A Split-Luciferase Reporter Recognizing GFP and mCherry Tags to Facilitate Studies of Protein–Protein Interactions

 ,
,
Abstract
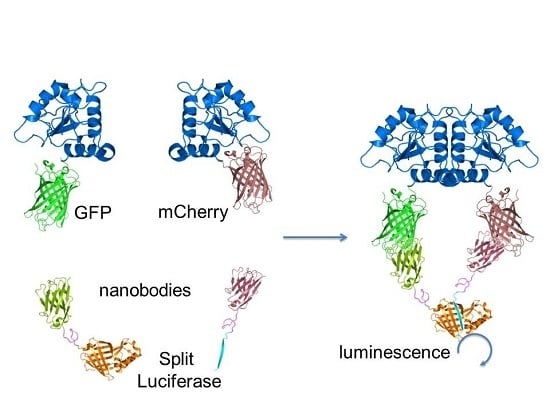
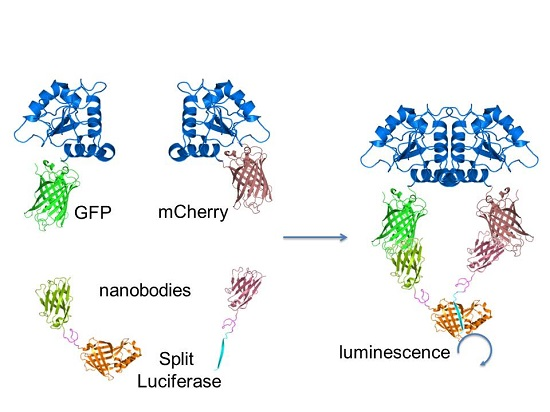
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
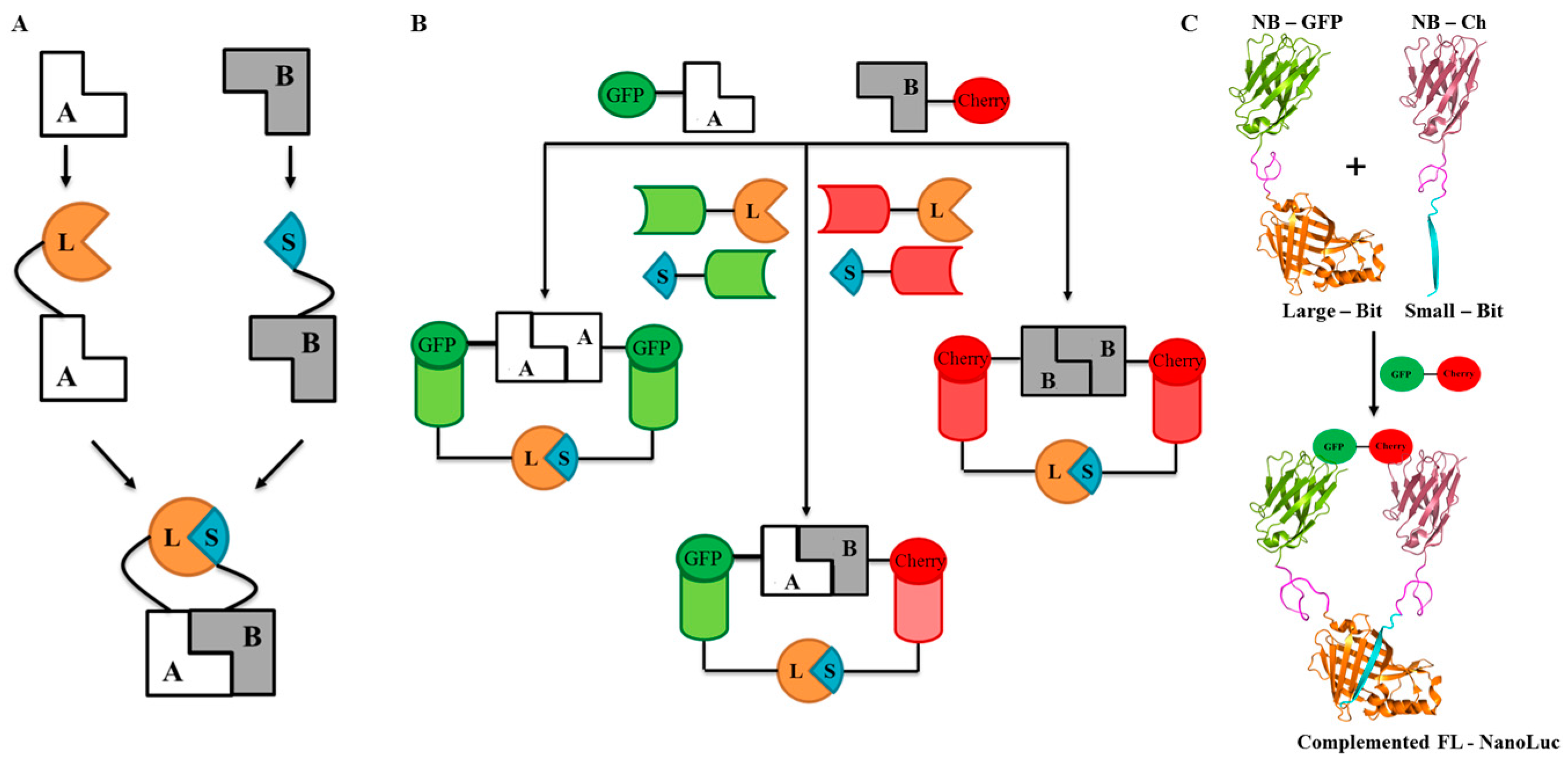
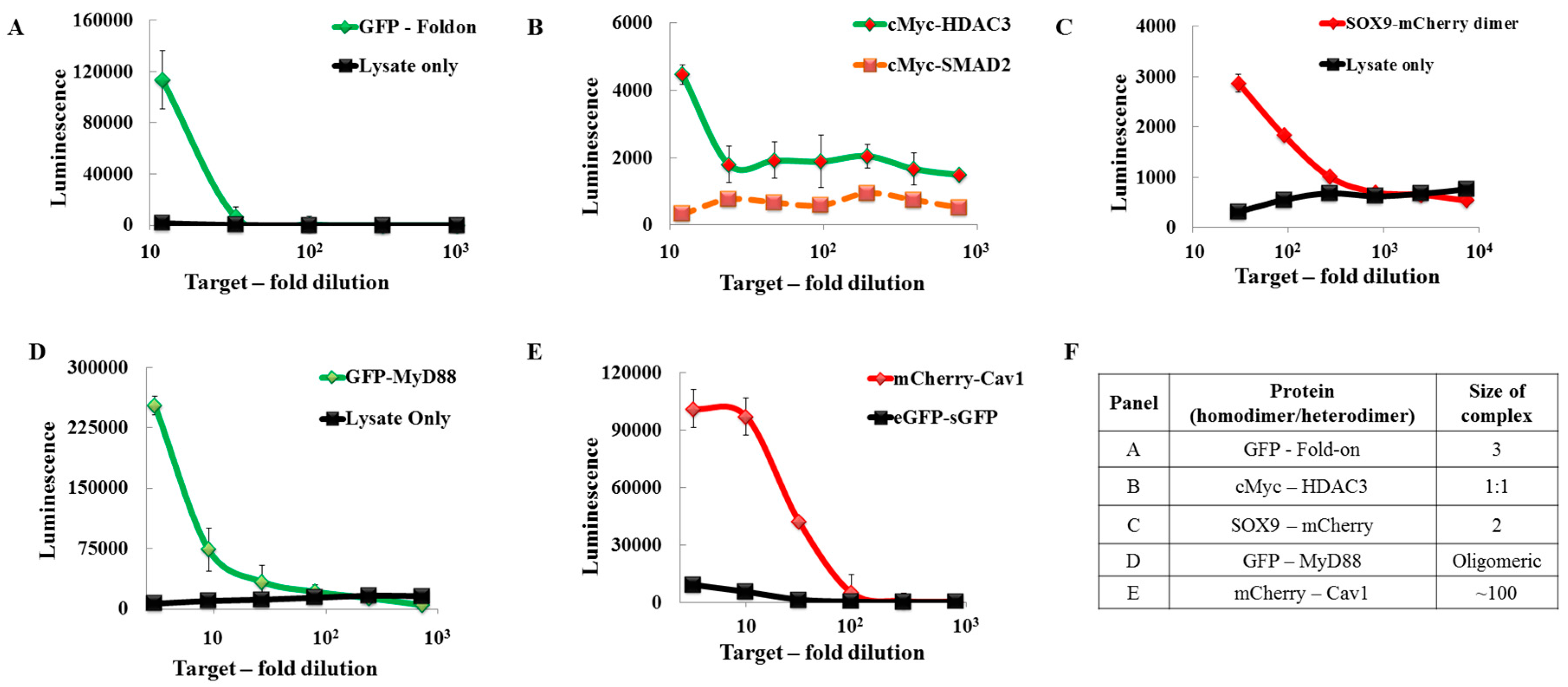
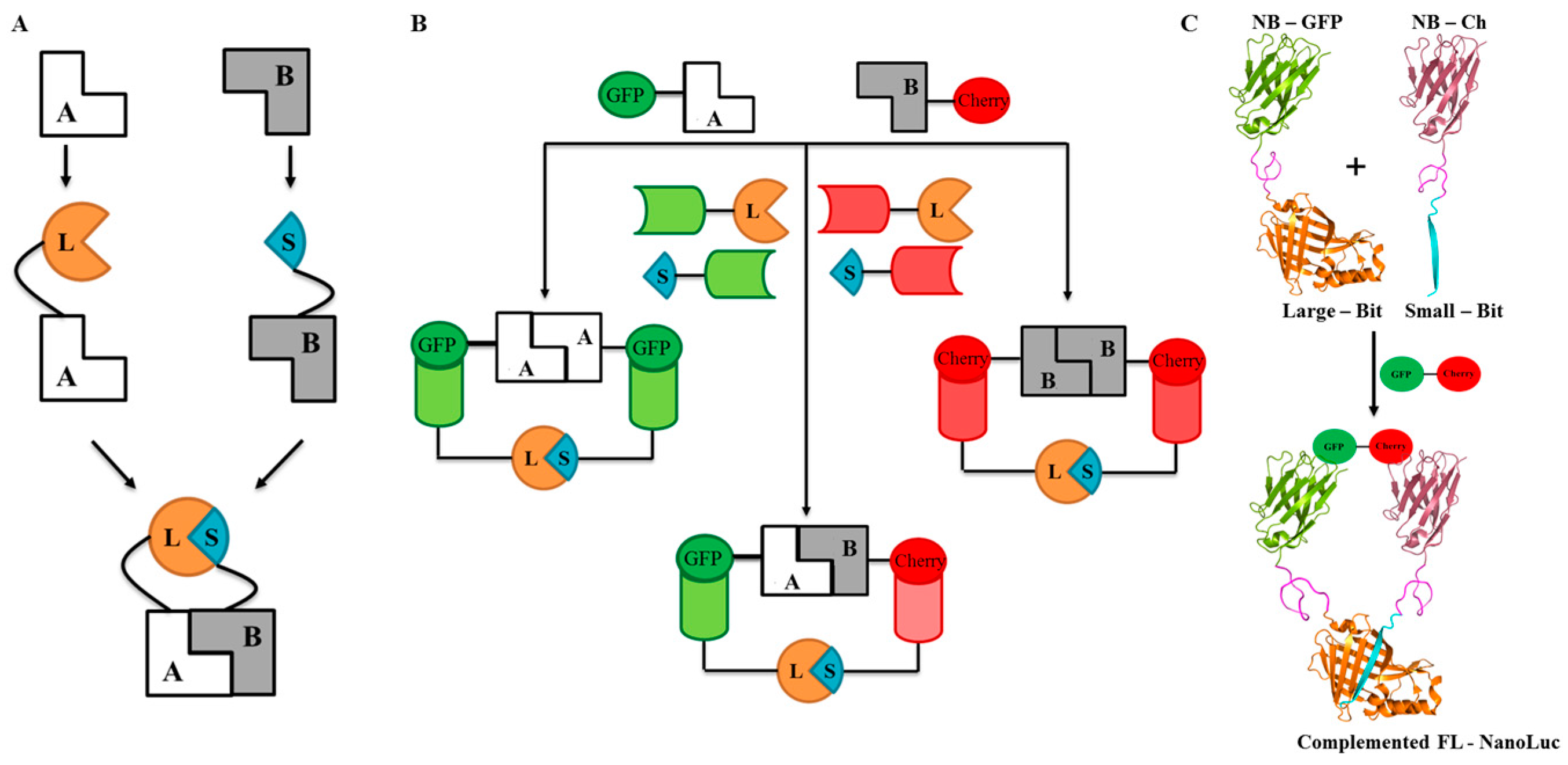
2.1. Design and Universal Application
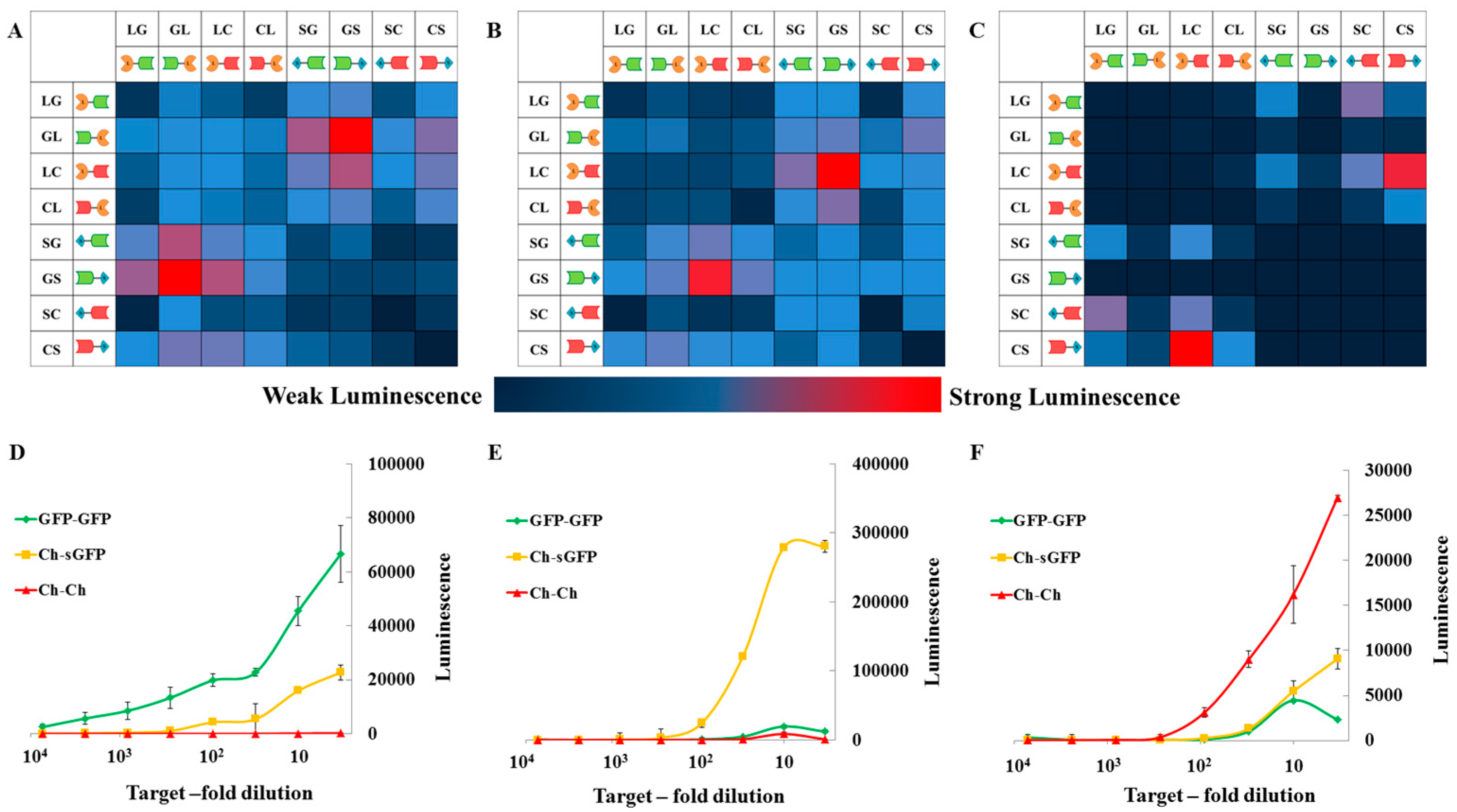
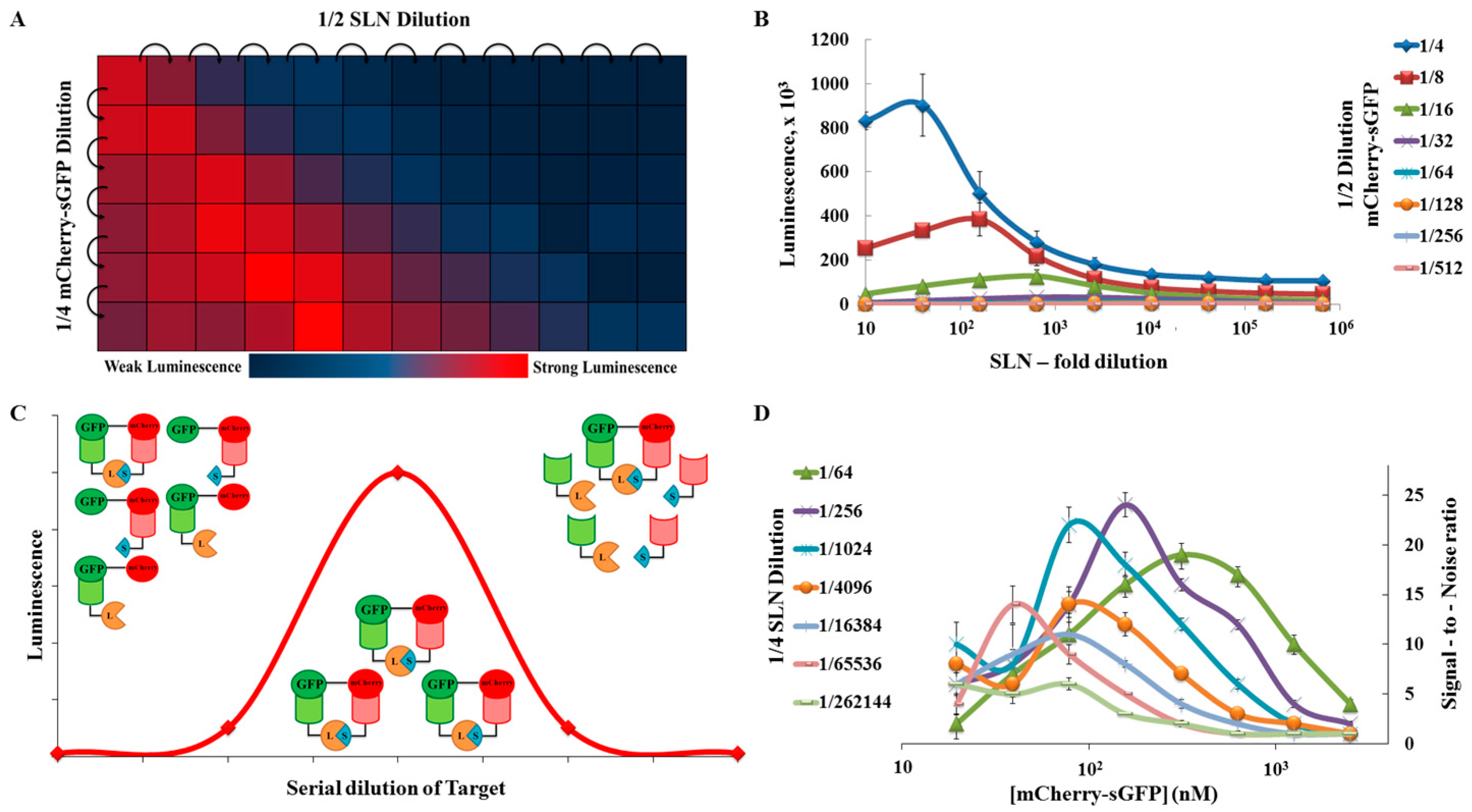
2.2. Proof of Principle and Calibration of the PPI Assay
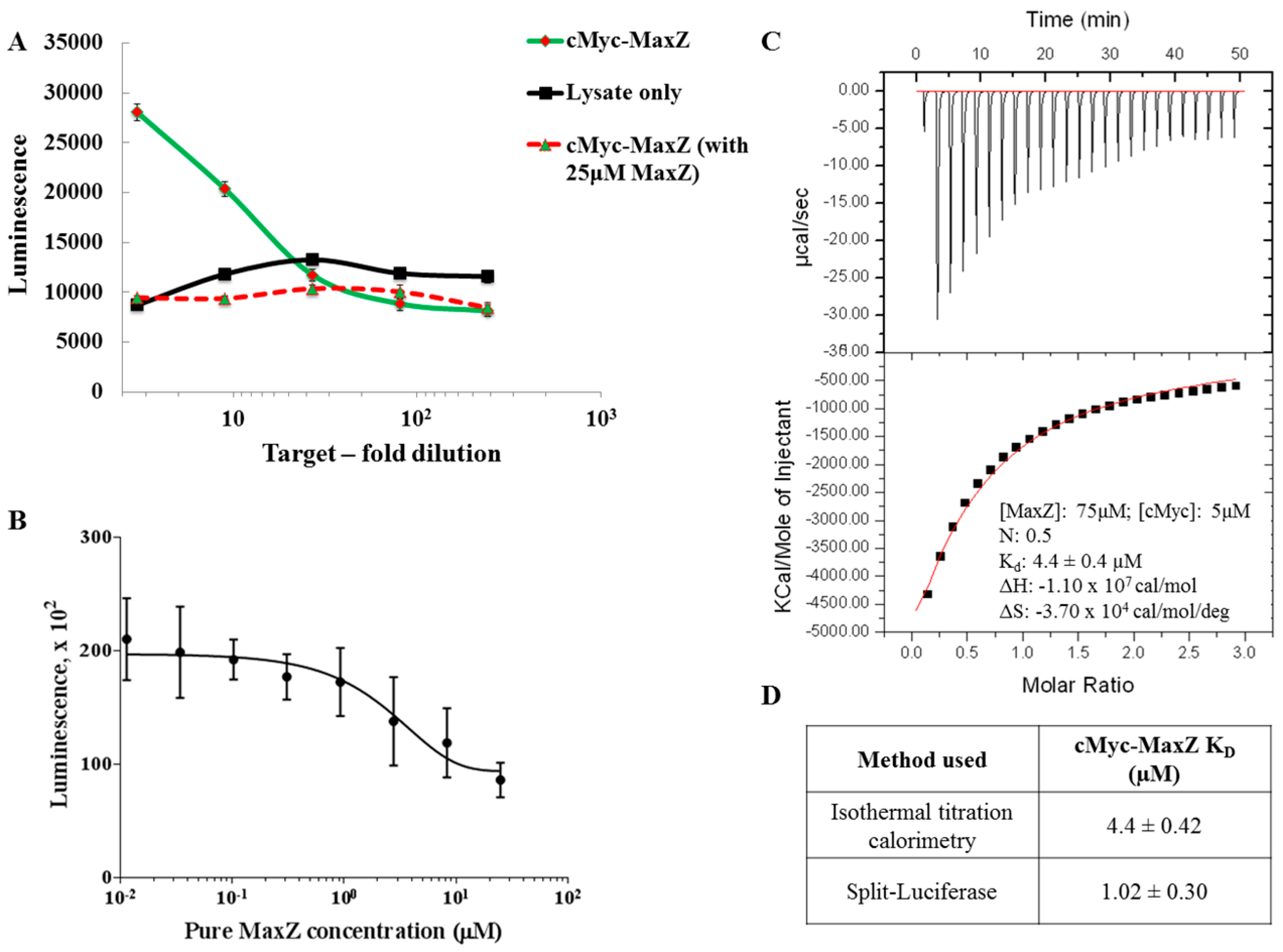
2.3. Application to Measure PPI Disruption
3. Discussion
4. Materials and Methods
4.1. Gateway Cloning and Protein Expression
4.2. Split-Luciferase Assay for Direct Detection Studies
4.3. Split-Luciferase Assay for Indirect Detection Studies
4.4. Split-Luciferase Assay for Apparent KD Calculation
4.5. Split Luciferase Assay for PPI, Homodimerisation, and Oligomerisation Detection
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| PPI | Protein–Protein Interaction |
| sGFP | superfolder GFP |
| eGFP | enhanced GFP |
| mCherry | monomeric Cherry |
| PCA | Protein Complementation Assays |
| DHFR | Dihydrofolate Reductase |
| nLuc | nanoLuc luciferase |
| L | large bit of the nanoLuc 18kDa |
| S | small bit of the nanoLuc 1.3kDa |
| G | anti-GFP nanobody |
| C | anti-Cherry nanobody |
| SLN | Split Luc Nanobody fusion peptide |
| GL | anti-GFP–Large bit |
| LG | Large bit–anti-GFP |
| CL | anti-Cherry–Large bit |
| LC | Large bit–anti-Cherry |
| GS | anti-GFP–Small bit |
| SG | small bit–anti-GFP |
| CS | anti-Cherry–Small bit |
| SC | Small bit–anti-Cherry |
| CFRS | anti-Cherry flexible-rigid linker small bit |
| LTE | Leishmania tarentolae Extract |
References
