
Splice Variants of the RTK Family: Their Role in Tumour Progression and Response to Targeted Therapy

Abstract
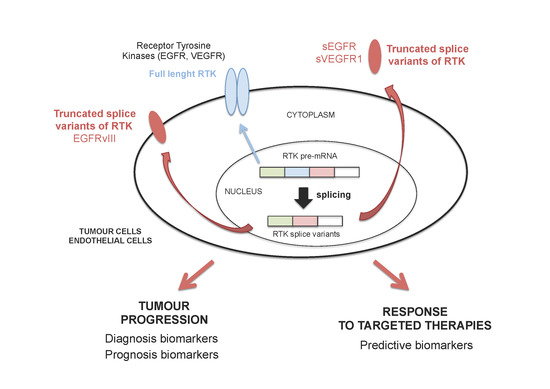
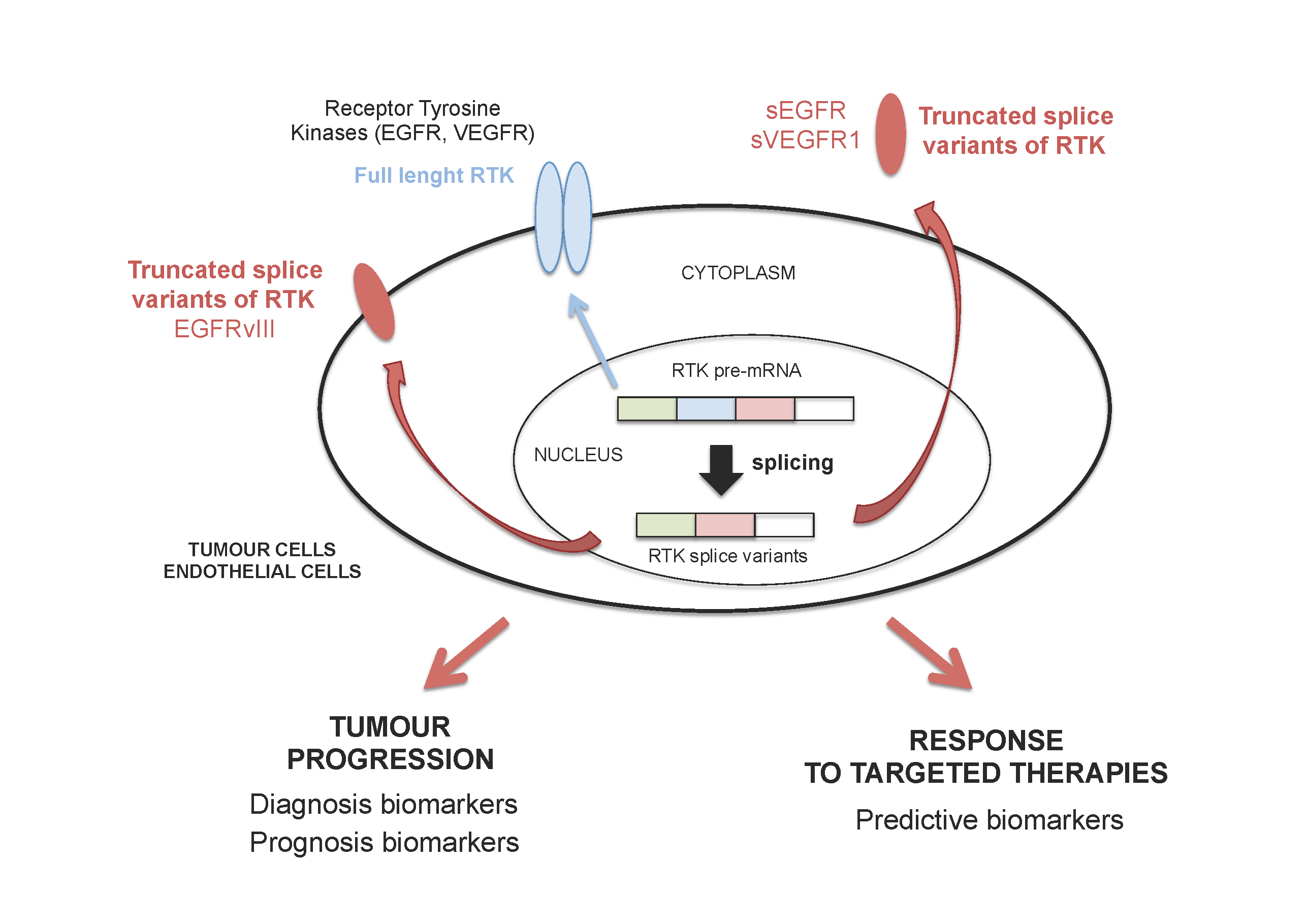
:
1. Introduction
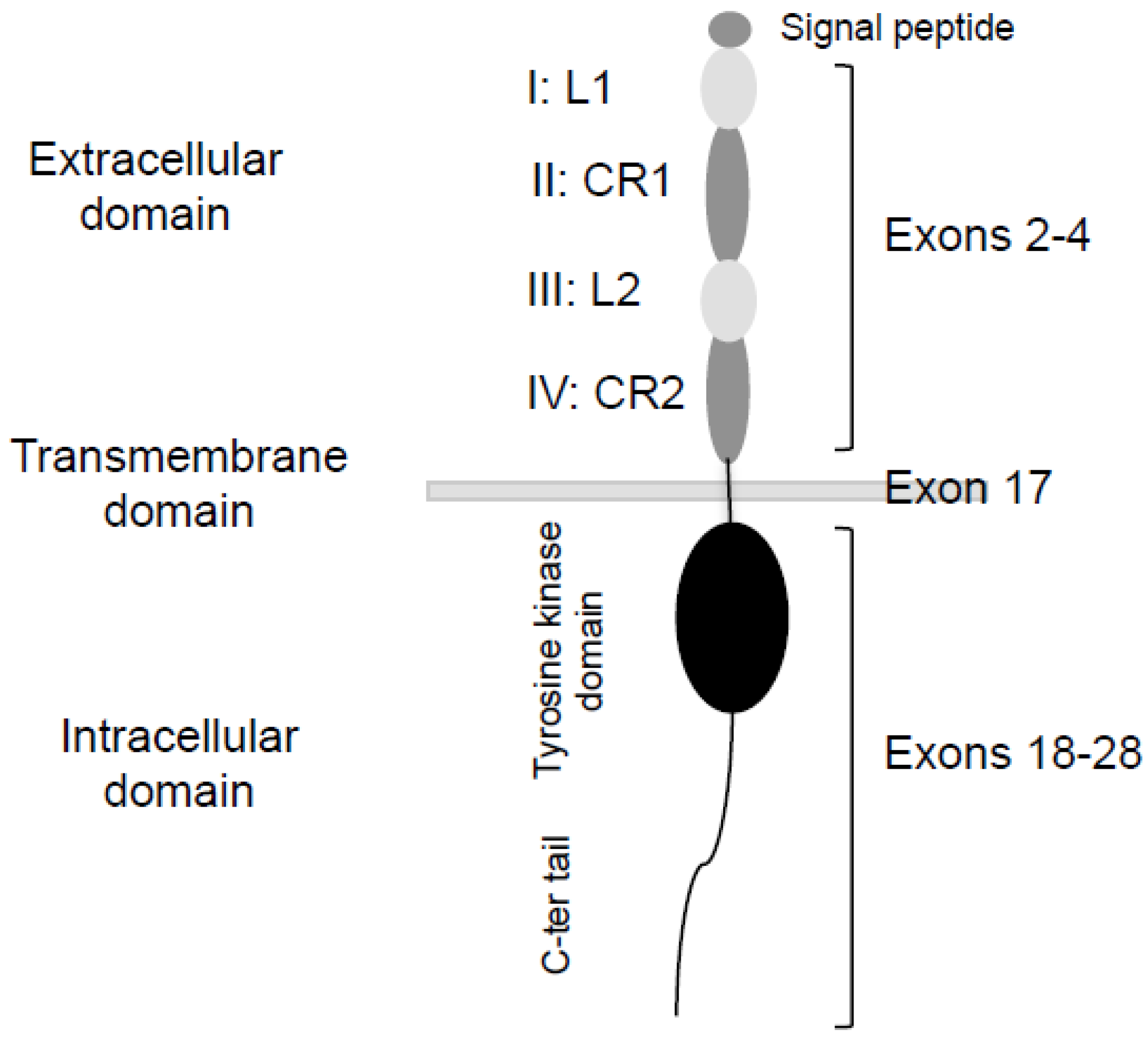
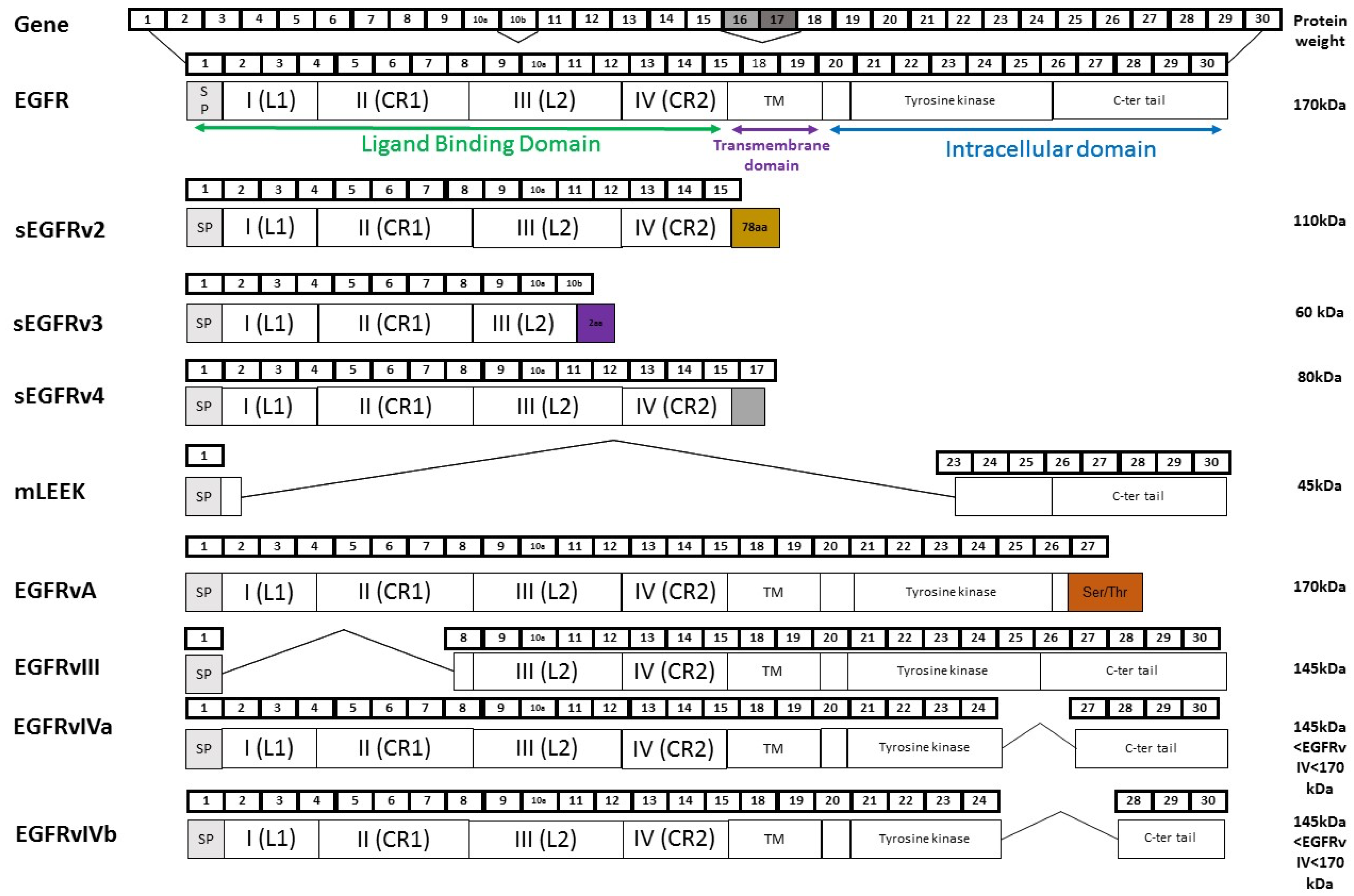
2. Splicing of EGFR: An Alternative Method to Control Tumour Progression
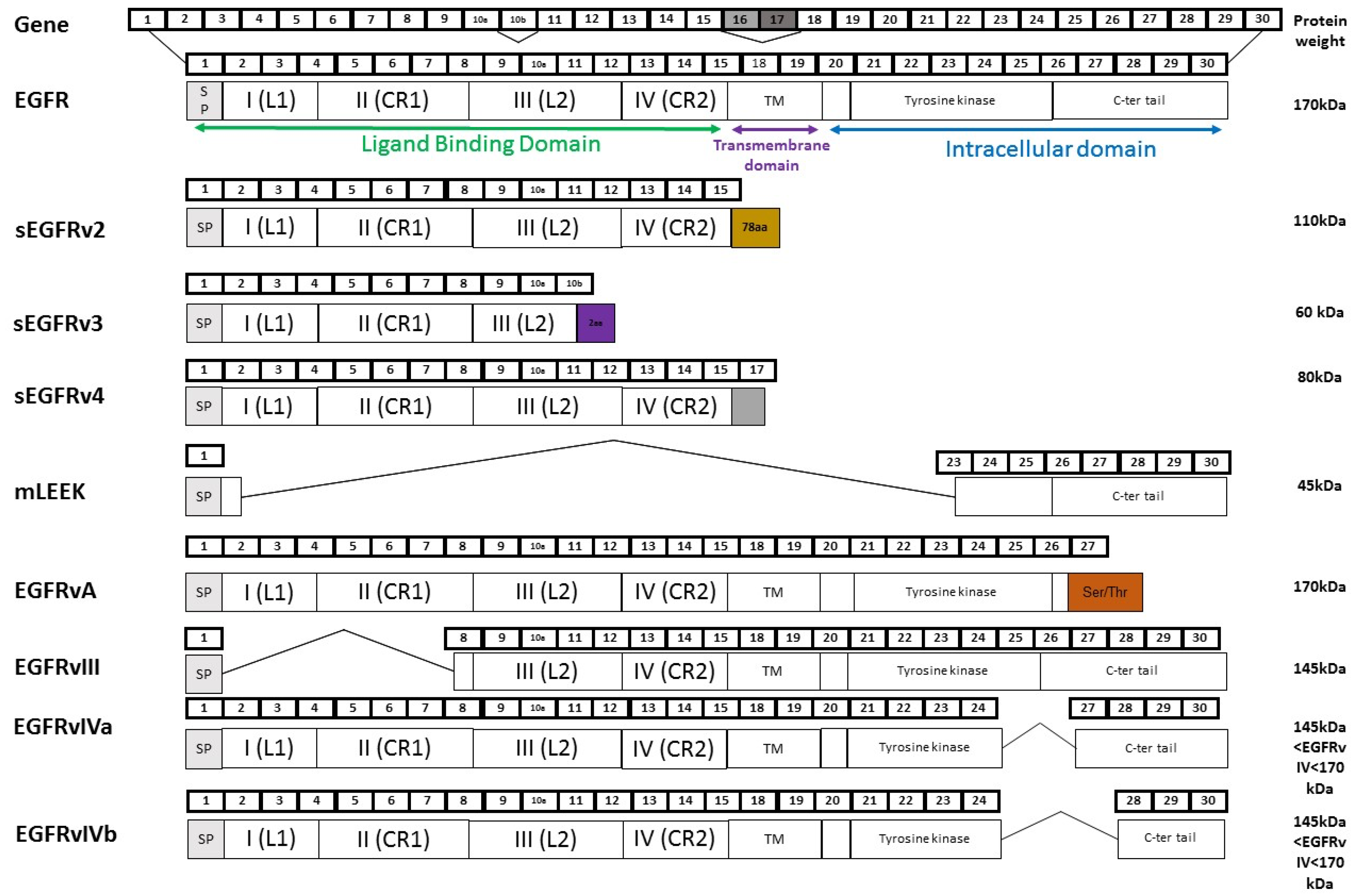
2.1. Soluble EGFR Variants
2.2. EGFRvIII
2.3. EGFRvIV
2.4. EGFRvA
2.5. mLEEK
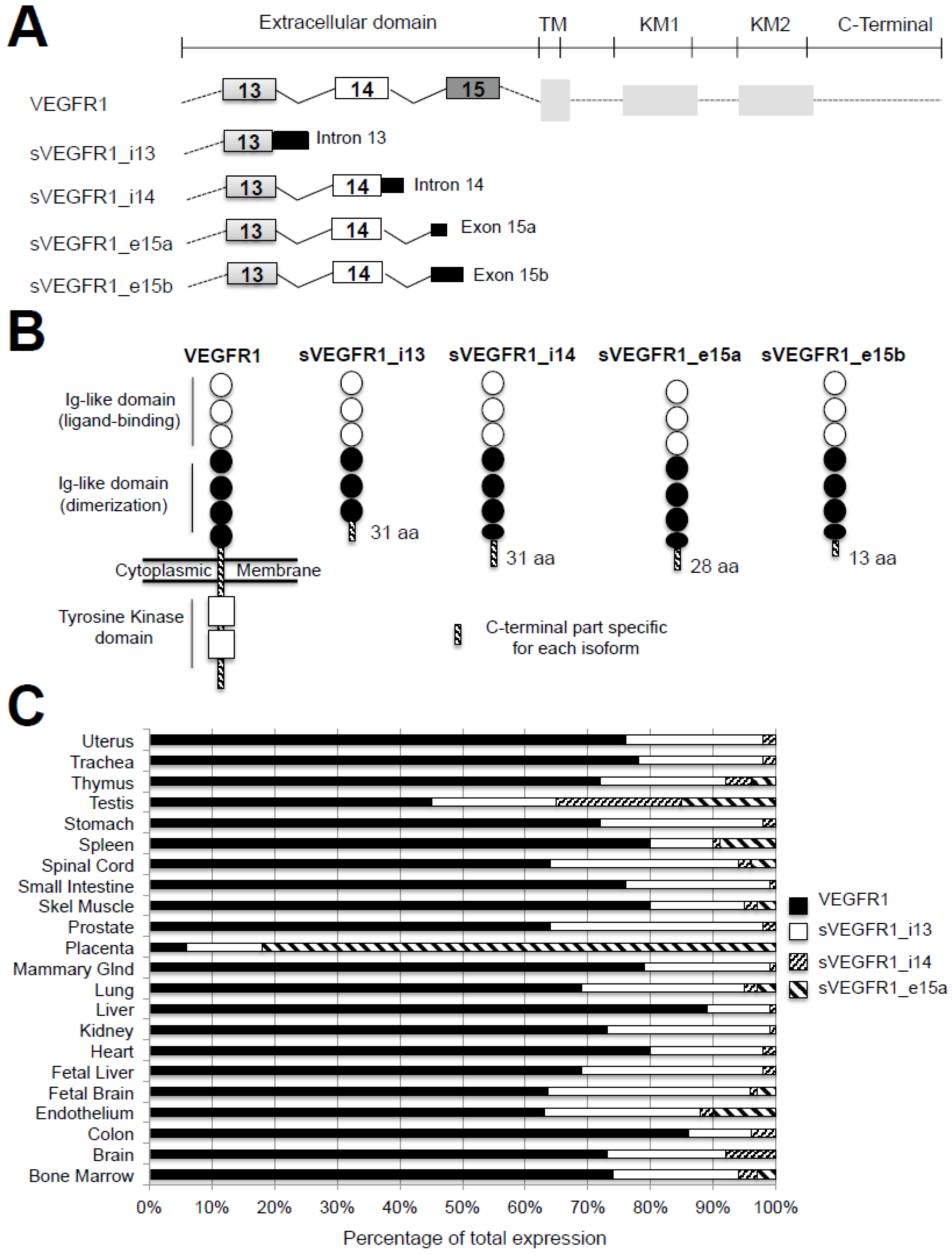
3. Alternative Splicing of VEGFR1: From Anti-Angiogenic to Pro-Angiogenic Factors
3.1. VEGFR1 Splice Variants
3.2. Expression and Regulation of sVEGFR1 in Tissues
3.3. Role of Vascular Functions of sVEGFR1s in Physiological and Pathological Conditions
3.4. Role of sVEGFR1 in Tumour Progression
3.5. sVEGFR1 as a Prognostic Biomarker in Cancer
3.6. sVEGFR1 as a Biomarker of Tumour Response to Therapies
4. Conclusions
Acknowledgements
Conflicts of Interest
References
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef]
- Brivanlou, A.H.; Darnell, J.E., Jr. Signal transduction and the control of gene expression. Science 2002, 295, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signalling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, A.; Schlessinger, J. Signal transduction by receptors with tyrosine kinase activity. Cell 1990, 61, 203–212. [Google Scholar] [CrossRef]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Sultan, M.; Schulz, M.H.; Richard, H.; Magen, A.; Klingenhoff, A.; Scherf, M.; Seifert, M.; Borodina, T.; Soldatov, A.; Parkhomchuk, D.; et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 2008, 321, 956–960. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Manley, J.L. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes Dev. 2010, 24, 2343–2364. [Google Scholar]
- Chen, G.; Wang, J.; Liu, Z.; Kornmann, M. Exon III splicing of fibroblast growth factor receptor 1 is modulated by growth factors and cyclin D1. Pancreas 2008, 37, 159–164. [Google Scholar] [CrossRef] [PubMed]
- De Figueiredo-Pontes, L.L.; Wong, D.W.; Tin, V.P.; Chung, L.P.; Yasuda, H.; Yamaguchi, N.; Nakayama, S.; Janne, P.A.; Wong, M.P.; Kobayashi, S.S.; et al. Identification and characterization of ALK kinase splicing isoforms in non-small-cell lung cancer. J. Thorac. Oncol. 2014, 9, 248–253. [Google Scholar] [CrossRef] [PubMed]
- O’Bryan, J.P.; Frye, R.A.; Cogswell, P.C.; Neubauer, A.; Kitch, B.; Prokop, C.; Espinosa, R.; Le Beau, M.M.; Earp, H.S.; Liu, E.T. AXL, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol. Cell. Biol. 1991, 11, 5016–5031. [Google Scholar] [CrossRef] [PubMed]
- Vogel, W.; Gish, G.D.; Alves, F.; Pawson, T. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol. Cell 1997, 1, 13–23. [Google Scholar] [CrossRef]
- Guillaudeau, A.; Durand, K.; Bessette, B.; Chaunavel, A.; Pommepuy, I.; Projetti, F.; Robert, S.; Caire, F.; Rabinovitch-Chable, H.; Labrousse, F. EGFR soluble isoforms and their transcripts are expressed in meningiomas. PLoS ONE 2012, 7, e37204. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Cvrljevic, A.N.; Johns, T.G. The epidermal growth factor receptor variant III (EGFRvIII): Where wild things are altered. FEBS J. 2013, 280, 5350–5370. [Google Scholar] [CrossRef] [PubMed]
- Padfield, E.; Ellis, H.P.; Kurian, K.M. Current therapeutic advances targeting EGFR and EGFRvIII in glioblastoma. Front. Oncol. 2015, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Piccione, E.C.; Lieu, T.J.; Gentile, C.F.; Williams, T.R.; Connolly, A.J.; Godwin, A.K.; Koong, A.C.; Wong, A.J. A novel epidermal growth factor receptor variant lacking multiple domains directly activates transcription and is overexpressed in tumors. Oncogene 2012, 31, 2953–2967. [Google Scholar] [CrossRef] [PubMed]
- Sundvall, M.; Korhonen, A.; Paatero, I.; Gaudio, E.; Melino, G.; Croce, C.M.; Aqeilan, R.I.; Elenius, K. Isoform-specific monoubiquitination, endocytosis, and degradation of alternatively spliced ErbB4 isoforms. Proc. Natl. Acad. Sci. USA 2008, 105, 4162–4167. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Greendorfer, J.S.; Jiao, J.; Kelpke, S.C.; Thompson, J.A. Alternatively spliced FGFR-1 isoforms differentially modulate endothelial cell activation of c-YES. Arch. Biochem. Biophys. 2006, 450, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Eswarakumar, V.P.; Lax, I.; Schlessinger, J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005, 16, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Malakar, P.; Chartarifsky, L.; Hija, A.; Leibowitz, G.; Glaser, B.; Dor, Y.; Karni, R. Insulin receptor alternative splicing is regulated by insulin signaling and modulates β cell survival. Sci. Rep. 2016, 6, 31222. [Google Scholar] [CrossRef] [PubMed]
- Frampton, G.M.; Ali, S.M.; Rosenzweig, M.; Chmielecki, J.; Lu, X.; Bauer, T.M.; Akimov, M.; Bufill, J.A.; Lee, C.; Jentz, D.; et al. Activation of met via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to met inhibitors. Cancer Discov. 2015, 5, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Wang, R.; Ye, T.; Yu, S.; Hu, H.; Shen, X.; Li, Y.; Ji, H.; Sun, Y.; Chen, H. MET exon 14 skipping defines a unique molecular class of non-small cell lung cancer. Oncotarget 2016, 7, 41691–41702. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.S.; Rodrigues, D.M.; Hyndman, B.D.; Crupi, M.J.; Nicolescu, A.C.; Mulligan, L.M. Alternative splicing results in RET isoforms with distinct trafficking properties. Mol. Biol. Cell 2012, 23, 3838–3850. [Google Scholar] [CrossRef] [PubMed]
