DFT Study of the Structure, Reactivity, Natural Bond Orbital and Hyperpolarizability of Thiazole Azo Dyes

Abstract
:
1. Introduction
2. Results and Discussion
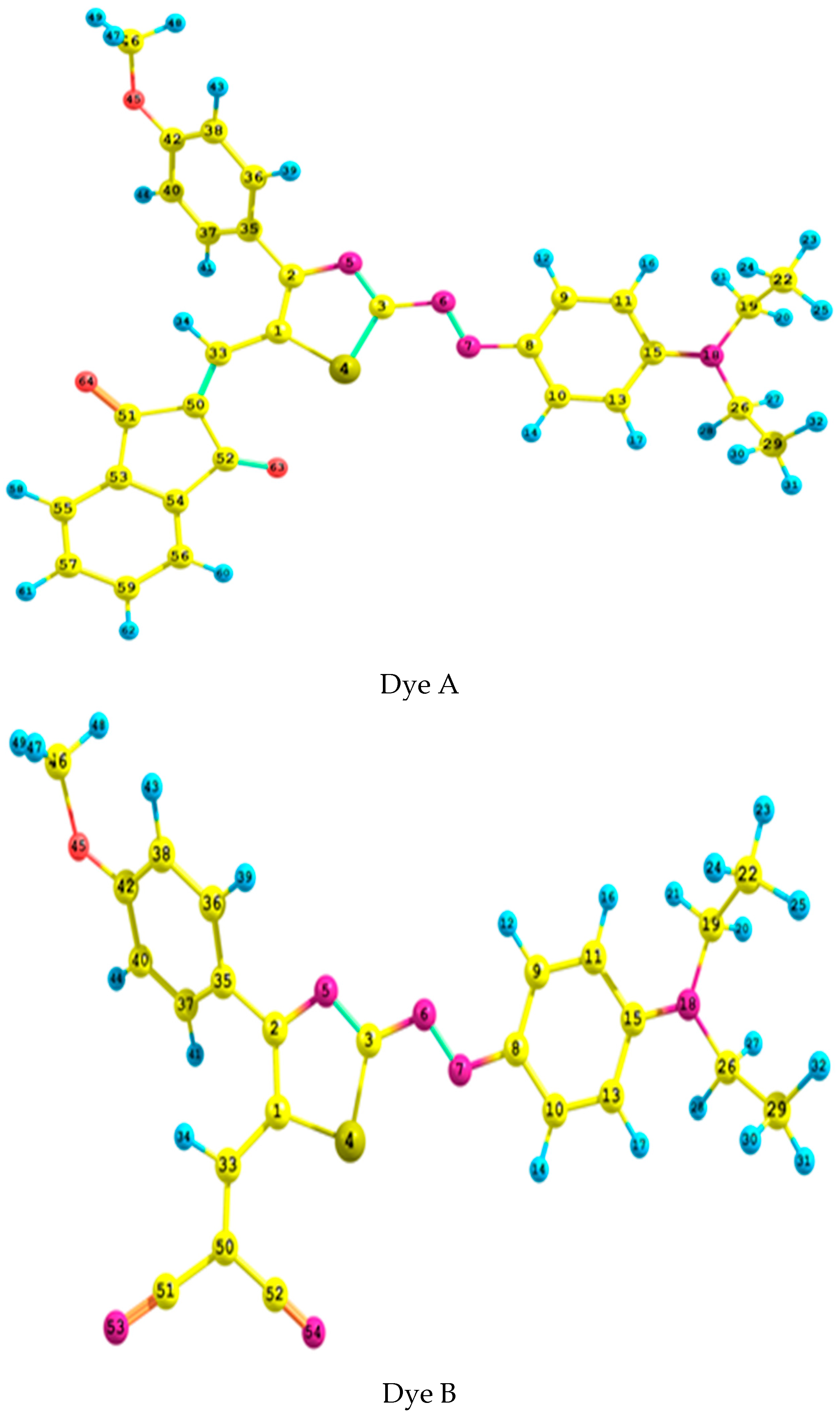
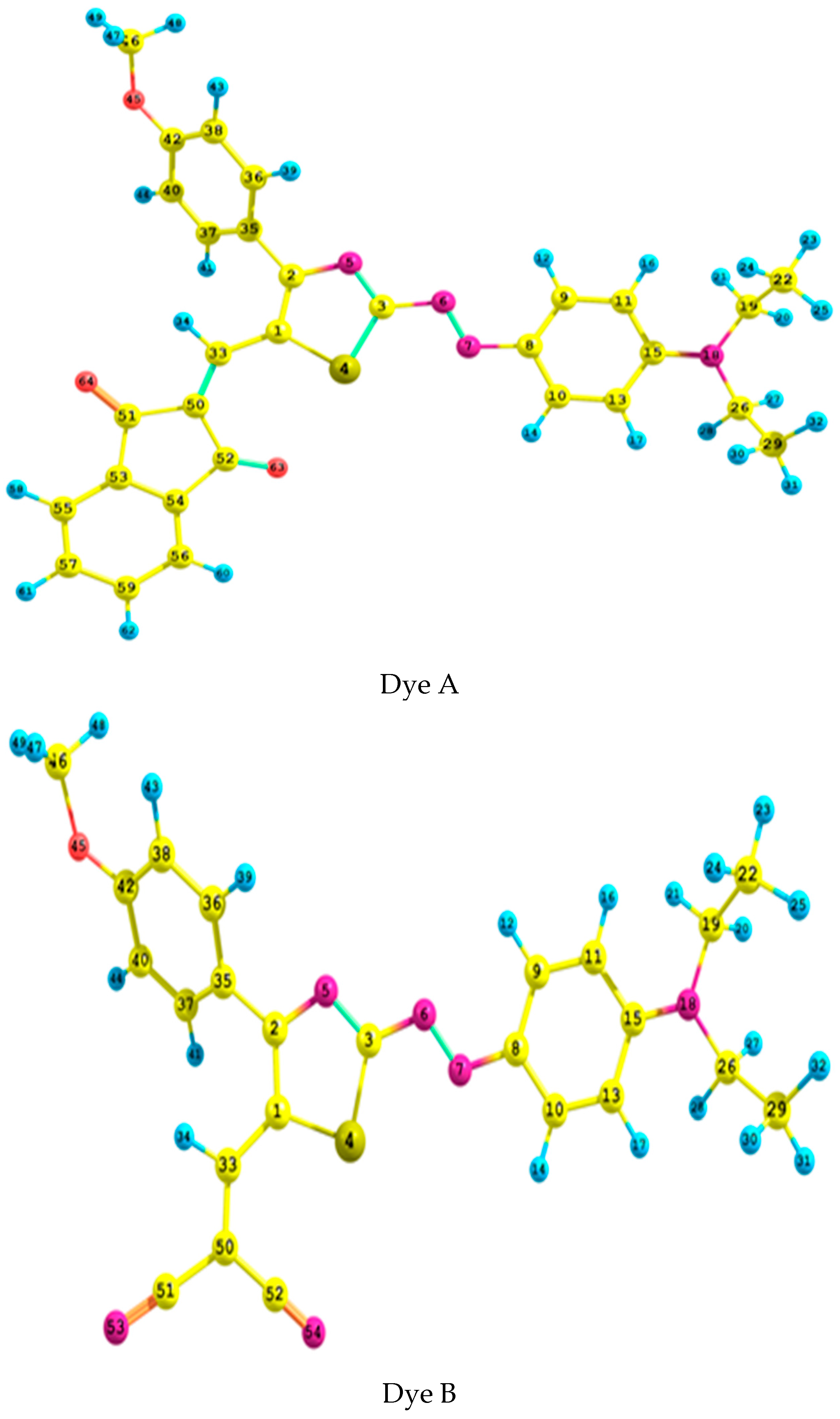
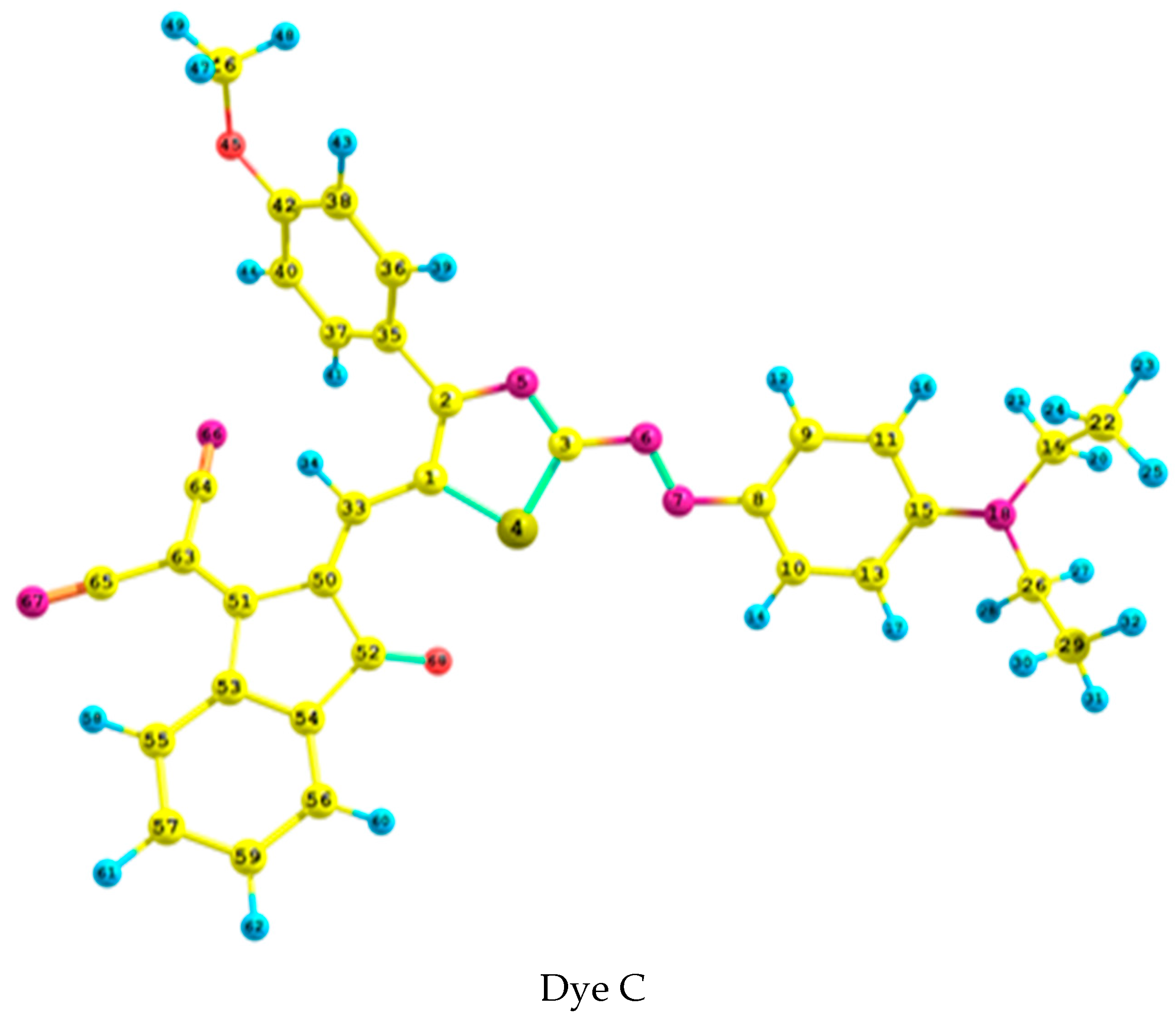
2.1. Geometrical Analysis
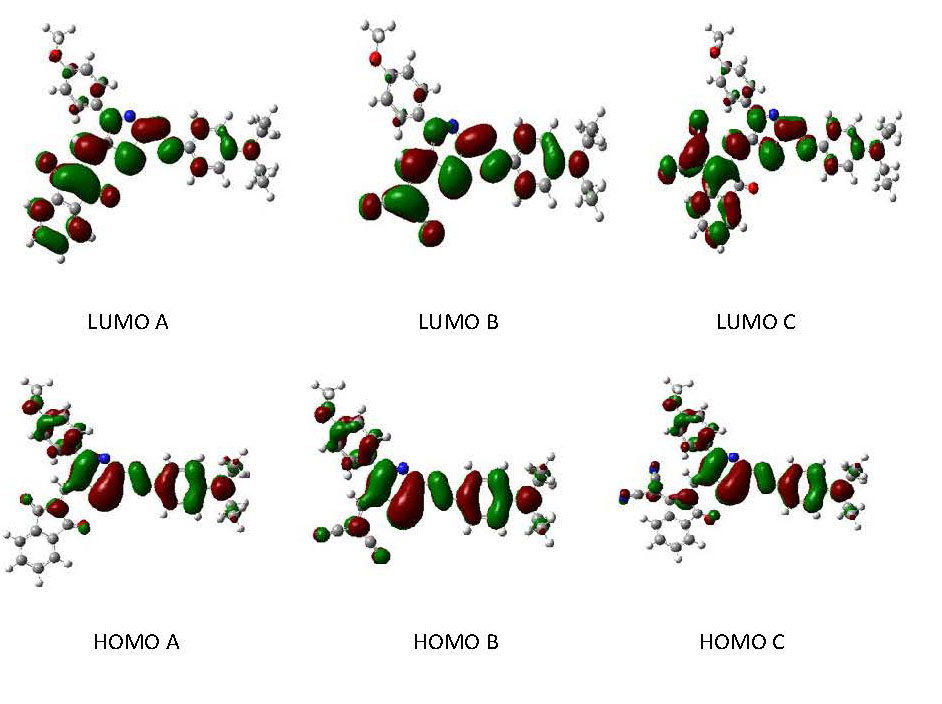
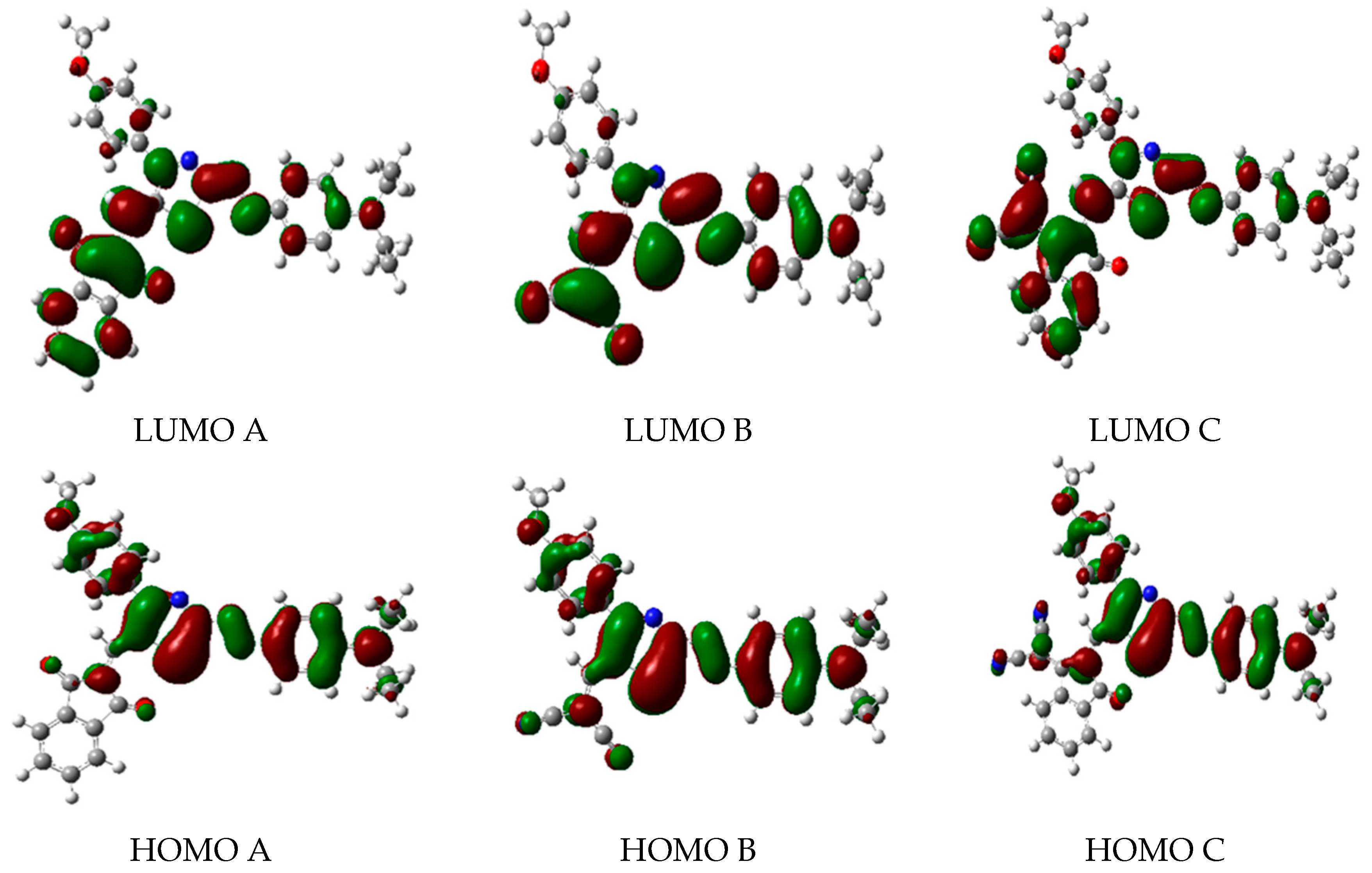
2.2. Frontier Molecular Orbitals (FMOs)
2.3. UV–Visible Spectral Analysis
2.4. Nonlinear Optical (NLO) Properties
2.5. Natural Bond Orbital (NBO) Analysis
3. Computational Details
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Xie, X.N.; Chung, H.J.; Sow, C.H.; Wee, A.T.S. Oxide growth and its dielectrical properties on alkylsilated native-SiO2/Si surface. Chem. Phys. Lett. 2004, 388, 446–451. [Google Scholar] [CrossRef]
- Sanz, N.; Terech, P.; Djurado, D.; Deme, B.; Ibanez, A. Structural Characterization of New Nanocomposite Hybrid Materials: Organic Nanocrystals Grown in Gel−Glasses. Langmuir 2003, 19, 3493–3498. [Google Scholar] [CrossRef]
- Gulino, A.; Bazzano, S.; Condorelli, G.G.; Giuffrida, S.; Mineo, P.; Satriano, C.; Scamporrino, E.; Ventimiglia, G.; Vitalini, D.; Fragala, I. Engineered Silica Surfaces with an Assembled C60 Fullerene Monolayer. Chem. Mater. 2005, 17, 1079–1084. [Google Scholar] [CrossRef]
- Enami, Y.; Meredith, G.; Peyghambarian, N.; Jen, A.K.-Y. Hybrid electro-optic polymer/sol–gel waveguide modulator fabricated by all-wet etching process. Appl. Phys. Lett. 2003, 83, 4692–4694. [Google Scholar] [CrossRef]
- Shirahata, N.; Shin, W.; Murayama, N.; Hozumi, A.; Yokogawa, Y.; Kameyama, T.; Masuda, Y.; Koumoto, K. Reliable Monolayer-Template Patterning of SnO2 Thin Films from Aqueous Solution and Their Hydrogen-Sensing Properties. Adv. Funct. Mater. 2004, 14, 580–588. [Google Scholar] [CrossRef]
- Gershewitz, O.; Grinstein, M.; Sukenik, C.N.; Regev, K.; Ghabboun, J.; Cahen, D. Effect of Molecule−Molecule Interaction on the Electronic Properties of Molecularly Modified Si/SiOx Surfaces. J. Phys. Chem. B 2004, 108, 664–672. [Google Scholar] [CrossRef]
- Moreno-Yruela, C.; Garin, J.; Orduna, J.; Franco, S.; Quintero, E.; Lopez Navarrete, J.T.; Diosdado, B.E.; Villacampa, B.; Casado, J.; Andreu, R. D-π-A Compounds with Tunable Intramolecular Charge Transfer Achieved by Incorporation of Butenolide Nitriles as Acceptor Moieties. J. Org. Chem. 2015, 80, 12115–12128. [Google Scholar] [CrossRef] [PubMed]
- Kulhanek, J.; Bures, F. Imidazole as a parent π-conjugated backbone in charge-transfer chromophores. Beilstein J. Org. Chem. 2012, 8, 25–49. [Google Scholar] [CrossRef] [PubMed]
- Spraul, B.K.; Suresh, S.; Sassa, T.; Herranz, M.A.; Echegoyen, L.; Wada, T.; Perahiaa, D.; Smith, D.W., Jr. Thermally stable triaryl amino chromophores with high molecular hyperpolarizabilities. Tetrahedron Lett. 2004, 45, 3253–3256. [Google Scholar] [CrossRef]
