M.; Frago, L.M.; Argente, J.; Chowen, J.A. Early postnatal overnutrition increases adipose tissue accrual in response to a sucrose-enriched diet. Am. J. Physiol. Endocrinol. Metab. 2012, 302, 1586–1598. [Google Scholar] [CrossRef] [PubMed]
- Balland, E.; Dam, J.; Langlet, F.; Caron, E.; Steculorum, S.; Messina, A.; Rasika, S.; Falluel-Morel, A.; Anouar, Y.; Dehouck, B.; et al. Hypothalamic tanycytes are an ERK-gated conduit for leptin into the brain. Cell Metab. 2014, 19, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Collden, G.; Balland, E.; Parkash, J.; Caron, E.; Langlet, F.; Prevot, V.; Bouret, S.G. Neonatal overnutrition causes early alterations in the central response to peripheral ghrelin. Mol. Metab. 2015, 4, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Qi, Y.; Yang, Y. Astrocytes control food intake by inhibiting AGRP neuron activity via adenosine A1 receptors. Cell Rep. 2015, 11, 798–807. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.-L.; Zou, S.; Chen, J.-J.; Zhao, J.; Li, S. The neuroprotective effect of the association of aquaporin-4/glutamate transporter-1 against Alzheimer’s disease. Neural Plast. 2016, 2016, 4626593. [Google Scholar] [CrossRef] [PubMed]
- Glowatzki, E.; Cheng, N.; Hiel, H.; Yi, E.; Tanaka, K.; Ellis-Davies, G.C.R.; Rothstein, J.D.; Bergles, D.E. The glutamate-aspartate transporter GLAST mediates glutamate uptake at inner hair cell afferent synapses in the mammalian cochlea. J. Neurosci. 2006, 26, 7659. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.K.; Yasui, M. The transmembrane transporter domain of glutamate transporters is a process tip localizer. Sci. Rep. 2015, 5, 9032. [Google Scholar] [CrossRef] [PubMed]
- Aponte, Y.; Atasoy, D.; Sternson, S.M. AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat. Neurosci. 2011, 14, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Atasoy, D.; Su, H.H.; Sternson, S.M. Hunger states switch a flip-flop memory circuit via a synaptic AMPK-dependent positive feedback loop. Cell 2011, 146, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Denis, R.; Joly-Amado, A.; Webber, E.; Langlet, F.; Schaeffer, M.; Padilla, S.L.; Cansell, C.; Dehouck, B.; Castel, J.; Delbès, A.-S.; et al. Palatability can drive feeding independent of AGRP neurons. Cell Metab. 2015, 22, 646–657. [Google Scholar] [CrossRef] [PubMed]
- Luquet, S.; Phillips, C.T.; Palmiter, R.D. NPY/AGRP neurons are not essential for feeding responses to glucoprivation. Peptides 2007, 28, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Rubin, A.; Chowen, J.A.; Argente, J.; Frago, L.M. Growth hormone-releasing peptide 6 protection of hypothalamic neurons from glutamate excitotoxicity is caspase independent and not mediated by insulin-like growth factor I. Eur. J. Neurosci. 2009, 29, 2115–2124. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Rubin de Celix, A.; Chowen, J.A.; Argente, J.; Frago, L.M. Growth hormone releasing peptide-6 acts as a survival factor in glutamate-induced excitotoxicity. J. Neurochem. 2006, 99, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, Y.; Li, E.; Park, S. Ghrelin protects spinal cord motoneurons against chronic glutamate excitotoxicity by inhibiting microglial activation. Korean J. Physiol. Pharmacol. 2012, 16, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.; Lee, S.; Li, E.; Kim, Y.; Park, S. Ghrelin protects spinal cord motoneurons against chronic glutamate-induced excitotoxicity via ERK1/2 and phosphatidylinositol-3-kinase/AKT/glycogen synthase kinase-3β pathways. Exp. Neurol. 2011, 230, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Pellerin, L. Astrocytes couple synaptic activity to glucose utilization in the brain. Physiology 1999, 14, 177–182. [Google Scholar]