- Ghigna, C.; de Toledo, M.; Bonomi, S.; Valacca, C.; Gallo, S.; Apicella, M.; Eperon, I.; Tazi, J.; Biamonti, G. Pro-metastatic splicing of ron proto-oncogene mrna can be reversed: Therapeutic potential of bifunctional oligonucleotides and indole derivatives. RNA Biol. 2010, 7, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Krishnaswamy, S.; Mohammed, A.K.; Amer, O.E.; Tripathi, G.; Alokail, M.S.; Al-Daghri, N.M. Novel splicing variants of recepteur d’origine nantais (RON) tyrosine kinase involving exons 15–19 in lung cancer. Lung Cancer 2016, 92, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Vaishnavi, A.; Le, A.T.; Doebele, R.C. TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov. 2015, 5, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Kendall, R.L.; Wang, G.; Thomas, K.A. Identification of a natural soluble form of the vascular endothelial growth factor receptor, FLT-1, and its heterodimerization with KDR. Biochem. Biophys. Res. Commun. 1996, 226, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Vorlova, S.; Rocco, G.; Lefave, C.V.; Jodelka, F.M.; Hess, K.; Hastings, M.L.; Henke, E.; Cartegni, L. Induction of antagonistic soluble decoy receptor tyrosine kinases by intronic polyA activation. Mol. Cell 2011, 43, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Meyerhardt, J.A.; Ancukiewicz, M.; Abrams, T.A.; Schrag, D.; Enzinger, P.C.; Chan, J.A.; Kulke, M.H.; Wolpin, B.M.; Goldstein, M.; Blaszkowsky, L.; et al. Phase I study of cetuximab, irinotecan, and vandetanib (ZD6474) as therapy for patients with previously treated metastastic colorectal cancer. PLoS ONE 2012, 7, e38231. [Google Scholar] [CrossRef] [PubMed]
- Heist, R.S.; Duda, D.G.; Sahani, D.V.; Ancukiewicz, M.; Fidias, P.; Sequist, L.V.; Temel, J.S.; Shaw, A.T.; Pennell, N.A.; Neal, J.W.; et al. Improved tumor vascularization after anti-VEGF therapy with carboplatin and nab-paclitaxel associates with survival in lung cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 1547–1552. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ibrahimi, O.A.; Olsen, S.K.; Umemori, H.; Mohammadi, M.; Ornitz, D.M. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J. Biol. Chem. 2006, 281, 15694–15700. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Fukabori, Y.; McBride, G.; Nikolaropolous, S.; McKeehan, W.L. Exon switching and activation of stromal and embryonic fibroblast growth factor (FGF)-FGF receptor genes in prostate epithelial cells accompany stromal independence and malignancy. Mol. Cell. Biol. 1993, 13, 4513–4522. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.Y.; Lambert, Q.T.; Reuther, G.W.; Der, C.J. Involvement of fibroblast growth factor receptor 2 isoform switching in mammary oncogenesis. Mol. Cancer Res. 2008, 6, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.Q.; He, C.; Chen, Y.Q.; Wang, D.; Wang, M.H. Altered expression of the RON receptor tyrosine kinase in primary human colorectal adenocarcinomas: Generation of different splicing RON variants and their oncogenic potential. Oncogene 2003, 22, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Mitra, D.; Brumlik, M.J.; Okamgba, S.U.; Zhu, Y.; Duplessis, T.T.; Parvani, J.G.; Lesko, S.M.; Brogi, E.; Jones, F.E. An oncogenic isoform of HER2 associated with locally disseminated breast cancer and trastuzumab resistance. Mol. Cancer Ther. 2009, 8, 2152–2162. [Google Scholar] [CrossRef] [PubMed]
- Jorissen, R.N.; Walker, F.; Pouliot, N.; Garrett, T.P.; Ward, C.W.; Burgess, A.W. Epidermal growth factor receptor: Mechanisms of activation and signalling. Exp. Cell Res. 2003, 284, 31–53. [Google Scholar] [CrossRef]
- Ferguson, K.M. Structure-based view of epidermal growth factor receptor regulation. Annu. Rev. Biophys. 2008, 37, 353–373. [Google Scholar] [CrossRef] [PubMed]
- Perez-Torres, M.; Valle, B.L.; Maihle, N.J.; Negron-Vega, L.; Nieves-Alicea, R.; Cora, E.M. Shedding of epidermal growth factor receptor is a regulated process that occurs with overexpression in malignant cells. Exp. Cell Res. 2008, 314, 2907–2918. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, M.P.; Keller, S.; Alonso, A.; Riedle, S.; Dempsey, P.J.; Altevogt, P. Generation of novel, secreted epidermal growth factor receptor (EGFR/ErbB1) isoforms via metalloprotease-dependent ectodomain shedding and exosome secretion. J. Cell. Biochem. 2008, 103, 1783–1797. [Google Scholar] [CrossRef] [PubMed]
- Reiter, J.L.; Maihle, N.J. A 1.8 kb alternative transcript from the human epidermal growth factor receptor gene encodes a truncated form of the receptor. Nucleic Acids Res. 1996, 24, 4050–4056. [Google Scholar] [CrossRef] [PubMed]
