Cellular and Oxidative Mechanisms Associated with Interleukin-6 Signaling in the Vasculature

Departments of Pharmacology and Neurology, The University of Mississippi Medical Center, Arthur C. Guyton Laboratory Research Building, Jackson, MS 39216, USA
Int. J. Mol. Sci. 2017, 18(12), 2563; https://doi.org/10.3390/ijms18122563
Submission received: 20 October 2017
/
Revised: 17 November 2017
/
Accepted: 19 November 2017
/
Published: 29 November 2017
(This article belongs to the Special Issue Oxidative Stress in Vascular Diseases)
Abstract
:Reactive oxygen species, particularly superoxide, promote endothelial dysfunction and alterations in vascular structure. It is increasingly recognized that inflammatory cytokines, such as interleukin-6 (IL-6), contribute to endothelial dysfunction and vascular hypertrophy and fibrosis. IL-6 is increased in a number of cardiovascular diseases, including hypertension. IL-6 is also associated with a higher incidence of future cardiovascular events and all-cause mortality. Both immune and vascular cells produce IL-6 in response to a number of stimuli, such as angiotensin II. The vasculature is responsive to IL-6 produced from vascular and non-vascular sources via classical IL-6 signaling involving a membrane-bound IL-6 receptor (IL-6R) and membrane-bound gp130 via Jak/STAT as well as SHP2-dependent signaling pathways. IL-6 signaling is unique because it can also occur via a soluble IL-6 receptor (sIL-6R) which allows for IL-6 signaling in tissues that do not normally express IL-6R through a process referred to as IL-6 trans-signaling. IL-6 signaling mediates a vast array of effects in the vascular wall, including endothelial activation, vascular permeability, immune cell recruitment, endothelial dysfunction, as well as vascular hypertrophy and fibrosis. Many of the effects of IL-6 on vascular function and structure are representative of loss or reductions in nitric oxide (NO) bioavailability. IL-6 has direct effects on endothelial nitric oxide synthase activity and expression as well as increasing vascular superoxide, which rapidly inactivates NO thereby limiting NO bioavailability. The goal of this review is to highlight both the cellular and oxidative mechanisms associated with IL-6-signaling in the vascular wall in general, in hypertension, and in response to angiotensin II.
1. Linking Oxidative Stress with Hypertension and Vascular Dysfunction
It is generally well accepted that oxidative stress plays an important role in hypertension and related vascular sequelae, including endothelial dysfunction and vascular hypertrophy and fibrosis [1,2,3,4]. Oxidative stress is defined as an increase in reactive oxygen species, such as superoxide, due to increased production or reduced metabolism. Increases in vascular superoxide have several negative consequences on vascular biology, including reductions in nitric oxide (NO) bioavailability. NO, especially that derived from endothelial nitric oxide synthase (eNOS), normally exerts many protective effects on the vessel wall such as limiting endothelial activation, inflammation, vascular growth, and fibrosis [5,6,7,8,9]. Nitric oxide derived from the enzymatic activity of eNOS also plays an important role in endothelial responses in numerous blood vessels [10,11,12,13]. eNOS expression and activity is regulated at the transcriptional and translation level as well as by multiple post-translational mechanisms, including acetylation, phosphorylation, and sumolyation [14,15,16]. In addition, increases in vascular superoxide can greatly limit NO bioavailability thus negatively impacting endothelial function.
Some of the first evidence to implicate superoxide in the impairment of NO-mediated endothelial function came from studies in which exogenous superoxide dismutase, a scavenger of superoxide, was demonstrated to improve endothelial function in hypertensive blood vessels [17]. Infusion of exogenous SOD also was found to lower blood pressure in spontaneously hypertensive rats implicating a role for superoxide in hypertension [18,19]. Thus, pharmacological interventions aimed at reducing superoxide result in increased NO bioavailability, blood pressure lowering, and improvement of endothelial function.