- Stumpf, M.P.; Thorne, T.; de Silva, E.; Stewart, R.; An, H.J.; Lappe, M.; Wiuf, C. Estimating the size of the human interactome. Proc. Natl. Acad. Sci. USA 2008, 105, 6959–6964. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.A.; McClendon, C.L. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 2007, 450, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Braisted, A.C.; Oslob, J.D.; Delano, W.L.; Hyde, J.; McDowell, R.S.; Waal, N.; Yu, C.; Arkin, M.R.; Raimundo, B.C. Discovery of a potent small molecule IL-2 inhibitor through fragment assembly. J. Am. Chem. Soc. 2003, 125, 3714–3715. [Google Scholar] [CrossRef] [PubMed]
- Chin, J.W.; Schepartz, A. Design and evolution of a miniature Bcl-2 binding protein. Angew. Chem. Int. Ed. 2001, 40, 3806–3809. [Google Scholar] [CrossRef]
- Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Qiu, S.; Ding, Y.; Gao, W.; Stuckey, J.; Krajewski, K.; Roller, P.P.; Tomita, Y. Structure-based design of potent non-peptide MDM2 inhibitors. J. Am. Chem. Soc. 2005, 127, 10130–10131. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, S.; Meyer, S.C.; Goldman, A.; Zhou, M.; Ghosh, I. A minimalist approach toward protein recognition by epitope transfer from functionally evolved β-sheet surfaces. J. Am. Chem. Soc. 2006, 128, 14356–14363. [Google Scholar] [CrossRef] [PubMed]
- Engvall, E.; Perlmann, P. Enzyme-linked immunosorbent assay, ELISA. J. Immunol. 1972, 109, 129–135. [Google Scholar] [PubMed]
- Homola, J.; Yee, S.S.; Gauglitz, G. Surface plasmon resonance sensors. Sens. Actuators B Chem. 1999, 54, 3–15. [Google Scholar] [CrossRef]
- Lea, W.A.; Simeonov, A. Fluorescence Polarization Assays in Small Molecule Screening. Expert Opin. Drug Discov. 2011, 6, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Fields, S.; Song, O.-K. A novel genetic system to detect protein-protein interactions. Nature 1989, 340, 245–246. [Google Scholar] [CrossRef] [PubMed]
- Fields, S. High-throughput two-hybrid analysis. FEBS J. 2005, 272, 5391–5399. [Google Scholar] [CrossRef] [PubMed]
- Michnick, S.W.; Ear, P.H.; Manderson, E.N.; Remy, I.; Stefan, E. Universal strategies in research and drug discovery based on protein-fragment complementation assays. Nat. Rev. Drug Discov. 2007, 6, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Richards, F.M.; Vithayathil, P.J. The preparation of subtilisin-modified ribonuclease and the separation of the peptide and protein components. J. Biol. Chem. 1959, 234, 1459–1465. [Google Scholar] [PubMed]
