Next-Generation Sequencing in Oncology: Genetic Diagnosis, Risk Prediction and Cancer Classification



Abstract
:1. Introduction
2. NGS—Next-Generation Sequencing Technology
2.1. NGS Technology, Historical Perspective and State-of-the-Art
2.2. NGS Methods
- (a)
- Libray preparation or sample processing. The material is first fragmented mechanically or enzymatically to yield fragments whose size is compatible with the sequencer (small fragments of 200–300 nucleotides for short-read sequencing, longer for the long-read sequencing). This material can be enriched to analyse a limited number of genetic regions (e.g., disease gene-panels or microbes [10]) or all coding exons of the human genome (from approximately 21,000 genes; Whole-Exome-Sequencing, WES). The complete genomic DNA can also be sequenced (Whole-Genome-Sequencing, WGS) and it does not require any enrichment step (see Scetion 2.2.3). The regions that are intended to be analysed are defined regions of interest (ROIs). An amplification step through PCR with 4–12 cycles is perfomed in most cases. During this step, proper linkers and barcodes are attached to the DNA fragments and are necessary for subsequent analyses by the sequencer. DNA barcodes, which are unique nucleotide tags (6–8 nt), allow pooling samples together in one single flowcell for the sequencing reaction.
- (b)
- (c)
- Initial quality and raw data analyses. General quality control about the read quality is done mostly with FastQC [12]. Many pre-processing tools are available for removal of bad quality reads, trimming, etc. After mapping, specificity is determined, i.e., the fraction (%) of the total number of predefined ROIs, which are correctly enriched and sequenced.
- (d)
- Variant calling and data interpretation. This last step is dependent on the specific application. In this review, some methods and bioinformatics tools relevant to data interpretation in the field of oncogenomics will be given.
2.2.1. Gene-Panels
2.2.2. WES—Whole-Exome-Sequencing
2.2.3. WGS—Whole-Genome-Sequencing
2.3. Data Analyses and Interpretation
2.4. Conclusions
3. Inherited Cancer Syndromes
3.1. Historical Perspective in Inherited Cancer Syndromes
3.2. Gene-Panels in Cancer Syndromes
3.2.1. Technical Validity
3.2.2. Clinical Relevance
3.2.3. Cancer Gene-Panels: In Conclusion
3.3. WES and WGS
3.4. Identification of Pathogenic Germline RNA-Splice Mutations Using RNA-Seq
3.5. Risk Modifiers
3.6. Prevention of Inherited Cancer Syndromes
4. Cancer Somatic Mutation Analysis
4.1. WES and WGS: Somatic Mutation Analyses, Cancer Classifiers and Diagnosis
4.2. The Use of Gene-Panels in Somatic Mutation Detection: Towards Clinical Applications
4.2.1. Technical Validity
4.2.2. Clinical Utility
4.2.3. Clinical Trials
4.2.4. Conclusive Remarks on Gene-Panels for Somatic Mutations Analysis
4.3. Liquid Biopsy Analyses
5. Pharmacogenetics
6. Other Applications and Future Directions
7. Limitations and Complications of NGS in Genetic Diagnostics and Ethical Considerations
8. Conclusive Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ADR | Adverse drug reaction |
| BRCAx | Test negative to BRCA1/2 mutation |
| BWA | Burrows-wheeler-alignment |
| Bp | Base pair |
| ChIP-seq | Chromatin immunoprecipitation |
| CPIC | Clinical pharmacogenetics implementation consortium |
| CTC | Circulating tumour cell |
| ctDNA | Cell-free tumour DNA |
| DNA-seq | DNA sequencing |
| DPWG | Dutch pharmacogenetics working group |
| FAP | Familial adenomatous polyposis coli |
| FFPE | Formalin fixed paraffin embedded |
| Gb | Giga base |
| GWAS | Genome wide association studies |
| HBOC | Hereditary breast and ovarian cancer |
| ICSI | Intra cytoplasmic sperm injection |
| Indels | Insertion or deletion (of DNA fragments) |
| IVF | In vitro fertilisation |
| Kb | Kilo base pair |
| KNMP | Royal Dutch pharmacists association |
| MIP | Molecular inversion probe |
| MMR | Mismatch repair |
| NGS | Next-generation sequencing |
| PCR | Polymerase chain reaction |
| PGD | Preimplantation genetic diagnosis |
| PGS | Preimplantation genetic screening |
| PGH | Preimplantation genetic haplotyping |
| PGx | Pharmacogenetics |
| RNA-seq | RNA sequencing |
| ROI | Regions of interest |
| SAM | Sequence-Alignment-MAP tools |
| SBS | Sequencing by synthesis |
| SMaRT | Spliceosome-mediated RNA trans-splicing |
| SMRT | Single molecule real-time |
| SNP | Singe nucleotide polymorphism |
| VUS | Variant of undetermined (unknown) clinical relevance |
| WES | Whole exome sequencing |
| WGS | Whole genome sequencing |
| Important definitions | |
| Actionable mutation | A mutation that is potentially responsive to a targeted therapy |
| Coverage (in NGS) | (Or depth) the number of reads per nucleotide |
| Cistrome | The set of (cis-acting) DNA sequences targeted by a specific transcription factor |
| Depth | (Or coverage) the number of reads per nucleotide |
| Diagnostic yield | The likelihood that a given test will return information that helps establishing a diagnosis |
| Driver mutation | A somatic mutation OCCURRING in cancer that is implicated in oncogenesis |
| Driver gene | A gene where driver mutations are frequently found |
| Effect-size (of a gene variant) | The measurement of the magnitude of the effect of a gene variant |
| FastQC | Application handling raw-sequence data from high throughput NGS sequencers |
| Genetic risk modifier | A gene variant with no intrinsic pathogenic action and a small effect-size, but that can modify the penetrance of a co-existing pathogenic mutation |
| Passenger mutation | A somatic mutation (found in cancer but not only) that has no implication in oncogenesis and does not give growth advantage |
| Penetrance (genetics) | The proportion of INDIVIDUALS carrying a gene variant (or mutation) and expressing the phenotype associated to the variant |
| Sequencing by synthesis (in NGS) | method developed by Solexa® and currently used by Illumina® in which each dNTP with a fluorescent reversible terminator is added separately, then is cleaved to allow the incorporation of the next base |
| Single molecule real-time (in NGS) | In this method, several DNA polymerase enzymes are contained in separate nanophotonic structures where a single stranded DNA is also incorporated. DNA synthesis is made using differentially labeled nucleotides |
| Specificity (in NGS) | The amount of regions of interest (ROI) theoretically captured for a NGS analyses that are correctly enriched and sequenced |
| Spliceogenic variant | A gene variant causing a splice alteration |
| Targeted re-sequencing | Isolation and sequencing of a subset of genes or regions of interest. Different approaches can be used (see the text) |
| Theranostic | This is a sort of diagnostic test that can be used to select a targeted therapy |
| Translatome | All mRNA fragments that are translated in a moment or in a condition. |
| Variant of undetermined/unknown clinical significance (VUS) | A rare genetic variant for which pathogenicity was neither confirmed nor excluded |
References
- Marziali, A.; Akeson, M. New DNA sequencing methods. Annu. Rev. Biomed. Eng. 2001, 3, 195–223. [Google Scholar] [CrossRef] [PubMed]
- Next-Gen-Field-Guid. Available online: http://www.molecularecologist.com/next-gen-fieldguide-2014 (accessed on 9 January 2016).
- Illumina®. Available online: http://www.illumina.com (accessed on 10 January 2017).
- Ion-Torrent®. Available online: http://www.thermofisher.com (accessed on 7 November 2016).
- PacBio®. Available online: http://www.pacb.com/ (accessed on 11 November 2016).
- Roche®. Available online: http://www.roche.com/ (accessed on 11 November 2016).
- Nanopore®. Available online: https://www.nanoporetech.com/ (accessed on 11 November 2016).
- GeneReader. Available online: https://www.qiagen.com/nl/resources/technologies/ngs/ (accessed on 10 January 2017).
- X-Genomics®. Available online: http://www.10xgenomics.com/technology/ (accessed on 10 December 2016).
- Duncavage, E.J.; Magrini, V.; Becker, N.; Armstrong, J.R.; Demeter, R.T.; Wylie, T.; Abel, H.J.; Pfeifer, J.D. Hybrid Capture and Next-Generation Sequencing Identify Viral Integration Sites from Formalin-Fixed, Paraffin-Embedded Tissue. J. Mol. Diagn. 2011, 13, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High-Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 10 January 2017).
- RainDance®. Available online: http://www.raindancetech.com/ (accessed on 11 November 2016).
- Niedzicka, M.; Fijarczyk, A.; Dudek, K.; Stuglik, M.; Babik, W. Molecular Inversion Probes for targeted resequencing in non-model organisms. Sci. Rep. 2016, 6, 24051. [Google Scholar] [CrossRef] [PubMed]
- Bao, R.; Huang, L.; Andrade, J.; Tan, W.; Kibbe, W.A.; Jiang, H.; Feng, G. Review of current methods, applications, and data management for the bioinformatics analysis of whole exome sequencing. Cancer Inform. 2014, 13, 67–82. [Google Scholar] [PubMed]
- Leggett, R.M.; Ramirez-Gonzalez, R.H.; Clavijo, B.J.; Waite, D.; Davey, R.P. Sequencing quality assessment tools to enable data-driven informatics for high throughput genomics. Front. Genet. 2013, 4, 288. [Google Scholar] [CrossRef] [PubMed]
- Ekblom, R.; Wolf, J.B. A field guide to whole-genome sequencing, assembly and annotation. Evol. Appl. 2014, 7, 1026–1042. [Google Scholar] [CrossRef] [PubMed]
- Chrystoja, C.C.; Diamandis, E.P. Whole genome sequencing as a diagnostic test: Challenges and opportunities. Clin. Chem. 2014, 60, 724–733. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- OMIM. Available online: http://omim/org/ (accessed on 10 November 2016).
- Fokkema, I.F.; Taschner, P.E.; Schaafsma, G.C.; Celli, J.; Laros, J.F.; den Dunnen, J.T. LOVD v.2.0: The next generation in gene variant databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Wallis, Y.; Payne, S.; McAnulty, C.; Bodmer, D.; Sister-mans, E.; Robertson, K.; Moore, D.; Abbs, S.; Deans, Z.; Devereau, A. Practice Guidelines for the Evaluation of Pathogenicity and the Reporting of Sequence Variants in Clinical Molecular Genetics. In Association for Clinical Genetic Science & Dutch Society of Clinical Genetic Laboratory Specialists; Association for Clinical Genetic Science: Birmingham, UK, 2013; pp. 1–16. [Google Scholar]
- Hoppman-Chaney, N.; Peterson, L.M.; Klee, E.W.; Middha, S.; Courteau, L.K.; Ferber, M.J. Evaluation of oligonucleotide sequence capture arrays and comparison of next-generation sequencing platforms for use in molecular diagnostics. Clin. Chem. 2010, 56, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.; Lee, M.K.; Casadei, S.; Thornton, A.M.; Stray, S.M.; Pennil, C.; Nord, A.S.; Mandell, J.B.; Swisher, E.M.; King, M.C. Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 12629–12633. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.; Casadei, S.; Lee, M.K.; Pennil, C.C.; Nord, A.S.; Thornton, A.M.; Roeb, W.; Agnew, K.J.; Stray, S.M.; Wickramanayake, A.; et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 18032–18037. [Google Scholar] [CrossRef] [PubMed]
- BROCA. Available online: http://www.tests.labmed.washington.edu/BROCA (accessed on 7 November 2016).
- Pritchard, C.C.; Smith, C.; Salipante, S.J.; Lee, M.K.; Thornton, A.M.; Nord, A.S.; Gulden, C.; Kupfer, S.S.; Swisher, E.M.; Bennett, R.L.; et al. ColoSeq provides comprehensive lynch and polyposis syndrome mutational analysis using massively parallel sequencing. J. Mol. Diagn. 2012, 14, 357–366. [Google Scholar] [CrossRef] [PubMed]
- ColoSeq. Available online: http://www.tests.labmed.washington.edu/COLOSEQ (accessed on 4 November 2016).
- Castera, L.; Krieger, S.; Rousselin, A.; Legros, A.; Baumann, J.J.; Bruet, O.; Brault, B.; Fouillet, R.; Goardon, N.; Letac, O.; et al. Next-generation sequencing for the diagnosis of hereditary breast and ovarian cancer using genomic capture targeting multiple candidate genes. Eur. J. Hum. Genet. 2014, 22, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.T.; Cheng, J.; Mitchell, T.N.; Syed, A.; Zehir, A.; Mensah, N.Y.; Oultache, A.; Nafa, K.; Levine, R.L.; Arcila, M.E.; et al. Detection of mutations in myeloid malignancies through paired-sample analysis of microdroplet-PCR deep sequencing data. J. Mol. Diagn. 2014, 16, 504–518. [Google Scholar] [CrossRef] [PubMed]
- LaDuca, H.; Stuenkel, A.J.; Dolinsky, J.S.; Keiles, S.; Tandy, S.; Pesaran, T.; Chen, E.; Gau, C.L.; Palmaer, E.; Shoaepour, K.; et al. Utilization of multigene panels in hereditary cancer predisposition testing: Analysis of more than 2,000 patients. Genet. Med. 2014, 16, 830–837. [Google Scholar] [CrossRef] [PubMed]
- Ambry-Genetics. Available online: http://www.ambrygen.com (accessed on 7 November 2016).
- Cragun, D.; Radford, C.; Dolinsky, J.S.; Caldwell, M.; Chao, E.; Pal, T. Panel-based testing for inherited colorectal cancer: A descriptive study of clinical testing performed by a US laboratory. Clin. Genet. 2014, 86, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Minion, L.E.; Dolinsky, J.S.; Chase, D.M.; Dunlop, C.L.; Chao, E.C.; Monk, B.J. Hereditary predisposition to ovarian cancer, looking beyond BRCA1/BRCA2. Gynecol. Oncol. 2015, 137, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Myriad-Genetics. Available online: http://www.myriadpro.com (accessed on 11 November 2016).
- Judkins, T.; Leclair, B.; Bowles, K.; Gutin, N.; Trost, J.; McCulloch, J.; Bhatnagar, S.; Murray, A.; Craft, J.; Wardell, B.; et al. Development and analytical validation of a 25-gene next generation sequencing panel that includes the BRCA1 and BRCA2 genes to assess hereditary cancer risk. BMC Cancer 2015, 15, 215. [Google Scholar] [CrossRef] [PubMed]
- Yurgelun, M.B.; Allen, B.; Kaldate, R.R.; Bowles, K.R.; Judkins, T.; Kaushik, P.; Roa, B.B.; Wenstrup, R.J.; Hartman, A.R.; Syngal, S. Identification of a Variety of Mutations in Cancer Predisposition Genes in Patients with Suspected Lynch Syndrome. Gastroenterology 2015, 149, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.; Battelli, C.; Allen, B.; Kaldate, R.; Bhatnagar, S.; Bowles, K.; Timms, K.; Garber, J.E.; Herold, C.; Ellisen, L.; et al. Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer 2015, 121, 25–33. [Google Scholar] [CrossRef]
- Schroeder, C.; Faust, U.; Sturm, M.; Hackmann, K.; Grundmann, K.; Harmuth, F.; Bosse, K.; Kehrer, M.; Benkert, T.; Klink, B.; et al. HBOC multi-gene panel testing: Comparison of two sequencing centers. Breast Cancer Res. Treat. 2015, 152, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Aloraifi, F.; McDevitt, T.; Martiniano, R.; McGreevy, J.; McLaughlin, R.; Egan, C.M.; Cody, N.; Meany, M.; Kenny, E.; Green, A.J.; et al. Detection of novel germline mutations for breast cancer in non-BRCA1/2 families. FEBS J. 2015, 282, 3424–3437. [Google Scholar] [CrossRef] [PubMed]
- Invitae. Available online: http://www.invitae.com (accessed on 11 November 2016).
- Lincoln, S.E.; Kobayashi, Y.; Anderson, M.J.; Yang, S.; Desmond, A.J.; Mills, M.A.; Nilsen, G.B.; Jacobs, K.B.; Monzon, F.A.; Kurian, A.W.; et al. A Systematic Comparison of Traditional and Multigene Panel Testing for Hereditary Breast and Ovarian Cancer Genes in More Than 1000 Patients. J. Mol. Diagn. 2015, 17, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Emory-Genetics-Laboratory. Available online: http://www.geneticslab.emory.edu/ (accessed on 11 November 2016).
- GENE-DX. Available online: http://www.genedx.com (accessed on 11 November 2016).
- Susswein, L.R.; Marshall, M.L.; Nusbaum, R.; Vogel Postula, K.J.; Weissman, S.M.; Yackowski, L.; Vaccari, E.M.; Bissonnette, J.; Booker, J.K.; Cremona, M.L.; et al. Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genet. Med. 2016, 18, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Fulgent-Diagnostics. Available online: http://www.fulgentgenetics.com (accessed on 11 November 2016).
- CentoGene. Available online: http://www.centogene.com (accessed on 7 November 2016).
