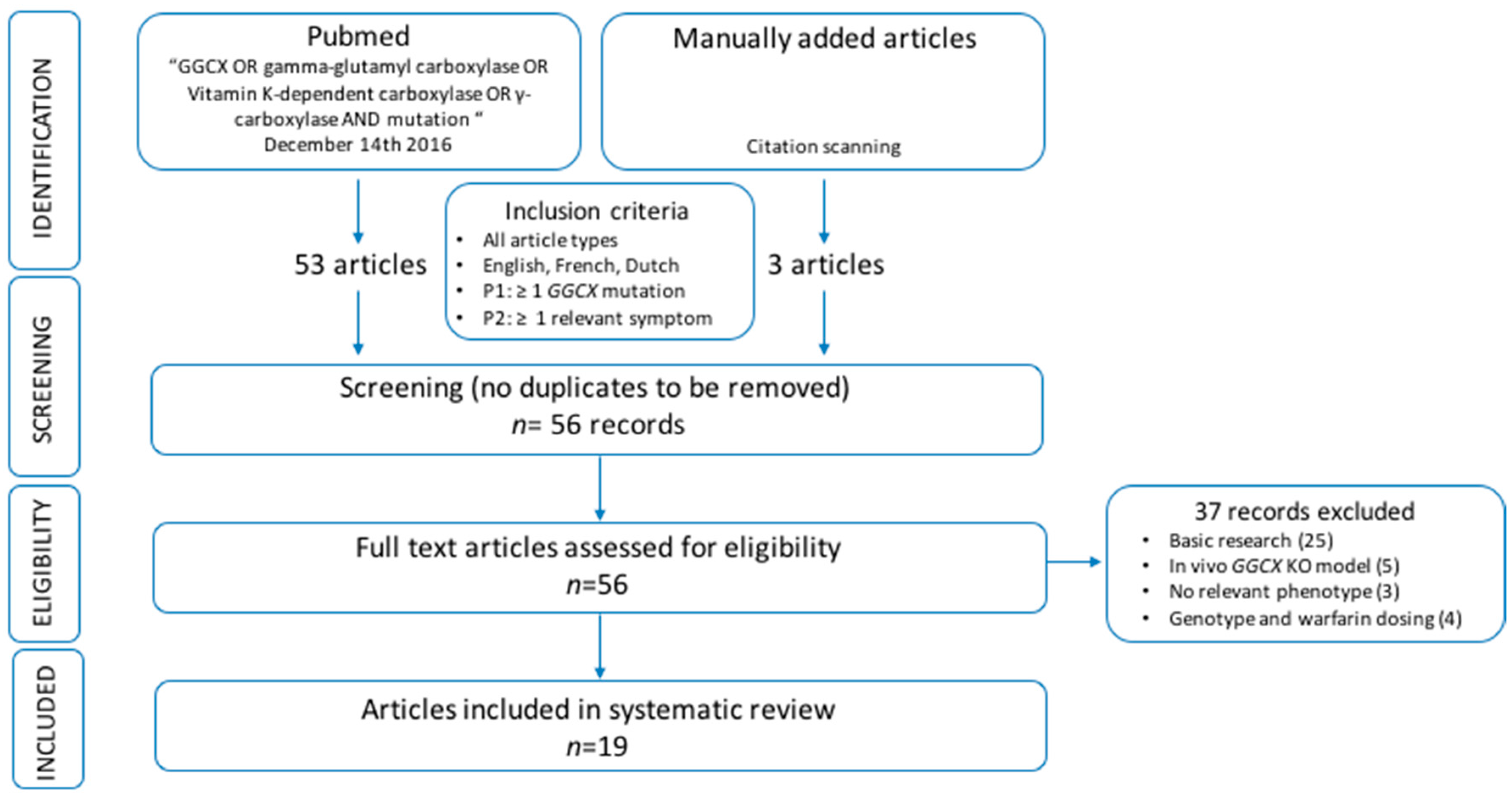
1. Introduction
The gamma-glutamyl carboxylase enzyme (GGCX) catalyzes the conversion of specific glutamate (Glu) residues to gamma-carboxyglutamate (Gla) residues, a process called gamma-carboxylation [
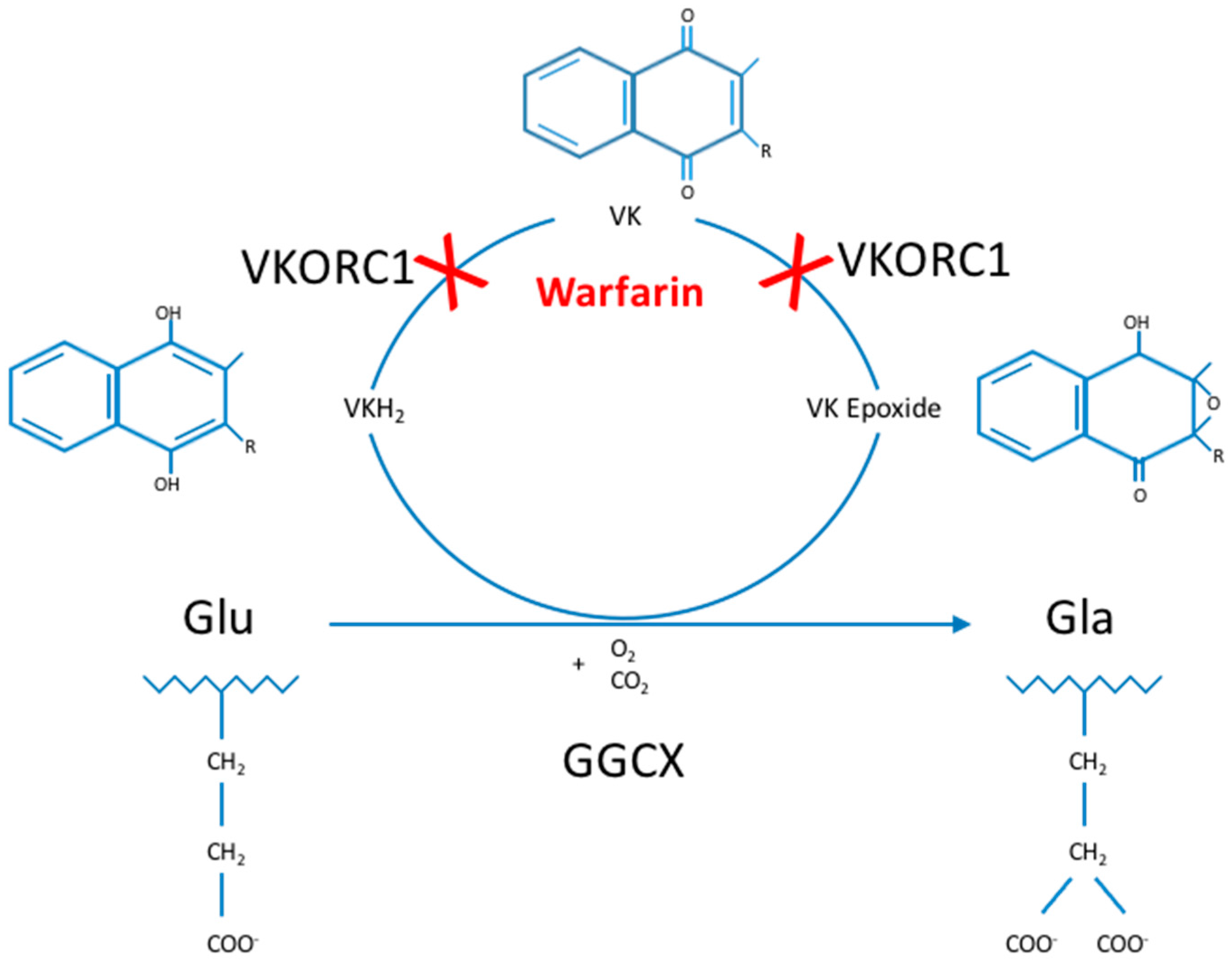
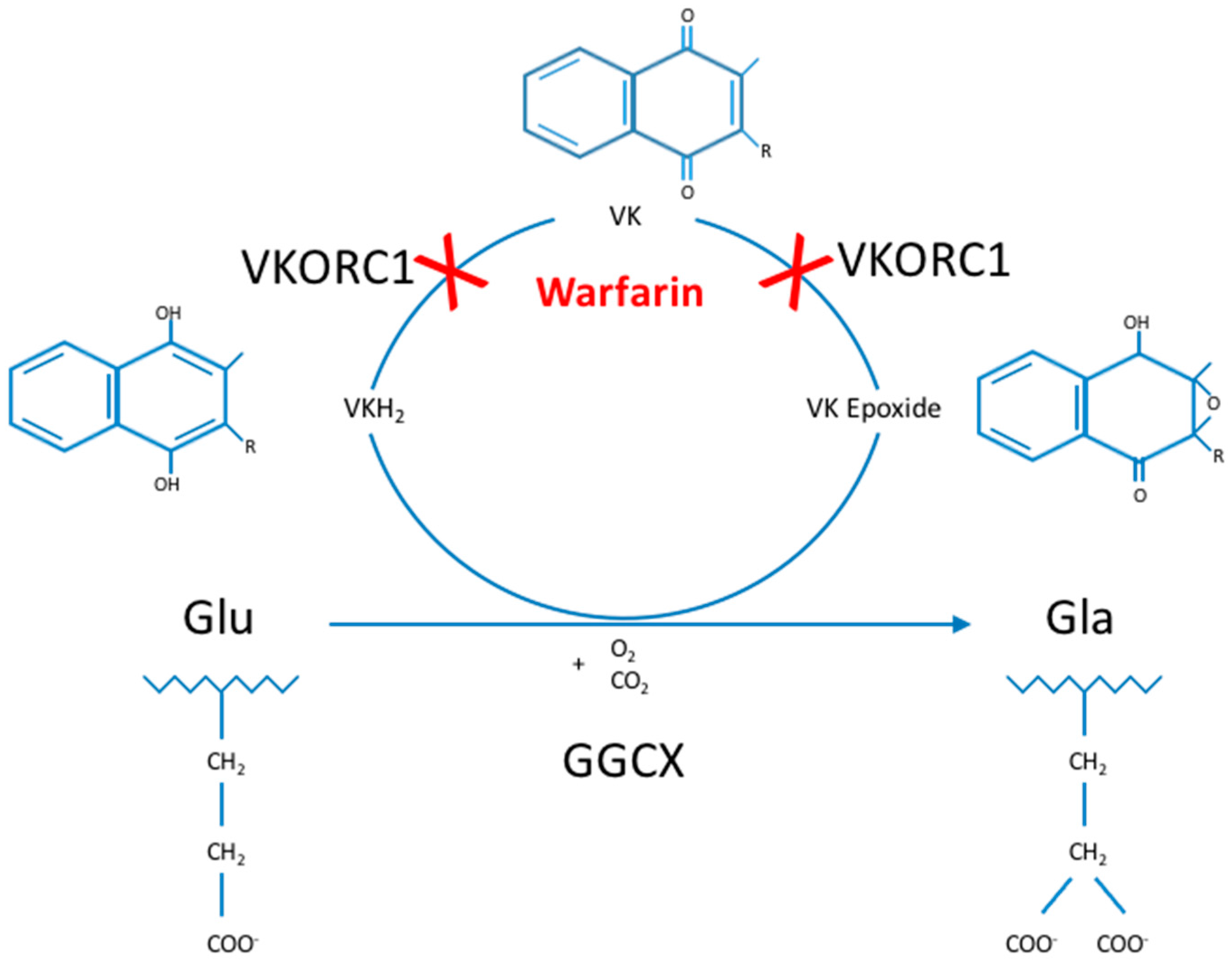
1]. This posttranslational modification process uses vitamin K (VK) as an essential cofactor and is part of the so-called VK cycle (
Figure 1) [
2]. Gamma-carboxylation is essential in the activation and proper functioning of multiple VK-dependent proteins (VKDP), the most well-known of which are involved in blood clotting, including coagulation factors (FII, FVII, FIX and FX) and natural anti-clotting agents (protein C, protein S (ProS; OMIM*176880) and protein Z). Moreover, GGCX catalyzes gamma-carboxylation of other VKDP, involved in various biological processes such as inflammation (e.g., ProS, and gla-rich protein (GRP)), bone formation (osteocalcin (OC; OMIM*112260)), cell proliferation (growth arrest-specific 6 (Gas6; OMIM*600441)) and soft tissue mineralization (matrix gla protein (MGP; OMIM*154870)) [
3,
4]. Finally, several VKDP have a currently unknown function (proline-rich gla proteins (PRGP), and transmembrane gla proteins (TMG)) [
1,
2,
5,
6,
7].