- Haam, J.; Halmos, K.C.; Di, S.; Tasker, J.G. Nutritional state-dependent ghrelin activation of vasopressin neurons via retrograde trans-neuronal-glial stimulation of excitatory GABA circuits. J. Neurosci. 2014, 34, 6201–6213. [Google Scholar] [CrossRef] [PubMed]
- Violante, I.R.; Anastasovska, J.; Sanchez-Canon, G.J.; Rodrigues, T.B.; Righi, V.; Nieto-Charques, L.; Parkinson, J.R.C.; Bloom, S.R.; Bell, J.D.; Cerdán, S. Cerebral activation by fasting induces lactate accumulation in the hypothalamus. Magn. Reson. Med. 2009, 62, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Fuente-Martin, E.; Granado, M.; Garcia-Caceres, C.; Sanchez-Garrido, M.A.; Frago, L.M.; Tena-Sempere, M.; Argente, J.; Chowen, J.A. Early nutritional changes induce sexually dimorphic long-term effects on body weight gain and the response to sucrose intake in adult rats. Metabolism 2012, 61, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Milanski, M.; Degasperi, G.; Coope, A.; Morari, J.; Denis, R.; Cintra, D.E.; Tsukumo, D.M.L.; Anhe, G.; Amaral, M.E.; Takahashi, H.K. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: Implications for the pathogenesis of obesity. J. Neurosci. 2009, 29, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Tang, J.; Yang, T.; Ma, H.; Yi, D.; Gu, C.; Yu, S. Cardioprotective effect of ghrelin in cardiopulmonary bypass involves a reduction in inflammatory response. PLoS ONE 2013, 8, e55021. [Google Scholar] [CrossRef] [PubMed]
- Dixit, V.D.; Schaffer, E.M.; Pyle, R.S.; Collins, G.D.; Sakthivel, S.K.; Palaniappan, R.; Lillard, J.W.; Taub, D.D. Ghrelin inhibits leptin-and activation-induced proinflammatory cytokine expression by human monocytes and T cells. J. Clin. Investig. 2004, 114, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Mulholland, M.; Zhang, W. Ghrelin O-acyltransferase (goat) and energy metabolism. Sci. China Life Sci. 2016, 59, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Zigman, J.M.; Bouret, S.G.; Andrews, Z.B. Obesity impairs the action of the neuroendocrine ghrelin system. Trends Endocrinol. Metab. 2016, 27, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Briggs, D.I.; Lockie, S.H.; Benzler, J.; Wu, Q.; Stark, R.; Reichenbach, A.; Hoy, A.J.; Lemus, M.B.; Coleman, H.A.; Parkington, H.C. Evidence that diet-induced hyperleptinemia, but not hypothalamic gliosis, causes ghrelin resistance in NPY/AGRP neurons of male mice. Endocrinology 2014, 155, 2411–2422. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Swanson, R.A. Astrocytes and brain injury. J. Cereb. Blood Flow Metab. 2003, 23, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lundkvist, A.; Andersson, D.; Wilhelmsson, U.; Nagai, N.; Pardo, A.C.; Nodin, C.; Ståhlberg, A.; Aprico, K.; Larsson, K. Protective role of reactive astrocytes in brain ischemia. J. Cereb. Blood Flow Metab. 2008, 28, 468–481. [Google Scholar] [CrossRef] [PubMed]
- Theodoric, N.; Bechberger, J.F.; Naus, C.C.; Sin, W.-C. Role of gap junction protein connexin43 in astrogliosis induced by brain injury. PLoS ONE 2012, 7, e47311. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, J.W.; Asher, R.A. The glial scar and central nervous system repair. Brain Res. Bull. 1999, 49, 377–391. [Google Scholar] [CrossRef]
- Wang, C.Y.; Yang, S.H.; Tzeng, S.F. MicroRNA-145 as one negative regulator of astrogliosis. Glia 2015, 63, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Liberto, C.M.; Albrecht, P.J.; Herx, L.M.; Yong, V.W.; Levison, S.W. Pro-regenerative properties of cytokine-activated astrocytes. J. Nurochem. 