- LeCours, S.M.; Guan, H.-W.; DiMagno, S.G.; Wang, C.H.; Therien, M.J. Push−Pull Arylethynyl Porphyrins: New Chromophores That Exhibit Large Molecular First-Order Hyperpolarizabilities. J. Am. Chem. Soc. 1996, 118, 1497–1503. [Google Scholar] [CrossRef]
- Di Bella, S.; Fragala, I.; Ledoux, I.; Diaz-Garcia, M.A.; Marks, T.J. Synthesis, Characterization, Optical Spectroscopic, Electronic Structure, and Second-Order Nonlinear Optical (NLO) Properties of a Novel Class of Donor−Acceptor Bis(salicylaldiminato)nickel(II) Schiff Base NLO Chromophores. J. Am. Chem. Soc. 1997, 119, 9550–9557. [Google Scholar] [CrossRef]
- Miller, R.D.; Lee, V.Y.; Moylan, C.R. Substituted Azole Derivatives as Nonlinear Optical Chromophores. Chem. Mater. 1994, 6, 1023–1032. [Google Scholar] [CrossRef]
- Wang, Y.-K.; Shu, C.-F.; Breitung, E.M.; McMahon, R.J. Synthesis and characterization of thiazole-containing chromophores for second-order nonlinear optics. J. Mater. Chem. 1999, 9, 1449–1452. [Google Scholar] [CrossRef]
- Hrobarik, P.; Zahradnik, P.; Fabian, W.M.F. Computational design of benzothiazole-derived push–pull dyes with high molecular quadratic hyperpolarizabilities. Phys. Chem. Chem. Phys. 2004, 6, 495–502. [Google Scholar] [CrossRef]
- Hrobarik, P.; Sigmundova, I.; Zahradnik, P.; Kasak, P.; Arion, V.; Franz, E.; Clays, K. Molecular Engineering of Benzothiazolium Salts with Large Quadratic Hyperpolarizabilities: Can Auxiliary Electron-Withdrawing Groups Enhance Nonlinear Optical Responses? J. Phys. Chem. C 2010, 114, 22289–22302. [Google Scholar] [CrossRef]
- Hrobarik, P.; Hrobarikova, V.; Sigmundova, I.; Zahradnik, P.; Fakis, M.; Polyzos, I.; Persephonis, P. Benzothiazoles with Tunable Electron-Withdrawing Strength and Reverse Polarity: A Route to Triphenylamine-Based Chromophores with Enhanced Two-Photon Absorption. J. Org. Chem. 2011, 76, 8726–8736. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, M.; Teshome, A.; Kay, A.J.; Gainsford, G.J.; Bhuiyan, M.D.H.; Asselberghs, I.; Clays, K. Synthesis and optical properties of NLO chromophores containing an indoline donor and azo linker. Dyes Pigments 2012, 95, 455–464. [Google Scholar] [CrossRef]
- Castro, M.C.R.; Schellenberg, P.; Belsley, M.; Fonseca, A.M.C.; Fernandes, S.S.M.; Raposo, M.M.M. Design, synthesis and evaluation of redox, second order nonlinear optical properties and theoretical DFT studies of novel bithiophene azo dyes functionalized with thiadiazole acceptor groups. Dyes Pigments 2012, 95, 392–399. [Google Scholar] [CrossRef] [Green Version]
- Satam, M.A.; Raut, R.K.; Sekar, N. Fluorescent azo disperse dyes from 3-(1,3-benzothiazol-2-yl)naphthalen-2-ol and comparison with 2-naphthol analogs. Dyes Pigments 2013, 96, 92–103. [Google Scholar] [CrossRef]