The GGCX enzyme is encoded by the
GGCX gene, located on the reverse strand of chromosome 2p11.2 (chromosomal position in assembly GRCh38.p7: 85,544,723–85,561,509). The gene is not considered to be polymorphic, as, according to the gnomAD database (combining data from the ExAc and 1000 genomes databases), of the 409 exonic variants (missense and loss-of-function) that have been identified in the
GGCX gene, only two variants, i.e., rs699664 and rs6173310, have an allele frequency of >0.0001 (
Table S1) [
8].
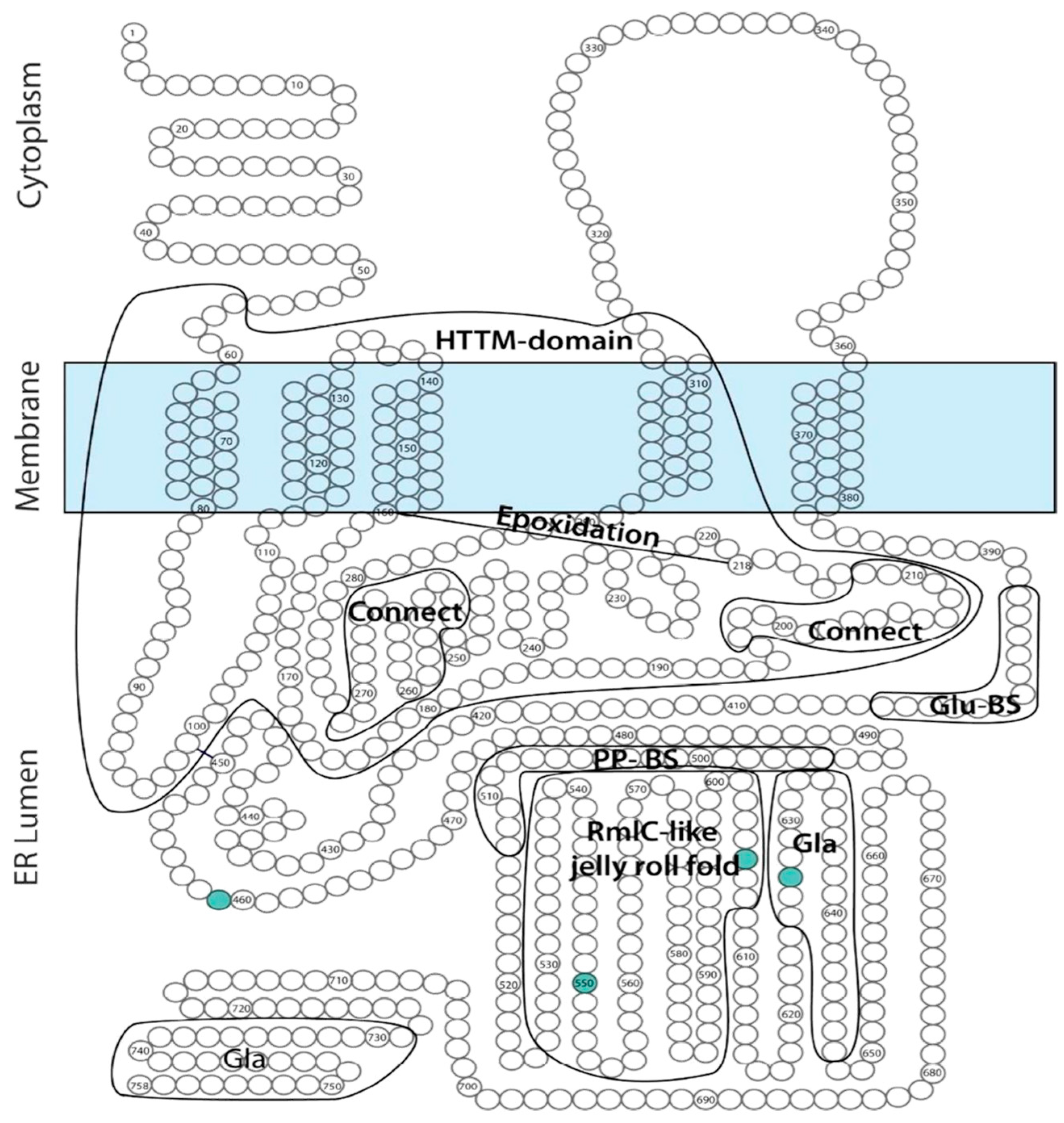
GGCX has 10 transcripts, of which the longest is NM000821.6 (ENST00000233838.8; Uniprot P38435), comprising 15 exons and 7569 nucleotides. The protein encoded by this transcript is a 94 kDa, 758 amino acid (AA) transmembrane protein, expressed ubiquitously throughout the body and localized on the lipid membrane of the endoplasmic reticulum (ER). The N-terminal part of GGCX is localized in the cytoplasm, followed by 5 transmembrane domains (TMD), and the C-terminal portion is localized in the ER lumen (
Figure 2). To date, the crystal structure of the GGCX enzyme is still incompletely resolved [
9].