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Renwick, A.; Thompson, D.; Seal, S.; Kelly, P.; Chagtai, T.; Ahmed, M.; North, B.; Jayatilake, H.; Barfoot, R.; Spanova, K.; et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat. Genet. 2006, 38, 873–875. [Google Scholar] [CrossRef]
- Rahman, N.; Seal, S.; Thompson, D.; Kelly, P.; Renwick, A.; Elliott, A.; Reid, S.; Spanova, K.; Barfoot, R.; Chagtai, T.; et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat. Genet. 2007, 39, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, K.; Hall, P.; Gonzalez-Neira, A.; Ghoussaini, M.; Dennis, J.; Milne, R.L.; Schmidt, M.K.; Chang-Claude, J.; Bojesen, S.E.; Bolla, M.K.; et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat. Genet. 2013, 45, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Sakoda, L.C.; Jorgenson, E.; Witte, J.S. Turning of COGS moves forward findings for hormonally mediated cancers. Nat. Genet. 2013, 45, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Ghoussaini, M.; Fletcher, O.; Michailidou, K.; Turnbull, C.; Schmidt, M.K.; Dicks, E.; Dennis, J.; Wang, Q.; Humphreys, M.K.; Luccarini, C.; et al. Genome-wide association analysis identifies three new breast cancer susceptibility loci. Nat. Genet. 2012, 44, 312–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Study, C.; Houlston, R.S.; Webb, E.; Broderick, P.; Pittman, A.M.; di Bernardo, M.C.; Lubbe, S.; Chandler, I.; Vijayakrishnan, J.; Sullivan, K.; et al. Meta-analysis of genome-wide association data identifies four new susceptibility loci for colorectal cancer. Nat. Genet. 2008, 40, 1426–1435. [Google Scholar]
- Jori, B.; Kamps, R.; Xanthoulea, S.; Delvoux, B.; Blok, M.J.; van de Vijver, K.K.; de Koning, B.; Oei, F.T.; Tops, C.M.; Speel, E.J.; et al. Germ-line variants identified by next generation sequencing in a panel of estrogen and cancer associated genes correlate with poor clinical outcome in Lynch syndrome patients. Oncotarget 2015, 6, 41108–41122. [Google Scholar] [PubMed]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, K.D.; Noltner, K.A.; Buzin, C.H.; Gu, D.; Wen-Fong, C.Y.; Nguyen, V.Q.; Han, J.H.; Lowstuter, K.; Longmate, J.; Sommer, S.S.; et al. Beyond Li Fraumeni Syndrome: Clinical characteristics of families with p53 germline mutations. J. Clin. Oncol. 2009, 27, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Hearle, N.; Schumacher, V.; Menko, F.H.; Olschwang, S.; Boardman, L.A.; Gille, J.J.; Keller, J.J.; Westerman, A.M.; Scott, R.J.; Lim, W.; et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin. Cancer Res. 2006, 12, 3209–3215. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Beeghly-Fadiel, A.; Long, J.; Zheng, W. Genetic variants associated with breast-cancer risk: Comprehensive research synopsis, meta-analysis, and epidemiological evidence. Lancet Oncol. 2011, 12, 477–488. [Google Scholar] [CrossRef]
- Peng, S.; Lu, B.; Ruan, W.; Zhu, Y.; Sheng, H.; Lai, M. Genetic polymorphisms and breast cancer risk: Evidence from meta-analyses, pooled analyses, and genome-wide association studies. Breast Cancer Res. Treat. 2011, 127, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Lerner-Ellis, J.; Khalouei, S.; Sopik, V.; Narod, S.A. Genetic risk assessment and prevention: The role of genetic testing panels in breast cancer. Expert Rev. Anticancer Ther. 2015, 15, 1315–1326. [Google Scholar] [CrossRef] [PubMed]
- D’Argenio, V.; Esposito, M.V.; Telese, A.; Precone, V.; Starnone, F.; Nunziato, M.; Cantiello, P.; Iorio, M.; Evangelista, E.; D’Aiuto, M.; et al. The molecular analysis of BRCA1 and BRCA2: Next-generation sequencing supersedes conventional approaches. Clin. Chim. Acta 2015, 446, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Breast-Health-UK. Available online: https://www.breasthealthuk.com (accessed on 15 November 2016).
- Easton, D.F.; Pharoah, P.D.; Antoniou, A.C.; Tischkowitz, M.; Tavtigian, S.V.; Nathanson, K.L.; Devilee, P.; Meindl, A.; Couch, F.J.; Southey, M.; et al. Gene-Panel Sequencing and the Prediction of Breast-Cancer Risk. N. Engl. J. Med. 2015, 372, 2243–2257. [Google Scholar] [CrossRef] [PubMed]
- Rainville, I.R.; Rana, H.Q. Next-generation sequencing for inherited breast cancer risk: Counseling through the complexity. Curr. Oncol. Rep. 2014, 16, 371. [Google Scholar] [CrossRef] [PubMed]
- Mannan, A.U.; Singh, J.; Lakshmikeshava, R.; Thota, N.; Singh, S.; Sowmya, T.S.; Mishra, A.; Sinha, A.; Deshwal, S.; Soni, M.R.; et al. Detection of high frequency of mutations in a breast and/or ovarian cancer cohort: Implications of embracing a multi-gene panel in molecular diagnosis in India. J. Hum. Genet. 2016, 61, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Underhill, M.L.; Germansky, K.A.; Yurgelun, M.B. Advances in Hereditary Colorectal and Pancreatic Cancers. Clin. Ther. 2016, 38, 1600–1621. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, L.C.; Kerkhof, J.; Stuart, A.; Reilly, J.; Eng, B.; Woodside, C.; Levstik, A.; Howlett, C.J.; Rupar, A.C.; Knoll, J.H.; et al. Clinical Next-Generation Sequencing Pipeline Outperforms a Combined Approach Using Sanger Sequencing and Multiplex Ligation-Dependent Probe Amplification in Targeted Gene Panel Analysis. J. Mol. Diagn. 2016, 18, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Pinto, P.; Paulo, P.; Santos, C.; Rocha, P.; Pinto, C.; Veiga, I.; Pinheiro, M.; Peixoto, A.; Teixeira, M.R. Implementation of next-generation sequencing for molecular diagnosis of hereditary breast and ovarian cancer highlights its genetic heterogeneity. Breast Cancer Res. Treat. 2016, 159, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Kraus, C.; Hoyer, J.; Vasileiou, G.; Wunderle, M.; Lux, M.P.; Fasching, P.A.; Krumbiegel, M.; Uebe, S.; Reuter, M.; Beckmann, M.W.; et al. Gene panel sequencing in familial Breast/Ovarian Cancer patients identifies multiple novel mutations also in genes others than BRCA1/2. Int. J. Cancer 2017, 140, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Mavaddat, N.; Peock, S.; Frost, D.; Ellis, S.; Platte, R.; Fineberg, E.; Evans, D.G.; Izatt, L.; Eeles, R.A.; Adlard, J.; et al. Embrace, Cancer risks for BRCA1 and BRCA2 mutation carriers: Results from prospective analysis of EMBRACE. J. Natl. Cancer Inst. 2013, 105, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Canto, M.I.; Harinck, F.; Hruban, R.H.; Offerhaus, G.J.; Poley, J.W.; Kamel, I.; Nio, Y.; Schulick, R.S.; Bassi, C.; Kluijt, I.; et al. International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut 2013, 62, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.C.; Domchek, S.; Parmigiani, G.; Chen, S. Breast cancer risk among male BRCA1 and BRCA2 mutation carriers. J. Natl. Cancer Inst. 2007, 99, 1811–1814. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Yano, K.; Matsuo, F.; Saito, H.; Katagiri, T.; Kurumizaka, H.; Yoshimoto, M.; Kasumi, F.; Akiyama, F.; Sakamoto, G.; et al. Identification of Rad51 alteration in patients with bilateral breast cancer. J. Hum. Genet. 2000, 45, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.H. Recognition, signaling, and repair of DNA double-strand breaks produced by ionizing radiation in mammalian cells: The molecular choreography. Mutat. Res. 2012, 751, 158–246. [Google Scholar] [CrossRef] [PubMed]
- Provenzale, D.; Gupta, S.; Ahnen, D.J.; Bray, T.; Cannon, J.A.; Cooper, G.; David, D.S.; Early, D.S.; Erwin, D.; Ford, J.M.; et al. Genetic/Familial High-Risk Assessment: Colorectal Version 1.2016, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2016, 14, 1010–1030. [Google Scholar]
- Van der Post, R.S.; Vogelaar, I.P.; Carneiro, F.; Guilford, P.; Huntsman, D.; Hoogerbrugge, N.; Caldas, C.; Schreiber, K.E.; Hardwick, R.H.; Ausems, M.G.; et al. Hereditary diffuse gastric cancer: Updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J. Med. Genet. 2015, 52, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Vasen, H.F.; Gruis, N.A.; Frants, R.R.; van der Velden, P.A.; Hille, E.T.; Bergman, W. Risk of developing pancreatic cancer in families with familial atypical multiple mole melanoma associated with a specific 19 deletion of p16 (p16-Leiden). Int. J. Cancer 2000, 87, 809–811. [Google Scholar] [CrossRef]
- Van Lier, M.G.; Wagner, A.; Mathus-Vliegen, E.M.; Kuipers, E.J.; Steyerberg, E.W.; van Leerdam, M.E. High cancer risk in Peutz-Jeghers syndrome: A systematic review and surveillance recommendations. Am. J. Gastroenterol. 2010, 105, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, E.; Leedham, S.; Lewis, A.; Segditsas, S.; Becker, M.; Cuadrado, P.R.; Davis, H.; Kaur, K.; Heinimann, K.; Howarth, K.; et al. Hereditary mixed polyposis syndrome is caused by a 40-kb upstream duplication that leads to increased and ectopic expression of the BMP antagonist GREM1. Nat. Genet. 2012, 44, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Loveday, C.; Turnbull, C.; Ramsay, E.; Hughes, D.; Ruark, E.; Frankum, J.R.; Bowden, G.; Kalmyrzaev, B.; Warren-Perry, M.; Snape, K.; et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat. Genet. 2011, 43, 879–882. [Google Scholar] [CrossRef]
- Liu, W.; Dong, X.; Mai, M.; Seelan, R.S.; Taniguchi, K.; Krishnadath, K.K.; Halling, K.C.; Cunningham, J.M.; Boardman, L.A.; Qian, C.; et al. Mutations in AXIN2 cause colorectal cancer with defective mismatch repair by activating beta-catenin/TCF signalling. Nat. Genet. 2000, 26, 146–147. [Google Scholar] [CrossRef] [PubMed]
- Wautot, V.; Vercherat, C.; Lespinasse, J.; Chambe, B.; Lenoir, G.M.; Zhang, C.X.; Porchet, N.; Cordier, M.; Beroud, C.; Calender, A. Germline mutation profile of MEN1 in multiple endocrine neoplasia type 1: search for correlation between phenotype and the functional domains of the MEN1 protein. Hum. Mutat. 2002, 20, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Frey, M.K.; Kim, S.H.; Bassett, R.Y.; Martineau, J.; Dalton, E.; Chern, J.Y.; Blank, S.V. Rescreening for genetic mutations using multi-gene panel testing in patients who previously underwent non-informative genetic screening. Gynecol. Oncol. 2015, 139, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Hruban, R.H.; Kamiyama, M.; Borges, M.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Palmisano, E.; Brune, K.; Jaffee, E.M.; et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science 2009, 324, 217. [Google Scholar] [CrossRef] [PubMed]
- Comino-Mendez, I.; Gracia-Aznarez, F.J.; Schiavi, F.; Landa, I.; Leandro-Garcia, L.J.; Leton, R.; Honrado, E.; Ramos-Medina, R.; Caronia, D.; Pita, G.; et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat. Genet. 2011, 43, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Palles, C.; Cazier, J.B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Salguero, I.; et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013, 45, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Aznarez, F.J.; Fernandez, V.; Pita, G.; Peterlongo, P.; Dominguez, O.; de la Hoya, M.; Duran, M.; Osorio, A.; Moreno, L.; Gonzalez-Neira, A.; et al. Whole exome sequencing suggests much of non-BRCA1/BRCA2 familial breast cancer is due to moderate and low penetrance susceptibility alleles. PLoS ONE 2013, 8, e55681. [Google Scholar] [CrossRef] [PubMed]
- Hilbers, F.S.; Meijers, C.M.; Laros, J.F.; van Galen, M.; Hoogerbrugge, N.; Vasen, H.F.; Nederlof, P.M.; Wijnen, J.T.; van Asperen, C.J.; Devilee, P. Exome sequencing of germline DNA from non-BRCA1/2 familial breast cancer cases selected on the basis of aCGH tumor profiling. PLoS ONE 2013, 8, e55734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, D.J.; Odefrey, F.A.; Hammet, F.; Giles, G.G.; Baglietto, L.; ABCFS; MCCS; Hopper, J.L.; Schmidt, D.F.; Makalic, E.; et al. FAN1 variants identified in multiple-case early-onset breast cancer families via exome sequencing: No evidence for association with risk for breast cancer. Breast Cancer Res. Treat. 2011, 130, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Snape, K.; Ruark, E.; Tarpey, P.; Renwick, A.; Turnbull, C.; Seal, S.; Murray, A.; Hanks, S.; Douglas, J.; Stratton, M.R.; et al. Predisposition gene identification in common cancers by exome sequencing: Insights from familial breast cancer. Breast Cancer Res. Treat. 2012, 134, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.R.; Doyle, M.A.; Ryland, G.L.; Rowley, S.M.; Choong, D.Y.; Tothill, R.W.; Thorne, H.; kConFab; Barnes, D.R.; Li, J.; et al. Exome sequencing identifies rare deleterious mutations in DNA repair genes FANCC and BLM as potential breast cancer susceptibility alleles. PLoS Genet. 2012, 8, e1002894. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Directors, A.B.O. Points to consider in the clinical application of genomic sequencing. Genet. Med. 2012, 14, 759–761. [Google Scholar]
- Fecteau, H.; Vogel, K.J.; Hanson, K.; Morrill-Cornelius, S. The evolution of cancer risk assessment in the era of next generation sequencing. J. Genet. Couns. 2014, 23, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Luco, R.F.; Allo, M.; Schor, I.E.; Kornblihtt, A.R.; Misteli, T. Epigenetics in Alternative Pre-mRNA Splicing. Cell 2011, 144, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Brandão, R.D.; van Roozendaal, K.; Tserpelis, D.; Gomez Garcia, E.; Blok, M.J. Characterisation of unclassified variants in the BRCA1/2 genes with a putative effect on splicing. Breast Cancer Res. Treat. 2011, 129, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Anczuków, O.; Buisson, M.; Salles, M.-J.; Triboulet, S.; Longy, M.; Lidereau, R.; Sinilnikova, O.M.; Mazoyer, S. Unclassified variants identified in BRCA1 exon 11: Consequences on splicing. Genes Chromosomes Cancer 2008, 47, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, C.; Krieger, S.; Vezain, M.; Rousselin, A.; Tournier, I.; Martins, A.; Berthet, P.; Chevrier, A.; Dugast, C.; Layet, V.; et al. Screening BRCA1 and BRCA2 unclassified variants for splicing mutations using reverse transcription PCR on patient RNA and an ex vivo assay based on a splicing reporter minigene. J. Med. Genet. 2008, 45, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Campos, B.; Díez, O.; Domènech, M.; Baena, M.; Balmaña, J.; Sanz, J.; Ramírez, A.; Alonso, C.; Baiget, M. RNA analysis of eight BRCA1 and BRCA2 unclassified variants identified in breast/ovarian cancer families from Spain. Hum. Mutat. 2003, 22, 337. [Google Scholar] [CrossRef]
- Caux-Moncoutier, V.; Pages-Berhouet, S.; Michaux, D.; Asselain, B.; Castera, L.; de Pauw, A.; Buecher, B.; Gauthier-Villars, M.; Stoppa-Lyonnet, D.; Houdayer, C. Impact of BRCA1 and BRCA2 variants on splicing: Clues from an allelic imbalance study. Eur. J. Hum. Genet. 2009, 17, 1471–1480. [Google Scholar] [CrossRef] [PubMed]
- Fackenthal, J.D.; Cartegni, L.; Krainer, A.R.; Olopade, O.I. BRCA2 T2722R is a deleterious allele that causes exon skipping. Am. J. Hum. Genet. 2002, 71, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.M. Familial melanoma, pancreatic cancer and germline CDKN2A mutations. Hum. Mutat. 2004, 23, 630. [Google Scholar] [CrossRef] [PubMed]
- Harland, M.; Mistry, S.; Bishop, D.T.; Newton Bishop, J.A. A deep intronic mutation in CDKN2A is associated with disease in a subset of melanoma pedigrees. Hum. Mol. Genet. 2001, 10, 2679–2686. [Google Scholar] [CrossRef] [PubMed]
- Wadt, K.A.W.; Aoude, L.G.; Golmard, L.; Hansen, T.V.O.; Sastre-Garau, X.; Hayward, N.K.; Gerdes, A.-M. Germline RAD51B truncating mutation in a family with cutaneous melanoma. Fam. Cancer 2015, 14, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Rutter, J.L.; Goldstein, A.M.; Davila, M.R.; Tucker, M.A.; Struewing, J.P. CDKN2A point mutations D153spl(c.457G>T) and IVS2+1G>T result in aberrant splice products affecting both p16INK4a and p14ARF. Oncogene 2003, 22, 4444–4448. [Google Scholar] [CrossRef] [PubMed]
- Colapietro, P.; Gervasini, C.; Natacci, F.; Rossi, L.; Riva, P.; Larizza, L. NF1 exon 7 skipping and sequence alterations in exonic splice enhancers (ESEs) in a neurofibromatosis 1 patient. Hum. Genet. 2003, 113, 551–554. [Google Scholar] [PubMed]
- Cloonan, N.; Forrest, A.R.R.; Kolle, G.; Gardiner, B.B.A.; Faulkner, G.J.; Brown, M.K.; Taylor, D.F.; Steptoe, A.L.; Wani, S.; Bethel, G.; et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat. Methods 2008, 5, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Sultan, M.; Schulz, M.H.; Richard, H.; Magen, A.; Klingenhoff, A.; Scherf, M.; Seifert, M.; Borodina, T.; Soldatov, A.; Parkhomchuk, D.; et al. A Global View of Gene Activity and Alternative Splicing by Deep Sequencing of the Human Transcriptome. Science 2008, 321, 956–960. [Google Scholar] [CrossRef] [PubMed]
- Morin, R.D.; Bainbridge, M.; Fejes, A.; Hirst, M.; Krzywinski, M.; Pugh, T.J.; McDonald, H.; Varhol, R.; Jones, S.J.M.; Marra, M.A. Profiling the HeLa S3 transcriptome using randomly primed cDNA and massively parallel short-read sequencing. BioTechniques 2008, 45, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Ramaswami, G.; Lin, W.; Piskol, R.; Tan, M.H.; Davis, C.; Li, J.B. Accurate identification of human Alu and non-Alu RNA editing sites. Nat. Methods 2012, 9, 579–581. [Google Scholar] [CrossRef] [PubMed]
- DARNED. Available online: http://www.darned.ucc.ie (accessed on 7 November 2016).