2004, 89, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Lopez, N.E.; Krzyzaniak, M.J.; Blow, C.; Putnam, J.; Ortiz-Pomales, Y.; Hageny, A.M.; Eliceiri, B.; Coimbra, R.; Bansal, V. Ghrelin prevents disruption of the blood–brain barrier after traumatic brain injury. J. Neurotrauma 2012, 29, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Lopez, N.E.; Gaston, L.; Lopez, K.R.; Coimbra, R.C.; Hageny, A.; Putnam, J.; Eliceiri, B.; Coimbra, R.; Bansal, V. Early ghrelin treatment attenuates disruption of the blood–brain barrier and apoptosis after traumatic brain injury through a UCP-2 mechanism. Brain Res. 2012, 1489, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Ku, J.M.; Taher, M.; Chin, K.Y.; Barsby, T.; Austin, V.; Wong, C.H.; Andrews, Z.B.; Spencer, S.J.; Miller, A.A. Protective actions of des-acylated ghrelin on brain injury and blood–brain barrier disruption after stroke in mice. Clin. Sci. 2016, 130, 1545–1558. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Li, E.; Kim, Y.; Kim, S.; Park, S. Multiple signaling pathways mediate ghrelin-induced proliferation of hippocampal neural stem cells. J. Endocrinol. 2013, 218, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Seo, S.; Moon, M.; Park, S. Phosphatidylinositol-3-kinase/Akt/glycogen synthase kinase-3β and ERK1/2 pathways mediate protective effects of acylated and unacylated ghrelin against oxygen–glucose deprivation-induced apoptosis in primary rat cortical neuronal cells. J. Endocrinol. 2008, 198, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Ren, Y.; Liu, X.; Li, W.G.; Yang, J.; Geng, B.; Weintraub, N.L.; Tang, C. Protective effects of ghrelin on ischemia/reperfusion injury in the isolated rat heart. J. Cardiovasc. Pharmacol. 2004, 43, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Konturek, P.C.; Brzozowski, T.; Walter, B.; Burnat, G.; Hess, T.; Hahn, E.G.; Konturek, S.J. Ghrelin-induced gastroprotection against ischemia-reperfusion injury involves an activation of sensory afferent nerves and hyperemia mediated by nitric oxide. Eur. J. Pharmacol. 2006, 536, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Íşeri, S.Ö.; Şener, G.; Yüksel, M.; Contuk, G.; Çetinel, Ş.; Gedik, N.; Yeğen, B.Ç. Ghrelin against alendronate-induced gastric damage in rats. J. Endocrinol. 2005, 187, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, S.; Park, J.; Yoo, Y. TNF-α-induced ROS production triggering apoptosis is directly linked to Romo1 and Bcl-XL. Cell Death Differ. 2010, 17, 1420–1434. [Google Scholar] [CrossRef] [PubMed]
- Baldanzi, G.; Filigheddu, N.; Cutrupi, S.; Catapano, F.; Bonissoni, S.; Fubini, A.; Malan, D.; Baj, G.; Granata, R.; Broglio, F. Ghrelin and des-acyl ghrelin inhibit cell death in cardiomyocytes and endothelial cells through ERK1/2 and PI3-kinase/Akt. J. Cell Biol. 2002, 159, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.G.; Jiang, H.; Sun, Y. Developments in ghrelin biology and potential clinical relevance. Trends Endocrinol. Metab. 2005, 16, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Frago, L.M.; Paneda, C.; Argente, J.; Chowen, J.A. Growth hormone-releasing peptide-6 increases insulin-like growth factor-I mRNA levels and activates AKT in RCA-6 cells as a model of neuropeptide y neurones. J. Neuroendocrinol. 2005, 17, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Frago, L.M.; Paneda, C.; Dickson, S.L.; Hewson, A.K.; Argente, J.; Chowen, J.A. Growth hormone (GH) and GH-releasing peptide-6 increase brain insulin-like growth factor-I expression and activate intracellular signaling pathways involved in neuroprotection. Endocrinology 2002, 143, 4113–4122. [Google Scholar] [CrossRef] [PubMed]