Since these initial findings, a number of pharmacological and genetic approaches have provided additional evidence implicating an important role for oxidative stress, particularly NADPH oxidase-derived superoxide, in the development of hypertension and vascular dysfunction [20,21,22,23,24,25,26,27,28,29,30,31]. Increases in vascular superoxide may be an important link between activation of inflammatory molecules and pathways and endothelial dysfunction in cardiovascular disease. Emerging evidence suggests inflammatory cytokines, such as interleukin-6 (IL-6), affect expression and activity of both eNOS and NADPH oxidase thereby influencing NO and superoxide levels and contributing to oxidative stress [32,33,34,35,36,37]. The identification of vascular oxidases distinct from that expressed in neutrophils and macrophages was a major advance in the field of vascular biology [38,39]. A seminal discovery in this regard was the finding that angiotensin II stimulated NADH and NAPDH oxidase activity in cultured vascular smooth muscle [38]. Subsequent studies identified several isoforms of Nox, the main catalytic NADPH oxidase, and accessory proteins, such as p22phox, p47phox, p67phox and Rac1, which enhance oxidase activity and superoxide generation. There are several excellent reviews on the role of NADPH oxidase in the vasculature to which the author would refer the reader [40,41,42,43]. In addition to NADPH oxidase, other important enzymatic sources of superoxide in the vasculature include cyclooxygenase, cytochrome P450, uncoupled eNOS, and xanthine oxidase [44].
Hypertension or high blood pressure is the product of central, renal, and vascular mechanisms. Oxidative stress in the brain, kidney, and vasculature have been linked to hypertension as strategies aimed at lowering superoxide in all three organs has been shown to limit blood pressure and hypertension-related tissue injury [45,46,47,48,49,50,51]. The goal of this review is to highlight the effects of the proinflammatory cytokine IL-6 on oxidative and inflammatory mechanisms in general, in hypertension, and in response to angiotensin II [52,53,54,55,56,57,58,59,60,61]. As IL-6 has many important effects on blood vessels, including endothelial activation [62,63,64], immune cell recruitment [65,66,67,68,69,70], vascular permeability [71,72], vascular hypertrophy [73,74], and vascular fibrosis [75,76], and endothelial dysfunction [57,58] the discussion of this review will be limited to the effects of IL-6 on the vasculature (Figure 1).
2. Interleukin-6
IL-6 is a pleiotropic cytokine best recognized as a primary mediator of the acute phase response [77]. IL-6 also plays key roles in host defense, inflammation, cancer, as well as cellular growth and hypertrophy [78,79]. A number of stimuli, including angiotensin II as well as other inflammatory cytokines and growth factors, such as IL-1, TNFα and PDGF, are associated with increases in vascular cell-derived IL-6 [53,58,59,71,74,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115] (Table 1). IL-6 levels are increased in cardiovascular disease, including atherosclerosis and hypertension, where IL-6 is thought to promote alterations in vascular function and structure [55,57,58]. For example, plasma IL-6 concentrations correlate with blood pressure, plasma angiotensin II levels, and vascular hypertrophy, suggesting an important role of IL-6 in the development and maintenance of hypertension, particularly that mediated by angiotensin II [58]. Thus, there is substantial data implicating IL-6 in the initiation as well as progression and maintenance of cardiovascular disease through reductions in NO, increases in vascular superoxide, and alterations in vascular function.
3. Interleukin-6 Signaling
Although reviewed in greater depth in several excellent reviews on the subject, IL-6 signaling will be briefly discussed in terms of general mechanisms [117,118,119]. Classical IL-6 signaling involves IL-6 binding to its cognate membrane-bound IL-6 receptor (IL-6R) [78]. Expression of IL-6R is normally very low and expressed in a limited number of cell types, mainly immune cells and hepatocytes [118,119]. Although IL-6R has no intrinsic signaling capability interaction of the IL-6/IL-6R complex induces dimerization of the membrane-bound gp130 (the signaling molecule for IL-6R and related cytokine receptors) an important step in the transduction of the IL-6 signal [120,121,122,123]. Unlike IL-6R, gp130 is ubiquitously expressed and possesses intrinsic signaling capability. IL-6/IL-6R complex association and binding of gp130 initiates cellular signaling through several pathways including Janus-associated kinases (Jak; including Jak1, Jak2 and Tyk1), signal transducer and activator of transcription-1, -3, and -5 (STAT1, STAT3, and STAT5), SH2-domain containing phosphatase (SHP2), and extracellular signal related kinase 1/2, mitogen-activated protein kinase and phosphatidylinositol-3-kinase (PI3K/Akt) signaling pathways [78,119,124]. gp130-mediated activation of Jak family members and subsequent phosphorylation of specific tyrosine residues contained in the cytoplasmic portion of gp130, which serve as docking sites for recruitment of STAT molecules, results in phosphorylation and dimerization of phosphorylated STAT [78,124]. Phosphorylated STAT molecules migrate to the nucleus where they are involved in transcriptional activation of IL-6-dependent genes [124] (Figure 2).