- Zhang, Q.; Xiao, X. Genome Sequence-Independent Identification of RNA Editing Sites. Nat. Methods 2015, 12, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Cummings, B.B.; Marshall, J.L.; Tukiainen, T.; Lek, M.; Donkervoort, S.; Foley, A.R.; Bolduc, V.; Waddell, L.; Sandaradura, S.; O’Grady, G.L.; et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. bioRxiv 2016. [Google Scholar] [CrossRef]
- Lin, L.; Park, J.W.; Ramachandran, S.; Zhang, Y.; Tseng, Y.-T.; Shen, S.; Waldvogel, H.J.; Curtis, M.A.; Faull, R.L.M.; Troncoso, J.C.; et al. Transcriptome sequencing reveals aberrant alternative splicing in Huntington’s disease. Hum. Mol. Genet. 2016, 25, 3454–3466. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Sahashi, K.; Rigo, F.; Hung, G.; Horev, G.; Bennett, C.F.; Krainer, A.R. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 2011, 478, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Lentz, J.J.; Jodelka, F.M.; Hinrich, A.J.; McCaffrey, K.E.; Farris, H.E.; Spalitta, M.J.; Bazan, N.G.; Duelli, D.M.; Rigo, F.; Hastings, M.L. Rescue of hearing and vestibular function by antisense oligonucleotides in a mouse model of human deafness. Nat. Med. 2013, 19, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Wally, V.; Murauer, E.M.; Bauer, J.W. Spliceosome-Mediated Trans-Splicing: The Therapeutic Cut and Paste. J. Investig. Dermatol. 2012, 132, 1959–1966. [Google Scholar] [CrossRef] [PubMed]
- Tockner, B.; Kocher, T.; Hainzl, S.; Reichelt, J.; Bauer, J.W.; Koller, U.; Murauer, E.M. Construction and validation of a RNA trans-splicing molecule suitable to repair a large number of COL7A1 mutations. Gene Ther. 2016, 23, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Weissman, S.M.; Burt, R.; Church, J.; Erdman, S.; Hampel, H.; Holter, S.; Jasperson, K.; Kalady, M.F.; Haidle, J.L.; Lynch, H.T.; et al. Identification of individuals at risk for Lynch syndrome using targeted evaluations and genetic testing: National Society of Genetic Counselors and the Collaborative Group of the Americas on Inherited Colorectal Cancer joint practice guideline. J. Genet. Couns. 2012, 21, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, A.C.; Chenevix-Trench, G. Common genetic variants and cancer risk in Mendelian cancer syndromes. Curr. Opin. Genet. Dev. 2010, 20, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Milne, R.L.; Antoniou, A.C. Genetic modifiers of cancer risk for BRCA1 and BRCA2 mutation carriers. Ann. Oncol. 2011, 22, i11–i17. [Google Scholar] [CrossRef] [PubMed]
- Ao, A.; Wells, D.; Handyside, A.H.; Winston, R.M.; Delhanty, J.D. Preimplantation genetic diagnosis of inherited cancer: Familial adenomatous polyposis coli. J. Assist. Reprod. Genet. 1998, 15, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Abou-Sleiman, P.M.; Apessos, A.; Harper, J.C.; Serhal, P.; Winston, R.M.; Delhanty, J.D. First application of preimplantation genetic diagnosis to neurofibromatosis type 2 (NF2). Prenat. Diagn. 2002, 22, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Verlinsky, Y.; Rechitsky, S.; Verlinsky, O.; Chistokhina, A.; Sharapova, T.; Masciangelo, C.; Levy, M.; Kaplan, B.; Lederer, K.; Kuliev, A. Preimplantation diagnosis for neurofibromatosis. Reprod. Biomed. Online 2002, 4, 218–222. [Google Scholar] [CrossRef]
- Offit, K.; Sagi, M.; Hurley, K. Preimplantation genetic diagnosis for cancer syndromes: A new challenge for preventive medicine. JAMA 2006, 296, 2727–2730. [Google Scholar] [CrossRef] [PubMed]
- Handyside, A.H.; Kontogianni, E.H.; Hardy, K.; Winston, R.M. Pregnancies from biopsied human preimplantation embryos sexed by Y-specific DNA amplification. Nature 1990, 344, 768–770. [Google Scholar] [CrossRef] [PubMed]
- Dreesen, J.C.; Jacobs, L.J.; Bras, M.; Herbergs, J.; Dumoulin, J.C.; Geraedts, J.P.; Evers, J.L.; Smeets, H.J. Multiplex PCR of polymorphic markers flanking the CFTR gene; a general approach for preimplantation genetic diagnosis of cystic fibrosis. Mol. Hum. Reprod. 2000, 6, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Renwick, P.; Trussler, J.; Lashwood, A.; Braude, P.; Ogilvie, C.M. Preimplantation genetic haplotyping: 127 diagnostic cycles demonstrating a robust, efficient alternative to direct mutation testing on single cells. Reprod. Biomed. Online 2010, 20, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Drusedau, M.; Dreesen, J.C.; Derks-Smeets, I.; Coonen, E.; van Golde, R.; van Echten-Arends, J.; Kastrop, P.M.; Blok, M.J.; Gomez-Garcia, E.; Geraedts, J.P.; et al. PGD for hereditary breast and ovarian cancer: The route to universal tests for BRCA1 and BRCA2 mutation carriers. Eur. J. Hum. Genet. 2013, 21, 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- Altarescu, G.; Zeevi, D.A.; Zeligson, S.; Perlberg, S.; Eldar-Geva, T.; Margalioth, E.J.; Levy-Lahad, E.; Renbaum, P. Familial haplotyping and embryo analysis for Preimplantation genetic diagnosis (PGD) using DNA microarrays: A proof of principle study. J. Assist. Reprod. Genet. 2013, 30, 1595–1603. [Google Scholar] [CrossRef] [PubMed]
- Natesan, S.A.; Bladon, A.J.; Coskun, S.; Qubbaj, W.; Prates, R.; Munne, S.; Coonen, E.; Dreesen, J.C.; Stevens, S.J.; Paulussen, A.D.; et al. Genome-wide karyomapping accurately identifies the inheritance of single-gene defects in human preimplantation embryos in vitro. Genet. Med. 2014, 16, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Zamani Esteki, M.; Dimitriadou, E.; Mateiu, L.; Melotte, C.; van der Aa, N.; Kumar, P.; Das, R.; Theunis, K.; Cheng, J.; Legius, E.; et al. Concurrent whole-genome haplotyping and copy-number profiling of single cells. Am. J. Hum. Genet. 2015, 96, 894–912. [Google Scholar] [CrossRef]
- Treff, N.R.; Forman, E.J.; Scott, R.T., Jr. Next-generation sequencing for preimplantation genetic diagnosis. Fertil. Steril. 2013, 99, e17–e18. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, L.; Wang, H.; Ma, M.; Wang, X.; Li, Y.; Zhang, W.; Zhang, J.; Cram, D.S.; Yao, Y. The Performance of Whole Genome Amplification Methods and Next-Generation Sequencing for Pre-Implantation Genetic Diagnosis of Chromosomal Abnormalities. J. Genet. Genom. 2015, 42, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Huang, L.; Xu, L.; Huang, J.; Ma, F.; Zhu, X.; Tang, Y.; Liu, M.; Lian, Y.; Liu, P.; et al. Live births after simultaneous avoidance of monogenic diseases and chromosome abnormality by next-generation sequencing with linkage analyses. Proc. Natl. Acad. Sci. USA 2015, 112, 15964–15969. [Google Scholar] [CrossRef]
- Huang, L.; Ma, F.; Chapman, A.; Lu, S.; Xie, X.S. Single-Cell Whole-Genome Amplification and Sequencing: Methodology and Applications. Annu. Rev. Genom. Hum. Genet. 2015, 16, 79–102. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.; Geraedts, J.; Borry, P.; Cornel, M.C.; Dondorp, W.J.; Gianaroli, L.; Harton, G.; Milachich, T.; Kaariainen, H.; Liebaers, I.; et al. EuroGentest, Current issues in medically assisted reproduction and genetics in Europe: Research, clinical practice, ethics, legal issues and policy. Hum. Reprod. 2014, 29, 1603–1609. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.J.; Stephens, P.J.; Pleasance, E.D.; O’Meara, S.; Li, H.; Santarius, T.; Stebbings, L.A.; Leroy, C.; Edkins, S.; Hardy, C.; et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat. Genet. 2008, 40, 722–729. [Google Scholar] [CrossRef]
- Ley, T.J.; Mardis, E.R.; Ding, L.; Fulton, B.; McLellan, M.D.; Chen, K.; Dooling, D.; Dunford-Shore, B.H.; McGrath, S.; Hickenbotham, M.; et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 2008, 456, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.P.; Morin, R.D.; Khattra, J.; Prentice, L.; Pugh, T.; Burleigh, A.; Delaney, A.; Gelmon, K.; Guliany, R.; Senz, J.; et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature 2009, 461, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; McBride, D.J.; Lin, M.L.; Varela, I.; Pleasance, E.D.; Simpson, J.T.; Stebbings, L.A.; Leroy, C.; Edkins, S.; Mudie, L.J.; et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature 2009, 462, 1005–1010. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature, 2012; 490, 61–70. [Google Scholar]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Kriegsmann, M.; Endris, V.; Wolf, T.; Pfarr, N.; Stenzinger, A.; Loibl, S.; Denkert, C.; Schneeweiss, A.; Budczies, J.; Sinn, P.; et al. Mutational profiles in triple-negative breast cancer defined by ultradeep multigene sequencing show high rates of PI3K pathway alterations and clinically relevant entity subgroup specific differences. Oncotarget 2014, 5, 9952–9965. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Cheung, L.W.; Li, J.; Ju, Z.; Yu, S.; Stemke-Hale, K.; Dogruluk, T.; Lu, Y.; Liu, X.; Gu, C.; et al. Whole-exome sequencing combined with functional genomics reveals novel candidate driver cancer genes in endometrial cancer. Genome Res. 2012, 22, 2120–2129. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [PubMed]
- Ab Mutalib, N.S.; Syafruddin, S.E.; Md Zain, R.R.; Mohd Dali, A.Z.; Mohd Yunos, R.I.; Saidin, S.; Jamal, R.; Mokhtar, N.M. Molecular characterization of serous ovarian carcinoma using a multigene next generation sequencing cancer panel approach. BMC Res. Notes 2014, 7, 805. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [Green Version]
- Mackenzie, R.; Kommoss, S.; Winterhoff, B.J.; Kipp, B.R.; Garcia, J.J.; Voss, J.; Halling, K.; Karnezis, A.; Senz, J.; Yang, W.; et al. Targeted deep sequencing of mucinous ovarian tumors reveals multiple overlapping RAS-pathway activating mutations in borderline and cancerous neoplasms. BMC Cancer 2015, 15, 415. [Google Scholar] [CrossRef] [PubMed]
- Muller, E.; Brault, B.; Holmes, A.; Legros, A.; Jeannot, E.; Campitelli, M.; Rousselin, A.; Goardon, N.; Frebourg, T.; Krieger, S.; et al. Genetic profiles of cervical tumors by high-throughput sequencing for personalized medical care. Cancer Med. 2015, 4, 1484–1493. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, F.C.; Kipp, B.R.; Voss, J.S.; Campion, M.B.; Minot, D.M.; Tu, Z.J.; Klee, E.W.; Sciallis, A.P.; Graham, R.P.; Lazaridis, K.N.; et al. Endoscopic ultrasound fine-needle aspiration cytology mutation profiling using targeted next-generation sequencing: Personalized care for rectal cancer. Am. J. Clin. Pathol. 2015, 143, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Zhang, L.; Chen, L.; Sah, S.; Latham, G.J.; Patel, R.; Song, Q.; Koeppen, H.; Tam, R.; Schleifman, E.; Mashhedi, H.; et al. Profiling cancer gene mutations in clinical formalin-fixed, paraffin-embedded colorectal tumor specimens using targeted next-generation sequencing. Oncologist 2014, 19, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Malapelle, U.; Vigliar, E.; Sgariglia, R.; Bellevicine, C.; Colarossi, L.; Vitale, D.; Pallante, P.; Troncone, G. Ion Torrent next-generation sequencing for routine identification of clinically relevant mutations in colorectal cancer patients. J. Clin. Pathol. 2015, 68, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Tinhofer, I.; Budach, V.; Saki, M.; Konschak, R.; Niehr, F.; Johrens, K.; Weichert, W.; Linge, A.; Lohaus, F.; Krause, M.; et al. Targeted next-generation sequencing of locally advanced squamous cell carcinomas of the head and neck reveals druggable targets for improving adjuvant chemoradiation. Eur. J. Cancer 2016, 57, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar]
- Saba, N.F.; Wilson, M.; Doho, G.; DaSilva, J.; Benjamin Isett, R.; Newman, S.; Chen, Z.G.; Magliocca, K.; Rossi, M.R. Mutation and Transcriptional Profiling of Formalin-Fixed Paraffin Embedded Specimens as Companion Methods to Immunohistochemistry for Determining Therapeutic Targets in Oropharyngeal Squamous Cell Carcinoma (OPSCC): A Pilot of Proof of Principle. Head Neck Pathol. 2015, 9, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.T.; Chen, S.J.; Lee, L.Y.; Hsueh, C.; Yang, L.Y.; Lin, C.Y.; Fan, K.H.; Wang, H.M.; Ng, S.H.; Lin, C.H.; et al. An Ultra-Deep Targeted Sequencing Gene Panel Improves the Prognostic Stratification of Patients with Advanced Oral Cavity Squamous Cell Carcinoma. Medicine 2016, 95, e2751. [Google Scholar] [CrossRef] [PubMed]
- Fassan, M.; Simbolo, M.; Bria, E.; Mafficini, A.; Pilotto, S.; Capelli, P.; Bencivenga, M.; Pecori, S.; Luchini, C.; Neves, D.; et al. High-throughput mutation profiling identifies novel molecular dysregulation in high-grade intraepithelial neoplasia and early gastric cancers. Gastric Cancer 2014, 17, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Huo, X.; Ye, H.; Tang, C.; Nandakumar, V.; Lou, F.; Zhang, D.; Dong, H.; Sun, H.; Jiang, S.; et al. Genetic mutation analysis of human gastric adenocarcinomas using ion torrent sequencing platform. PLoS ONE 2014, 9, e100442. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Lohr, J.M.; Nilsson, M.; Segersvard, R.; Matsson, H.; Verbeke, C.; Heuchel, R.; Kere, J.; Iafrate, A.J.; Zheng, Z.; et al. Variant Profiling of Candidate Genes in Pancreatic Ductal Adenocarcinoma. Clin. Chem. 2015, 61, 1408–1416. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.J.; Yachida, S.; Mudie, L.J.; Stephens, P.J.; Pleasance, E.D.; Stebbings, L.A.; Morsberger, L.A.; Latimer, C.; McLaren, S.; Lin, M.L.; et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 2010, 467, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.C.; Basturk, O.; Brannon, A.R.; Bhanot, U.; Scott, S.N.; Bouvier, N.; LaFemina, J.; Jarnagin, W.R.; Berger, M.F.; Klimstra, D.; et al. GNAS and KRAS Mutations Define Separate Progression Pathways in Intraductal Papillary Mucinous Neoplasm-Associated Carcinoma. J. Am. Coll. Surg. 2015, 220, 845–e1. [Google Scholar] [CrossRef] [PubMed]
- Ku, B.M.; Jung, H.A.; Sun, J.M.; Ko, Y.H.; Jeong, H.S.; Son, Y.I.; Baek, C.H.; Park, K.; Ahn, M.J. High-throughput profiling identifies clinically actionable mutations in salivary duct carcinoma. J. Transl. Med. 2014, 12, 299. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, J.; Hong, M.E.; Do, I.G.; Kang, S.Y.; Ha, S.Y.; Kim, S.T.; Park, S.H.; Kang, W.K.; Choi, M.G.; et al. High-throughput sequencing and copy number variation detection using formalin fixed embedded tissue in metastatic gastric cancer. PLoS ONE 2014, 9, e111693. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [Green Version]
- Fonseca, I.; Bell, A.; Wani, K.; Bell, D. Global transcriptome and sequenome analysis of formalin-fixed salivary epithelial-myoepithelial carcinoma specimens. Genes Chromosomes Cancer 2015, 54, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Li-Chang, H.H.; Kasaian, K.; Ng, Y.; Lum, A.; Kong, E.; Lim, H.; Jones, S.J.; Huntsman, D.G.; Schaeffer, D.F.; Yip, S. Retrospective review using targeted deep sequencing reveals mutational differences between gastroesophageal junction and gastric carcinomas. BMC Cancer 2015, 15, 32. [Google Scholar] [CrossRef] [PubMed]
- Salomao, M.; Luna, A.M.; Sepulveda, J.L.; Sepulveda, A.R. Mutational analysis by next generation sequencing of gastric type dysplasia occurring in hyperplastic polyps of the stomach: Mutations in gastric hyperplastic polyps. Exp. Mol. Pathol. 2015, 99, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, F.C.; Kipp, B.R.; Kerr, S.E.; Voss, J.S.; Graham, R.P.; Campion, M.B.; Minot, D.M.; Tu, Z.J.; Klee, E.W.; Lazaridis, K.N.; et al. Kinase genotype analysis of gastric gastrointestinal stromal tumor cytology samples using targeted next-generation sequencing. Clin. Gastroenterol. Hepatol. 2015, 13, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar]
- Scarpa, A.; Sikora, K.; Fassan, M.; Rachiglio, A.M.; Cappellesso, R.; Antonello, D.; Amato, E.; Mafficini, A.; Lambiase, M.; Esposito, C.; et al. Molecular typing of lung adenocarcinoma on cytological samples using a multigene next generation sequencing panel. PLoS ONE 2013, 8, e80478. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar]