- Reiter, J.L.; Threadgill, D.W.; Eley, G.D.; Strunk, K.E.; Danielsen, A.J.; Sinclair, C.S.; Pearsall, R.S.; Green, P.J.; Yee, D.; Lampland, A.L.; et al. Comparative genomic sequence analysis and isolation of human and mouse alternative EGFR transcripts encoding truncated receptor isoforms. Genomics 2001, 71, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Flickinger, T.W.; Maihle, N.J.; Kung, H.J. An alternatively processed mRNA from the avian c-erbB gene encodes a soluble, truncated form of the receptor that can block ligand-dependent transformation. Mol. Cell. Biol. 1992, 12, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Baron, A.T.; Cora, E.M.; Lafky, J.M.; Boardman, C.H.; Buenafe, M.C.; Rademaker, A.; Liu, D.; Fishman, D.A.; Podratz, K.C.; Maihle, N.J. Soluble epidermal growth factor receptor (sEGFR/sErbB1) as a potential risk, screening, and diagnostic serum biomarker of epithelial ovarian cancer. Cancer Epidemiol. Biomark. Prev. 2003, 12, 103–113. [Google Scholar]
- Asgeirsson, K.S.; Agrawal, A.; Allen, C.; Hitch, A.; Ellis, I.O.; Chapman, C.; Cheung, K.L.; Robertson, J.F. Serum epidermal growth factor receptor and HER2 expression in primary and metastatic breast cancer patients. Breast Cancer Res. 2007, 9, R75. [Google Scholar] [CrossRef] [PubMed]
- Maramotti, S.; Paci, M.; Manzotti, G.; Rapicetta, C.; Gugnoni, M.; Galeone, C.; Cesario, A.; Lococo, F. Soluble epidermal growth factor receptors (sEGFRS) in cancer: Biological aspects and clinical relevance. Int. J. Mol. Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Lemos-Gonzalez, Y.; Rodriguez-Berrocal, F.J.; Cordero, O.J.; Gomez, C.; de la Cadena, M.P. Alteration of the serum levels of the epidermal growth factor receptor and its ligands in patients with non-small cell lung cancer and head and neck carcinoma. Br. J. Cancer 2007, 96, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Oh, J.Y.; Ryu, S.K.; Kim, S.J.; Lee, N.Y.; Kim, Y.S.; Yi, S.Y.; Shim, K.S.; Han, W.S. Detection of epidermal growth factor receptor in the serum of gastric carcinoma patients. Cancer 1997, 79, 1879–1883. [Google Scholar] [CrossRef]
- Jantus-Lewintre, E.; Sirera, R.; Cabrera, A.; Blasco, A.; Caballero, C.; Iranzo, V.; Rosell, R.; Camps, C. Analysis of the prognostic value of soluble epidermal growth factor receptor plasma concentration in advanced non-small-cell lung cancer patients. Clin. Lung Cancer 2011, 12, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Lococo, F.; Paci, M.; Rapicetta, C.; Rossi, T.; Sancisi, V.; Braglia, L.; Cavuto, S.; Bisagni, A.; Bongarzone, I.; Noonan, D.M.; et al. Preliminary evidence on the diagnostic and molecular role of circulating soluble EGFR in non-small cell lung cancer. Int. J. Mol. Sci. 2015, 16, 19612–19630. [Google Scholar] [CrossRef] [PubMed]
- Zampino, M.G.; Magni, E.; Santoro, L.; Zorzino, L.; Dell’Orto, P.; Sonzogni, A.; Fazio, N.; Monfardini, L.; Chiappa, A.; Biffi, R.; et al. Epidermal growth factor receptor serum (sEGFR) level may predict response in patients with EGFR-positive advanced colorectal cancer treated with gefitinib? Cancer Chemother. Pharmacol. 2008, 63, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Muller, V.; Witzel, I.; Pantel, K.; Krenkel, S.; Luck, H.J.; Neumann, R.; Keller, T.; Dittmer, J.; Janicke, F.; Thomssen, C. Prognostic and predictive impact of soluble epidermal growth factor receptor (sEGFR) protein in the serum of patients treated with chemotherapy for metastatic breast cancer. Anticancer Res. 2006, 26, 1479–1487. [Google Scholar] [PubMed]
- Huang, P.H.; Xu, A.M.; White, F.M. Oncogenic EGFR signaling networks in glioma. Sci. Signal. 2009, 2, re6. [Google Scholar] [CrossRef] [PubMed]
- Nagane, M.; Coufal, F.; Lin, H.; Bogler, O.; Cavenee, W.K.; Huang, H.J. A common mutant epidermal growth factor receptor confers enhanced tumorigenicity on human glioblastoma cells by increasing proliferation and reducing apoptosis. Cancer Res. 1996, 56, 5079–5086. [Google Scholar] [PubMed]
- Ekstrand, A.J.; James, C.D.; Cavenee, W.K.; Seliger, B.; Pettersson, R.F.; Collins, V.P. Genes for epidermal growth factor receptor, transforming growth factor α, and epidermal growth factor and their expression in human gliomas in vivo. Cancer Res. 1991, 51, 2164–2172. [Google Scholar] [PubMed]
- Moscatello, D.K.; Holgado-Madruga, M.; Godwin, A.K.; Ramirez, G.; Gunn, G.; Zoltick, P.W.; Biegel, J.A.; Hayes, R.L.; Wong, A.J. Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Res. 1995, 55, 5536–5539. [Google Scholar] [PubMed]
- Feldkamp, M.M.; Lala, P.; Lau, N.; Roncari, L.; Guha, A. Expression of activated epidermal growth factor receptors, Ras-guanosine triphosphate, and mitogen-activated protein kinase in human glioblastoma multiforme specimens. Neurosurgery 1999, 45, 1442–1453. [Google Scholar] [CrossRef] [PubMed]
- Nieto, Y.; Nawaz, F.; Jones, R.B.; Shpall, E.J.; Nawaz, S. Prognostic significance of overexpression and phosphorylation of epidermal growth factor receptor (EGFR) and the presence of truncated EGFRvIII in locoregionally advanced breast cancer. J. Clin. Oncol. 2007, 25, 4405–4413. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Gong, X.; Tang, C.K. Evidence of high incidence of EGFRvIII expression and coexpression with EGFR in human invasive breast cancer by laser capture microdissection and immunohistochemical analysis. Int. J. Cancer 2002, 98, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Pillay, V.; Allaf, L.; Wilding, A.L.; Donoghue, J.F.; Court, N.W.; Greenall, S.A.; Scott, A.M.; Johns, T.G. The plasticity of oncogene addiction: Implications for targeted therapies directed to receptor tyrosine kinases. Neoplasia 2009, 11, 448–458, 442 p following 458. [Google Scholar] [CrossRef] [PubMed]