GGCX has multiple highly conserved domains (
Figure 2), including the horizontally transferred transmembrane domain (HTTM—AA 56–315), spanning the first four TMDs, the function of which is currently unclear in humans [
10]. Interestingly, in multiple species such as eukaryotes, bacteria and archae, the HTTM-domain seems to play an important role in VK-dependent carboxylation [
11]. Within the TMD, the proline-residue at position 378 in TMD5 is proposed to play an important role in the correct orientation of GGCX; replacing that proline by a leucine leads to an important decrease in the formation of a disulfide bond in the protein, which is an important posttranslational modification step (see below) [
12]. However, apart from the disulfide bond other factors must play a role in GGCX orientation, as even with a removal of the disulfide bond and a complete cleavage of GGCX between TMD4 and TMD5, the GGCX protein domains remain close together. Tie et al. suggested that an interaction between TMD2 and TMD5 could play an important role in this process [
12]. Other important functional domains in the GGCX enzyme are the propeptide binding site (most recently proposed to be localized at AA 491–507), suggested as the primary location of interaction between GGCX and its substrates, and the glutamate binding site (AA 393–404), which interacts with the Glu-containing regions of VKDP, a necessary step for gamma-carboxylation. Interestingly, L394 and W399, which are localized in this predicted glutamate binding region, seem to play a role in polypeptide binding by GGCX, hereby stimulating the connection between the propeptide binding sites and the glutamate binding sites, thus facilitating gamma-carboxylation [
9,
13,
14,
15]. GGCX is further predicted to contain an RmlC (deoxythymidine-6-deoxy-
d-xylo-4-hexulose 3,5 epimerase; EC5.1.3.13)-like jelly roll fold, comprising a double-stranded beta-helix jelly roll fold as is identified in RmlC, from AA 526 until 607 [
11]. The function of this domain is however currently unclear.
Finally, GGCX also undergoes posttranslational modifications, such as glycosylation of 4 asparagine residues (AA 459, 550, 605 and 627) and the formation of a disulfide bond (between cysteine-residue 99 and 450), which stabilizes the protein leading to a more efficient enzymatic function [
16,
17,
18]. GGCX further has 2 autocarboxylating Gla-domains, suggested to be localized at AA 625–647 and 729–758 in the C-terminal region of the enzyme in the ER lumen. Possibly, these Gla-domains have a yet undiscovered role in other processes than VKDP gamma-carboxylation [
19].
In 1998,
GGCX mutations were first linked to human disease by Brenner et al. in four patients with a combined deficiency of all VK-dependent blood coagulation factors (factor II, VII, IX, X and ProS and protein C) due to a homozygous missense mutation in the