- Johnsson, N.; Varshavsky, A. Split ubiquitin as a sensor of protein interactions in vivo. Proc. Natl. Acad. Sci. USA 1994, 91, 10340–10344. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, I.; Hamilton, A.D.; Regan, L. Antiparallel leucine zipper-directed protein reassembly: Application to the green fluorescent protein. J. Am. Chem. Soc. 2000, 122, 5658–5659. [Google Scholar] [CrossRef]
- MacDonald, M.L.; Lamerdin, J.; Owens, S.; Keon, B.H.; Bilter, G.K.; Shang, Z.; Huang, Z.; Yu, H.; Dias, J.; Minami, T. Identifying off-target effects and hidden phenotypes of drugs in human cells. Nat. Chem. Biol. 2006, 2, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Jach, G.; Pesch, M.; Richter, K.; Frings, S.; Uhrig, J.F. An improved mRFP1 adds red to bimolecular fluorescence complementation. Nat. Methods 2006, 3, 597–600. [Google Scholar] [CrossRef] [PubMed]
- Rossi, F.; Charlton, C.A.; Blau, H.M. Monitoring protein-protein interactions in intact eukaryotic cells by β-galactosidase complementation. Proc. Natl. Acad. Sci. USA 1997, 94, 8405–8410. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.N.; Campbell-Valois, F.-X.; Michnick, S.W. Oligomerization domain-directed reassembly of active dihydrofolate reductase from rationally designed fragments. Proc. Natl. Acad. Sci. USA 1998, 95, 12141–12146. [Google Scholar] [CrossRef] [PubMed]
- Galarneau, A.; Primeau, M.; Trudeau, L.-E.; Michnick, S.W. β-Lactamase protein fragment complementation assays as in vivo and in vitro sensors of protein-protein interactions. Nat. Biotechnol. 2002, 20, 619–622. [Google Scholar] [CrossRef] [PubMed]
- Luker, K.E.; Smith, M.C.; Luker, G.D.; Gammon, S.T.; Piwnica-Worms, H.; Piwnica-Worms, D. Kinetics of regulated protein-protein interactions revealed with firefly luciferase complementation imaging in cells and living animals. Proc. Natl. Acad. Sci. USA 2004, 101, 12288–12293. [Google Scholar] [CrossRef] [PubMed]