In addition to classical IL-6R signaling, IL-6 signaling can also occur in non-IL-6R expressing cells via IL-6 trans-signaling involving a soluble IL-6R (sIL-6R) [124,125,126,127]. sIL-6R derived primarily from cleavage of membrane-bound IL-6R through ADAM17 and ADAM10 activation and to a lesser degree through alternate IL-6R splice variant [128,129]. ADAM17 and ADAM10 appear to promote release of sIL-6R in humans, whereas only ADAM10 has been found to be involved in production and release of sIL-6R in mice [129]. Binding of IL-6 to sIL-6R and subsequent binding to tissue bound gp130 IL-6/sIL-6R signaling transduction proceeds in a similar manner as classical IL-6R signaling (Figure 2). sIL-6R has the added advantage that it also serves to bind circulating IL-6 thereby greatly extending the half-life of IL-6 [130]. sIL-6R also regulates leukocyte interactions with endothelium, for example sIL-6R promotes endothelial production of monocyte chemoattractant protein-1 (MCP-1 aka CCL2), a key chemokine that regulates monocyte/macrophage infiltration to the vascular wall [64].
In order to limit the proinflammatory effects of IL-6, IL-6 signaling is regulated by a number of inhibitory molecules and processes. Inhibition of IL-6 signaling is important to limit inflammation and the untoward effects, such as tissue injury, associated with chronic IL-6 signaling. Therefore, IL-6 signaling is tightly regulated and highly specific and involves several different inhibitory mechanisms. For example, STAT3-dependent transcription is rapidly inactivated by suppressor of cytokine signaling 3 (SOCS3), which is itself an IL-6 target gene [131,132,133,134]. SOCS3 serves to functionally inhibit IL-6 signaling by binding to gp130 and Jak by targeting the complex for ubiquitination and degradation [132]. SOCS3 also interacts with SHP2 domain thereby reducing SHP2 signaling [132]. Thus, IL-6-induced SOCS3 expression serves as a major negative feedback loop for IL-6 signaling.
In addition to sIL-6R, a soluble form of gp130 (sgp130) has also been identified in plasma [135,136]. sgp130 serves as a functional antagonist of IL-6 trans-signaling by binding to the IL-6/sIL-6R complex thereby preventing the IL-6/IL-6R complex from binding with membrane-bound gp130 [137]. The antagonistic effect of sgp130 is greatly enhanced in the presence of sIL-6R and it has been postulated that IL-6R/sgp130 stoichiometry may be an important determinant of sIL-6R activity [138,139,140,141]. The potential therapeutic use of sgp130 has been exploited in the development of a recombinant form of sgp130, which contains an Fc moiety, sgp130-Fc [78,142].
A third mechanism of IL-6 signaling inhibition involves protein inhibitor of activated STAT-3 (PIAS3), which binds elements in IL-6 target genes thereby inhibiting phosphorylated STAT3/DNA interaction and gene transcription [142]. Whether PIAS3 is functionally important in the vasculature has yet to be determined. Nonetheless, exploitation of PIAS3 may represent a novel mechanism to limit IL-6 and IL-6-related injury in the vascular wall.
4. Vascular Sources of Interleukin-6
Several immune cell types, including macrophages, monocytes, B cells and T cells, produce IL-6 [125,143]. In addition, major components of the vascular wall, including endothelial cells, vascular smooth muscle cells, and fibroblasts are also important sources of IL-6 [92,93,94,95,96,97,98,144]. Thus, increased vascular expression of IL-6 most likely reflects that derived from both resident and infiltrating immune cells as well as vascular cells themselves. Increases in IL-6 originating within the vascular wall can act in either an autocrine or paracrine fashion that promote vascular dysfunction through increases in reactive oxygen species and reductions in NO which then serves to promote immune cell activation and vascular infiltration. Aging also appears to influence vascular IL-6 production as aged aortic vascular smooth cells exhibit higher basal secretion of IL-6 than young vascular smooth muscle cells [145]. Thus, vascular cells as well as resident and infiltrating immune cells have the capacity to produce IL-6 in response to a vast array of stimuli (Table 1). In addition, vascular cells are responsive to IL-6 via classical IL-6- as well as IL-6-trans-signaling, the latter of which appears to be the main mechanism of IL-6 signaling in vascular cells. Such a dual signaling pathway has been proposed to endow vascular and immune cells with specific cellular responses to various IL-6-producing stimuli [146]. The remaining discussion will be devoted to the effects of IL-6 on the three major vascular cell types followed by the effects of IL-6 on endothelial function and blood pressure.