- Emlet, D.R.; Gupta, P.; Holgado-Madruga, M.; del Vecchio, C.A.; Mitra, S.S.; Han, S.Y.; Li, G.; Jensen, K.C.; Vogel, H.; Xu, L.W.; et al. Targeting a glioblastoma cancer stem-cell population defined by EGF receptor variant III. Cancer Res. 2014, 74, 1238–1249. [Google Scholar] [CrossRef] [PubMed]
- Halatsch, M.E.; Gehrke, E.E.; Vougioukas, V.I.; Botefur, I.C.; A-Borhani, F.; Efferth, T.; Gebhart, E.; Domhof, S.; Schmidt, U.; Buchfelder, M. Inverse correlation of epidermal growth factor receptor messenger RNA induction and suppression of anchorage-independent growth by OSI-774, an epidermal growth factor receptor tyrosine kinase inhibitor, in glioblastoma multiforme cell lines. J. Neurosurg. 2004, 100, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; Kaye, A.H.; Luwor, R.B. The EGFRvIII variant in glioblastoma multiforme. J. Clin. Neurosci. 2009, 16, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Eller, J.L.; Longo, S.L.; Hicklin, D.J.; Canute, G.W. Activity of anti-epidermal growth factor receptor monoclonal antibody c225 against glioblastoma multiforme. Neurosurgery 2002, 51, 1005–1014. [Google Scholar] [PubMed]
- Li, L.; Quang, T.S.; Gracely, E.J.; Kim, J.H.; Emrich, J.G.; Yaeger, T.E.; Jenrette, J.M.; Cohen, S.C.; Black, P.; Brady, L.W. A phase II study of anti-epidermal growth factor receptor radioimmunotherapy in the treatment of glioblastoma multiforme. J. Neurosurg. 2010, 113, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.S.; Zhang, Z.Y.; Jia, Z.F.; Wang, G.X.; Qiu, M.Z.; Zhou, H.X.; Yu, S.Z.; Chang, J.; Jiang, H.; Pu, P.Y. Suppression of EGFR expression by antisense or small interference RNA inhibits U251 glioma cell growth in vitro and in vivo. Cancer Gene Ther. 2006, 13, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Moscatello, D.K.; Ramirez, G.; Wong, A.J. A naturally occurring mutant human epidermal growth factor receptor as a target for peptide vaccine immunotherapy of tumors. Cancer Res. 1997, 57, 1419–1424. [Google Scholar] [PubMed]
- Sampson, J.H.; Aldape, K.D.; Archer, G.E.; Coan, A.; Desjardins, A.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Herndon, J.E.; McLendon, R.E.; et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol. 2011, 13, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Ekstrand, A.J.; Sugawa, N.; James, C.D.; Collins, V.P. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc. Natl. Acad. Sci. USA 1992, 89, 4309–4313. [Google Scholar] [CrossRef] [PubMed]
- Pines, G.; Huang, P.H.; Zwang, Y.; White, F.M.; Yarden, Y. EGFRvIV: A previously uncharacterized oncogenic mutant reveals a kinase autoinhibitory mechanism. Oncogene 2010, 29, 5850–5860. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Wang, H.; Zhou, K.; Luo, X.; Pan, X.; Shi, B.; Jiang, H.; Zhang, J.; Li, K.; Wang, H.M.; et al. A novel EGFR isoform confers increased invasiveness to cancer cells. Cancer Res. 2013, 73, 7056–7067. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Zhou, M.; Wang, B.; Shi, B.; Jiang, H.; Zhang, J.; Li, Z. Weak binding to E3 ubiquitin ligase c-Cbl increases EGFRvA protein stability. FEBS Lett. 2016, 590, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Eichmann, A.; Simons, M. VEGF signaling inside vascular endothelial cells and beyond. Curr. Opin. Cell Biol. 2012, 24, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Arcondeguy, T.; Lacazette, E.; Millevoi, S.; Prats, H.; Touriol, C. VEGF-A mRNA processing, stability and translation: A paradigm for intricate regulation of gene expression at the post-transcriptional level. Nucleic Acids Res. 2013, 41, 7997–8010. [Google Scholar] [CrossRef] [PubMed]
- Oltean, S.; Bates, D.O. Hallmarks of alternative splicing in cancer. Oncogene 2014, 33, 5311–5318. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. Vascular endothelial growth factor: Basic science and clinical progress. Endocr. Rev. 2004, 25, 581–611. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Vascular endothelial growth factor (VEGF) signaling in tumor progression. Crit. Rev. Oncol. Hematol. 2007, 62, 179–213. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.O.; Cui, T.G.; Doughty, J.M.; Winkler, M.; Sugiono, M.; Shields, J.D.; Peat, D.; Gillatt, D.; Harper, S.J. VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 2002, 62, 4123–4131. [Google Scholar] [PubMed]
- Cebe Suarez, S.; Pieren, M.; Cariolato, L.; Arn, S.; Hoffmann, U.; Bogucki, A.; Manlius, C.; Wood, J.; Ballmer-Hofer, K. A VEGF-A splice variant defective for heparan sulfate and neuropilin-1 binding shows attenuated signaling through VEGFR-2. Cell. Mol. Life Sci. 2006, 63, 2067–2077. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, H.; Li, X.; Harper, S.J.; Bates, D.O.; Claesson-Welsh, L. Vascular endothelial growth factor (VEGF)-A165b is a weak in vitro agonist for VEGF receptor-2 due to lack of coreceptor binding and deficient regulation of kinase activity. Cancer Res. 2008, 68, 4683–4692. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, R.J.; Hayashi, T.; Cho, W.G.; Kleinman, M.E.; Dridi, S.; Takeda, A.; Baffi, J.Z.; Yamada, K.; Kaneko, H.; Green, M.G.; et al. Alternatively spliced vascular endothelial growth factor receptor-2 is an essential endogenous inhibitor of lymphatic vessel growth. Nat. Med. 2009, 15, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.P.; Andrews, J.I.; Raikwar, N.S.; Kelley, E.A.; Herse, F.; Dechend, R.; Golos, T.G.; Liu, K.Z. A recently evolved novel trophoblast-enriched secreted form of fms-like tyrosine kinase-1 variant is up-regulated in hypoxia and preeclampsia. J. Clin. Endocrinol. Metab. 2009, 94, 2524–2530. [Google Scholar] [CrossRef] [PubMed]