- Paulmurugan, R.; Gambhir, S. Monitoring protein-protein interactions using split synthetic renilla luciferase protein-fragment-assisted complementation. Anal. Chem. 2003, 75, 1584–1589. [Google Scholar] [CrossRef] [PubMed]
- Massoud, T.F.; Paulmurugan, R.; Gambhir, S.S. A molecularly engineered split reporter for imaging protein-protein interactions with positron emission tomography. Nat. Med. 2010, 16, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.M.; Kries, H.; Csuhai, E.; Kast, P.; Hilvert, D. Design, selection, and characterization of a split chorismate mutase. Protein Sci. 2010, 19, 1000–1010. [Google Scholar] [CrossRef] [PubMed]
- Remy, I.; Michnick, S.W. Application of protein-fragment complementation assays in cell biology. Biotechniques 2007, 42, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Shyu, Y.J.; Liu, H.; Deng, X.; Hu, C. Identification of new fluorescent protein fragments for biomolecular fluorescence complementation analysis under physiological conditions. Biotechniques 2006, 40, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Ooi, A.T.; Stains, C.I.; Ghosh, I.; Segal, D.J. Sequence-enabled reassembly of β-lactamase (SEER-LAC): A sensitive method for the detection of double-stranded DNA. Biochemistry 2006, 45, 3620–3625. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.L. Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Remy, I.; Campbell-Valois, F.; Michnick, S.W. Detection of protein-protein interactions using a simple survival protein-fragment complementation assay based on the enzyme dihydrofolate reductase. Nat. Protoc. 2007, 2, 2120–2125. [Google Scholar] [CrossRef] [PubMed]
- Rackham, O.; Brown, C.M. Visualization of RNA-protein interactions in living cells: FMRP and IMP1 interact on mRNAs. EMBO J. 2004, 23, 3346–3355. [Google Scholar] [CrossRef] [PubMed]