- Preusser, M.; Berghoff, A.S.; Koller, R.; Zielinski, C.C.; Hainfellner, J.A.; Liebmann-Reindl, S.; Popitsch, N.; Geier, C.B.; Streubel, B.; Birner, P. Spectrum of gene mutations detected by next generation exome sequencing in brain metastases of lung adenocarcinoma. Eur. J. Cancer 2015, 51, 1803–1811. [Google Scholar] [CrossRef] [PubMed]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, I.S.; Devarakonda, S.; Lockwood, C.M.; Spencer, D.H.; Guebert, K.; Bredemeyer, A.J.; Al-Kateb, H.; Nguyen, T.T.; Duncavage, E.J.; Cottrell, C.E.; et al. Clinical next-generation sequencing in patients with non-small cell lung cancer. Cancer 2015, 121, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Sahm, F.; Schrimpf, D.; Jones, D.T.; Meyer, J.; Kratz, A.; Reuss, D.; Capper, D.; Koelsche, C.; Korshunov, A.; Wiestler, B.; et al. Next-generation sequencing in routine brain tumor diagnostics enables an integrated diagnosis and identifies actionable targets. Acta Neuropathol. 2016, 131, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Zacher, A.; Kaulich, K.; Stepanow, S.; Wolter, M.; Kohrer, K.; Felsberg, J.; Malzkorn, B.; Reifenberger, G. Molecular diagnostics of gliomas using next generation sequencing of a glioma-tailored gene panel. Brain Pathol. 2016. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Brat, D.J.; Verhaak, R.G.; Aldape, K.D.; Yung, W.K.; Salama, S.R.; Cooper, L.A.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Tabone, T.; Abuhusain, H.J.; Nowak, A.K.; Australian, G.; Clinical Outcome of Glioma Network; Erber, W.N.; McDonald, K.L. Multigene profiling to identify alternative treatment options for glioblastoma: A pilot study. J. Clin. Pathol. 2014, 67, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Virk, S.M.; Gibson, R.M.; Quinones-Mateu, M.E.; Barnholtz-Sloan, J.S. Identification of variants in primary and recurrent glioblastoma using a cancer-specific gene panel and whole exome sequencing. PLoS ONE 2015, 10, e0124178. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar]
- Cancer Genome Atlas Research Network; Linehan, W.M.; Spellman, P.T.; Ricketts, C.J.; Creighton, C.J.; Fei, S.S.; Davis, C.; Wheeler, D.A.; Murray, B.A.; Schmidt, L.; et al. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 135–145. [Google Scholar] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar]
- Davis, C.F.; Ricketts, C.J.; Wang, M.; Yang, L.; Cherniack, A.D.; Shen, H.; Buhay, C.; Kang, H.; Kim, S.C.; Fahey, C.C.; et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell 2014, 26, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.R.; Eble, J.N.; Amin, M.B.; Gupta, N.S.; Smith, S.C.; Sholl, L.M.; Montironi, R.; Hirsch, M.S.; Hornick, J.L. Succinate dehydrogenase-deficient renal cell carcinoma: Detailed characterization of 11 tumors defining a unique subtype of renal cell carcinoma. Mod. Pathol. 2015, 28, 80–94. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, M.; Gruppioni, E.; Massari, F.; Giunchi, F.; Altimari, A.; Ciccarese, C.; Bimbatti, D.; Scarpa, A.; Iacovelli, R.; Porta, C.; et al. Wide spetcrum mutational analysis of metastatic renal cell cancer: A retrospective next generation sequencing approach. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar]
- Nikiforov, Y.E.; Carty, S.E.; Chiosea, S.I.; Coyne, C.; Duvvuri, U.; Ferris, R.L.; Gooding, W.E.; Hodak, S.P.; LeBeau, S.O.; Ohori, N.P.; et al. Highly accurate diagnosis of cancer in thyroid nodules with follicular neoplasm/suspicious for a follicular neoplasm cytology by ThyroSeq v2 next-generation sequencing assay. Cancer 2014, 120, 3627–3634. [Google Scholar] [CrossRef] [PubMed]
- Picarsic, J.L.; Buryk, M.A.; Ozolek, J.; Ranganathan, S.; Monaco, S.E.; Simons, J.P.; Witchel, S.F.; Gurtunca, N.; Joyce, J.; Zhong, S.; et al. Molecular Characterization of Sporadic Pediatric Thyroid Carcinoma with the DNA/RNA ThyroSeq v2 Next-Generation Sequencing Assay. Pediatr. Dev. Pathol. 2016, 19, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar]
- Patel, K.P.; Newberry, K.J.; Luthra, R.; Jabbour, E.; Pierce, S.; Cortes, J.; Singh, R.; Mehrotra, M.; Routbort, M.J.; Luthra, M.; et al. Correlation of mutation profile and response in patients with myelofibrosis treated with ruxolitinib. Blood 2015, 126, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Cazzola, M.; Boultwood, J.; Malcovati, L.; Vyas, P.; Bowen, D.; Pellagatti, A.; Wainscoat, J.S.; Hellstrom-Lindberg, E.; Gambacorti-Passerini, C.; et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N. Engl. J. Med. 2011, 365, 1384–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Lopez, J.; Lahuerta, J.J.; Pepin, F.; Gonzalez, M.; Barrio, S.; Ayala, R.; Puig, N.; Montalban, M.A.; Paiva, B.; Weng, L.; et al. Prognostic value of deep sequencing method for minimal residual disease detection in multiple myeloma. Blood 2014, 123, 3073–3079. [Google Scholar] [CrossRef]
- Faham, M.; Zheng, J.; Moorhead, M.; Carlton, V.E.; Stow, P.; Coustan-Smith, E.; Pui, C.H.; Campana, D. Deep-sequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood 2012, 120, 5173–5180. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar]
- Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar]
- Churi, C.R.; Shroff, R.; Wang, Y.; Rashid, A.; Kang, H.C.; Weatherly, J.; Zuo, M.; Zinner, R.; Hong, D.; Meric-Bernstam, F.; et al. Mutation profiling in cholangiocarcinoma: Prognostic and therapeutic implications. PLoS ONE 2014, 9, e115383. [Google Scholar] [CrossRef]
- Shitara, M.; Okuda, K.; Suzuki, A.; Tatematsu, T.; Hikosaka, Y.; Moriyama, S.; Sasaki, H.; Fujii, Y.; Yano, M. Genetic profiling of thymic carcinoma using targeted next-generation sequencing. Lung Cancer 2014, 86, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Lo Iacono, M.; Monica, V.; Righi, L.; Grosso, F.; Libener, R.; Vatrano, S.; Bironzo, P.; Novello, S.; Musmeci, L.; Volante, M.; et al. Targeted next-generation sequencing of cancer genes in advanced stage malignant pleural mesothelioma: A retrospective study. J. Thorac. Oncol. 2015, 10, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Cherniack, A.D.; Dewal, N.; Moffitt, R.A.; Danilova, L.; Murray, B.A.; Lerario, A.M.; Else, T.; Knijnenburg, T.A.; Ciriello, G.; et al. Comprehensive Pan-Genomic Characterization of Adrenocortical Carcinoma. Cancer Cell 2016, 29, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Harms, P.W.; Collie, A.M.; Hovelson, D.H.; Cani, A.K.; Verhaegen, M.E.; Patel, R.M.; Fullen, D.R.; Omata, K.; Dlugosz, A.A.; Tomlins, S.A.; et al. Next generation sequencing of Cytokeratin 20-negative Merkel cell carcinoma reveals ultraviolet-signature mutations and recurrent TP53 and RB1 inactivation. Mod. Pathol. 2016, 29, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Ballester, L.Y.; Sarabia, S.F.; Sayeed, H.; Patel, N.; Baalwa, J.; Athanassaki, I.; Hernandez, J.A.; Fang, E.; Quintanilla, N.M.; Roy, A.; et al. Integrating Molecular Testing in the Diagnosis and Management of Children with Thyroid Lesions. Pediatr. Dev. Pathol. 2016, 19, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.H.; DuBois, S.G.; Glade Bender, J.L.; Kim, A.; Crompton, B.D.; Parker, E.; Dumont, I.P.; Hong, A.L.; Guo, D.; Church, A.; et al. Multicenter Feasibility Study of Tumor Molecular Profiling to Inform Therapeutic Decisions in Advanced Pediatric Solid Tumors: The Individualized Cancer Therapy (iCat) Study. JAMA Oncol. 2016. [Google Scholar] [CrossRef]
- Frampton, G.M.; Fichtenholtz, A.; Otto, G.A.; Wang, K.; Downing, S.R.; He, J.; Schnall-Levin, M.; White, J.; Sanford, E.M.; An, P.; et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol. 2013, 31, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Chevrier, S.; Arnould, L.; Ghiringhelli, F.; Coudert, B.; Fumoleau, P.; Boidot, R. Next-generation sequencing analysis of lung and colon carcinomas reveals a variety of genetic alterations. Int. J. Oncol. 2014, 45, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Liebers, M.; Zhelyazkova, B.; Cao, Y.; Panditi, D.; Lynch, K.D.; Chen, J.; Robinson, H.E.; Shim, H.S.; Chmielecki, J.; et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat. Med. 2014, 20, 1479–1484. [Google Scholar] [CrossRef] [PubMed]
- Haraldsdottir, S.; Hampel, H.; Tomsic, J.; Frankel, W.L.; Pearlman, R.; de la Chapelle, A.; Pritchard, C.C. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology 2014, 147, 1308e1–1316e1. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Lee, J.; Hong, M.; Park, K.; Park, J.O.; Ahn, T.; Park, S.H.; Park, Y.S.; Lim, H.Y.; Sun, J.M.; et al. The NEXT-1 (Next generation pErsonalized tX with mulTi-omics and preclinical model) trial: Prospective molecular screening trial of metastatic solid cancer patients, a feasibility analysis. Oncotarget 2015, 6, 33358–33368. [Google Scholar] [PubMed]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Dahlman, K.H.; Knol, J.; Gilbert, J.; Puzanov, I.; Means-Powell, J.; Balko, J.M.; Lovly, C.M.; Murphy, B.A.; Goff, L.W.; et al. Enabling a genetically informed approach to cancer medicine: A retrospective evaluation of the impact of comprehensive tumor profiling using a targeted next-generation sequencing panel. Oncologist 2014, 19, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.R.; Patel, K.P.; Routbort, M.J.; Aldape, K.; Lu, X.; Manekia, J.; Abraham, R.; Reddy, N.G.; Barkoh, B.A.; Veliyathu, J.; et al. Clinical massively parallel next-generation sequencing analysis of 409 cancer-related genes for mutations and copy number variations in solid tumours. Br. J. Cancer 2014, 111, 2014–2023. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.R.; Patel, K.P.; Routbort, M.J.; Reddy, N.G.; Barkoh, B.A.; Handal, B.; Kanagal-Shamanna, R.; Greaves, W.O.; Medeiros, L.J.; Aldape, K.D.; et al. Clinical validation of a next-generation sequencing screen for mutational hotspots in 46 cancer-related genes. J. Mol. Diagn. 2013, 15, 607–622. [Google Scholar] [CrossRef]
- Parker, B.C.; Zhang, W. Fusion genes in solid tumors: An emerging target for cancer diagnosis and treatment. Chin. J. Cancer 2013, 32, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Cancer-Genome-Project. Available online: http://www.sanger.ac.uk/science/groups/cancer-genome-project (accessed on November 2016).
- Pleasance, E.D.; Stephens, P.J.; O’Meara, S.; McBride, D.J.; Meynert, A.; Jones, D.; Lin, M.L.; Beare, D.; Lau, K.W.; Greenman, C.; et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature 2010, 463, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Bignell, G.R.; Greenman, C.D.; Davies, H.; Butler, A.P.; Edkins, S.; Andrews, J.M.; Buck, G.; Chen, L.; Beare, D.; Latimer, C.; et al. Signatures of mutation and selection in the cancer genome. Nature 2010, 463, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Tang, G.; Bindal, N.; Bamford, S.; Dawson, E.; Cole, C.; Kok, C.Y.; Jia, M.; Ewing, R.; Menzies, A.; et al. COSMIC (the Catalogue of Somatic Mutations in Cancer): A resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 2010, 38, D652–D657. [Google Scholar] [CrossRef]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012, 483, 570–575. [Google Scholar] [CrossRef] [PubMed]
- The-Cancer-Genome-Atlas. Available online: https://cancergenome.nih.gov/ (accessed on November 2016).
- Giordano, T.J. The cancer genome atlas research network: A sight to behold. Endocr. Pathol. 2014, 25, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Zardavas, D.; Maetens, M.; Irrthum, A.; Goulioti, T.; Engelen, K.; Fumagalli, D.; Salgado, R.; Aftimos, P.; Saini, K.S.; Sotiriou, C.; et al. The AURORA initiative for metastatic breast cancer. Br. J. Cancer 2014, 111, 1881–1887. [Google Scholar] [CrossRef] [PubMed]
- International Cancer Genome Consortium; Hudson, T.J.; Anderson, W.; Artez, A.; Barker, A.D.; Bell, C.; Bernabe, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; et al. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.J.; Pleasance, E.D.; Stephens, P.J.; Dicks, E.; Rance, R.; Goodhead, I.; Follows, G.A.; Green, A.R.; Futreal, P.A.; Stratton, M.R. Subclonal phylogenetic structures in cancer revealed by ultra-deep sequencing. Proc. Natl. Acad. Sci. USA 2008, 105, 13081–13086. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Lin, Y.; Liu, J.; Wan, L.; Liu, Z.; Cheng, S.; Fei, L.; Deng, R.; Wang, J.; Chen, X.; et al. A targeted next-generation sequencing method for identifying clinically relevant mutation profiles in lung adenocarcinoma. Sci. Rep. 2016, 6, 22338. [Google Scholar] [CrossRef] [PubMed]
- Tsongalis, G.J.; Peterson, J.D.; de Abreu, F.B.; Tunkey, C.D.; Gallagher, T.L.; Strausbaugh, L.D.; Wells, W.A.; Amos, C.I. Routine use of the Ion Torrent AmpliSeq Cancer Hotspot Panel for identification of clinically actionable somatic mutations. Clin. Chem. Lab. Med. 2014, 52, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Bourgon, R.; Lu, S.; Yan, Y.; Lackner, M.R.; Wang, W.; Weigman, V.; Wang, D.; Guan, Y.; Ryner, L.; Koeppen, H.; et al. High-throughput detection of clinically relevant mutations in archived tumor samples by multiplexed PCR and next-generation sequencing. Clin. Cancer Res. 2014, 20, 2080–2091. [Google Scholar] [CrossRef] [PubMed]
- Portier, B.P.; Kanagal-Shamanna, R.; Luthra, R.; Singh, R.; Routbort, M.J.; Handal, B.; Reddy, N.; Barkoh, B.A.; Zuo, Z.; Medeiros, L.J.; et al. Quantitative assessment of mutant allele burden in solid tumors by semiconductor-based next-generation sequencing. Am. J. Clin. Pathol. 2014, 141, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Boleij, A.; Tops, B.B.; Rombout, P.D.; Dequeker, E.M.; Ligtenberg, M.J.; van Krieken, J.H.; Dutch RAS EQA Initiative. RAS testing in metastatic colorectal cancer: Excellent reproducibility amongst 17 Dutch pathology centers. Oncotarget 2015, 6, 15681–15689. [Google Scholar] [CrossRef] [PubMed]
- Haslam, K.; Catherwood, M.A.; Dobbin, E.; Sproul, A.; Langabeer, S.E.; Mills, K.I. Inter-Laboratory Evaluation of a Next-Generation Sequencing Panel for Acute Myeloid Leukemia. Mol. Diagn. Ther. 2016, 20, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.Q.; Fellowes, A.; Doig, K.; Ellul, J.; Bosma, T.J.; Irwin, D.; Vedururu, R.; Tan, A.Y.; Weiss, J.; Chan, K.S.; et al. Assessing the clinical value of targeted massively parallel sequencing in a longitudinal, prospective population-based study of cancer patients. Br. J. Cancer 2015, 112, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Kotoula, V.; Lyberopoulou, A.; Papadopoulou, K.; Charalambous, E.; Alexopoulou, Z.; Gakou, C.; Lakis, S.; Tsolaki, E.; Lilakos, K.; Fountzilas, G. Evaluation of two highly-multiplexed custom panels for massively parallel semiconductor sequencing on paraffin DNA. PLoS ONE 2015, 10, e0128818. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Mosier, S.L.; Thiess, M.; Beierl, K.F.; Debeljak, M.; Tseng, L.H.; Chen, G.; Yegnasubramanian, S.; Ho, H.; Cope, L.; et al. Clinical validation of KRAS, BRAF, and EGFR mutation detection using next-generation sequencing. Am. J. Clin. Pathol. 2014, 141, 856–866. [Google Scholar] [CrossRef] [PubMed]
- De Leng, W.W.; Gadellaa-van Hooijdonk, C.G.; Barendregt-Smouter, F.A.; Koudijs, M.J.; Nijman, I.; Hinrichs, J.W.; Cuppen, E.; van Lieshout, S.; Loberg, R.D.; de Jonge, M.; et al. Targeted Next Generation Sequencing as a Reliable Diagnostic Assay for the Detection of Somatic Mutations in Tumours Using Minimal DNA Amounts from Formalin Fixed Paraffin Embedded Material. PLoS ONE 2016, 11, e0149405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burghel, G.J.; Hurst, C.D.; Watson, C.M.; Chambers, P.A.; Dickinson, H.; Roberts, P.; Knowles, M.A. Towards a Next-Generation Sequencing Diagnostic Service for Tumour Genotyping: A Comparison of Panels and Platforms. Biomed. Res. Int. 2015, 2015, 478017. [Google Scholar] [CrossRef] [PubMed]
- Drmanac, R.; Sparks, A.B.; Callow, M.J.; Halpern, A.L.; Burns, N.L.; Kermani, B.G.; Carnevali, P.; Nazarenko, I.; Nilsen, G.B.; Yeung, G.; et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 2010, 327, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Al-Kateb, H.; Nguyen, T.T.; Steger-May, K.; Pfeifer, J.D. Identification of major factors associated with failed clinical molecular oncology testing performed by next generation sequencing (NGS). Mol. Oncol. 2015, 9, 1737–1743. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, F.C.; Kipp, B.R.; Kerr, S.E.; Voss, J.S.; Lazaridis, K.N.; Katzka, D.A.; Levy, M.J. Characterization of endoscopic ultrasound fine-needle aspiration cytology by targeted next-generation sequencing and theranostic potential. Clin. Gastroenterol. Hepatol. 2015, 13, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Schwaederle, M.; Daniels, G.A.; Piccioni, D.E.; Fanta, P.T.; Schwab, R.B.; Shimabukuro, K.A.; Parker, B.A.; Kurzrock, R. On the Road to Precision Cancer Medicine: Analysis of Genomic Biomarker Actionability in 439 Patients. Mol. Cancer Ther. 2015, 14, 1488–1494. [Google Scholar] [CrossRef] [PubMed]
- Tran, B.; Brown, A.M.; Bedard, P.L.; Winquist, E.; Goss, G.D.; Hotte, S.J.; Welch, S.A.; Hirte, H.W.; Zhang, T.; Stein, L.D.; et al. Feasibility of real time next generation sequencing of cancer genes linked to drug response: Results from a clinical trial. Int. J. Cancer 2013, 132, 1547–1555. [Google Scholar] [CrossRef]
- Boland, G.M.; Piha-Paul, S.A.; Subbiah, V.; Routbort, M.; Herbrich, S.M.; Baggerly, K.; Patel, K.P.; Brusco, L.; Horombe, C.; Naing, A.; et al. Clinical next generation sequencing to identify actionable aberrations in a phase I program. Oncotarget 2015, 6, 20099–20110. [Google Scholar] [CrossRef] [PubMed]