- Heydarian, M.; McCaffrey, T.; Florea, L.; Yang, Z.; Ross, M.M.; Zhou, W.; Maynard, S.E. Novel splice variants of sFlt1 are upregulated in preeclampsia. Placenta 2009, 30, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Szalai, G.; Romero, R.; Chaiworapongsa, T.; Xu, Y.; Wang, B.; Ahn, H.; Xu, Z.; Chiang, P.J.; Sundell, B.; Wang, R.; et al. Full-length human placental sFlt-1-e15a isoform induces distinct maternal phenotypes of preeclampsia in mice. PLoS ONE 2015, 10, e0119547. [Google Scholar]
- Hornig, C.; Barleon, B.; Ahmad, S.; Vuorela, P.; Ahmed, A.; Weich, H.A. Release and complex formation of soluble VEGFR-1 from endothelial cells and biological fluids. Lab. Investig. 2000, 80, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Jebbink, J.; Keijser, R.; Veenboer, G.; van der Post, J.; Ris-Stalpers, C.; Afink, G. Expression of placental FLT1 transcript variants relates to both gestational hypertensive disease and fetal growth. Hypertension 2011, 58, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Karumanchi, S.A.; Bdolah, Y. Hypoxia and sFlt-1 in preeclampsia: The “chicken-and-egg” question. Endocrinology 2004, 145, 4835–4837. [Google Scholar] [CrossRef] [PubMed]
- Sela, S.; Itin, A.; Natanson-Yaron, S.; Greenfield, C.; Goldman-Wohl, D.; Yagel, S.; Keshet, E. A novel human-specific soluble vascular endothelial growth factor receptor 1: Cell-type-specific splicing and implications to vascular endothelial growth factor homeostasis and preeclampsia. Circ. Res. 2008, 102, 1566–1574. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.H.; Oh, J.H.; Seo, J.A.; Lee, K.W.; Kim, S.G.; Choi, K.M.; Baik, S.H.; Choi, D.S.; Kang, Y.S.; Han, S.Y.; et al. Vascular endothelial growth factor (VEGF) and soluble VEGF receptor FLT-1 in diabetic nephropathy. Kidney Int. 2005, 67, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Bando, H.; Mori, T.; Takahashi, K.; Matsumoto, H.; Yasutome, M.; Weich, H.; Toi, M. Overexpression of soluble vascular endothelial growth factor receptor 1 in colorectal cancer: Association with progression and prognosis. Cancer Sci. 2007, 98, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Ahmed, A. Elevated placental soluble vascular endothelial growth factor receptor-1 inhibits angiogenesis in preeclampsia. Circ. Res. 2004, 95, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Hewett, P.W.; Al-Ani, B.; Sissaoui, S.; Fujisawa, T.; Cudmore, M.J.; Ahmed, A. Autocrine activity of soluble FLT-1 controls endothelial cell function and angiogenesis. Vasc. Cell. 2011, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Dong, Z.; Zhang, Y.; Zhang, X.; Song, X.; Sun, M.; Hu, Y.; Liu, S.; Wang, K.; Qu, X.; et al. Interleukin-4 and granulocyte-macrophage colony-stimulating factor mediates the upregulation of soluble vascular endothelial growth factor receptor-1 in RAW264.7 cells-a process in which p38 mitogen-activated protein kinase signaling has an important role. J. Microbiol. Immunol. Infect. 2016, 49, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Liebermann, D.A.; Tront, J.S.; Holtzman, E.J.; Huang, Y.; Hoffman, B.; Geifman-Holtzman, O. Gadd45a stress signaling regulates sFlt-1 expression in preeclampsia. J. Cell. Physiol. 2009, 220, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Anton, L.; Olarerin-George, A.O.; Hogenesch, J.B.; Elovitz, M.A. Placental expression of miR-517a/b and miR-517c contributes to trophoblast dysfunction and preeclampsia. PLoS ONE 2015, 10, e0122707. [Google Scholar] [CrossRef] [PubMed]
- Boeckel, J.N.; Guarani, V.; Koyanagi, M.; Roexe, T.; Lengeling, A.; Schermuly, R.T.; Gellert, P.; Braun, T.; Zeiher, A.; Dimmeler, S. Jumonji domain-containing protein 6 (Jmjd6) is required for angiogenic sprouting and regulates splicing of VEGF-receptor 1. Proc. Natl. Acad. Sci. USA 2011, 108, 3276–3281. [Google Scholar] [CrossRef] [PubMed]
- Kangsamaksin, T.; Murtomaki, A.; Kofler, N.M.; Cuervo, H.; Chaudhri, R.A.; Tattersall, I.W.; Rosenstiel, P.E.; Shawber, C.J.; Kitajewski, J. NOTCH decoys that selectively block DLL/NOTCH or JAG/NOTCH disrupt angiogenesis by unique mechanisms to inhibit tumor growth. Cancer Discov. 2015, 5, 182–197. [Google Scholar] [CrossRef] [PubMed]
- Kendall, R.L.; Thomas, K.A. Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 10705–10709. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.T.; Stefanini, M.O.; Mac Gabhann, F.; Kontos, C.D.; Annex, B.H.; Popel, A.S. A systems biology perspective on sVEGFR1: Its biological function, pathogenic role and therapeutic use. J. Cell. Mol. Med. 2010, 14, 528–552. [Google Scholar] [CrossRef] [PubMed]