- Mureev, S.; Kovtun, O.; Nguyen, U.T.; Alexandrov, K. Species-independent translational leaders facilitate cell-free expression. Nat. Biotechnol. 2009, 27, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Gambin, Y.; Ariotti, N.; McMahon, K.A.; Bastiani, M.; Sierecki, E.; Kovtun, O.; Polinkovsky, M.E.; Magenau, A.; Jung, W.; Okano, S.; et al. Single-molecule analysis reveals self assembly and nanoscale segregation of two distinct cavin subcomplexes on caveolae. eLife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Dixon, A.S.; Schwinn, M.K.; Hall, M.P.; Zimmerman, K.; Otto, P.; Lubben, T.H.; Butler, B.L.; Binkowski, B.F.; Machleidt, T.; Kirkland, T.A.; et al. NanoLuc Complementation Reporter Optimized for Accurate Measurement of Protein Interactions in Cells. ACS Chem. Biol. 2016, 11, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Kubala, M.H.; Kovtun, O.; Alexandrov, K.; Collins, B.M. Structural and thermodynamic analysis of the GFP:GFP-nanobody complex. Protein Sci. 2010, 19, 2389–2401. [Google Scholar] [CrossRef] [PubMed]
- Fridy, P.C.; Li, Y.; Keegan, S.; Thompson, M.K.; Nudelman, I.; Scheid, J.F.; Oeffinger, M.; Nussenzweig, M.C.; Fenyo, D.; Chait, B.T.; et al. A robust pipeline for rapid production of versatile nanobody repertoires. Nat. Methods 2014, 11, 1253–1260. [Google Scholar] [CrossRef] [PubMed]
- Sierecki, E.; Stevers, L.M.; Giles, N.; Polinkovsky, M.E.; Moustaqil, M.; Mureev, S.; Johnston, W.A.; Dahmer-Heath, M.; Skalamera, D.; Gonda, T.J.; et al. Rapid mapping of interactions between Human SNX-BAR proteins measured in vitro by AlphaScreen and single-molecule spectroscopy. Mol. Cell. Proteom. 2014, 13, 2233–2245. [Google Scholar] [CrossRef] [PubMed]
- Robin, R.; Kainz, B.; Stadlmayr, G.; Schuster, H.; Pum, D.; Messner, P.; Obinger, C.; Schäffer, C. A Fusion Tag to Fold on: The S-Layer Protein SgsE Confers Improved Folding Kinetics to Translationally Fused Enhanced Green Fluorescent Protein. J. Microbiol. Biotechnol. 2012, 22, 1271–1278. [Google Scholar]