5. Effect of Interleukin-6 on Endothelial Cells
Exposure of endothelial cells to hypoxia is associated with increases in oxidative stress and IL-6 as well as increases in endothelial cell permeability [59,71,72]. Anti-IL-6 neutralizing antibodies reduce both oxidative stress and endothelial permeability as measured by trans-endothelial electrical resistance, whereas addition of exogenous IL-6 has the opposite effect. Interestingly, antioxidants are very effective in limiting increases in reactive oxygen species, endothelial permeability, as well as IL-6 produced in response to hypoxia [59,71]. These results suggest that increases in reactive oxygen species, most likely functioning as signaling molecules, occurs upstream of both the increase in IL-6 and endothelial permeability produced by hypoxia.
Addition of IL-1 or IL-4 alone or in combination with INF-γ or TNF-α to cultured human aortic endothelial cells is associated with oxidative stress, as assessed using dihydroethidium staining, as well as marked increases in IL-6 expression [100]. Antioxidants or inhibitors of NADPH oxidase are very effective in limiting increases in IL-6 expression produced by IL-4 [100]. Consistent with these findings, IL-4 was unable to increase IL-6 levels in blood vessels from Nox2-deficient mice [100]. These findings provide pharmacological and genetic evidence linking NADPH oxidase-derived increases in oxidative stress and endothelial IL-6 production in response to inflammatory stimuli.
IL-6 can stimulate endothelial expression of adhesion molecules, including ICAM-1, VCAM-1 as well as E-selectin, thereby enhancing immune cell adherence and extravasation into the vascular wall [65,147,148]. For example, IL-6 has been shown to elevate ICAM-1 expression in endothelial cells via a STAT3-dependent mechanism involving both increases in oxidative stress and activation of Rac1 [148]. Reductions in NO bioavailability have been suggested to contribute in part to an increase in ICAM-1 expression as NO donors have been found to effectively suppress ICAM-1 expression and STAT3 phosphorylation in endothelial cells [147]. IL-6 mediated increases in adhesion molecule expression is thought to play an important role in the early phase of inflammation where lymphocytes such as neutrophils predominate, whereas in latter phases infiltration of monocytic cells, such as macrophages, predominate [67]. Indeed, IL-6 mediated reductions in NO bioavailability and increases in oxidative stress are associated with increased endothelial production of MCP-1 [149].
IL-6 is associated with dose-dependent reductions in eNOS expression in cultured endothelial cells [150,151]. The reduction in eNOS expression appears to occur independently of any effect on eNOS mRNA stability [150]. Instead, IL-6 appears to inhibit eNOS promoter activity via STAT3-mediated inhibition of SIE at amino acid residue-1024 [150]. In addition, IL-6 is also associated with reduced eNOS activity due in part to reductions in eNOS phosphorylation at Ser1177 (human eNOS), a phosphorylation site associated with enhanced eNOS activity [151]. The observed reduction in eNOS phosphorylation is reflective of reductions in upstream Akt activity, a key kinase involved with phosphorylation of eNOS at Ser1177. Thus, IL-6 appears to directly regulate eNOS expression and activity in cultured endothelial cells resulting in decreased NO bioavailability, which would be predicted to impact endothelial function in vivo [152].
IL-6 also inhibits eNOS activity through increases in expression of caveolin-1 [151]. Caveolin-1 binds eNOS through protein:protein interaction thereby effectively suppressing eNOS activity [151]. eNOS activity is suppressed by caveolin-1 as the eNOS:caveolin-1 complex is trafficked to caveolae rather than the cytosol where eNOS activity is most functional. The effect of IL-6 stimulated increases in caveolin-1 is not limited to functional suppression of basal eNOS activity but also includes suppression of agonist-, such as bradykinin-, induced increases in eNOS activity and NO levels [151].