- Lane, B.R.; Bissonnette, J.; Waldherr, T.; Ritz-Holland, D.; Chesla, D.; Cottingham, S.L.; Alberta, S.; Liu, C.; Thompson, A.B.; Graveel, C.; et al. Development of a Center for Personalized Cancer Care at a Regional Cancer Center: Feasibility Trial of an Institutional Tumor Sequencing Advisory Board. J. Mol. Diagn. 2015, 17, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Uzilov, A.V.; Ding, W.; Fink, M.Y.; Antipin, Y.; Brohl, A.S.; Davis, C.; Lau, C.Y.; Pandya, C.; Shah, H.; Kasai, Y.; et al. Development and clinical application of an integrative genomic approach to personalized cancer therapy. Genome Med. 2016, 8, 62. [Google Scholar] [CrossRef] [PubMed]
- Lih, C.J.; Sims, D.J.; Harrington, R.D.; Polley, E.C.; Zhao, Y.; Mehaffey, M.G.; Forbes, T.D.; Das, B.; Walsh, W.D.; Datta, V.; et al. Analytical Validation and Application of a Targeted Next-Generation Sequencing Mutation-Detection Assay for Use in Treatment Assignment in the NCI-MPACT Trial. J. Mol. Diagn. 2016, 18, 51–67. [Google Scholar] [CrossRef]
- Ananda, G.; Mockus, S.; Lundquist, M.; Spotlow, V.; Simons, A.; Mitchell, T.; Stafford, G.; Philip, V.; Stearns, T.; Srivastava, A.; et al. Development and validation of the JAX Cancer Treatment Profile for detection of clinically actionable mutations in solid tumors. Exp. Mol. Pathol. 2015, 98, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, C.C.; Salipante, S.J.; Koehler, K.; Smith, C.; Scroggins, S.; Wood, B.; Wu, D.; Lee, M.K.; Dintzis, S.; Adey, A.; et al. Validation and implementation of targeted capture and sequencing for the detection of actionable mutation, copy number variation, and gene rearrangement in clinical cancer specimens. J. Mol. Diagn. 2014, 16, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, C.E.; Al-Kateb, H.; Bredemeyer, A.J.; Duncavage, E.J.; Spencer, D.H.; Abel, H.J.; Lockwood, C.M.; Hagemann, I.S.; O’Guin, S.M.; Burcea, L.C.; et al. Validation of a next-generation sequencing assay for clinical molecular oncology. J. Mol. Diagn. 2014, 16, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; O’Toole, S.A.; Trent, R.J. Somatic DNA mutation analysis in targeted therapy of solid tumours. Transl. Pediatr. 2015, 4, 125–138. [Google Scholar] [PubMed]
- Damodaran, S.; Berger, M.F.; Roychowdhury, S. Clinical tumor sequencing: Opportunities and challenges for precision cancer medicine. Am. Soc. Clin. Oncol. Educ. Book 2015, e175–e182. [Google Scholar] [CrossRef] [PubMed]
- Tafe, L.J.; Gorlov, I.P.; de Abreu, F.B.; Lefferts, J.A.; Liu, X.; Pettus, J.R.; Marotti, J.D.; Bloch, K.J.; Memoli, V.A.; Suriawinata, A.A.; et al. Implementation of a Molecular Tumor Board: The Impact on Treatment Decisions for 35 Patients Evaluated at Dartmouth-Hitchcock Medical Center. Oncologist 2015, 20, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Kidess, E.; Jeffrey, S.S. Circulating tumor cells versus tumor-derived cell-free DNA: Rivals or partners in cancer care in the era of single-cell analysis? Genome Med. 2013, 5, 70. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Janku, F.; Zhan, Q.; Fan, J.B. Accessing Genetic Information with Liquid Biopsies. Trends Genet. 2015, 31, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Hayashi, N.; Iguchi, T.; Ito, S.; Eguchi, H.; Mimori, K. Clinical and biological significance of circulating tumor cells in cancer. Mol. Oncol. 2016, 10, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Alconada, L.; Muinelo-Romay, L.; Madissoo, K.; Diaz-Lopez, A.; Krakstad, C.; Trovik, J.; Wik, E.; Hapangama, D.; Coenegrachts, L.; Cano, A.; et al. Molecular profiling of circulating tumor cells links plasticity to the metastatic process in endometrial cancer. Mol. Cancer 2014, 13, 223. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Auer, M.; Gasch, C.; Pichler, M.; Ulz, P.; Hoffmann, E.M.; Lax, S.; Waldispuehl-Geigl, J.; Mauermann, O.; Lackner, C.; et al. Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res. 2013, 73, 2965–2975. [Google Scholar] [CrossRef] [PubMed]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.; Kaper, F.; Dawson, S.J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci. Transl. Med. 2012, 4, 136ra68. [Google Scholar] [CrossRef] [PubMed]
- Couraud, S.; Vaca-Paniagua, F.; Villar, S.; Oliver, J.; Schuster, T.; Blanche, H.; Girard, N.; Tredaniel, J.; Guilleminault, L.; Gervais, R.; et al. Noninvasive diagnosis of actionable mutations by deep sequencing of circulating free DNA in lung cancer from never-smokers: A proof-of-concept study from BioCAST/IFCT-1002. Clin. Cancer Res. 2014, 20, 4613–4624. [Google Scholar] [CrossRef] [PubMed]
- Rothe, F.; Laes, J.F.; Lambrechts, D.; Smeets, D.; Vincent, D.; Maetens, M.; Fumagalli, D.; Michiels, S.; Drisis, S.; Moerman, C.; et al. Plasma circulating tumor DNA as an alternative to metastatic biopsies for mutational analysis in breast cancer. Ann. Oncol. 2014, 25, 1959–1965. [Google Scholar] [CrossRef] [PubMed]
- Lebofsky, R.; Decraene, C.; Bernard, V.; Kamal, M.; Blin, A.; Leroy, Q.; Rio Frio, T.; Pierron, G.; Callens, C.; Bieche, I.; et al. Circulating tumor DNA as a non-invasive substitute to metastasis biopsy for tumor genotyping and personalized medicine in a prospective trial across all tumor types. Mol. Oncol. 2015, 9, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Ward, D.G.; Baxter, L.; Gordon, N.S.; Ott, S.; Savage, R.S.; Beggs, A.D.; James, J.D.; Lickiss, J.; Green, S.; Wallis, Y.; et al. Multiplex PCR and Next Generation Sequencing for the Non-Invasive Detection of Bladder Cancer. PLoS ONE 2016, 11, e0149756. [Google Scholar] [CrossRef] [PubMed]
- Schwaederle, M.; Husain, H.; Fanta, P.T.; Piccioni, D.E.; Kesari, S.; Schwab, R.B.; Banks, K.C.; Lanman, R.B.; Talasaz, A.; Parker, B.A.; et al. Detection rate of actionable mutations in diverse cancers using a biopsy-free (blood) circulating tumor cell DNA assay. Oncotarget 2016, 7, 9707–9717. [Google Scholar] [PubMed]
- Schwaederle, M.; Husain, H.; Fanta, P.T.; Piccioni, D.E.; Kesari, S.; Schwab, R.B.; Patel, S.P.; Harismendy, O.; Ikeda, M.; Parker, B.A.; et al. Use of Liquid Biopsies in Clinical Oncology: Pilot Experience in 168 Patients. Clin. Cancer Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, A.; Del Grammastro, M.; Felicioni, L.; Malatesta, S.; Filice, G.; Centi, I.; de Pas, T.; Santoro, A.; Chella, A.; Brandes, A.A.; et al. Assessment of EGFR mutations in circulating tumor cell preparations from NSCLC patients by next generation sequencing: Toward a real-time liquid biopsy for treatment. PLoS ONE 2014, 9, e103883. [Google Scholar] [CrossRef] [PubMed]
- Sorber, L.; Zwaenepoel, K.; Deschoolmeester, V.; van Schil, P.E.; van Meerbeeck, J.; Lardon, F.; Rolfo, C.; Pauwels, P. Circulating cell-free nucleic acids and platelets as a liquid biopsy in the provision of personalized therapy for lung cancer patients. Lung Cancer 2016. [Google Scholar] [CrossRef] [PubMed]
- Braig, F.; Voigtlaender, M.; Schieferdecker, A.; Busch, C.J.; Laban, S.; Grob, T.; Kriegs, M.; Knecht, R.; Bokemeyer, C.; Binder, M. Liquid biopsy monitoring uncovers acquired RAS-mediated resistance to cetuximab in a substantial proportion of patients with head and neck squamous cell carcinoma. Oncotarget 2016, 7, 42988–42955. [Google Scholar] [CrossRef] [PubMed]
- Guttery, D.S.; Page, K.; Hills, A.; Woodley, L.; Marchese, S.D.; Rghebi, B.; Hastings, R.K.; Luo, J.; Pringle, J.H.; Stebbing, J.; et al. Noninvasive detection of activating estrogen receptor 1 (ESR1) mutations in estrogen receptor-positive metastatic breast cancer. Clin. Chem. 2015, 61, 974–982. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Huang, A.; Yang, X.R. Liquid Biopsy and its Potential for Management of Hepatocellular Carcinoma. J. Gastrointest. Cancer 2016, 47, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Lou, F.; Wu, Y.; Sun, D.Q.; Zhang, J.B.; Chen, W.; Ye, H.; Liu, J.H.; Wei, S.; Zhao, M.Y.; et al. Circulating tumor DNA identified by targeted sequencing in advanced-stage non-small cell lung cancer patients. Cancer Lett. 2016, 370, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, S.; Ueda, Y.; Onitake, Y.; Sueda, T.; Ohta, E.; Morihara, N.; Hirano, S.; Irisuna, F.; Hiyama, E. Circulating free DNA as non-invasive diagnostic biomarker for childhood solid tumors. J. Pediatr. Surg. 2015, 50, 2094–2097. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M.; Iguchi, T.; Masuda, T.; Nakahara, Y.; Hirata, H.; Uchi, R.; Niida, A.; Momose, K.; Sakimura, S.; Chiba, K.; et al. Somatic mutations in plasma cell-free DNA are diagnostic markers for esophageal squamous cell carcinoma recurrence. Oncotarget 2016, 7, 62280–62291. [Google Scholar] [CrossRef] [PubMed]
- Gasch, C.; Pantel, K.; Riethdorf, S. Whole Genome Amplification in Genomic Analysis of Single Circulating Tumor Cells. Methods Mol. Biol. 2015, 1347, 221–232. [Google Scholar] [PubMed]
- Kalow, W.; Tang, B.K.; Endrenyi, L. Hypothesis: Comparisons of inter- and intra-individual variations can substitute for twin studies in drug research. Pharmacogenetics 1998, 8, 283–289. [Google Scholar] [PubMed]
- Magdelijns, F.J.; Stassen, P.M.; Stehouwer, C.D.; Pijpers, E. Direct health care costs of hospital admissions due to adverse events in The Netherlands. Eur. J. Public Health 2014, 24, 1028–1033. [Google Scholar] [CrossRef] [PubMed]
- Brennan, P.; Wild, C.P. Genomics of Cancer and a New Era for Cancer Prevention. PLoS Genet. 2015, 11, e1005522. [Google Scholar] [CrossRef]
- Patel, J.N. Cancer pharmacogenomics, challenges in implementation, and patient-focused perspectives. Pharmgenomics Pers. Med. 2016, 9, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Amstutz, U.; Froehlich, T.K.; Largiader, C.R. Dihydropyrimidine dehydrogenase gene as a major predictor of severe 5-fluorouracil toxicity. Pharmacogenomics 2011, 12, 1321–1336. [Google Scholar] [CrossRef] [PubMed]
- Van Kuilenburg, A.B.; Dobritzsch, D.; Meijer, J.; Meinsma, R.; Benoist, J.F.; Assmann, B.; Schubert, S.; Hoffmann, G.F.; Duran, M.; de Vries, M.C.; et al. Dihydropyrimidinase deficiency: Phenotype, genotype and structural consequences in 17 patients. Biochim. Biophys. Acta 2010, 1802, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Pesenti, C.; Gusella, M.; Sirchia, S.M.; Miozzo, M. Germline oncopharmacogenetics, a promising field in cancer therapy. Cell. Oncol. (Dordr) 2015, 38, 65–89. [Google Scholar] [CrossRef] [PubMed]
- Whirl-Carrillo, M.; McDonagh, E.M.; Hebert, J.M.; Gong, L.; Sangkuhl, K.; Thorn, C.F.; Altman, R.B.; Klein, T.E. Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 2012, 92, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Froehlich, T.K.; Amstutz, U.; Aebi, S.; Joerger, M.; Largiader, C.R. Clinical importance of risk variants in the dihydropyrimidine dehydrogenase gene for the prediction of early-onset fluoropyrimidine toxicity. Int. J. Cancer 2015, 136, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Price, M.J.; Murray, S.S.; Angiolillo, D.J.; Lillie, E.; Smith, E.N.; Tisch, R.L.; Schork, N.J.; Teirstein, P.S.; Topol, E.J.; Investigators, G. Influence of genetic polymorphisms on the effect of high- and standard-dose clopidogrel after percutaneous coronary intervention: The GIFT (Genotype Information and Functional Testing) study. J. Am. Coll. Cardiol. 2012, 59, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
- Tammiste, A.; Jiang, T.; Fischer, K.; Magi, R.; Krjutskov, K.; Pettai, K.; Esko, T.; Li, Y.; Tansey, K.E.; Carroll, L.S.; et al. Whole-exome sequencing identifies a polymorphism in the BMP5 gene associated with SSRI treatment response in major depression. J. Psychopharmacol. 2013, 27, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.K.; Need, A.C.; Lohoff, F.W.; Zai, C.C.; Chowdhury, N.I.; Muller, D.J.; Putkonen, A.; Repo-Tiihonen, E.; Hallikainen, T.; Yagcioglu, A.E.; et al. Exome sequence analysis of Finnish patients with clozapine-induced agranulocytosis. Mol. Psychiatry 2014, 19, 403–405. [Google Scholar] [CrossRef] [PubMed]
- Apellaniz-Ruiz, M.; Lee, M.Y.; Sanchez-Barroso, L.; Gutierrez-Gutierrez, G.; Calvo, I.; Garcia-Estevez, L.; Sereno, M.; Garcia-Donas, J.; Castelo, B.; Guerra, E.; et al. Whole-exome sequencing reveals defective CYP3A4 variants predictive of paclitaxel dose-limiting neuropathy. Clin. Cancer Res. 2015, 21, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Ashley, E.A.; Butte, A.J.; Wheeler, M.T.; Chen, R.; Klein, T.E.; Dewey, F.E.; Dudley, J.T.; Ormond, K.E.; Pavlovic, A.; Morgan, A.A.; et al. Clinical assessment incorporating a personal genome. Lancet 2010, 375, 1525–1535. [Google Scholar] [CrossRef]
- Karageorgos, I.; Mizzi, C.; Giannopoulou, E.; Pavlidis, C.; Peters, B.A.; Zagoriti, Z.; Stenson, P.D.; Mitropoulos, K.; Borg, J.; Kalofonos, H.P.; et al. Identification of cancer predisposition variants in apparently healthy individuals using a next-generation sequencing-based family genomics approach. Hum. Genom. 2015, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Wu, G.; Broeckel, U.; Smith, C.A.; Turner, V.; Haidar, C.E.; Wang, S.; Carter, R.; Karol, S.E.; Neale, G.; et al. Comparison of genome sequencing and clinical genotyping for pharmacogenes. Clin. Pharmacol. Ther. 2016, 100, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Caudle, K.E.; Klein, T.E.; Hoffman, J.M.; Muller, D.J.; Whirl-Carrillo, M.; Gong, L.; McDonagh, E.M.; Sangkuhl, K.; Thorn, C.F.; Schwab, M.; et al. Incorporation of pharmacogenomics into routine clinical practice: The Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline development process. Curr. Drug Metab. 2014, 15, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Swen, J.J.; Nijenhuis, M.; de Boer, A.; Grandia, L.; Maitland-van der Zee, A.H.; Mulder, H.; Rongen, G.A.; van Schaik, R.H.; Schalekamp, T.; Touw, D.J.; et al. Pharmacogenetics: From bench to byte—An update of guidelines. Clin. Pharmacol. Ther. 2011, 89, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Mizzi, C.; Peters, B.; Mitropoulou, C.; Mitropoulos, K.; Katsila, T.; Agarwal, M.R.; van Schaik, R.H.; Drmanac, R.; Borg, J.; Patrinos, G.P. Personalized pharmacogenomics profiling using whole-genome sequencing. Pharmacogenomics 2014, 15, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Ross-Innes, C.S.; Stark, R.; Teschendorff, A.E.; Holmes, K.A.; Ali, H.R.; Dunning, M.J.; Brown, G.D.; Gojis, O.; Ellis, I.O.; Green, A.R.; et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 2012, 481, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Droog, M.; Nevedomskaya, E.; Kim, Y.; Severson, T.; Flach, K.D.; Opdam, M.; Schuurman, K.; Gradowska, P.; Hauptmann, M.; Dackus, G.; et al. Comparative Cistromics Reveals Genomic Cross-talk between FOXA1 and ERα in Tamoxifen-Associated Endometrial Carcinomas. Cancer Res. 2016, 76, 3773–3784. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, G.; Yan, P.; Maharry, K.; Frankhouser, D.; Nicolet, D.; Metzeler, K.H.; Kohlschmidt, J.; Mrozek, K.; Wu, Y.Z.; Bucci, D.; et al. Epigenetics meets genetics in acute myeloid leukemia: Clinical impact of a novel seven-gene score. J. Clin. Oncol. 2014, 32, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Guo, X.; Wu, Y.; Li, S.; Yan, J.; Peng, L.; Xiao, Z.; Wang, S.; Deng, Z.; Dai, L.; et al. Methylation profiling of 48 candidate genes in tumor and matched normal tissues from breast cancer patients. Breast Cancer Res. Treat. 2015, 149, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Mannoor, K.; Gao, L.; Tan, A.; Guarnera, M.A.; Zhan, M.; Shetty, A.; Stass, S.A.; Xing, L.; Jiang, F. Characterization of microRNA transcriptome in lung cancer by next-generation deep sequencing. Mol. Oncol. 2014, 8, 1208–1219. [Google Scholar] [CrossRef] [PubMed]
- Smircich, P.; Eastman, G.; Bispo, S.; Duhagon, M.A.; Guerra-Slompo, E.P.; Garat, B.; Goldenberg, S.; Munroe, D.J.; Dallagiovanna, B.; Holetz, F.; et al. Ribosome profiling reveals translation control as a key mechanism generating differential gene expression in Trypanosoma cruzi. BMC Genom. 2015, 16, 443. [Google Scholar] [CrossRef]
- King, H.A.; Gerber, A.P. Translatome profiling: Methods for genome-scale analysis of mRNA translation. Brief Funct. Genom. 2016, 15, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Salipante, S.J.; Scroggins, S.M.; Hampel, H.L.; Turner, E.H.; Pritchard, C.C. Microsatellite instability detection by next generation sequencing. Clin. Chem. 2014, 60, 1192–1199. [Google Scholar] [CrossRef] [PubMed]
- Platt, J.; Cox, R.; Enns, G.M. Points to consider in the clinical use of NGS panels for mitochondrial disease: An analysis of gene inclusion and consent forms. J. Genet. Couns. 2014, 23, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Cimino, P.J.; Zhao, G.; Wang, D.; Sehn, J.K.; Lewis, J.S., Jr.; Duncavage, E.J. Detection of viral pathogens in high grade gliomas from unmapped next-generation sequencing data. Exp. Mol. Pathol. 2014, 96, 310–315. [Google Scholar] [CrossRef] [PubMed]
- AWMF. Available online: http://www.awmf.org/awmf-online-das-portal-der-wissenschaftlichen-medizin/awmf-aktuell.html (accessed on 7 November 2016).