GGCX gene [
13]. The disease was coined VK-dependent clotting factor deficiency-1 (VKCFD1, OMIM#277450), an autosomal recessive disorder, characterized by a mild to severe bleeding tendency and a moderate predisposition to thrombotic events [
13,
20]. VKCFD1 was shown to be associated with skeletal (midfacial hypoplasia, reduced bone mass, chondrodysplasia punctata) or cardiac abnormalities (patent ductus arteriosus Botalli, septal closure defects) in some patients [
13,
21,
22,
23,
24,
25,
26,
27,
28,
29,
30]. Next to VKCFD1, a second autosomal recessive coagulation factor deficiency exists, VKCFD2 (OMIM#607473), caused by
VKORC1 (vitamin K epoxide reductase complex, subunit 1; OMIM*608547) mutations and is also characterized by a deficiency of all VK-dependent clotting factors. In contrast to VKCFD1, this general deficiency can usually be reversed completely using low doses of VK (ca. 5–10 mg/week) [
31,
32]. Skeletal abnormalities (in particular osteoporosis) have been described in VKCFD2 patients [
33], but no cardiac involvement has been identified yet.
More recently, biallelic
GGCX mutations were shown to cause a phenotype characterized by not only VKCFD1 but also elastic fiber (EF) mineralization and fragmentation, leading to loss of skin elasticity and loosening of the skin with a cutis laxa appearance. In the original seven patients, the phenotype was demonstrated to be similar to but more severe than the skin features in pseudoxanthoma elasticum (PXE; OMIM#264800), an autosomal recessive ectopic mineralization disorder. The disease was therefore called PXE-like disorder with combined coagulation factor deficiency (OMIM#610842). Classic PXE is caused by EF mineralization in soft tissues due to biallelic
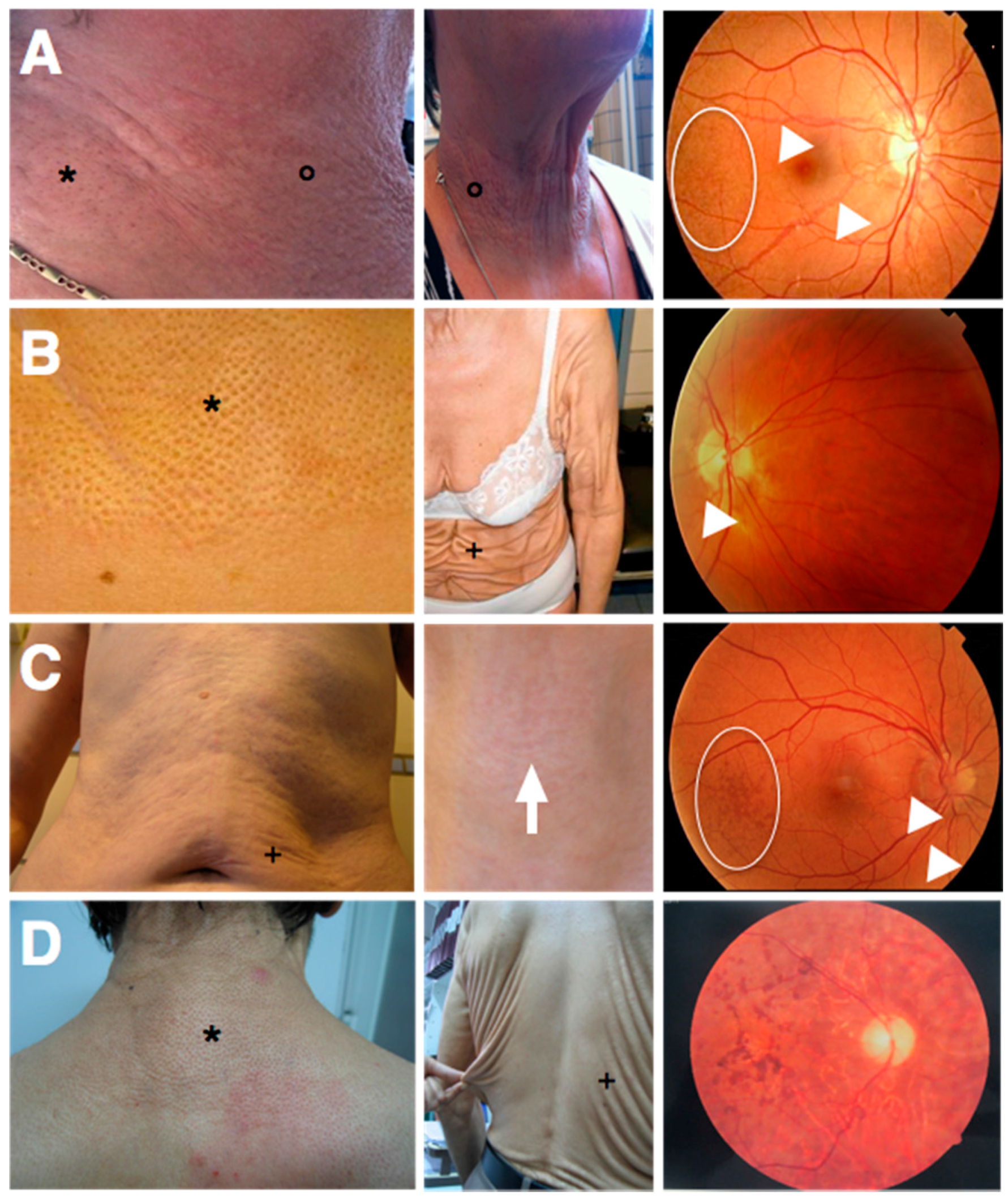
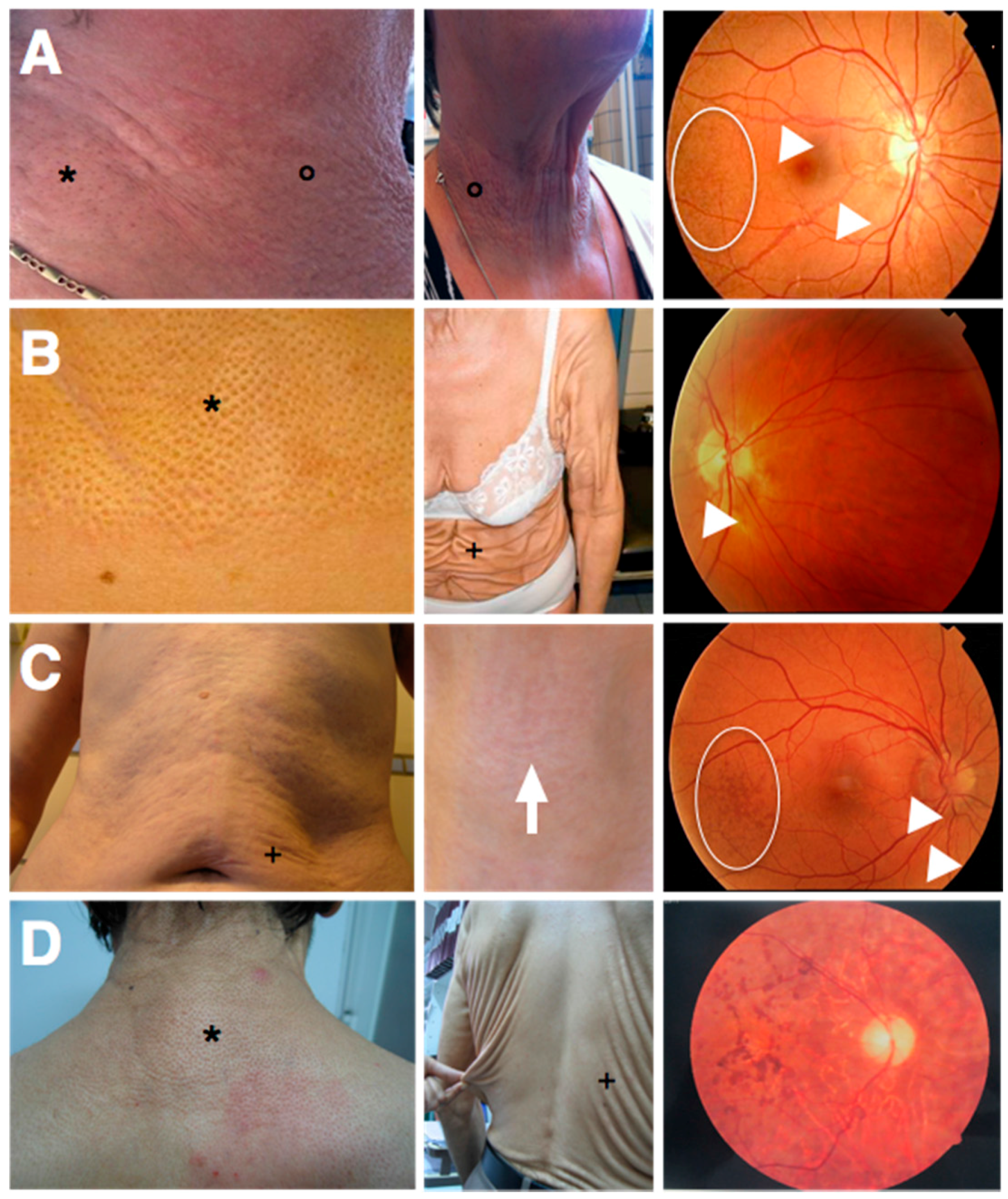
ABCC6 (ATP-binding cassette, subfamily C, member 6; OMIM*603234) mutations and features yellowish skin papules and plaques in flexural areas (although in more severe cases an increase in skin laxity may occur), ophthalmological symptoms (asymptomatic peau d’orange and angioid streaks and in more advanced stages subretinal neovascularization, bleeding and scarring leading to legal blindness when untreated) and cardiovascular symptoms (peripheral artery disease, cardiac diastolic dysfunction) (
Figure 3A) [
34,
35]. Strikingly, at first the seven patients with the PXE-like syndrome had typical dermal EF calcifications leading to yellowish papules but later progressively developed excessive skin folds, not only confined to flexural areas (
Figure 3B) [
36]. In contrast to PXE, cardiovascular symptoms were absent and the retinopathy much milder, with mainly asymptomatic lesions (peau d’orange and angioid streaks) [
36]. Since the original report, additional patients have been identified with a similar phenotype [
21,
37].