- Hrobarik, P.; Hrobarikova, V.; Semak, V.; Kasak, P.; Rakovsky, E.; Polyzos, I.; Fakis, M.; Persephonis, P. Quadrupolar Benzobisthiazole-Cored Arylamines as Highly Efficient Two-Photon Absorbing Fluorophores. Org. Lett. 2014, 16, 6358–6361. [Google Scholar] [CrossRef] [PubMed]
- Varanasi, P.R.; Jen, A.K.-Y.; Chandrasekhar, J.; Namboothiri, I.N.N.; Rathna, A. The Important Role of Heteroaromatics in the Design of Efficient Second-Order Nonlinear Optical Molecules: Theoretical Investigation on Push−Pull Heteroaromatic Stilbenes. J. Am. Chem. Soc. 1996, 118, 12443–12448. [Google Scholar] [CrossRef]
- Albert, I.D.L.; Marks, T.J.; Ratner, M.A. Large Molecular Hyperpolarizabilities. Quantitative Analysis of Aromaticity and Auxiliary Donor−Acceptor Effects. J. Am. Chem. Soc. 1997, 119, 6575–6782. [Google Scholar] [CrossRef]
- Breitung, E.M.; Shu, C.-F.; McMahon, R.J. Thiazole and Thiophene Analogues of Donor−Acceptor Stilbenes: Molecular Hyperpolarizabilities and Structure−Property Relationships. J. Am. Chem. Soc. 2000, 122, 1154–1160. [Google Scholar] [CrossRef]
- Popli, S.; Patel, U.D. Destruction of azo dyes by anaerobic–aerobic sequential biological treatment: A review. Int. J. Environ. Sci. Technol. 2015, 12, 405–442. [Google Scholar] [CrossRef]
- Klaus, G.K.; Kopps, S.; Myslak, Z.W. Carcinogenicity of azo colorants: Influence of solubility and bioavailability. Toxicol. Lett. 2004, 151, 203–210. [Google Scholar]
- Salvador, M.A.; Almeida, P.; Reis, L.V.; Santos, P.F. Near-infrared absorbing delocalized cationic azo dyes. Dyes Pigments 2009, 82, 118–123. [Google Scholar] [CrossRef]
- Chen, L.; Cui, Y.; Qian, G.; Wang, M. Synthesis and spectroscopic characterization of an alkoxysilane dye containing azo-benzothiazole chromophore for nonlinear optical applications. Dyes Pigments 2007, 73, 338–343. [Google Scholar] [CrossRef]
- Seferoglu, Z.; Ertan, N.; Kickelbick, G.; Hokelek, T. Single crystal X-ray structure analysis for two thiazolylazo indole dyes. Dyes Pigments 2009, 82, 20–25. [Google Scholar] [CrossRef]
- Yen, M.S.; Wang, I.J. Synthesis and absorption spectra of hetarylazo dyes derived from coupler 4-aryl-3-cyano-2-aminothiophenes. Dyes Pigments 2004, 61, 243–250. [Google Scholar]
- Towns, A.D. Developments in azo disperse dyes derived from heterocyclic diazo components. Dyes Pigments 1999, 42, 3–28. [Google Scholar] [CrossRef]
- El-Shishtawy, R.M.; Borbone, F.; Al-amshany, Z.M.; Tuzi, A.; Barsella, A.; Asiri, A.M.; Roviello, A. Thiazole azo dyes with lateral donor branch: Synthesis, structure and second order NLO properties. Dyes Pigments 2013, 96, 45–51. [Google Scholar] [CrossRef]