- Ve, T.; Vajjhala, P.R.; Hedger, A.; Croll, T.; DiMaio, F.; Horsefield, S.; Yu, X.; Lavrencic, P.; Hassan, Z.; Morgan, G.P.; et al. Structural basis of TIR-domain-assembly formation in MAL- and MyD88-dependent TLR4 signaling. Nat. Struct. Mol. Biol. 2017, 24, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Elahi, A.; Ajidahun, A.; Clark, W.; Hernandez, J.; Achille, A.; Hao, J.-H.; Seto, E.; Shibata, D. The interplay between histone deacetylases and c-Myc in the transcriptional suppression of HPP1 in colon cancer. Cancer Biol. Ther. 2014, 15, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Coustry, F.; Oh, C.-D.; Hattori, T.; Maity, S.N.; de Crombrugghe, B.; Yasuda, H. The dimerization domain of SOX9 is required for transcription activation of a chondrocyte-specific chromatin DNA template. Nucleic Acids Res. 2010, 38, 6018–6028. [Google Scholar] [CrossRef] [PubMed]
- Walhout, A.J.; Gubbels, J.M.; Bernards, R.; van der Vliet, P.C.; Timmers, H.T. c-Myc/Max heterodimers bind cooperatively to the E-box sequences located in the first intron of the rat ornithine decarboxylase (ODC) gene. Nucleic Acids Res. 1997, 25, 1493–1501. [Google Scholar] [CrossRef] [PubMed]
- Overman, J.; Fontaine, F.; Moustaqil, M.; Mittal, D.; Sierecki, E.; Sacilotto, N.; Zuegg, J.; Robertson, A.A.B.; Holmes, K.; Salim, A.A.; et al. Pharmacological targeting of the transcription factor SOX18 delays breast cancer in mice. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Cummins, T.D.; Sapkota, G.P. Characterization of Protein Complexes Using Chemical Cross-Linking Coupled Electrospray Mass Spectrometry. Methods Mol. Biol. 2017. [Google Scholar] [CrossRef]
- Grinkevich, P.; Iermak, I.; Luedtke, N.A.; Mesters, J.R.; Ettrich, R.; Ludwig, J. pHluorin-assisted expression, purification, crystallization and X-ray diffraction data analysis of the C-terminal domain of the HsdR subunit of the Escherichia coli type I restriction-modification system EcoR124I. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2016, 72, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Weise, C.; Bottcher, C.; Fan, H.; Yin, J. Expression, purification and structural analysis of functional GABA transporter 1 using the baculovirus expression system. Beilstein J. Org. Chem. 2017, 13, 874–882. [Google Scholar] [CrossRef] [PubMed]