- Ambati, B.K.; Nozaki, M.; Singh, N.; Takeda, A.; Jani, P.D.; Suthar, T.; Albuquerque, R.J.; Richter, E.; Sakurai, E.; Newcomb, M.T.; et al. Corneal avascularity is due to soluble VEGF receptor-1. Nature 2006, 443, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Ambati, B.K.; Patterson, E.; Jani, P.; Jenkins, C.; Higgins, E.; Singh, N.; Suthar, T.; Vira, N.; Smith, K.; Caldwell, R. Soluble vascular endothelial growth factor receptor-1 contributes to the corneal antiangiogenic barrier. Br. J. Ophthalmol. 2007, 91, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Rai, A.; Kambham, N.; Sung, J.F.; Singh, N.; Petitt, M.; Dhal, S.; Agrawal, R.; Sutton, R.E.; Druzin, M.L.; et al. Endometrial VEGF induces placental sFLT1 and leads to pregnancy complications. J. Clin. Investig. 2014, 124, 4941–4952. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.K.; Dimberg, A.; Kreuger, J.; Claesson-Welsh, L. VEGF receptor signalling—In control of vascular function. Nat. Rev. Mol. Cell Biol. 2006, 7, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Kumai, Y.; Ooboshi, H.; Ibayashi, S.; Ishikawa, E.; Sugimori, H.; Kamouchi, M.; Kitazono, T.; Egashira, K.; Iida, M. Postischemic gene transfer of soluble FLT-1 protects against brain ischemia with marked attenuation of blood-brain barrier permeability. J. Cereb. Blood Flow Metab. 2007, 27, 1152–1160. [Google Scholar] [CrossRef] [PubMed]
- Tsao, P.N.; Chan, F.T.; Wei, S.C.; Hsieh, W.S.; Chou, H.C.; Su, Y.N.; Chen, C.Y.; Hsu, W.M.; Hsieh, F.J.; Hsu, S.M. Soluble vascular endothelial growth factor receptor-1 protects mice in sepsis. Crit. Care Med. 2007, 35, 1955–1960. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.J.; Maynard, S.E.; Qian, C.; Lim, K.H.; England, L.J.; Yu, K.F.; Schisterman, E.F.; Thadhani, R.; Sachs, B.P.; Epstein, F.H.; et al. Circulating angiogenic factors and the risk of preeclampsia. N. Engl. J. Med. 2004, 350, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.E.; Min, J.Y.; Merchan, J.; Lim, K.H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Investig. 2003, 111, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Eming, S.A.; Lauer, G.; Cole, M.; Jurk, S.; Christ, H.; Hornig, C.; Krieg, T.; Weich, H.A. Increased levels of the soluble variant of the vascular endothelial growth factor receptor VEGFR-1 are associated with a poor prognosis in wound healing. J. Invest. Dermatol. 2004, 123, 799–802. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, R.; Paskin-Flerlage, S.; Zamanian, R.T.; Zimmerman, P.; Schmidt, J.W.; Deng, D.Y.; Southwood, M.; Spencer, R.; Lai, C.S.; Parker, W.; et al. Circulating angiogenic modulatory factors predict survival and functional class in pulmonary arterial hypertension. Pulm. Circ. 2013, 3, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Jesmin, S.; Gando, S.; Yanagida, Y.; Mizugaki, A.; Sultana, S.N.; Zaedi, S.; Yokota, H. The role of angiogenic factors and their soluble receptors in acute lung injury (ALI)/acute respiratory distress syndrome (ARDS) associated with critical illness. J. Inflamm. 2013, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- Goldman, C.K.; Kendall, R.L.; Cabrera, G.; Soroceanu, L.; Heike, Y.; Gillespie, G.Y.; Siegal, G.P.; Mao, X.; Bett, A.J.; Huckle, W.R.; et al. Paracrine expression of a native soluble vascular endothelial growth factor receptor inhibits tumor growth, metastasis, and mortality rate. Proc. Natl. Acad. Sci. USA 1998, 95, 8795–8800. [Google Scholar] [CrossRef] [PubMed]
- Verrax, J.; Defresne, F.; Lair, F.; Vandermeulen, G.; Rath, G.; Dessy, C.; Preat, V.; Feron, O. Delivery of soluble VEGF receptor 1 (sFlt1) by gene electrotransfer as a new antiangiogenic cancer therapy. Mol. Pharm. 2011, 8, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Ueno, H.; Nakanishi, Y.; Sakamoto, T.; Inoue, K.; Shimizu, K.; Oohashi, H.; Hara, N. Suppression of tumor angiogenesis and growth by gene transfer of a soluble form of vascular endothelial growth factor receptor into a remote organ. Cancer Res. 2000, 60, 2169–2177. [Google Scholar] [PubMed]
- Shiose, S.; Sakamoto, T.; Yoshikawa, H.; Hata, Y.; Kawano, Y.; Ishibashi, T.; Inomata, H.; Takayama, K.; Ueno, H. Gene transfer of a soluble receptor of VEGF inhibits the growth of experimental eyelid malignant melanoma. Investig. Ophthalmol. Vis. Sci. 2000, 41, 2395–2403. [Google Scholar]
- Kearney, J.B.; Kappas, N.C.; Ellerstrom, C.; DiPaola, F.W.; Bautch, V.L. The VEGF receptor Flt-1 (VEGFR-1) is a positive modulator of vascular sprout formation and branching morphogenesis. Blood 2004, 103, 4527–4535. [Google Scholar] [CrossRef] [PubMed]
- Orecchia, A.; Lacal, P.M.; Schietroma, C.; Morea, V.; Zambruno, G.; Failla, C.M. Vascular endothelial growth factor receptor-1 is deposited in the extracellular matrix by endothelial cells and is a ligand for the α5β1 integrin. J. Cell Sci. 2003, 116, 3479–3489. [Google Scholar] [CrossRef] [PubMed]
- Miyake, T.; Kumasawa, K.; Sato, N.; Takiuchi, T.; Nakamura, H.; Kimura, T. Soluble VEGF receptor 1 (sFlt1) induces non-apoptotic death in ovarian and colorectal cancer cells. Sci. Rep. 2016, 6, 24853. [Google Scholar] [CrossRef] [PubMed]