- Watts, J.D.; Watts, D.J.; Huang, M.-L. Theoretical Study of the Tautomerism, Structures, and Vibrational Frequencies of the Phosphaalkenes XP = C(CH3)2 (X = H, F, Cl, Br, OH, ArF (ArF = 2,6-(CF3)2C6H3)). J. Phys. Chem. A 2009, 113, 1886–1891. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, M.; Kuze, N.; Fujiwara, H.; Takeuchi, H.; Konaka, S. Molecular structure of 2-methylthiophene studied by gas electron diffraction combined with microwave spectroscopic data. J. Mol. Struct. THEOCHEM 1995, 372, 173–180. [Google Scholar] [CrossRef]
- Garza, A.J.; Osman, O.I.; Wazzan, N.A.; Khan, S.B.; Gustavo, G.E.; Asiri, A.M. Photochromic and nonlinear optical properties of fulgides: A density functional theory study. Comput. Theor. Chem. 2013, 1022, 82–85. [Google Scholar] [CrossRef]
- Fleming, I. Frontier Orbitals and Organic Chemical Reactions, 1st ed.; Wiley: London, UK, 1978; pp. 879–880. [Google Scholar]
- Kavitha, E.; Sandaraganesan, N.; Sebastian, S. Molecular structure, vibrational spectroscopic and HOMO, LUMO studies of 4-nitroaniline by density functional method. Indian J. Pure Appl. Phys. 2010, 48, 20–30. [Google Scholar]
- Kim, K.H.; Han, Y.K.; Jung, J. Basis set effects on relative energies and HOMO–LUMO energy gaps of fullerene C36. Theor. Chem. Acc. 2005, 113, 233–237. [Google Scholar] [CrossRef]
- Aihara, J. Reduced HOMO−LUMO Gap as an Index of Kinetic Stability for Polycyclic Aromatic Hydrocarbons. J. Phys. Chem. A 1999, 103, 7487–7495. [Google Scholar] [CrossRef]
- Kaatz, P.; Donley, E.A.; Shelton, D.P. A comparison of molecular hyperpolarizabilities from gas and liquid phase measurements. J. Chem. Phys. 1998, 108, 849–856. [Google Scholar] [CrossRef]
- Pearson, R.G. Chemical hardness and density functional theory. J. Chem. Sci. 2005, 117, 369–377. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Maiti, B. HSAB Principle Applied to the Time Evolution of Chemical Reactions. J. Am. Chem. Soc. 2003, 125, 2705–2710. [Google Scholar] [CrossRef] [PubMed]
- Parr, P.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Mebi, A.C. DFT study on structure, electronic properties, and reactivity of cis-isomers of [(NC5H4-S)2Fe(CO)2]. J. Chem. Sci. 2011, 123, 727–731. [Google Scholar] [CrossRef]
- Skoog, D.A.; Holler, F.J.; Crouch, S.R. Principles of Instrumental Analysis, 6th ed.; Brooks/Cole, Thomson Learning: Melbourne, Australia, 2007; pp. 335–398. [Google Scholar]
- Stratmann, R.E.; Scuseria, G.E.; Frisch, M.J. An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules. J. Chem. Phys. 1998, 109, 8218–8224. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Coe, B.J.; Harries, J.L.; Helliwell, M.; Jones, M.A.; Asselberghs, I.; Clays, K.; Brunschwig, B.S.; Harris, J.A.; Garin, J.; Orduna, J. Pentacyanoiron(II) as an Electron Donor Group for Nonlinear Optics: Medium-Responsive Properties and Comparisons with Related Pentaammineruthenium(II) Complexes. J. Am. Chem. Soc. 2006, 128, 12192–12204. [Google Scholar] [CrossRef] [PubMed]