- Sjostrand, D.; Diamanti, R.; Lundgren, C.A.K.; Wiseman, B.; Hogbom, M. A rapid expression and purification condition screening protocol for membrane protein structural biology. Protein Sci. Publ. Protein Soc. 2017, 26, 1653–1666. [Google Scholar] [CrossRef] [PubMed]
- Unzueta, U.; Seras-Franzoso, J.; Cespedes, M.V.; Saccardo, P.; Cortes, F.; Rueda, F.; Garcia-Fruitos, E.; Ferrer-Miralles, N.; Mangues, R.; Vazquez, E.; et al. Engineering tumor cell targeting in nanoscale amyloidal materials. Nanotechnology 2017, 28. [Google Scholar] [CrossRef] [PubMed]
- Bond, T.E.H.; Sorenson, A.E.; Schaeffer, P.M. A green fluorescent protein-based assay for high-throughput ligand-binding studies of a mycobacterial biotin protein ligase. Microbiol. Res. 2017, 205, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Garg, H.; Sedano, M.; Plata, G.; Punke, E.B.; Joshi, A. Development of Virus-Like-Particle Vaccine and Reporter Assay for Zika Virus. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Hwang, G.H.; Park, S.M.; Han, H.J.; Kim, J.S.; Yun, S.P.; Ryu, J.M.; Lee, H.J.; Chang, W.; Lee, S.J.; Choi, J.H.; et al. Purification of small molecule-induced cardiomyocytes from human induced pluripotent stem cells using a reporter system. J. Cell. Physiol. 2017, 232, 3384–3395. [Google Scholar] [CrossRef] [PubMed]
- Gambin, Y.; Polinkovsky, M.; Francois, B.; Giles, N.; Bhumkar, A.; Sierecki, E. Confocal Spectroscopy to Study Dimerization, Oligomerization and Aggregation of Proteins: A Practical Guide. Int. J. Mol. Sci. 2016, 17, 655. [Google Scholar] [CrossRef] [PubMed]