- National-Comprehensive-Cancer-Network. Available online: https://www.nccn.org (accessed on 11 November 2016).
- Sie, A.S.; Prins, J.B.; van Zelst-Stams, W.A.; Veltman, J.A.; Feenstra, I.; Hoogerbrugge, N. Patient experiences with gene panels based on exome sequencing in clinical diagnostics: High acceptance and low distress. Clin. Genet. 2015, 87, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Yorczyk, A.; Robinson, L.S.; Ross, T.S. Use of panel tests in place of single gene tests in the cancer genetics clinic. Clin. Genet. 2015, 88, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.J.; Rubinstein, W.S.; Facio, F.M.; Ng, D.; Singh, L.N.; Teer, J.K.; Mullikin, J.C.; Biesecker, L.G. Secondary variants in individuals undergoing exome sequencing: Screening of 572 individuals identifies high-penetrance mutations in cancer-susceptibility genes. Am. J. Hum. Genet. 2012, 91, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Van El, C.G.; Cornel, M.C.; Borry, P.; Hastings, R.J.; Fellmann, F.; Hodgson, S.V.; Howard, H.C.; Cambon-Thomsen, A.; Knoppers, B.M.; Meijers-Heijboer, H.; et al. Whole-genome sequencing in health care. Recommendations of the European Society of Human Genetics. Eur. J. Hum. Genet. 2013, 21, S1–S5. [Google Scholar] [CrossRef] [PubMed]
- Eurogentest. Available online: http://www.eurogentest.org (accessed on 11 November 2016).
- Hens, K.; Dondorp, W.; Handyside, A.H.; Harper, J.; Newson, A.J.; Pennings, G.; Rehmann-Sutter, C.; de Wert, G. Dynamics and ethics of comprehensive preimplantation genetic testing: A review of the challenges. Hum. Reprod. Update 2013, 19, 366–375. [Google Scholar] [CrossRef] [PubMed]
- De Wert, G.; Dondorp, W.; Shenfield, F.; Devroey, P.; Tarlatzis, B.; Barri, P.; Diedrich, K.; Provoost, V.; Pennings, G. ESHRE task force on ethics and Law22: Preimplantation genetic diagnosis. Hum. Reprod. 2014, 29, 1610–1617. [Google Scholar] [CrossRef] [PubMed]
- KNMP. Available online: https://www.knmp.nl/patientenzorg/medicatiebewaking/farmacogenetica (accessed on 11 November 2016).
- Gallego, C.J.; Shirts, B.H.; Bennette, C.S.; Guzauskas, G.; Amendola, L.M.; Horike-Pyne, M.; Hisama, F.M.; Pritchard, C.C.; Grady, W.M.; Burke, W.; et al. Next-Generation Sequencing Panels for the Diagnosis of Colorectal Cancer and Polyposis Syndromes: A Cost-Effectiveness Analysis. J. Clin. Oncol. 2015, 33, 2084–2091. [Google Scholar] [CrossRef] [PubMed]
- Gallego, C.J.; Perez, M.L.; Burt, A.; Amendola, L.M.; Shirts, B.H.; Pritchard, C.C.; Hisama, F.M.; Bennett, R.L.; Veenstra, D.L.; Jarvik, G.P. Next Generation Sequencing in the Clinic: A Patterns of Care Study in a Retrospective Cohort of Subjects Referred to a Genetic Medicine Clinic for Suspected Lynch Syndrome. J. Genet. Couns. 2016, 25, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Shirts, B.H.; Casadei, S.; Jacobson, A.L.; Lee, M.K.; Gulsuner, S.; Bennett, R.L.; Miller, M.; Hall, S.A.; Hampel, H.; Hisama, F.M.; et al. Improving performance of multigene panels for genomic analysis of cancer predisposition. Genet. Med. 2016, 18, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Amendola, L.M.; Dorschner, M.O.; Robertson, P.D.; Salama, J.S.; Hart, R.; Shirts, B.H.; Murray, M.L.; Tokita, M.J.; Gallego, C.J.; Kim, D.S.; et al. Actionable exomic incidental findings in 6503 participants: Challenges of variant classification. Genome Res. 2015, 25, 305–315. [Google Scholar] [CrossRef] [PubMed]
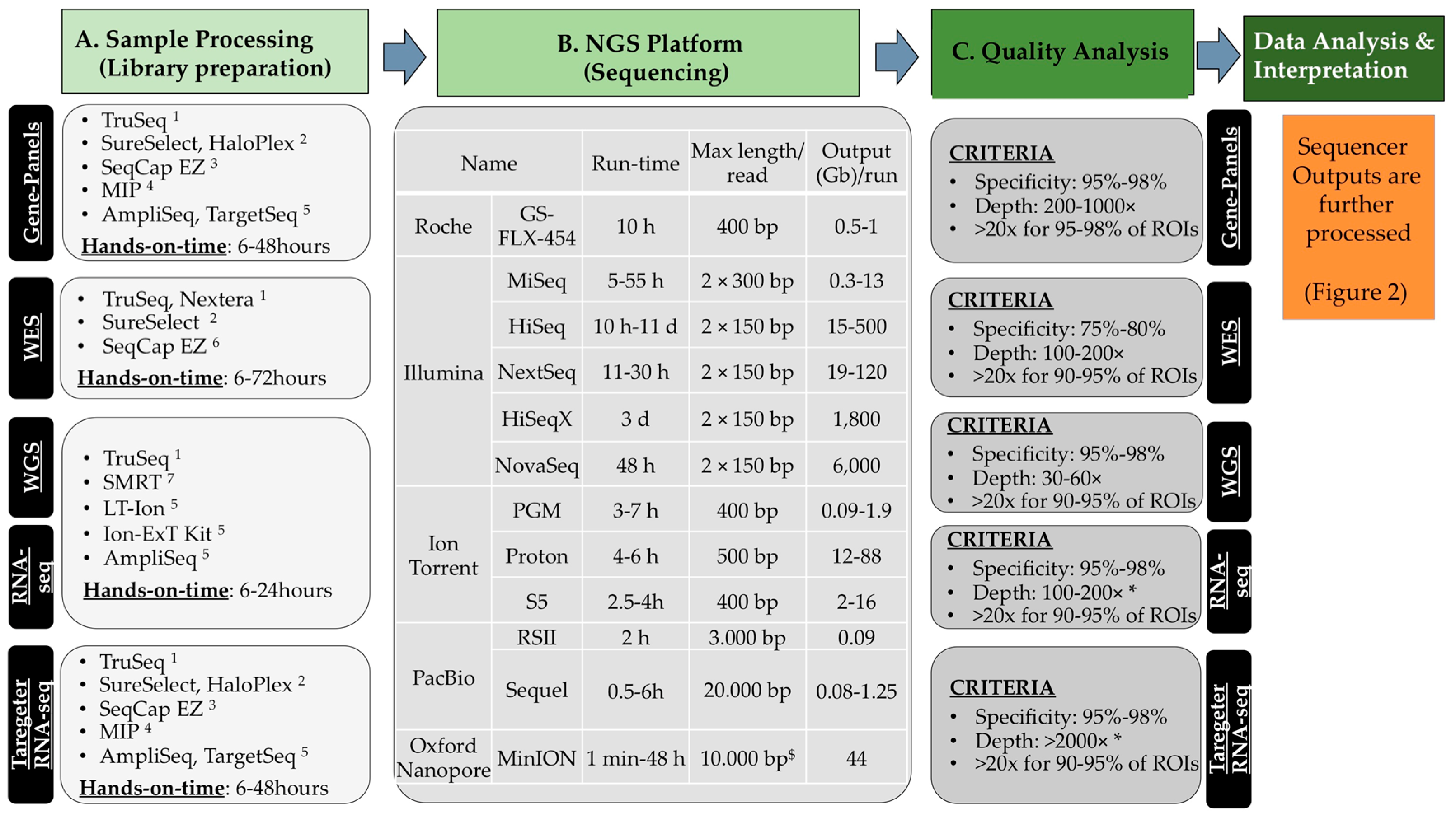
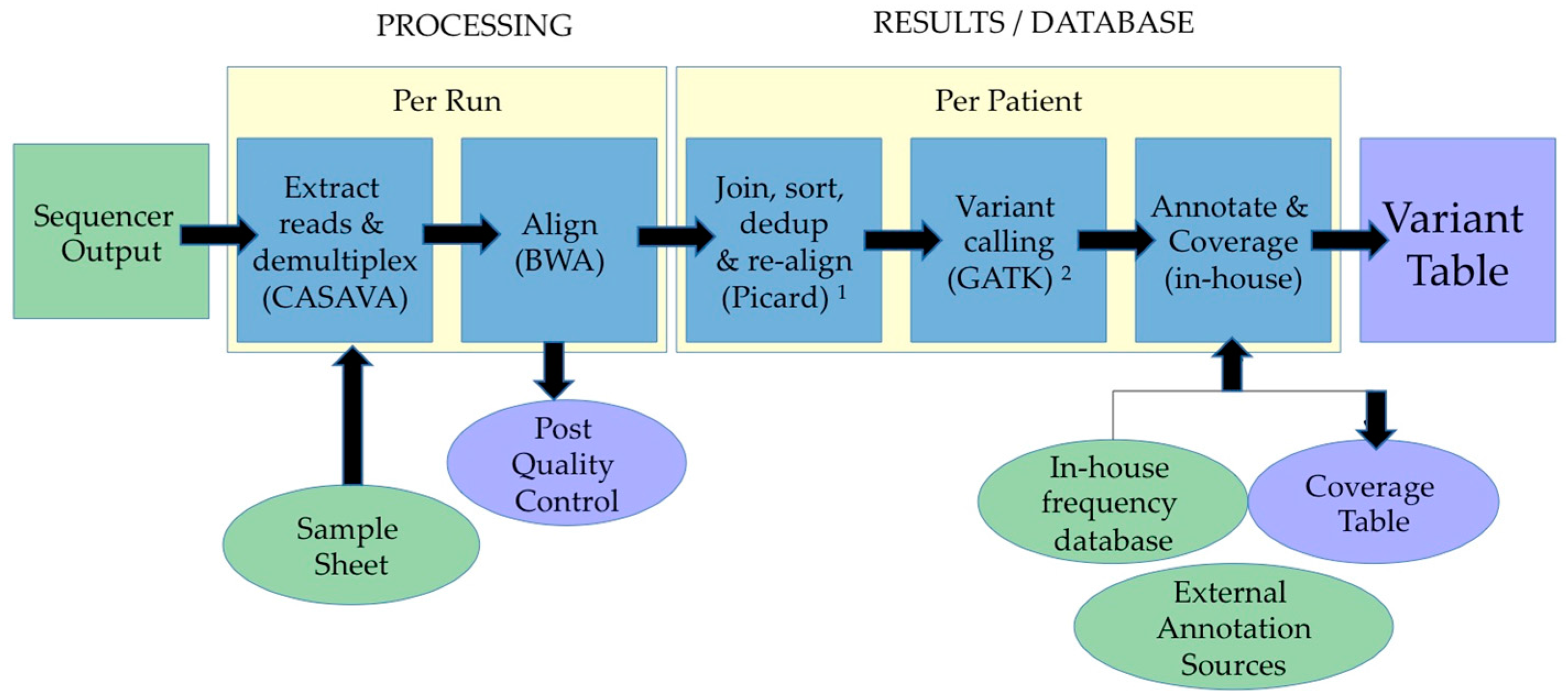

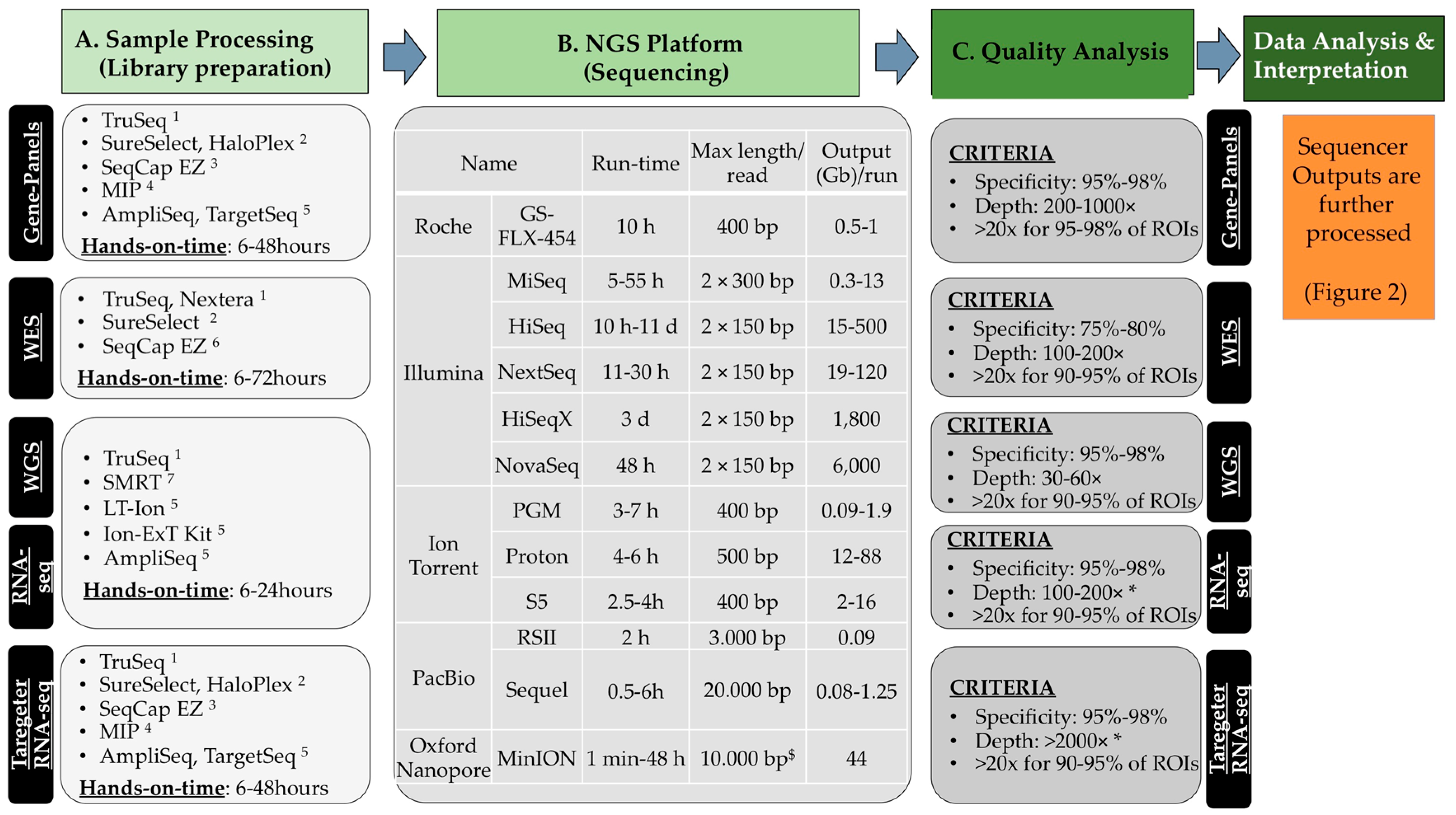{kind=link}
{kind=link}
| Developer | Name | ROIs 1DNA Capture Method and FeaturesKind of Study/Purpose | No. Genes 2 | Reference |
|---|---|---|---|---|
| Mayo Clinic | No specific name was given | - ROIs: exonic regions, 272 Kb - Enrichment: a. solid-phase (NimbleGen 385K)—PCR (ligation mediated) amplification; b. BamH1 digestion of library for Illumina sequencing (fragmentation) - Sequencer: a. GS-FLX Roche 454 (average depth: 30 reads/bp); b. Illumina (Genome Analyzer II; average depth: 266 reads/bp) - Preclinical: 5 patients with Lynch syndrome | 22 | [23] |
| University of Washington | BROCA | - ROIs: exons, introns, 10 Kb at the 3′ and 5′s genomic region, 1 Mb - Enrichment: oligo based hybridisation in solution - a. Sequencer: Illumina (Genome Analyzer IIX; depth > 1200 reads/bp) - Preclinical: 20 women with HBOC | 21 | [24] |
| - b. Sequencer: Illumina (Genome Analyzer IIX; depth: 449 reads/bp) - Preclinical: 360 women with ovarian cancer | 21 | [25] | ||
| FROM 2012—Clinical use - ROIs: variable - Enrichment: oligo based hybridisation in solution - Sequencer: Illumina (HiSeq2000; depth: 320–1000) | Up to 66 | [26] | ||
| ColoSeq | - ROIs: exons, introns, 10 Kb at the 3′ and 5′s genomic region, 1.1 Mb - Enrichment: oligo based hybridisation in solution - Sequencer: Illumina (HiSeq2000; depth: 320) - Preclinical: 82 DNA specimens, 7 genes (209 Kb) analysed | 31 (7) | [27] | |
| FROM 2012—Clinical use - Sequencer: Illumina (HiSeq2000; depth: 320–1000) | [28] | |||
| François Baclesse, France | (Custom Design Castera et al.) | - ROIs: exons, introns - Enrichment: oligo based hybridisation in solution - Sequencer: Illumina (Genome Analyzer IIX) - Clinical utility: 708 suspected HBOC patients | 16–24 | [29] |
| Memorial Sloan-Kettering Cancer Center | (Custom design) | - ROIs: exons - Enrichment: RainDance technology - Sequencer: Illumina (MiSeq; depth: 1765×) - Feasibility: 30 patients with myeloid malignancies | 28 | [30] |
| Ambry Genetics | BreastNext, OvaNext, ColoNext, CancerNext | - ROIs: exons - Enrichment: RainDance technology - Sequencer: Illumina (HiSeq, MiSeq NGS, 2000) - Centre experience: 2079 patients | 14–21 | [31,32] |
| ColoNext | - Centre experience: 586 patients, personal history of colorectal cancer indicated for genetic test; subject mutations status was unknown. Pathogenic mutation identified in 10% of the patients | 14 | [33] | |
| OvaNext | - Centre experience: 911 patients referred for gene testing for HBOC | 21 | [34] | |
| Myriad | Multiple Hereditary Cancer Panel | - ROIs: exons - Enrichment: RainDance technology - Sequencer: Illumina (MiSeq or HiSeq2500) | [35] | |
| - Clinical feasibility: 1964 patients with or suspected hereditable cancer syndrome | 25 | [36] | ||
| - Clinical feasibility: 1260 patients with suspected Lynch syndrome | 25 | [37] | ||
| - Clinical experience: 2158 patients with suspected hereditable cancer syndrome | 25 | [38] | ||
| University of Tubingen | - ROIs: exons - Enrichment: enrichment by hybridisation and amplification by a. in solution HaloPlex technology (56–58 genes) b. oligo based hybridisation in solution (94 genes) - Sequencer: Illumina (MiSeq) - Clinical feasibility: 620 patients with high-risk HBOC family profile indicated for genetic testing | 56–94 (10) | [39] | |
| Trinity College Dublin | - ROIs: exons, 1.6 Mb - Enrichment: oligo based hybridisation in solution - Sequencer: Illumina (Genome Analyzer II/HiSeq2000; depth: 89×) - Clinical feasibility: 104 patients with no BRCA1/2 mutation and 101 controls | 320 | [40] | |
| Invitae | Various gene-panels available | - ROIs: exons, 95 Kb - Enrichment: oligo based hybridisation in solution; integrated for low-covered regions with DNA Technologies (Coral, IL, USA) xGen Lockdown probes - Sequencer: Illumina (MiSeq, HiSeq2500; depth: 450×) | 42–80 | [41] |
| Colorectal Cancer Panel | - Clinical feasibility/experience: 1062 HBOC subjects | 29 | [42] | |
| Emory Genetics Laboratory 3 | Several gene-panels available | - ROIs: exons - Enrichment: oligo based hybridisation in solution - Sequencer: Illumina (HiSeq 2500) | 19–60 | [43] |
| GeneDX 4 | Various gene-panels available | - ROIs: exons - Enrichment: oligo based hybridisation in solution - Sequencer: Illumina (MiSeq or HiSeq; depth: 100×) | 20–32 | [44] |
| - Clinical experience: 8 panels applied to 10030 patients between 2013 and 2014 | Up to 29 | [45] | ||
| Fulgent Diagnostics 5 | - ROIs: exons - Enrichment: oligo based hybridisation in solution - Sequencer: Illumina (MiSeq or HiSeq) | 21–38 | [46] | |
| CentoGene 6 | - ROIs: exons - Enrichment: oligo based hybridisation in solution - Sequencer: Illumina (Depth > 20×) | 4–31 | [47] | |
| Qiagen (GeneReader technology) | - ROIs: exons - Enrichment: amplicon-based PCR - Sequencer: Qiagen (Ultra depth) | Up to 20 | [8] |
| Risk-category | GENE NAME | Breast and Ovarian Cancer | Colorectal Cancer | All Types | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BROCA 1 | Custom Design 2 | BreastGene 3 | BreastNext 4 | HBC High-Risk 5 | High Risk BC 6 | CentoBreast 7 | B/OC Gene-Panel 5 | B/OC NGS Pan el 8 | BOC 6 | OvaNext 4 | ColoSeq 9 | ColoNext 4 | CRC 10 | CRC 5 | CentoColon 7 | CC NGS Panel 8 | G/CRC 6 | CancerNext 4 | Cross-Cancer 10 | Comprehensive 5 | H-M Risk 5 | myRisk 11 | HCS 6 | CentoCancer 7 | ||
| High-risk genes | APC | ◉ | ◉ | ○ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | |||||||||||
| BMPR1A | ◉ | A | ◉ | ○ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | |||||||||
| BRCA1 | ○ | A | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ○ | ○ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | |||||||
| BRCA2 | ○ | A | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ○ | ○ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ||||||
| CDH1 | ○ | A | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ○ | ○ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | |
| CDKN2A | ◉ | ◉ | ○ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ||||||||||||||
| EPCAM | ○ | ◉ | ◉ | ◉ | ○ | ○ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ||||||||||
| MLH1 | ○ | B | ◉ | ◉ | ◉ | ○ | ○ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ||||||||
| MSH2 | ○ | B | ◉ | ◉ | ◉ | ○ | ○ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | |||||||
| MSH6 | ○ | B | ◉ | ◉ | ◉ | ○ | ○ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | |||||||
| MUTYH | ○ | ◉ | ◉ | ◉ | ◉ | ○ | ○ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ||||||
| PMS2 | ○ | B | ◉ | ◉ | ◉ | ◉ | ○ | ○ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ||||||
| PTEN | ○ | A | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ○ | ○ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | |
| SMAD4 | ◉ | ◉ | ○ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ||||||||||
| STK11 | ○ | A | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ○ | ○ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | |||
| TP53 | ○ | B | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ○ | ○ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | |
| VHL | ◉ | ◉ | ○ | ◉ | ◉ | ◉ | ◉ | |||||||||||||||||||
| Moderate-risk genes | ATM | ○ | A | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ○ | ○ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ||||
| BRIP1 * | ○ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ○ | ○ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | |||||||||
| CHEK2 | ○ | A | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ○ | ○ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ||
| MRE11A | ○ | A | ◉ | ◉ | ◉ | ◉ | ○ | ○ | ◉ | ◉ | ||||||||||||||||
| PALB2 ** | ○ | A | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ○ | ○ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | |||||||
| POLD1 | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | |||||||||||||
| POLE | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | |||||||||||||||
| AXIN2 | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ||||||||||||||||||
| BARD1 | ○ | A | ◉ | ◉ | ◉ | ◉ | ◉ | ○ | ○ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | |||||||||||
| CDK4 | ◉ | ◉ | ○ | ◉ | ◉ | ◉ | ◉ | ◉ | ||||||||||||||||||
| FANCC | ◉ | ◉ | ◉ | ◉ | ||||||||||||||||||||||
| NBN | ○ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ○ | ○ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | |||||||||||