- Reis, H. Problems in the comparison of theoretical and experimental hyperpolarizabilities revisited. J. Chem. Phys. 2006, 125, 014506. [Google Scholar] [CrossRef] [PubMed]
- Sim, F.; Chin, S.; Dupuis, M.; Rice, J.E. Electron correlation effects in hyperpolarizabilities of p-nitroaniline. J. Phys. Chem. 1993, 97, 1158–1163. [Google Scholar] [CrossRef]
- Garza, A.J.; Osman, O.I.; Asiri, A.M.; Gustavo, G.E. Can Gap Tuning Schemes of Long-Range Corrected Hybrid Functionals Improve the Description of Hyperpolarizabilities? J. Phys. Chem. B 2015, 119, 1202–1212. [Google Scholar] [CrossRef] [PubMed]
- Thanthiriwatte, K.S.; Nalin de Silva, K.M. Non-linear optical properties of novel fluorenyl derivatives-ab initio quantum chemical calculations. J. Mol. Struct. THEOCHEM 2002, 617, 169–175. [Google Scholar] [CrossRef]
- Sriyanka Mendis, B.A.; Nalin de Silva, K.M. A comprehensive study of non-linear optical properties of novel charge transfer molecular systems. J. Mol. Struct. THEOCHEM 2004, 678, 31–38. [Google Scholar] [CrossRef]
- Asiri, A.M.; Khan, S.A.; Al-Amoudi, M.S.; Alamry, K.A. Synthesis, characterization, absorbance, fluorescence and non-linear optical properties of some donor acceptor chromophores. Bull. Korean Chem. Soc. 2012, 33, 1900–1906. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinhold, F. Natural localized molecular orbitals. J. Chem. Phys. 1985, 83, 1736–1740. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinhold, F. Natural Population Analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinhold, F. Natural bond orbital analysis of near-Hartree–Fock water dimer. J. Chem. Phys. 1983, 78, 4066–4073. [Google Scholar] [CrossRef]
- Foster, J.P.; Weinhold, F. Natural hybrid orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; et al. Gaussian 09, Revision A.02, Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Dennington, R.; Keith, T.; Millam, J. GaussView, Version 5, Semichem Inc.: Shawnee Mission, KS, USA, 2009.
- Geomodeling in GeoGraphix. Available online: http://www.chemcraftprog.com (accessed on 8 July 2016).
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis-sets for anion calculations. III. † The 3–21+G basis set for 1st-row elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-Consistent Molecular Orbital Methods. 25. Supplementary Functions for Gaussian Basis Sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C.A. New hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Glendenning, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO Version 3.1, Gaussian Inc.: Pittsburg, PA, USA, 2001.
- Gross, E.K.U.; Kohn, W. Time-Dependent Density-Functional Theory. Adv. Quantum Chem. 1990, 21, 255–291. [Google Scholar]