- Ruffini, F.; Failla, C.M.; Orecchia, A.; Bani, M.R.; Dorio, A.S.; Fortes, C.; Zambruno, G.; Graziani, G.; Giavazzi, R.; D’Atri, S.; et al. Expression of the soluble vascular endothelial growth factor receptor-1 in cutaneous melanoma: Role in tumour progression. Br. J. Dermatol. 2011, 164, 1061–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thielemann, A.; Baszczuk, A.; Kopczynski, Z.; Kopczynski, P.; Grodecka-Gazdecka, S. Clinical usefulness of assessing VEGF and soluble receptors sVEGFR-1 and sVEGFR-2 in women with breast cancer. Ann. Agric. Environ. Med. 2013, 20, 293–297. [Google Scholar] [PubMed]
- Lamszus, K.; Ulbricht, U.; Matschke, J.; Brockmann, M.A.; Fillbrandt, R.; Westphal, M. Levels of soluble vascular endothelial growth factor (VEGF) receptor 1 in astrocytic tumors and its relation to malignancy, vascularity, and VEGF-A. Clin. Cancer Res. 2003, 9, 1399–1405. [Google Scholar] [PubMed]
- Wierzbowska, A.; Robak, T.; Wrzesien-Kus, A.; Krawczynska, A.; Lech-Maranda, E.; Urbanska-Rys, H. Circulating VEGF and its soluble receptors sVEGFR-1 and sVEGFR-2 in patients with acute leukemia. Eur. Cytokine Netw. 2003, 14, 149–153. [Google Scholar] [PubMed]
- Toi, M.; Bando, H.; Ogawa, T.; Muta, M.; Hornig, C.; Weich, H.A. Significance of vascular endothelial growth factor (VEGF)/soluble VEGF receptor-1 relationship in breast cancer. Int. J. Cancer 2002, 98, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L.; Reusch, P.; Barleon, B.; Hang, C.; Dobbs, N.; Marme, D. Soluble Tie2 and Flt1 extracellular domains in serum of patients with renal cancer and response to antiangiogenic therapy. Clin. Cancer Res. 2001, 7, 1992–1997. [Google Scholar] [PubMed]
- Nagaoka, S.; Yoshida, T.; Akiyoshi, J.; Akiba, J.; Hisamoto, T.; Yoshida, Y.; Abe, M.; Koga, H.; Toirimura, T.; Ueno, T.; et al. The ratio of serum placenta growth factor to soluble vascular endothelial growth factor receptor-1 predicts the prognosis of hepatocellular carcinoma. Oncol. Rep. 2010, 23, 1647–1654. [Google Scholar] [PubMed]
- Kulapaditharom, B.; Boonkitticharoen, V.; Sritara, C. Plasma vascular endothelial growth factor dysregulation in defining aggressiveness of head and neck squamous cell carcinoma. J. Oncol. 2012, 2012, 687934. [Google Scholar] [CrossRef] [PubMed]
- Ilhan, N.; Ilhan, N.; Deveci, F. Functional significance of vascular endothelial growth factor and its receptor (receptor-1) in various lung cancer types. Clin. Biochem. 2004, 37, 840–845. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.T.; Chang, M.C.; Wei, S.C.; Tien, Y.W.; Hsu, C.; Liang, P.C.; Tsao, P.N.; Jan, I.S.; Wong, J.M. Serum vascular endothelial growth factor/soluble vascular endothelial growth factor receptor 1 ratio is an independent prognostic marker in pancreatic cancer. Pancreas 2008, 37, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Bando, H.; Weich, H.A.; Brokelmann, M.; Horiguchi, S.; Funata, N.; Ogawa, T.; Toi, M. Association between intratumoral free and total VEGF, soluble VEGFR-1, VEGFR-2 and prognosis in breast cancer. Br. J. Cancer 2005, 92, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Aref, S.; El Sherbiny, M.; Goda, T.; Fouda, M.; Al Askalany, H.; Abdalla, D. Soluble VEGF/sFlt1 ratio is an independent predictor of aml patient out come. Hematology 2005, 10, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, D.; Lenz, H.J.; de Haas, S.; Carmeliet, P.; Scherer, S.J. Markers of response for the antiangiogenic agent bevacizumab. J. Clin. Oncol. 2013, 31, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Tolaney, S.M.; Boucher, Y.; Duda, D.G.; Martin, J.D.; Seano, G.; Ancukiewicz, M.; Barry, W.T.; Goel, S.; Lahdenrata, J.; Isakoff, S.J.; et al. Role of vascular density and normalization in response to neoadjuvant bevacizumab and chemotherapy in breast cancer patients. Proc. Natl. Acad. Sci. USA 2015, 112, 14325–14330. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Finn, R.S.; Mulcahy, M.; Gurtler, J.; Sun, W.; Schwartz, J.D.; Dalal, R.P.; Joshi, A.; Hozak, R.R.; Xu, Y.; et al. A phase II and biomarker study of ramucirumab, a human monoclonal antibody targeting the VEGF receptor-2, as first-line monotherapy in patients with advanced hepatocellular cancer. Clin. Cancer Res. 2013, 19, 6614–6623. [Google Scholar] [CrossRef] [PubMed]
- Willett, C.G.; Duda, D.G.; di Tomaso, E.; Boucher, Y.; Ancukiewicz, M.; Sahani, D.V.; Lahdenranta, J.; Chung, D.C.; Fischman, A.J.; Lauwers, G.Y.; et al. Efficacy, safety, and biomarkers of neoadjuvant bevacizumab, radiation therapy, and fluorouracil in rectal cancer: A multidisciplinary phase II study. J. Clin. Oncol. 2009, 27, 3020–3026. [Google Scholar] [CrossRef] [PubMed]
- Thomas-Schoemann, A.; Blanchet, B.; Boudou-Rouquette, P.; Golmard, J.L.; Noe, G.; Chenevier-Gobeaux, C.; Lebbe, C.; Pages, C.; Durand, J.P.; Alexandre, J.; et al. Soluble VEGFR-1: A new biomarker of sorafenib-related hypertension (i.e., sorafenib-related is the compound adjective?). J. Clin. Pharmacol. 2015, 55, 478–479. [Google Scholar] [CrossRef] [PubMed]