- Pañeda, C.; Arroba, A.I.; Frago, L.M.; Holm, A.M.; Rømer, J.; Argente, J.; Chowen, J.A. Growth hormone-releasing peptide-6 inhibits cerebellar cell death in aged rats. NeuroReport 2003, 14, 1633–1635. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, S.; Lin, Y.; Cao, L.; Wang, R.; Chi, Z. Ghrelin protects against cell death of hippocampal neurons in pilocarpine-induced seizures in rats. Neurosci. Lett. 2009, 453, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Jousilahti, P.; Bidel, S.; Antikainen, R.; Tuomilehto, J. Type 2 diabetes and the risk of parkinson’s disease. Diabetes Care 2007, 30, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Jousilahti, P.; Nissinen, A.; Antikainen, R.; Kivipelto, M.; Tuomilehto, J. Body mass index and the risk of parkinson disease. Neurology 2006, 67, 1955–1959. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Simpson, K.A.; Minnion, J.S.; Shillito, J.C.; Bloom, S.R. The role of gut hormones and the hypothalamus in appetite regulation. Endocr. J. 2010, 57, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, D. A life course of adiposity and dementia. Eur. J. Pharmacol. 2008, 585, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Kalmijn, S. Fatty acid intake and the risk of dementia and cognitive decline: A review of clinical and epidemiological studies. J. Nutr. Health Aging 2000, 4, 202–207. [Google Scholar] [PubMed]
- Sellbom, K.S.; Gunstad, J. Cognitive function and decline in obesity. J. Alzheimers Dis. 2012, 30, S89–S95. [Google Scholar] [PubMed]
- Spagnuolo, M.S.; Mollica, M.P.; Maresca, B.; Cavaliere, G.; Cefaliello, C.; Trinchese, G.; Scudiero, R.; Crispino, M.; Cigliano, L. High fat diet and inflammation—Modulation of haptoglobin level in rat brain. Front. Cell. Neurosci. 2015, 9, 479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, C.-X.; Heppner, K.M.; Kirchner, H.; Tong, J.; Bielohuby, M.; Gaylinn, B.D.; Müller, T.D.; Bartley, E.; Davis, H.W.; Zhao, Y. The goat-ghrelin system is not essential for hypoglycemia prevention during prolonged calorie restriction. PLoS ONE 2012, 7, e32100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Lluch, G.; Hunt, N.; Jones, B.; Zhu, M.; Jamieson, H.; Hilmer, S.; Cascajo, M.V.; Allard, J.; Ingram, D.K.; Navas, P.; et al. Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc. Natl. Acad. Sci. USA 2006, 103, 1768–1773. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Mattson, M.P. Dietary restriction and 2-deoxyglucose administration improve behavioral outcome and reduce degeneration of dopaminergic neurons in models of Parkinson’s disease. J. Neurosci. Res. 1999, 57, 195–206. [Google Scholar] [CrossRef]
- Maswood, N.; Young, J.; Tilmont, E.; Zhang, Z.; Gash, D.M.; Gerhardt, G.A.; Grondin, R.; Roth, G.S.; Mattison, J.; Lane, M.A. Caloric restriction increases neurotrophic factor levels and attenuates neurochemical and behavioral deficits in a primate model of parkinson’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 18171–18176. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Kim, E.; Lee, D.H.; Seo, S.; Ju, S.; Lee, D.; Kim, H.; Park, S. Ghrelin inhibits apoptosis in hypothalamic neuronal cells during oxygen-glucose deprivation. Endocrinology 2007, 148, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Li, L.-J.; Wang, J.; Xie, J.-X. Ghrelin antagonizes MPTP-induced neurotoxicity to the dopaminergic neurons in mouse substantia nigra. Exp. Neurol. 2008, 212, 532–537. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Song, N.; Xie, J.; Jiang, H. Ghrelin antagonized 1-methyl-4-phenylpyridinium (MPP+)-induced apoptosis in MES23.5 cells. J. Mol. Neurosci. 2009, 37, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Andrews, Z.B.; Diano, S.; Horvath, T.L. Mitochondrial uncoupling proteins in the CNS: In support of function and survival. Nat. Rev. Neurosci. 2005, 6, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Conti, B.; Sugama, S.; Lucero, J.; Winsky-Sommerer, R.; Wirz, S.A.; Maher, P.; Andrews, Z.; Barr, A.M.; Morale, M.C.; Paneda, C. Uncoupling protein 2 protects dopaminergic neurons from acute 1,2,3,6-methyl-phenyl-tetrahydropyridine toxicity. J. Neurochem. 