- Cances, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | N6–N7 | N5–C2 | N5–C3 | C1–C2 | C33–C50 | S4–C1 | C1–C33 | C33–C1–C2 | C15–N18–C26–C29 | C50–C33–C1–C2 | C41–C40–C2–N5 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| B3LYP | 6-311++G** | 1.269 | 1.357 | 1.314 | 1.412 | 1.366 | 1.760 | −82.8 | −36.3 | |||
| Aug-cc-pvdz | 1.274 | 1.360 | 1.318 | 1.416 | 1.372 | 1.766 | 1.423 | 124.3 | −83.0 | 176.5 | −35.9 | |
| CAM-B3LYP | 6-311++G** | 1.251 | 1.358 | 1.302 | 1.394 | 1.352 | 1.747 | 1.427 | 123.8 | −81.7 | 175.7 | −37.8 |
| Aug-cc-pvdz | 1.256 | 1.361 | 1.306 | 1.399 | 1.358 | 1.753 | 1.429 | 124.0 | −81.9 | 176.0 | −37.4 | |
| ωB97XD | 6-311++G** | 1.252 | 1.359 | 1.303 | 1.395 | 1.353 | 1.745 | 1.429 | 123.8 | −78.2 | 173.6 | −38.1 |
| Aug-cc-pvdz | 1.257 | 1.362 | 1.307 | 1.399 | 1.358 | 1.751 | 1.431 | 123.9 | −77.4 | 173.9 | −37.9 | |
| B3LYP | 6-311++G** | 1.272 | 1.358 | 1.311 | 1.408 | 1.374 | 1.759 | 1.419 | 125.0 | −83.2 | 176.0 | 37.0 |
| Aug-cc-pvdz | 1.277 | 1.362 | 1.316 | 1.412 | 1.378 | 1.764 | 1.422 | 125.1 | −83.1 | 176.4 | 36.5 | |
| CAM-B3LYP | 6-311++G** | 1.254 | 1.359 | 1.300 | 1.391 | 1.358 | 1.746 | 1.424 | 124.7 | −81.3 | 174.8 | 38.6 |
| Aug-cc-pvdz | 1.259 | 1.362 | 1.304 | 1.396 | 1.363 | 1.752 | 1.427 | 124.8 | −81.3 | 175.4 | 38.1 | |
| ωB97XD | 6-311++G** | 1.254 | 1.360 | 1.301 | 1.392 | 1.359 | 1.744 | 1.428 | 124.4 | −76.0 | 173.7 | 39.6 |
| Aug-cc-pvdz | 1.259 | 1.363 | 1.306 | 1.396 | 1.364 | 1.750 | 1.430 | 124.6 | −76.0 | 174.2 | 39.3 | |
| Expert. 1 | 1.297 | 1.368 | 1.318 | 1.382 | 1.361 | 1.744 | - | - | −88.8 | - | 34.6 | |
| B3LYP | 6-311++G** | 1.272 | 1.353 | 1.316 | 1.423 | 1.381 | 1.767 | 1.419 | 123.6 | −82.3 | 170.6 | −39.3 |
| Aug-cc-pvdz | 1.276 | 1.356 | 1.320 | 1.427 | 1.387 | 1.773 | 1.421 | 123.7 | −82.4 | 171.3 | −37.8 | |
| CAM-B3LYP | 6-311++G** | 1.253 | 1.354 | 1.303 | 1.403 | 1.365 | 1.753 | 1.426 | 123.1 | −81.2 | 170.9 | −41.6 |
| Aug-cc-pvdz | - | - | - | - | - | - | - | - | - | - | - | |
| ωB97XD | 6-311++G** | 1.253 | 1.356 | 1.305 | 1.402 | 1.364 | 1.750 | 1.428 | 122.7 | −77.0 | 171.2 | −44.0 |
| Aug-cc-pvdz | - | - | - | - | - | - | - | - | - | - | - | |
| Expert. 1 | 1.301 | 1.351 | 1.311 | 1.421 | 1.361 | 1.738 | - | - | 80 | - | −63 | |
| Parameter | N6–N7 | N5–C2 | N5–C3 | C1–C2 | C33–C50 | S4–C1 | S4–C3 | C15–N18–C26–C29 | C15–N18–C19–C22 | C41–C40–C2–N5 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| B3LYP | 6-311++G** | 0.005 | 0.010 | 0.007 | 0.026 | 0.013 | 0.015 | 0.033 | 5.6 | 8.4 | 2.4 |
| Aug-cc-pvdz | 0.020 | 0.006 | 0.002 | 0.030 | 0.017 | 0.020 | 0.037 | 5.7 | 8.4 | 1.9 | |
| CAM-B3LYP | 6-311++G** | 0.043 | 0.009 | 0.018 | 0.009 | 0.003 | 0.002 | 0.010 | 7.5 | 8.7 | 4.0 |
| Aug-cc-pvdz | 0.038 | 0.006 | 0.014 | 0.014 | 0.002 | 0.008 | 0.015 | 7.5 | 8.7 | 3.5 | |
| ωB97XD | 6-311++G** | 0.043 | 0.008 | 0.017 | 0.010 | 0.002 | 0.000 | 0.010 | 12.8 | 8.1 | 5.0 |