- Bowes, J.; Brown, A.J.; Hamon, J.; Jarolimek, W.; Sridhar, A.; Waldron, G.; Whitebread, S. Reducing safety-related drug attrition: The use of in vitro pharmacological profiling. Nat. Rev. Drug Discov. 2012, 11, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Gagoski, D.; Polinkovsky, M.E.; Mureev, S.; Kunert, A.; Johnston, W.; Gambin, Y.; Alexandrov, K. Performance benchmarking of four cell-free protein expression systems. Biotechnol. Bioeng. 2016, 113, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Sierecki, E.; Giles, N.; Bowden, Q.; Polinkovsky, M.E.; Steinbeck, J.; Arrioti, N.; Rahman, D.; Bhumkar, A.; Nicovich, P.R.; Ross, I.; et al. Nanomolar oligomerization and selective co-aggregation of α-synuclein pathogenic mutants revealed by single-molecule fluorescence. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Chung, S.; Kim, K.-M.; Jung, K.-C.; Park, C.; Hahm, E.-R.; Yang, C.-H. Determination of binding constant of transcription factor myc-max/max-max and E-box DNA: The effect of inhibitors on the binding. Biochim. Biophys. Acta Gen. Subj. 2004, 1670, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Duff, M.R.; Grubbs, J.; Howell, E.E. Isothermal Titration Calorimetry for Measuring Macromolecule-Ligand Affinity. J. Vis. Exp. 2011. [Google Scholar] [CrossRef]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moustaqil, M.; Bhumkar, A.; Gonzalez, L.; Raoul, L.; Hunter, D.J.B.; Carrive, P.; Sierecki, E.; Gambin, Y. A Split-Luciferase Reporter Recognizing GFP and mCherry Tags to Facilitate Studies of Protein–Protein Interactions. Int. J. Mol. Sci. 2017, 18, 2681. https://doi.org/10.3390/ijms18122681
Moustaqil M, Bhumkar A, Gonzalez L, Raoul L, Hunter DJB, Carrive P, Sierecki E, Gambin Y. A Split-Luciferase Reporter Recognizing GFP and mCherry Tags to Facilitate Studies of Protein–Protein Interactions. International Journal of Molecular Sciences. 2017; 18(12):2681. https://doi.org/10.3390/ijms18122681
Chicago/Turabian StyleMoustaqil, Mehdi, Akshay Bhumkar, Laura Gonzalez, Lisa Raoul, Dominic J. B. Hunter, Pascal Carrive, Emma Sierecki, and Yann Gambin. 2017. "A Split-Luciferase Reporter Recognizing GFP and mCherry Tags to Facilitate Studies of Protein–Protein Interactions" International Journal of Molecular Sciences 18, no. 12: 2681. https://doi.org/10.3390/ijms18122681