2005, 93, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Carret-Rebillat, A.-S.; Pace, C.; Gourmaud, S.; Ravasi, L.; Montagne-Stora, S.; Longueville, S.; Tible, M.; Sudol, E.; Chang, R.C.-C.; Paquet, C. Neuroinflammation and Aβ accumulation linked to systemic inflammation are decreased by genetic PKR down-regulation. Sci. Rep. 2015, 5, 8489. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Maier, O. Interrelation of oxidative stress and inflammation in neurodegenerative disease: Role of TNF. Oxid. Med. Cell. Longev. 2015, 2015, 610813. [Google Scholar] [CrossRef] [PubMed]
- Spencer, S.J.; Miller, A.A.; Andrews, Z.B. The role of ghrelin in neuroprotection after ischemic brain injury. Brain Sci. 2013, 3, 344–359. [Google Scholar] [CrossRef] [PubMed]
- Gyoneva, S.; Ransohoff, R.M. Inflammatory reaction after traumatic brain injury: Therapeutic potential of targeting cell–cell communication by chemokines. Trends Pharmacol. Sci. 2015, 36, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Baatar, D.; Patel, K.; Taub, D.D. The effects of ghrelin on inflammation and the immune system. Mol. Cell. Endocrinol. 2011, 340, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Cheyuo, C.; Wu, R.; Zhou, M.; Jacob, A.; Coppa, G.; Wang, P. Ghrelin suppresses inflammation and neuronal nitric oxide synthase in focal cerebral ischemia via the vagus nerve. Shock 2011, 35, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Stevanovic, D.; Starcevic, V.; Vilimanovich, U.; Nesic, D.; Vucicevic, L.; Misirkic, M.; Janjetovic, K.; Savic, E.; Popadic, D.; Sudar, E.; et al. Immunomodulatory actions of central ghrelin in diet-induced energy imbalance. Brain Behav. Immun. 2012, 26, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Yune, T.Y. Ghrelin inhibits oligodendrocyte cell death by attenuating microglial activation. Endocrinol. Metab. 2014, 29, 371–378. [Google Scholar] [CrossRef]
- Kenny, R.; Cai, G.; Bayliss, J.A.; Clarke, M.; Choo, Y.L.; Miller, A.A.; Andrews, Z.B.; Spencer, S.J. Endogenous ghrelin’s role in hippocampal neuroprotection after global cerebral ischemia: Does endogenous ghrelin protect against global stroke? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R980–R990. [Google Scholar] [CrossRef] [PubMed]
- Bayliss, J.A.; Lemus, M.B.; Stark, R.; Santos, V.V.; Thompson, A.; Rees, D.J.; Galic, S.; Elsworth, J.D.; Kemp, B.E.; Davies, J.S.; et al. Ghrelin-AMPK signaling mediates the neuroprotective effects of calorie restriction in Parkinson’s disease. J. Neurosci. 2016, 36, 3049. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lim, E.; Kim, Y.; Li, E.; Park, S. Ghrelin attenuates kainic acid-induced neuronal cell death in the mouse hippocampus. J. Endocrinol. 2010, 205, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Hossienzadeh, F.; Babri, S.; Alipour, M.R.; Ebrahimi, H.; Mohaddes, G. Effect of ghrelin on brain edema induced by acute and chronic systemic hypoxia. Neurosci. Lett. 2013, 534, 47–51. [Google Scholar] [CrossRef]
- Ravizza, T.; Balosso, S.; Vezzani, A. Inflammation and prevention of epileptogenesis. Neurosci. Lett. 2011, 497, 223–230. [Google Scholar] [CrossRef]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frago, L.M.; Chowen, J.A. Involvement of Astrocytes in Mediating the Central Effects of Ghrelin. Int. J. Mol. Sci. 2017, 18, 536. https://doi.org/10.3390/ijms18030536
Frago LM, Chowen JA. Involvement of Astrocytes in Mediating the Central Effects of Ghrelin. International Journal of Molecular Sciences. 2017; 18(3):536. https://doi.org/10.3390/ijms18030536
Chicago/Turabian StyleFrago, Laura M., and Julie A. Chowen. 2017. "Involvement of Astrocytes in Mediating the Central Effects of Ghrelin" International Journal of Molecular Sciences 18, no. 3: 536. https://doi.org/10.3390/ijms18030536