In 2011, we described a patient with a phenotype intermediate to PXE and the PXE-like syndrome (
Figure 3C). This boy had developed excessive skin folds, typical for the PXE-like syndrome, around the age of 10, which at first were confined to the abdomen, but later progressively affected the axillae, upper arms and elbows. Prior to the development of the skin folds, the skin had appeared to have an inflamed, reddish aspect, but the skin aspect was not suggestive for an acquired post-inflammatory form of cutis laxa [
38]. Upon clinical inspection, a very mild yellowish reticular rash, typical for PXE, was identified in the frontal neck. Further clinical workup included fundoscopic imaging, which revealed peau d’orange and angioid streaks, abdominal ultrasound showing renal microcalcifications, and normal coagulation tests (normal activated partial thromboplastin time (aPTT) and prothrombin time (PT)). Moreover, histological, biochemical and immunohistochemical characteristics were intermediate to PXE and the PXE-like syndrome. This patient was identified with compound heterozygous
ABCC6 mutations as well as a functional single nucleotide polymorphism (SNP) in the
GGCX gene [
39].
In 2014, Kariminejad et al. described 14 patients from two unrelated families with a PXE-like skin phenotype (cutis laxa) and a pigmentary retinopathy, caused by biallelic mutations in the
GGCX gene (
Figure 3D). These patients all developed progressive vision loss with night blindness in early childhood and yellowish skin papules on the back, the lateral sides of the neck, the chest and the flexural body areas between the ages of 11 and 25 as well as an unusually loose skin on the trunk, which gradually worsened and in later stages also affected the upper limbs. Ophthalmological workup, including an electroretinogram, identified non-detectable rod responses or rod responses with reduced amplitude and prolonged implicit time, compatible with a pigmentary retinopathy, but failed to show any PXE-related eye symptoms (such as angioid streaks, peau d’orange or subretinal hemorrhage). A similar phenotype was seen in all affected patients, varying only in time of onset of the eye and skin symptoms. An echocardiography showed no abnormalities. Interestingly, none of these patients had VKCFD1 [
40].
Next to the presence of phenotypes caused by mutations in the
GGCX gene, the modifying effect of
GGCX variants on the response to warfarin treatment (amongst others on dose and time in therapeutic range) has been suggested repeatedly in the past and has been extensively studied. However, the results of these association studies are ambiguous, seem to be population-dependent and cannot be seen separately from variants in other genes, relevant for warfarin metabolism. Hence, the effects of these
GGCX variants fall beyond the scope of this systematic review [
41,
42,
43,
44].
The variability of the GGCX-related phenotypes, as illustrated above and in
Table 1, is striking while their correlation with the underlying genotypes has to date remained unclear. Therefore, we explored the possibility of genotype–phenotype correlations between
GGCX mutations and the different GGCX-related diseases.
3. Discussion
For this systematic review, we assessed all patients described in literature with cardiac, dermatological, ophthalmological, osseous and/or coagulation abnormalities, caused by GGCX mutations, and explored possible genotype–phenotype correlations. As the number of patients suffering from these orphan diseases is too small to allow valid statistical interpretation of the data, we did not perform a meta-analysis. However, based on the data, we identified a trend that the presence of at least one GGCX mutations in the HTTM domain may predispose for the occurrence of a cardiac and/or osseous phenotype. Further, there seems to be an association between aging and the occurrence of a skin and, less clearly, an ocular phenotype rather than a link with a specific genotype. However, most of the patients with an ophthalmological phenotype also had at least one HTTM domain mutation. Regarding the bleeding phenotype, severely affected patients seem to have homozygous mutations or compound heterozygous mutations affecting the same protein domain.
3.1. Cardiac Phenotype
To date, no direct pathophysiological link has been established between
GGCX mutations and cardiac abnormalities. However, Gas6—a VKDP gamma-carboxylated by GGCX—activates AXL (AXL receptor tyrosine kinase; OMIM*109135), a receptor tyrosine kinase (RTK) that interacts with non-muscle myosin IIB, an essential protein for normal development of the murine heart [
51,
52]. As all patients with cardiac involvement had at least one mutation located in the HTTM domain, possibly this domain plays a role in Gas6 carboxylation, which could explain the cardiac problems in patients. Cardiac anomalies were found in 8/47 patients (17%) with
GGCX mutations, in contrast to 4/63 patients (6%) with fetal warfarin syndrome, caused by inhibition of gamma-carboxylation by warfarin administration during pregnancy [
53]. A possible explanation for this discrepancy is that warfarin is a dose-dependent inhibitor of the VK cycle and
GGCX mutations lead to a permanent and persistent defect in gamma-carboxylation [
54]. Deficiency of another VKDP, MGP, has also been associated with cardiac involvement, including pulmonary artery stenosis and ventricular septal defects [
55,
56].
As in the study cohort all patients with cardiac problems have at least one mutation in the HTTM domain, patients with a
GGCX mutation in this domain might benefit from an echocardiography to rule out possible anomalies or to enable early initiation of treatment if a heart defect is identified. This is for example the case in patients with a small to moderate patent ductus arteriosus Botalli, who may remain asymptomatic during childhood, leaving the defect thus undetected. Some of these patients develop congestive heart failure in early adulthood due to a chronic, left heart overload [
57]. Early diagnosis can enable early surgical closure of the patent ductus arteriosus Botalli, hereby preventing the development of heart failure.
3.2. Dermatological Phenotype
The patients diagnosed with skin symptoms were often described with a less severe bleeding phenotype, suggesting the presence of a residual carboxylation capacity. Recently, an exon 3 deletion, present in P13 and P31–P43 was proven to completely inactivate the GGCX enzyme. In P31–P43, this mutation (c.373+3G>T), which is localized in TMD1 and -2, was present homozygously and led to cutis laxa, a pigmentary retinopathy but no VKCFD1. Interestingly, P13 harbored a heterozygous exon 3 deletion, caused by another splice site mutation c.215-1G>T, with the p.(R485P) mutation on the other allele and had VKCFD1 but no dermatological phenotype. Jin et al. showed that p.(R485P) led to a GGCX enzyme with some residual function, which according to the authors played an important role in the development of a bleeding phenotype, as a high dose of VK was necessary to ameliorate the phenotype [
58]. These data could indicate that TMD1 and -2 have no significant role in the carboxylation of VK-dependent coagulation factors. Indeed, only four patients with VKCFD1 (P13, P15, and P29) have a heterozygous mutation in TMD1 and/or TMD2, and all but one had a second mutation in another GGCX domain. P47 had a homozygous
GGCX mutation affecting the HTTM (TMD1) and only had VKCFD1 and no skin phenotype. However, the mutation also affected the N-terminal region of the protein, which may be an explanation for the VKCFD1 phenotype in this patient. Further, the absence of a skin phenotype could be due to the young age of the patient when it was described in literature (four years of age at last described follow-up).