| RAD51C | ○ | A | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ○ | ○ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | |||||||||
| RAD51D | ◉ | B | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | |||||||||||
| XRCC2 | ◉ | C | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | |||||||||||||||||
| Low-risk | AIP | ◉ | ||||||||||||||||||||||||
| AKT | ◉ | ◉ | ◉ | |||||||||||||||||||||||
| ALK | ◉ | |||||||||||||||||||||||||
| ATR | ◉ | ◉ | ||||||||||||||||||||||||
| BAP1 | ◉ | B | ◉ | ○ | ◉ | |||||||||||||||||||||
| Low-risk genes | BLM | ◉ | ◉ | ◉ | ◉ | ◉ | ||||||||||||||||||||
| BUB1B | ○ | ◉ | ◉ | ◉ | ||||||||||||||||||||||
| CDC77 | ◉ | |||||||||||||||||||||||||
| CTNNA1 | ◉ | ◉ | ||||||||||||||||||||||||
| CTNNB1 | ◉ | |||||||||||||||||||||||||
| DICER | ◉ | |||||||||||||||||||||||||
| EXO1 | ◉ | |||||||||||||||||||||||||
| FAM175A | ◉ | ◉ | ||||||||||||||||||||||||
| FH | ◉ | ◉ | ◉ | |||||||||||||||||||||||
| FLCN | ◉ | ◉ | ◉ | ◉ | ◉ | |||||||||||||||||||||
| GALNT12 | ◉ | ◉ | ◉ | ◉ | ||||||||||||||||||||||
| GEN1 | ◉ | |||||||||||||||||||||||||
| GPC3 | ◉ | ◉ | ||||||||||||||||||||||||
| GREM1 | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | ◉ | |||||||||||||||||
| HOXB13 | ◉ | ◉ | ◉ | |||||||||||||||||||||||
| KIT | ○ | ◉ | ||||||||||||||||||||||||
| MAX | ◉ | ◉ | ||||||||||||||||||||||||
| MEN1 | ◉ | ◉ | ◉ | ◉ | ||||||||||||||||||||||
| MET | ◉ | ◉ | ||||||||||||||||||||||||
| MGMT | ◉ | |||||||||||||||||||||||||
| MLH3 | B | ◉ | ||||||||||||||||||||||||
| NBS1 | A | |||||||||||||||||||||||||
| NF1 | ◉ | ◉ | ◉ | ◉ | ◉ | |||||||||||||||||||||
| NF2 | ◉ | |||||||||||||||||||||||||
| NTHL1 | ◉ | ◉ | ◉ | ◉ | ||||||||||||||||||||||
| PALLD | ◉ | |||||||||||||||||||||||||
| PDGFRA | ◉ | ◉ | ||||||||||||||||||||||||
| PHOX2B | ◉ | |||||||||||||||||||||||||
| PIK3CA | ◉ | ◉ | ◉ | |||||||||||||||||||||||
| PMS1 | ○ | B | ◉ | ◉ | ||||||||||||||||||||||
| PRKAR1A | ◉ | |||||||||||||||||||||||||
| PRSS1 | ◉ | |||||||||||||||||||||||||
| PTCH1 | ◉ | ◉ | ||||||||||||||||||||||||
| RAD50 | ○ | A | ◉ | ◉ | ◉ | ◉ | ○ | ○ | ◉ | ◉ | ◉ | |||||||||||||||
| RAD51 | C | ◉ | ||||||||||||||||||||||||
| RAD51B | ◉ | C | ||||||||||||||||||||||||
| RET | ◉ | ○ | ◉ | |||||||||||||||||||||||
| RINT1 | ◉ | ◉ | ||||||||||||||||||||||||
| SDHA | ◉ | |||||||||||||||||||||||||
| SDHAF2 | ◉ | |||||||||||||||||||||||||
| RINT1 | ◉ | ◉ | ||||||||||||||||||||||||
| SDHA | ◉ | |||||||||||||||||||||||||
| SDHAF2 | ◉ | |||||||||||||||||||||||||
| SDHB | ◉ | ◉ | ◉ | ◉ | ||||||||||||||||||||||
| SDHC | ◉ | ◉ | ◉ | |||||||||||||||||||||||
| SDHD | ◉ | ◉ | ◉ | ◉ | ||||||||||||||||||||||
| SMARCA4 | ◉ | ◉ | ◉ | ◉ | ◉ | |||||||||||||||||||||
| SMARCB1 | ◉ | |||||||||||||||||||||||||
| SUFU | ◉ | |||||||||||||||||||||||||
| TMEM127 | ◉ | |||||||||||||||||||||||||
| TSC1 | ◉ | ◉ | ||||||||||||||||||||||||
| TSC2 | ◉ | ◉ | ||||||||||||||||||||||||
| XRCC3 | C | ◉ | ||||||||||||||||||||||||
| Cancer Syndrome | Site at High-Risk of Cancer | Gene Mutated | Life-Time Risk of Cancer (%) Per Site by 70 (* 80 or ## 50) Years | References | |||||
|---|---|---|---|---|---|---|---|---|---|
| Breast | Endometrium | Ovary | Colon-Rectum | Prostate | Other Sites | ||||
| General Population | Non Applicable | 7.3 | 1.6 | 0.7 | 1.9 | 8.2 | 2.6–0.1 | [63,64,71,72,73] | |
| - Hereditary breast and ovarian cancer syndrome (HBOC) | Breast, Ovary Pancreas, Prostate | BRCA1 | 46–87 | 39–63 | Up to 16 | [63,64,71,72,73] | |||
| BRCA2 | 43–84 # | 16–27 | 20 | Pancreas: 7 * | [63,64,71,72,73] | ||||
| Breast, Pancreas | PALB2 | 20 | [63,64] | ||||||
| Breast | RAD51A | [74] | |||||||
| Ovary, Breast | RAD51C | [75] | |||||||
| Breast | BARD1 | ||||||||
| Ovary, Breast | BRIP1 | 20 | [63,64] | ||||||
| - Lynch syndrome | Ovary, Colon Rectum Endometrial Pancreas, Stomach | MLH1 | 25–60 | 4–12 | 52–82 | Stomach: 6–13; Small bowel: 3–6 | [76] | ||
| MSH2 | 25–61 | 4–13 | 52–83 | Stomach: 6–13 Small bowel: 3–6 | [76] | ||||
| Ovary, Colon Rectum Endometrium | MSH6 | 16–71 | 10–69 | [76] | |||||
| PMS2 | 15–20 | [76] | |||||||
| - Ataxia-telangiectasia | Breast, Pancreas Risk of Leukaemia Risk of Lymphoma | ATM | |||||||
| - Hereditary breast and colorectal cancer | Breast, Colon Rectum | CHEK2 | 25 | [63,64] | |||||
| - Cowden syndrome - PTEN hammartoma tumour syndrome - Bannayan–Riley–Ruvalcaba syndrome | Breast, Colon Rectum Endometrium Other sites Risk of Melanoma | PTEN | 77–85 | 19–28 | 9–16 | Melanoma: up to 6 Kidney: 15–35 Thyroid 21–38 | |||
| - Familial adenomatousus polyposis - Syndrome attenuated familial - Adenomatous polyposis gardner syndrome | Colon, Rectum Pancreas, Stomach | APC | Up to 15 | 93 ## | Brain | [76] | |||
| - Hereditary diffuse gastric cancer | Breast, Stomach | CDH1 | 39–52 * | Stomach: 56–83 * | [63,64,77] | ||||
| - Juvenile polyposis syndrome | Colon, rectum Stomach | BMPR1 | Stomach: up to 20 | [76] | |||||
| - Juvenile polyposis syndrome - Hereditary haemorrhagic telangiectasia | Colon, Rectum, Stomach, other sites | SMAD4 | 40–50 * | Stomach: up to 20 * | [76] | ||||
| - Li Fraumeni syndrome | Overall cancer risk at young age | TP53 | [63] | ||||||
| - Melanoma-pancreatic cancer syndrome - Melanoma cancer syndrome | Pancreas Risk of Melanoma | CDKN2A1 | Pancreas: up to 17 Melanoma: 28–76 * | [78] | |||||
| - MUTYH-associated polyposis syndrome - MUTYH-associated colon cancer risk | Colon, Rectum | MUTHY | 3–10 * | [76] | |||||
| - Peutz Jeghers syndromes | Breast, Colon Rectum Endometrium Pancreas, Stomach Ovary | STK11 | 45–50 | 9 | 18–21 | 39 | Cervix: 10 Stomach: 29 Pancreas 11–36 Lung: 15–17 Small bowel: 13 | [79] | |
| - Retinoblastoma | Retinoblastoma | RB | 26 * | [64] | |||||
| - Hereditary mixed polyposis syndrome | Colon, Rectum | SCG5 GREM1 | [80] | ||||||
| - Hereditary ovarian cancer risk | Ovary | RAD51D | [81] | ||||||
| - Melanoma-pancreatic cancer syndrome - Melanoma cancer syndrome | Pancreas, risk of Melanoma | CDK4 | |||||||
| - Nijmegen breakage syndrome | Breast, Prostate | NBN | |||||||
| - Neurofibromatosis | Risk of sarcomas | NF1 | |||||||
| - Oligodontia-colorectal cancer syndrome | Colon, rectum | AXIN2 | [82] | ||||||
| - Multiple endocrine neoplasia | Parathyroid gland, Pancreas Pituitary gland | MEN1 | [83] | ||||||
| - Polymerase proofreading-associated syndrome | Colon, Rectum | POLE, POLD1 | |||||||
| - Von Hippel-Lindau syndrome | Kidney, Pancreas, Genital tract | VHL | |||||||
| - Turcot syndrome | Brain | APC, MLH1, PMS2 | |||||||
| Disease | Cases | Somatic Mutation-Other Analyses* | Design and Samples (FFPE **) Sequencer Aim/Project/Trial | Reference |
|---|---|---|---|---|
| Breast cancer | ||||
| Breast Cancer | 15 | WES | - Samples: primary tumours - Sequencer: Illumina - Project: cosmic cancer genome project | [146] |
| 510 | WES -RNA-seq -miRNA -methylation | - Samples: frozen samples from 507 patients (invasive disease) with no prior treatment and with companion normal DNA (adjacent tissue, blood) - Sequencer: Illumina - Project: TCGA | [147] | |
| 21 | WES | - Samples: various BC (oestrogen-receptor-positive, HER2-positive, BRCA2-positive, triple negative, BRCA1-positive) - Sequencer: Illumina - Project: cancer genome project | [148] | |
| 100 | WES | - Samples: primary BC, 79 oestrogen-receptor-positive/21 negative - Sequencer: Illumina - Project: cancer genome project | [149] | |
| Invasive lobular Breast Cancer | 127 | WES | - Samples: Frozen tumours (817 BC in total)/matched normal - Sequencer: Illumina - Project: TCGA | [150] |
| Triple Negative Breast Cancer | 104 | 44-gene-panel | - Samples: FFPE specimens from archival triple negative tumours - Sequencer: Ion Torrent - Aim: diagnosis/classification of triple negative BC | [151] |
| Gynaecological cancer | ||||
| Endometrial Cancer | 13 | WES | - Samples: tumour/matched normal DNA, frozen specimens - Sequencer: SOLiD V3.0—Illumina - Aim: driver genes (including ARID1A, PI3K pathway) | [152] |
| 248 | WES (107 WGS) -RNA-seq -miRNA -methylation | - Samples: tumour/germline - Sequencer: Illumina - Project: TCGA | [153] | |
| Ovarian Cancer | 9 | 50-gene-panel | - Samples: FFPE from high grade tumour/10 normal ovary - Sequencer: Ion Torrent - Aim: proof of concept. TP53 frequently mutated | [154] |
| 316 | WES | - Sample: stage-II–IV high-grade serous samples/normal DNA - Sequencer: Illumina - Project: TCGA | [155] | |
| Mucinous Ovarian Tumours | 69 | 50-gene-panel | - Samples: FFPE from 37 tumours and 26 border line tumours - Sequencer: Ion Torrent - Aim: prognosis and actionable mutations (KRAS) | [156] |
| Cervical Cancer | 29 | 226-gene-panel | - Samples: 25 squamous cell, 4 adenocarcinoma and 7 normal cervix - Sequencer: Illumina - Aim: actionable mutations (PIK3CA, KRAS, FBXW7) | [157] |
| Colorectal cancer | ||||
| Rectal Cancer | 102 | 50-gene-panel | - Samples: fine-needle aspiration and lymph node cytology - Sequencer: Ion Torrent - Aim: feasibility, drug-resistance, theranostics | [158] |
| Colorectal Cancer | 276 | WES (97 WGS) -RNA-seq -miRNA -methylation | - Samples: tumour/normal pair - Sequencer: Illumina - Project: TCGA | [159] |
| Colorectal Cancer | 22 | 46-gene-panel | - Samples: FFPE, microdissected tumour/stroma surrounding the tumour - Sequencer: Ion Torrent - Aim: validation, performance, false positive rates | [160] |
| 114 | 50-gene-panel | - Samples: prospective metastatic samples - Sequencer: Ion Torrent - Aim: drug-resistance mutations (ERGR therapy) | [161] | |
| Head and Neck Cancer | ||||
| Squamous Cell Carcinomas | 208 | 45-gene-panel | - Samples: FFPE, locally advanced tumours, treated with adjuvant care - Sequencer: Ion Torrent - Aim: drug-resistance, actionable mutations (TP53, PI3K path), prognosis | [162] |
| 279 | WES (29 WGS) -RNA-seq -miRNA -methylation | - Samples: tumours (172 oral cavity, 33 oropharynx, 72 laryngeal sites) with known HPV status/normal DNA - Sequencer: Illumina - Project: TCGA | [163] | |
| Oropharyngeal Squamous Cell Carcinoma | 8 | 46-gene-panel | - Samples: FFPE specimens, 4 HPV-positive and 4 HPV-negative - Sequencer: Ion Torrent - Aim: proof of concept, therapeutic and actionable targets | [164] |
| Oral Cavity Squamous Cell Carcinoma | 345 | 10-gene-panel Ultra-deep seq | - Samples: retrospective node positive patient FFPE specimens - Sequencer: Ion Torrent - Aim: prognosis and target for drugs | [165] |
| Digestive system: stomach, salivary glands, pancreas | ||||
| Gastric Cancer | 15 | 50-gene-panel | - Samples: retrospective FFPE, high-grade intraepithelial-neoplasia (IP) associated cancer - Sequencer: Ion Torrent - Aim: marker for progression of IP to cancer | [166] |
| Gastric Adenocarc. 1 | 238 | 45-gene-panel | - Samples: retrospective FFPE - Sequencer: Ion Torrent - Aim: feasibility, actionable mutations | [167] |
| Pancreatic Ductal Adenocarc. 1 | 73 | 65-gene-panel | - Samples: fresh-frozen tissues from surgical resection samples - Sequencer: (anchored multiplex PCR)—Illumina - Aim: prognosis (KRAS-G12V associated with poor survival) | [168] |
| Pancreatic Adenocar. 1 | 13 | WES | - Samples: metastases from patients with multiple tumour lesions - Sequencer: Illumina - Project: cancer genome project | [169] |
| Pancreas Cancer | 38 | 275-gene-panel | - Samples: retrospective FFPE, intraductal-papillary-mucinous-neoplasms (IPMN)-associated invasive cancer (microdissected) - Sequencer: Illumina - Aim: markers for progression of IPMN to cancer | [170] |
| Salivary Duct Carcinoma | 37 | 50-gene-panel | - Samples: retrospective FFPE - Sequencer: Ion Torrent - Aim: diagnosis, actionable mutation (PIK3CA, ERBB2) | [171] |
| Gastric Cancer | 89 | 50-gene-panel | - Samples: retrospective FFPE tumour of patients with metastasis - Sequencer: Ion Torrent - Aim: NGS validation, feasibility, frequent mutations in TP53 (28%) | [172] |
| Gastric Adenocarc. 1 | 295 | WES (107 WGS) -RNA-seq -miRNA -methylation | - Samples: tumour/germline - Sequencer: Illumina - Project: TCGA | [173] |