| Aug-cc-pvdz | 0.038 | 0.005 | 0.012 | 0.014 | 0.003 | 0.006 | 0.015 | 12.8 | 8.0 | 4.7 | |
| Average | 0.031 | 0.007 | 0.012 | 0.017 | 0.007 | 0.009 | 0.020 | 8.7 | 8.4 | 3.6 | |
| B3LYP | 6-311++G** | 0.029 | 0.002 | 0.005 | 0.002 | 0.020 | 0.029 | 0.025 | 2.3 | 0.6 | 23.7 |
| Aug-cc-pvdz | 0.025 | 0.005 | 0.009 | 0.006 | 0.026 | 0.035 | 0.030 | 2.4 | 0.5 | 25.2 | |
| CAM-B3LYP | 6-311++G** | 0.048 | 0.003 | 0.008 | 0.018 | 0.004 | 0.015 | 0.004 | 1.2 | 0.1 | 21.4 |
| Aug-cc-pvdz | - | - | - | - | - | - | - | - | - | - | |
| ωB97XD | 6-311++G** | 0.048 | 0.005 | 0.006 | 0.019 | 0.003 | 0.012 | 0.006 | 3.0 | 1.1 | 19 |
| Aug-cc-pvdz | - | - | - | - | - | - | - | - | - | - | |
| Average | 0.038 | 0.004 | 0.007 | 0.011 | 0.013 | 0.023 | 0.016 | 2.2 | 0.6 | 22.3 | |
| Dye | Parameter | B3LYP | CAM-B3LYP | ωB97XD | |||
|---|---|---|---|---|---|---|---|
| 6-311++G** | aug-cc-pvdz | 6-311++G** | aug-cc-pvdz | 6-311++G** | aug-cc-pvdz | ||
| A | HOMO | −5.541 | −5.503 | −6.755 | −6.708 | −7.261 | −7.220 |
| LUMO | −3.115 | −3.103 | −2.040 | −2.030 | −1.459 | −1.457 | |
| E.G. | 2.426 | 2.400 | 4.715 | 4.678 | 5.802 | 5.763 | |
| D.M. | 8.66 | 8.72 | 7.03 | 7.13 | 6.77 | 6.88 | |
| η | 1.213 | 1.200 | 2.358 | 2.339 | 2.901 | 2.882 | |
| μ | 4.328 | 4.303 | 4.398 | 4.369 | 4.360 | 4.339 | |
| ω | 7.721 | 7.715 | 4.101 | 4.080 | 3.276 | 3.266 | |
| βtot | 24,750 | 24,596 | 19,938 | 20,324 | 17,363 | 18,326 | |
| B | HOMO | −5.826 | −5.786 | −7.035 | −6.989 | −7.536 | −7.497 |
| LUMO | −3.386 | −3.367 | −2.320 | −2.301 | −1.742 | −1.731 | |
| E.G. | 2.440 | 2.419 | 4.715 | 4.688 | 5.794 | 5.766 | |
| D.M. | 14.26 | 14.27 | 12.55 | 12.65 | 12.30 | 12.43 | |
| η | 1.220 | 1.210 | 2.358 | 2.344 | 2.897 | 2.883 | |
| μ | 4.606 | 4.577 | 4.678 | 4.645 | 4.639 | 4.614 | |
| ω | 8.695 | 8.657 | 4.640 | 4.602 | 3.714 | 3.692 | |
| βtot | 17,714 | 17,500 | 18,258 | 18,158 | 16,904 | 16,927 | |
| C | HOMO | −5.677 | −5.638 | −6.865 | −6.824 | −7.375 | - |
| LUMO | −3.442 | −3.427 | −2.427 | −2.371 | −1.852 | - | |
| E.G. | 2.235 | 2.211 | 4.438 | 4.453 | 5.523 | - | |
| D.M. | 14.05 | 14.11 | 11.92 | 11.76 | 11.65 | - | |
| η | 1.118 | 1.106 | 2.219 | 2.227 | 2.762 | - | |
| μ | 4.560 | 4.533 | 4.646 | 4.598 | 4.614 | - | |
| ω | 9.299 | 9.289 | 4.864 | 4.747 | 3.854 | - | |
| βtot | 39,756 | 39,063 | 34,274 | 30,424 | 27,619 | - | |
| pNA | D.M. | 7.17 | - | 7.23 | - | 7.16 | - |
| βtot | 1327 | - | 1350 | - | 1350 | - | |
| Expert. a β | 1072 ± 44 | ||||||
| Level of Theory | Parameter | A | B | C |
|---|---|---|---|---|
| B3LYP/6-311++G** | D.M. | 12.78 | 19.38 | 19.41 |
| E.G. | 2.290 | 2.301 | 2.139 | |
| βtot | 5.9890 | 4.4821 | 9.7969 | |
| λmax | 605 | 587 | 659 | |
| B3LYP/aug-cc-pvdz | D.M. | 12.91 | 19.52 | - |