Regarding the severity of the skin manifestations, PXE-like dermatological symptoms—mainly cutis laxa—are typically more severe compared to classic PXE patients with regard to their location (beyond flexural areas), number of additional skin folds and the time span during which they develop and aggravate [
36]. Interestingly, 21/24 patients discussed in this paper who are older than 18 years, have typical PXE-like skin manifestations (
Table 2 and
Table 5). This could indicate that skin symptoms may be due to an accumulation of certain substances which only leads to symptoms when a critical threshold is reached, irrespective of the patients’ genotypes. In this respect, Vanakker et al. showed an accumulation of uncarboxylated MGP and OC, two potent mineralization inhibitors, in patients with PXE-like disorder with multiple combined coagulation factor deficiency [
36]. Although the age of onset of skin problems is variable between the described patients (P3–P40), this would indicate that most, if not all, patients with biallelic
GGCX mutations develop skin lesions in the course of their disease.
For P13, at birth the putative diagnosis of Williams-Beuren syndrome was made based on his facial gestalt. Even though there is no direct link with VKDP and VKCFD1, this disease is caused by a deletion of the WBSCR (Williams–Beuren syndrome critical region), including the
ELN gene (elastin; OMIM*130160), responsible for the arteriopathy in Williams-Beuren syndrome. Further, cardiac involvement (most commonly supravalvular aortic stenosis) and a skin phenotype (soft loose skin) are features of this disease [
59]. Moreover, heterozygous
ELN mutations are associated with an autosomal dominant type of cutis laxa (ADCL1; OMIM#123700) [
60]. These clinical findings might indicate a possible link between these disease entities, although only one patient in the whole cohort was described with a facies resembling the Williams–Beuren syndrome, rendering the assumption less likely.
Of the remaining 24 patients without skin manifestations, 20 were 18 years old or younger and 21/24 had VKFCD1 (deficiency of the VK-dependent coagulation factors (
Table 2 and
Table 8). Hence, VKFCD1 and the PXE-like disorder with multiple coagulation factor deficiency could belong to a disease spectrum with partly overlapping etiopathogenetic mechanisms, but in whom different GGCX domains are important for the activation of the involved VKDP. However, the young age of these patients could also be an explanation for the absence of a skin phenotype. In severe cases, the additional skin folds may lead to restriction of normal physical activities and may be predilection sites for (severe) skin infections, which is important in patient counseling and follow-up.
Finally, in the analyzed literature, three patients are described with skin symptoms and a digenic inheritance of
ABCC6 and
GGCX mutations [
37,
46]. These patients were not included in this analysis because an unambiguous interpretation of the skin features is not possible, as both
GGCX and
ABCC6 mutation may lead to related skin phenotypes. Typically, the skin phenotype in these patients is less severe than in patients with biallelic
GGCX mutations, so it may be worthwhile to perform additional sequencing of the
ABCC6 gene in those patients in whom only one
GGCX mutation is withheld as it will influence genetic counseling and management.
3.3. Ophthalmological Phenotype
Regarding the eye symptoms, all patients with a homozygous skip of exon 3 were diagnosed with a pigmentary retinopathy, a disease mimicking retinitis pigmentosa. In mice, it has already been shown that absence of both ligands of the RTK MerTK (Mer tyrosine kinase proto-oncogene; OMIM*604705), Gas6 and ProS, leads to retinitis pigmentosa [
61]. In analogy, such a mechanism could play a role in the development of a pigmentary retinopathy in the two families with a homozygous skip of exon 3 in the
GGCX gene. As this mutation is located in the HTTM domain, this could again point to its putative roles in the carboxylation of both Gas6 and ProS, similar to what is observed in patients with a cardiac phenotype.
Furthermore, as in 17 of the 24 patients over 18 years of age an ophthalmological phenotype was identified and in only four out of 23 patients with cutis laxa no accompanying eye disease was reported, possibly, the major determinant of developing a GGCX-related retinopathy is also increasing age. Although this link seems less convincing compared to skin manifestations, it should be noted that (mild) angioid streaks are asymptomatic and can easily be overlooked during a routine ophthalmological checkup. In the spectrum, PXE-like skin and eye symptoms may be phenotypes that occur in most patients with increasing age, whereas VKCFD1 may be more variable, with neonatal complications in severe forms to apparent normal coagulation in very mildly affected patients, such as those patients described by Kariminejad et al. [
40] with PXE-like skin symptoms, an eye phenotype but no apparent VKCFD1. As mentioned above, possibly distinct GGCX domains play an important part role in the gamma-carboxylation of involved VKDP.
Overall, the PXE-like ophthalmological symptoms (P8, P10, P11, P16, P17, and P18) seem less severe compared to the dermatological features with no functional complications in the described patient cohort. However, as this cohort is small, we cannot exclude the possibility that in some patients with biallelic
GGCX mutations more severe PXE eye symptoms may occur with subretinal neovascularization and hence loss of vision if not treated immediately. Further, a pigmentary retinopathy with severe visual dysfunction may occur in some patients. It therefore seems appropriate to follow patients ophthalmologically, certainly from the time they start to develop skin symptoms. In mildly affected patients the typical PXE-like angioid streaks may be very small and thus only be picked up by very sensitive funduscopic imaging, for which confocal near-infrared reflectance imaging is superior compared to other techniques [
62,
63]. If symptoms are stable for a long time, follow-up may become less stringent; however, to our knowledge, there has thus far not been a comprehensive long-term follow-up study of eye symptoms in patients with biallelic
GGCX mutations.