| Salivary Epithelial-Myo Epithelial Carcinoma | 17 | 50-gene-panel -RNA-seq | - Samples: FFPE, tumours and 6 unmatched normal salivary glands - Sequencer: Ion Torrent - Aim: actionable mutations | [174] |
| Gastric Adenocarc. 1 | 167 | 46-gene-panel | - Samples: retrospective FFPE, 92 gastroesophageal junction and 75 lower stomach lesions - Sequencer: Ion Torrent - Aim: classification (gastroesophageal junction versus stomach) | [175] |
| Gastric Cancer | 8 | 48-gene-panel | - Samples: gastric hyperplastic polyps (GHP)/different grades of dysplasia - Sequencer: Illumina - Aim: markers for progression of GHP to cancer, mutation profile (TP53) | [176] |
| GastricGastro-Intestinal Stromal Tumours | 20 | 50-gene-panel | - Samples: retrospective endoscopic ultrasound-guided fine-needle aspiration specimens - Sequencer: Ion Torrent - Aim: feasibility, diagnosis | [177] |
| Non-Small-Lung-Cancer | ||||
| Squamous Lung Cancer | 178 | WES (19 WGS) -RNA-seq -miRNA -methylation | - Samples: frozen untreated stage I–IV/normal DNA - Sequencer: Illumina - Project: TCGA | [178] |
| Lung Adenocarc. 1 | 38 | 22-gene-panel | - Sample: retrospective trans-thoracic fine-needle aspiration cytology - Sequencer: Ion Torrent - Aim: proof of concept/actionable mutations | [179] |
| 230 | WES (93 WGS) -RNA-seq -miRNA -methylation | - Samples: tumour (untreated)/normal - Sequencer: Illumina - Project: TCGA | [180] | |
| 76 | 48-gene-panel | - Samples: FFPE neurosurgical brain metastasis analyses - Sequencer: Illumina - Aim: feasibility, actionable mutations, TP53 frequently mutated (46%) | [181] | |
| Lung Cancer | 183 | WES (23 WGS) | - Samples: tumour/normal - Sequencer: Illumina - Project: TCGA | [182] |
| Non-Small Cell Lung Cancer | 209 | 23-gene-panel | - Samples: retrospective FFPE tumour specimens - Sequencer: Illumina - Aim: feasibility, diagnostic yield, actionable mutations (KRAS, EGFR) | [183] |
| Neurological Cancers | ||||
| Brain Tumour | 150 | 130-gene-panel some introns and promoters | - Samples: FFPE, 79 retrospective (known mutations); glioblastomas (n = 47), pilocytic astrocytomas (n = 10), medulloblastomas (n = 14) - Sequencer: Illumina - Aim: validation NGS compared with standard, feasibility | [184] |
| Glioma | 820 | WES (42 WGS) -RNA-seq -miRNA -methylation | - Samples: frozen tumours (diffuse grade II-III-IV gliomas) - Sequencer: Illumina - Project: TCGA | [185] |
| Glioma | 121 | 20-gene-panel | - Samples: retrospective cases - Sequencer: Ion Torrent - Aim: validation, histological and molecular classification | [186] |
| Diffuse Lower-Grade Gliomas | 289 | WES (73 WGS) -RNA-seq -miRNA -methylation | - Samples: 100 astrocytomas, 77 oligoastrocytomas, and 116 oligodendrogliomas - Sequencer: Illumina - Project: TCGA | [187] |
| Glioblastoma | 291 | WES (163 WGS) -RNA-seq -miRNA -methylation | - Sample: tumour/germline - Sequencer: Illumina - Project: TCGA | [188] |
| 44 | 50-gene-panel | - Samples: Fresh-frozen (Australian Genomics and Clinical Outcome of Glioma Biospecimen Resource) - Sequencer: Ion Torrent - Aim: alternative treatment options | [189] | |
| 3 | WES and 409-gene-panel | - Samples: primary, recurrent tumour, blood DNA - Sequencer: Illumina (WES)/Ion Torrent (gene-panel) - Aim: compare NGS platforms, proof of concept | [190] | |
| Kidneys and urinary system | ||||
| Clear Cell Renal Carcinoma | 417 | WES -RNA-seq -miRNA -methylation | - Samples: Frozen tumour/matched normal kidney-blood DNA - Sequencer: Illumina - Project: TCGA | [191] |
| Papillary Renal-Cell Carcinoma | 157 | WES | - Samples: 75 type 1, 60 type 2 and 26 non-classified tumours - Sequencer: Illumina - Project: TCGA | [192] |
| Urothelial Bladder Carcinoma | 130 | WES (18 WGS) | - Samples: frozen tumours/matched blood or normal tissue - Sequencer: Illumina - Project: TCGA | [193] |
| Chromophobe Renal Cell Carcinoma | 66 | WES (50 WGS) | - Sample: tumour/germline - Sequencer: Illumina - Project: TCGA | [194] |
| Renal Cell Carcinoma | 10 | 275-gene-panel | - Samples: succinate dehydrogenase negative FFPE archive samples - Sequencer: Illumina - Aim: classification | [195] |
| 32 | 50-gene-panel | - Samples: Retrospective FFPE; 22 metastatic; treated with at least one tyrosine kinase or mTOR inhibitor - Sequencer: Ion Torrent - Aim: associations between histotype, mutation and response to therapy | [196] | |
| Thyroid cancer | ||||
| Papillary Thyroid Carcinoma | 402 | WES -RNA-seq -miRNA -methylation | - Samples: tumour/germline - Sequencer: Illumina - Project: TCGA | [197] |
| Thyroid Cancer | 143 | 13 genes (exons) and 42 gene fusions | - Samples: nodule fine-needle aspiration (known surgical outcome) - Sequencer: Ion Torrent - Aim: feasibility to make a diagnosis | [198] |
| Thyroid Cancer | 465 | 14 genes (exons) and 42 gene fusions | - Samples: frozen fine-needle aspiration of thyroid nodules with intermediate cytology (i.e., atypia or follicular lesions with undetermined significance); 98 samples had a definitive classification - Sequencer: Ion Torrent - Aim: diagnosis, performance | [199] |
| Haematological Cancers | ||||
| Acute Myeloid Leukaemia | 24 | WGS -RNA-seq | - Samples: tumour/normal skin - Sequencer: Illumina - Aim: Pilot, mutation landscape | [200] |
| Acute Myeloid Leukaemia | 150 | WES (50 WGS) -RNA-seq -miRNA -methylation | - Samples: tumours/normal skin - Sequencer: Illumina - Project: TCGA | [201] |
| Myelofibrosis | 95 | 28-gene-panel | - Samples: patients were treated in a trial with ruxolitinib in a phase 1/2 study - Sequencer: Illumina - Aim: response to therapy, frequent mutations (NRAS, KRAS, PTPN11, GATA2, TP53, and RUNX1). Multiple mutations correlated with outcome | [202] |
| Myelo-Dysplasia | 9 | WES | - Samples: fresh specimens: low-grade disease (bone marrow mononuclear cells or peripheral-blood granulocytes); normal DNA buccal swabs/T cells - Sequencer: Illumina - Project: cancer genome project | [203] |
| Multiple Myeloma | 133 | 5 | - Samples: application of NGS compared to standards - Sequencer: LymphoSIGHT (Sequenta) 2 - Aim: prognosis/persistence of minimal residual disease | [204] |
| Acute Lymphoblastic Leukaemia | 106 | 5 | - Samples: application of NGS compared to standards - Sequencer: LymphoSIGHT (Sequenta) 2 - Trial: NCT00137111 prognosis/persistence of minimal residual disease | [205] |
| Other types of cancer | ||||
| Cutaneous Melanoma | 320 | WES (157 WGS) -RNA-seq -miRNA -methylation | - Samples: tumour/blood DNA - Sequencer: Illumina - Project: TCGA | [206] |
| Prostate Cancer | 333 | WES -RNA-seq -miRNA -methylation | - Samples: frozen tumours/matched blood or normal tissue - Sequencer: Illumina - Project: TCGA | [207] |
| Cholangio-Carcinoma | 75 | a- 46-gene-panel b- 236-gene-panel (introns of 19 genes) | - Samples: archival FFPE material from patients with >3 months follow-up - Sequencer: Ion Torrent and Illumina - Aim: classification/diagnostics and prognostics | [208] |
| Thymic Carcinoma | 12 | 409-gene-panel | - Samples: Frozen squamous cell carcinoma/matched normal tissue (10 patients) - Sequencer: Ion Torrent - Aim: proof of concept, heterogeneous mutation landscape | [209] |
| Malignant Pleural Mesothelioma | 123 | 50-gene-panel | - Samples: macro(manual)dissected specimens from FFPE samples - Sequencer: Ion Torrent - Aim: proof of concept, frequent mutation (TP53, DNA repair), prognosis | [210] |
| Adrenocortical Carcinoma | 91 | WES (50 WGS) -RNA-seq -miRNA -methylation | - Samples: frozen tumours/matched blood or normal tissue - Sequencer: Illumina - Project: TCGA | [211] |
| Merkel Cell Carcinoma | 15 | 409-gene-panel | - Samples: polymavirus negative/CK20 negative FFPE specimens - Sequencer: Ion Torrent - Aim: driver genes exploration TP53, RB1, BAP1 | [212] |
| Paediatric oncology | ||||
| Thyroid Carcinoma | 27 | 50 | - Samples: FFPE specimens - Sequencer: Ion Torrent - Aim: proof of concept, frequent mutations in BRAF, RET and CTNNB1 | [213] |
| Thyroid Carcinomas | 18 | 60-gene-panel | - Samples: FFPE, differentiated cancer (sporadic)/previous molecular testing - Sequencer: Ion Torrent - Aim: proof of concept, classification, prognosis | [199] |
| Solid Tumours (extra cranial) | 100 | 275-gene-panel (introns of 30 genes) | - Samples: FFPE or fresh frozen (nonrhabdomyosarcoma soft-tissue, sarcoma, neuroblastoma, ewing sarcoma, osteosarcoma, rhabdomyosacroma, Wilms tumour, rare tumours) - Sequencer: Illumina - Trial (NCT01853345): actionable alterations, individualised cancer therapy | [214] |
| Studies focusing on multiple Cancers | ||||
| Various | 2221 | 287-gene-panel (introns of 19 genes) | - Samples: FFPE samples from consecutive clinical cases of which 249 previously characterised - Sequencer: Illumina - Aim: method applicability | [215] |
| Colorectal, Lung Cancers | 18 | 48-gene-panel | - Samples: archival FFPE material already genotyped - Sequencer: Illumina - Aim: NGS validation | [216] |
| Glioblastoma, Lung, Thyroid Cancers, Holangio - Carcinoma | 986 | 48-gene-panel -RNA seq | - Samples: FFPE samples (both for DNA-seq and RNA-seq) - Sequencer: (anchored multiplex PCR) Illumina or Ion Torrent - Aim: proof concept and clinical applicability | [217] |
| Colorectal and Endometrial Cancers | 32 | 19-gene-panel (ColoSeq) | - Samples: blood/tumour samples (MMR deficiency without hypermethylation of MLH1 or germline MMR mutations) - Sequencer: Illumina - Aim: diagnostic/surveillance of Lynch syndrome-like (sporadic) | [218] |
| Over 20 Cancer types | 407 | 50-gene-panel | - Samples: FFPE or fresh frozen (Gastric, lung, colorectal adenocarcinoma, soft tissue sarcoma, Hepatocellular carcinoma and other types) - Sequencer: Ion Torrent - Trial (NCT01853345): NEXT-1 trial. Genome-matched treatment | [219] |
| Various | 50 | WES | - Samples: 10 patients with chronic B cell lymphocytic leukaemia 20 patients with bone cancer (9 osteosarcoma, 11 chordoma). Tumour/germline DNA - Sequencer: Illumina - Project: cancer genome project | [220] |
| Breast, Head and Neck Cancers, Melanoma | 103 | 236-gene-panel DNA intronic sequences from 19 genes | - Samples: reviewed own experience on FFPE material - Sequencer: Illumina - Aim: validation of clinical potential | [221] |
| Various solid Tumour samples | 55 | 409-gene-panel | - Samples: FFPE from tumour (melanoma; gastrointestinal stromal, brain tumours; lung, breast, gynaecologic tract, gastrointestinal tract carcinomas) and matched normal tissues (from 20 samples). All samples previously tested for mutation landscape with a 46-gene-panel - Sequencer: Ion Torrent - Aim: validation: sensitivity, specificity, reproducibility, applicability | [222] |
| Various solid Tumour samples | 70 | 46-gene-panel | - Samples: FFPE specimens previously tested genetically (n = 22) and 48 tested in parallel. Melanoma (n = 36); colorectal (16), lung (5), gastrointestinal tract (5), papillary thyroid (4), endometrial serous (3) adenocarcinomas; squamous cell carcinoma (n = 1) - Sequencer: Ion Torrent - Aim: validation, sensitivity, applicability, feasibility | [223] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamps, R.; Brandão, R.D.; Bosch, B.J.v.d.; Paulussen, A.D.C.; Xanthoulea, S.; Blok, M.J.; Romano, A. Next-Generation Sequencing in Oncology: Genetic Diagnosis, Risk Prediction and Cancer Classification. Int. J. Mol. Sci. 2017, 18, 308. https://doi.org/10.3390/ijms18020308
Kamps R, Brandão RD, Bosch BJvd, Paulussen ADC, Xanthoulea S, Blok MJ, Romano A. Next-Generation Sequencing in Oncology: Genetic Diagnosis, Risk Prediction and Cancer Classification. International Journal of Molecular Sciences. 2017; 18(2):308. https://doi.org/10.3390/ijms18020308
Chicago/Turabian StyleKamps, Rick, Rita D. Brandão, Bianca J. van den Bosch, Aimee D. C. Paulussen, Sofia Xanthoulea, Marinus J. Blok, and Andrea Romano. 2017. "Next-Generation Sequencing in Oncology: Genetic Diagnosis, Risk Prediction and Cancer Classification" International Journal of Molecular Sciences 18, no. 2: 308. https://doi.org/10.3390/ijms18020308