- Duda, D.G.; Willett, C.G.; Ancukiewicz, M.; di Tomaso, E.; Shah, M.; Czito, B.G.; Bentley, R.; Poleski, M.; Lauwers, G.Y.; Carroll, M.; et al. Plasma soluble VEGFR-1 is a potential dual biomarker of response and toxicity for bevacizumab with chemoradiation in locally advanced rectal cancer. Oncologist 2010, 15, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Sahani, D.V.; Duda, D.G.; di Tomaso, E.; Ancukiewicz, M.; Catalano, O.A.; Sindhwani, V.; Blaszkowsky, L.S.; Yoon, S.S.; Lahdenranta, J.; et al. Efficacy, safety, and potential biomarkers of sunitinib monotherapy in advanced hepatocellular carcinoma: A phase II study. J. Clin. Oncol. 2009, 27, 3027–3035. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Hoff, P.M.; Morris, J.S.; Wolff, R.A.; Eng, C.; Glover, K.Y.; Adinin, R.; Overman, M.J.; Valero, V.; Wen, S.; et al. Phase II trial of infusional fluorouracil, irinotecan, and bevacizumab for metastatic colorectal cancer: Efficacy and circulating angiogenic biomarkers associated with therapeutic resistance. J. Clin. Oncol. 2010, 28, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Kim, C.; Yang, H.; Park, I.; Oh, N.; Hua, S.; Jeong, H.; An, H.J.; Kim, S.C.; Lee, G.M.; et al. Novel glycosylated VEGF decoy receptor fusion protein, VEGF-Grab, efficiently suppresses tumor angiogenesis and progression. Mol. Cancer Ther. 2015, 14, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Owen, L.A.; Uehara, H.; Cahoon, J.; Huang, W.; Simonis, J.; Ambati, B.K. Morpholino-mediated increase in soluble Flt-1 expression results in decreased ocular and tumor neovascularization. PLoS ONE 2012, 7, e33576. [Google Scholar] [CrossRef] [PubMed]
- Salton, M.; Kasprzak, W.K.; Voss, T.; Shapiro, B.A.; Poulikakos, P.I.; Misteli, T. Inhibition of vemurafenib-resistant melanoma by interference with pre-mRNA splicing. Nat. Commun. 2015, 6, 7103. [Google Scholar] [CrossRef] [PubMed]
- Salton, M.; Misteli, T. Small molecule modulators of pre-mRNA splicing in cancer therapy. Trends Mol. Med. 2016, 22, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Dvinge, H.; Kim, E.; Cho, H.; Micol, J.B.; Chung, Y.R.; Durham, B.H.; Yoshimi, A.; Kim, Y.J.; Thomas, M.; et al. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat. Med. 2016, 22, 672–678. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RTK | Splicing Events | Functional Consequences | References |
|---|---|---|---|
| ALK | Skipping of exons 2–3 Skipping of exons 4–11 | Truncated proteins with increased constitutive kinase activity and transformation potential in neuroblastoma | [8] |
| Skipping of exon 23 or exon 27 | Truncated proteins lacking the full kinase domain of ALK in Non Small Cell Lung Carcinoma | [9] | |
| AXL | Skipping of exon 10 | Shorter AXL protein with same transforming potential as full-length AXL | [10] |
| DDR | Exon skipping or inclusion | Distinct binding partners Differential activation by collagen | [11] |
| EGFR | Inclusion of exon 10, 9a, 16 or 17 | Soluble receptors acting as negative regulators of EGFR signalling | [12] |
| Skipping of exons 2–7 | Constitutively active receptor | [13] | |
| Enhanced signalling, survival, and tumourigenicity | [14] | ||
| Skipping of exons 2–22 | Enhanced migration and invasion Cancer stem cells marker | [15] | |
| ERB4 | N- and C-terminal alternative splicing generating four isoforms | Modulation of sub-cellular localization and partner binding | [16] |
| FGFR | Mutually exclusive exon 8 or 9 | Generation of distinct extracellular Ig-like domain III with distinct affinity for FGF ligands | [17] |
| Induction of Epithelial to Mesenchymal Transition (EMT), invasion and motility | [18] | ||
| INSR | Skipping or inclusion of exon 11 | Generation of INSR-A and INSR-B splice variants that respond differentially to IGF-II and insulin ligands and differentially activate the RAS/MAPK pathway | [19] |
| MET | Skipping of exon 14 | Activation of MET kinase activity Oncogenic transformation | [20] |
| Increased sensitivity to MET inhibitors | [21] | ||
| RET | 3′-end alternative splicing generating multiple isoforms that differ in their C-terminal domain | Modulation of signalling partner binding Distinct sub-cellular localization and trafficking properties Transforming capacity | [22] |
| RON | Skipping of exon 11 | Constitutively active receptor Enhanced signalling, invasion, motility | [23] |
| Skipping of exons 15–19, 16–19, 16–17 and 16 | Truncated protein lacking active kinase domain Dominant negative isoforms in lung cancers. | [24] | |
| NTRK | Skipping of exons 6, 7 and 9 | Constitutively active receptor Oncogenic function in neuroblastoma | [25] |
| VEGFR | Intron retention followed by premature polyadenylation | Soluble decoy receptor acting as negative regulator of VEGFR signalling | [26,27] |
| Increased resistance to anti-angiogenic therapies | [28,29] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abou-Fayçal, C.; Hatat, A.-S.; Gazzeri, S.; Eymin, B. Splice Variants of the RTK Family: Their Role in Tumour Progression and Response to Targeted Therapy. Int. J. Mol. Sci. 2017, 18, 383. https://doi.org/10.3390/ijms18020383
Abou-Fayçal C, Hatat A-S, Gazzeri S, Eymin B. Splice Variants of the RTK Family: Their Role in Tumour Progression and Response to Targeted Therapy. International Journal of Molecular Sciences. 2017; 18(2):383. https://doi.org/10.3390/ijms18020383
Chicago/Turabian StyleAbou-Fayçal, Cherine, Anne-Sophie Hatat, Sylvie Gazzeri, and Beatrice Eymin. 2017. "Splice Variants of the RTK Family: Their Role in Tumour Progression and Response to Targeted Therapy" International Journal of Molecular Sciences 18, no. 2: 383. https://doi.org/10.3390/ijms18020383