3.4. Osseous Phenotype
Bone symptoms in patients with
GGCX mutations show an important clinical overlap with other genetic syndromes, such as X-linked chondrodysplasia punctata (an autosomal recessive disorder caused by mutations in the
ARSE gene (arylsulphatase E; OMIM*123700), encoding ARSE), Keutel syndrome, due to
MGP gene mutations; and the fetal warfarin syndrome [
64]. Interestingly, Vanakker et al. confirmed an accumulation of uncarboxylated MGP and OC in patients with the PXE-like syndrome; similarly, an in vitro model for the mutation leading to the Keutel syndrome-like phenotype in P7 showed an abolishment of MGP carboxylation [
28,
36]. For ARSE, no data are available of a direct association with GGCX. However, it was shown that ARSE is specifically inhibited by warfarin, therefore having a possible role in warfarin embryopathy, thus ARSE could have a role in the VK cycle, which would explain the phenotypic overlap with patients with biallelic
GGCX mutations [
65]. Further investigation of this interaction proves a valid way to further unravel the association between the VK-dependent pathway and skeletal development.
Finally, all patients with skeletal and/or facial symptoms had at least one GGCX mutation in the HTTM-domain, so putatively this domain not only plays a role in Gas6 and ProS carboxylation but also in MGP (and OC) carboxylation. Because of the association with osteopenia and osteoporosis, patients with a mutation in the HTTM domain may benefit from an early densitometry (as P23 had already developed osteoporosis at 12 years old, first densitometry may be most valuable at a younger age) to facilitate early treatment when a decreased bone density is present. There is no genotypic difference between patients with a complete bone phenotype (chondrodysplasia punctata, reduced bone mass, facial dysmorphisms) or with only some of the osseous features. Therefore, early bone densitometry may better be advised to all patients with at least one mutation in the HTTM domain.
3.5. VKCFD1
Overall, VKCFD1 seems to be variable in severity, clinical presentation and response to VK supplementation in the analyzed patient cohort. However, there seems to be a trend that the genotypes of patients with very early onset of bleeding symptoms (<1 year of age) mainly comprise homozygous mutations or compound heterozygous mutations in or near the same protein domain. Possibly, biallelic hits in the same protein domain lead to a more severe malfunctioning of the carboxylase and thus more severe VKCFD1 symptoms occur in these patients at a younger age. However, this could not be confirmed by the activity of the coagulation factors in these patients, even though it is difficult to compare such values between patients whose coagulation factor function is measured in different labs, as different units and reference values are used, which cannot easily be compared to each other.
None of the patients with severe bleeding symptoms had a skin or eye phenotype. However, this could be a bias, as these severely affected patients are often described at a very young age and no follow-up studies are available, so putative development of cutis laxa or other PXE-like skin and/or eye manifestations at a later age remains a possibility, which is exemplified by the presence of skin symptoms in some of the patients who have a rather mild bleeding phenotype.
In a small subset of the patient cohort carboxylation status of extrahepatic VKDP were measured, and OC carboxylation seemed to respond (to a variable extent) to VK supplementation. Interestingly, in one of these patients, VK treatment did not influence the bleeding phenotype with no significant change in hemostatic parameters and recurrent bleeding episodes after initiation of the treatment. In contrast, another patient showed a distinct amelioration of the coagulation factor function on VK treatment, whereas MGP carboxylation did not increase. This variability in response to VK supplementation may result from different mechanisms in gamma-carboxylation of hemostatic and extrahepatic VKDP [
28]. Moreover, different
GGCX mutations affect different regions of the GGCX protein, which could interfere with propeptide binding of different VKDP [
21]. The cohort is however too small to make definite conclusions.
5. Conclusions
Gamma-carboxylation is an essential process in the activation of VKDP, which are important in numerous biological processes, such as blood clotting, inflammation, bone formation and cell proliferation. This posttranslational modification process is executed by the GGCX enzyme. Mutations in the gene encoding GGCX have been linked to multiple distinct phenotypes, affecting the heart, skin, eyes, blood clotting and bone metabolism. This review highlights the importance of mutations in the HTTM domain for at least the cardiac and bone phenotype, as all of the patients had at least one mutation in this domain, whereas multiple patients without cardiac or osseous manifestations had no mutations in the HTTM domain. Further, age was identified as the most important determinant of the development of PXE-like skin symptoms and to a lesser extent ophthalmological manifestations. Finally, distinct parts of the HTTM domain seem to have a specific role in the development of skin symptoms and not of VKCFD1. Based on our results, patients should be informed during genetic counseling about the possibility of skin lesions appearing in the course of their disease, taking into account that these lesions may be subtle at onset. In all, a detailed ophthalmological evaluation should be performed and adequate follow-up should be organized, as apart from a (mild) typical PXE-like retinopathy a pigmentary retinopathy with significant functional implications may also occur. Because of its association with reduced bone mass, a bone densitometry should be offered to all patients harboring at least one mutation in the HTTM domain of GGCX.
The main limitations of this systematic review are the low number of patients with GGCX-related phenotypes and the presence of consanguinity in some of the assessed families. There is a possibility that homozygosity at other loci could play a role in the occurrence of the other GGCX-related phenotypes in these patients. However, for most of these phenotypes, there are clinical discrepancies between siblings, which makes this less suggestive, even though the possibility of variable penetrance cannot be ruled out. These limitations are inherent to this type of autosomal recessive disorders, because most of them are rare hence making it impossible to have large cohorts with only individual probands. Another limitation is the incomplete information regarding the (non-)hemostatic parameters in some of the original papers, which do not mention reference values, making it impossible to correctly interpret the values in comparison to other patients, as the reference values tend to be highly laboratory-dependent. Further, the details of the VK treatment, with regards to mode of administration, VK dose, specific subtype of VK, and duration of the therapy, are often not or incompletely mentioned. These details could possibly influence the response to treatment in the described patients. However, the main purpose of this systematic review was to combine all known and described patients with GGCX-related phenotypes and to explore the possibility of genotype–phenotype correlations for the different phenotypes. This manuscript can be used as a guideline for future research on the GGCX protein structure and function, which then can lead to new insights in the VK cycle.





{kind=link}
{kind=link}
{kind=link}
{kind=link}