| E.G. | 2.269 | 2.286 | - | |
| βtot | 6.0588 | 4.4632 | - | |
| λmax | 612 | 593 | - | |
| CAM-B3LYP/6-311++G** | D.M. | 9.76 | 16.19 | 15.53 |
| E.G. | 4.515 | 4.496 | 4.298 | |
| βtot | 4.5591 | 4.4724 | 6.8587 | |
| λmax | 497 | 498 | 528 | |
| CAM-B3LYP/aug-cc-pvdz | D.M. | 9.97 | 16.46 | 15.85 |
| E.G. | 4.481 | 4.470 | 4.262 | |
| βtot | 4.7470 | 4.5521 | 7.1861 | |
| λmax | 505 | 504 | 539 | |
| ωB97XD/6-311++G** | D.M. | 9.29 | 15.72 | 15.08 |
| E.G. | 5.598 | 5.569 | 5.373 | |
| βtot | 4.0510 | 4.1197 | 5.9315 | |
| λmax | 484 | 487 | 511 | |
| ωB97XD/aug-cc-pvdz | D.M. | 9.54 | 16.08 | - |
| E.G. | 5.559 | 5.539 | - | |
| βtot | 4.2482 | 4.2476 | - | |
| λmax | 492 | 494 | - | |
| Expert. a | λmax (Expert) | 623 | 619 | 686 |
| μβO × 10−48/esu) | 740 | 800 | 1970 |
| Solvent | Parameter | A | B | C |
|---|---|---|---|---|
| CCl4 | D.M. | 8.971 | 15.120 | 14.469 |
| E.G. | 4.577 | 4.557 | 4.345 | |
| λmax | 490.50 | 491.12 | 523.50 | |
| Emax | 2.528 | 2.525 | 2.369 | |
| βtot | 3.2178 | 3.0893 | 4.9493 | |
| CHCl3 | D.M. | 9.761 | 16.186 | 15.530 |
| E.G. | 4.515 | 4.496 | 4.298 | |
| λmax | 496.90 | 497.92 | 528.02 | |
| Emax | 2.495 | 2.490 | 2.349 | |
| βtot | 4.5591 | 4.4724 | 6.8587 | |
| CH2Cl2 | D.M. | 10.160 | 16.706 | 16.052 |
| E.G. | 4.483 | 4.465 | 4.274 | |
| λmax | 499.60 | 500.70 | 529.80 | |
| Emax | 2.482 | 2.476 | 2.340 | |
| βtot | 5.4167 | 5.3578 | 8.0403 | |
| DMSO | D.M. | 10.564 | 17.222 | 16.570 |
| E.G. | 4.451 | 4.434 | 4.250 | |
| λmax | 503.10 | 504.10 | 532.20 | |
| Emax | 2.465 | 2.460 | 2.330 | |
| βtot | 6.4435 | 6.4101 | 9.4206 |
| Interaction | A | B | C |
|---|---|---|---|
| n2O45→π*C38–C42 | 44.70 | 45.65 | 44.95 |
| πC38–C42→π*C35–C36 | 58.80 | 54.83 | 55.10 |
| πC38–C42→π*C37–C40 | 30.69 | 30.83 | 30.08 |
| πC35–C36→π*C1–C2 | 23.31 | 23.69 | 22.83 |
| πC1–C2→π*C35–C50 | 40.80 | 43.29 | 49.30 |
| πC33–C50→π*C51–C63 | - | - | 37.99 |
| πC33–C50→π*C51–O64 | 34.05 | - | - |
| πC33–C50→π*C52–O63 | 33.30 | - | 31.75 |
| πC33–C50→π*C51–N53 | - | 30.50 | 30.79 |
| πC33–C50→π*C52–N54 | - | 28.67 | 29.99 |
| Total | 265.65 | 257.46 | 332.78 |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osman, O.I. DFT Study of the Structure, Reactivity, Natural Bond Orbital and Hyperpolarizability of Thiazole Azo Dyes. Int. J. Mol. Sci. 2017, 18, 239. https://doi.org/10.3390/ijms18020239
Osman OI. DFT Study of the Structure, Reactivity, Natural Bond Orbital and Hyperpolarizability of Thiazole Azo Dyes. International Journal of Molecular Sciences. 2017; 18(2):239. https://doi.org/10.3390/ijms18020239
Chicago/Turabian StyleOsman, Osman I. 2017. "DFT Study of the Structure, Reactivity, Natural Bond Orbital and Hyperpolarizability of Thiazole Azo Dyes" International Journal of Molecular Sciences 18, no. 2: 239. https://doi.org/10.3390/ijms18020239