Combined, it appears that IL-6 alone, or in combination with other inflammatory stimuli, can serve to promote and enhance not only endothelial permeability, but also recruitment and infiltration of the vascular wall by circulating immune cells, such as neutrophils, lymphocytes and macrophages, through upregulation of adhesion molecules and chemoattractants such as VCAM-1, ICAM-1 and MCP-1. Thus, endothelial recruitment of immune cells serve to enhance the level of inflammatory molecules. This is important as immune cells, such as neutrophils and macrophages, are also key sources of IL-6 and superoxide.
6. Effect of Interleukin-6 on Vascular Smooth Muscle
In addition to its many effects on endothelium, IL-6 has numerous effects on vascular smooth muscle, such as inducing proliferation, migration, and hypertrophy of vascular muscle [57,73,153]. A common mechanism linking IL-6 with vascular smooth muscle cell proliferation, migration, and hypertrophy is oxidative stress, specifically superoxide [56]. Angiotensin II is capable of producing increases in vascular superoxide in part through activation of the vascular NADPH oxidase [38,40,41]. Angiotensin II-induced increases in IL-6 can be inhibited by the addition of either Jak2, STAT1, or p47phox (an important component of vascular NADPH oxidase complex) antibodies [154]. Similarly, enhanced smooth muscle cell production of IL-6 in response to angiotensin II can be markedly reduced in the presence of DPI and AG490, pharmacologic inhibitors of flavin-containing enzymes such as NADPH oxidase and Jak2, respectively [154]. These findings are not limited to vascular smooth muscle as similar findings have been observed in renal mesangial cells, which share many properties and phenotypic markers as vascular smooth muscle [155,156,157]. Collectively, these studies suggest that increases in reactive oxygen species particularly increases in vascular superoxide occur upstream of angiotensin II-induced increases in IL-6 and involve NADPH oxidase-derived superoxide. Angiotensin II-induced increases in smooth muscle IL-6 levels were found to occur in an NFκB dependent manner [54,61]. Elevation of IL-6 produced by angiotensin II involve both activation and translocation of NFκB from the cytosol to the nucleus where it is involved in IL-6 gene transcription. These findings provide strong evidence linking angiotensin II signaling and NFκB pathways with increased IL-6 production in vascular smooth muscle.
Early studies found that IL-6 promotes both proliferation and migration of vascular smooth muscle cells [80,158,159]. In terms of proliferation, IL-6 is associated with increases in vascular smooth muscle cell number [98]. The increase in smooth muscle cell proliferation in response to IL-6 stimulation is associated with an increase in platelet-derived growth factor (PDGF, a potent inducer of cell growth) and incubation of vascular smooth muscle cells with anti-PDGF antibodies are very effective in limiting the increase in cell number produced by IL-6 [112,113]. In cell migration assays, IL-6 has been found to promote migration of vascular smooth muscle cells [159]. Inhibition of IL-6 signaling with IL-6 neutralizing antibodies is associated with reductions in cell migration. Interestingly, 12(S)HETE-induced migration can also be inhibited by neutralizing IL-6 antibodies, thereby implicating a role for IL-6 in 12(S)HETE-induced smooth muscle cell migration [80].
Consistent with the effects of angiotensin II on vascular smooth muscle in cell culture, angiotensin II is associated with vascular hypertrophy in vivo [57,65,73,160]. For example, angiotensin II infusion produces marked increases in vascular cross-sectional area, an index of hypertrophy whereas IL-6 deficiency limits the hypertrophic effect of angiotensin II [57]. Similarly, pharmacological inhibition of IL-6 signaling with small molecular inhibitors of STAT3 also result in marked attenuation of the hypertrophic response of the carotid artery to angiotensin II [160]. The reduction in hypertrophy, which occurs in response to IL-6 deficiency appears to occur independently of blood pressure, whereas the reduction in hypertrophy produced by STAT3 inhibitors may be blood pressure-dependent as STAT3 inhibition produces significant reductions in arterial pressure [160]. Taken together these findings suggest that angiotensin II-induced hypertrophy is dependent on IL-6 and downstream IL-6 signaling involving STAT3.
While such studies provide strong evidence linking angiotensin II and IL-6 with vascular hypertrophy, it was not known whether the effects of IL-6 were mediated by classical IL-6R signaling or those produced by IL-6-trans-signaling. An innovative study employing sgp130Fc (a soluble form of gp130 that binds IL-6/sIL-6R complexes and prevents them from signaling) inhibited the development of hypertension, but not vascular hypertrophy [73]. These findings indicate that vascular hypertrophy, but not hypertension, in response to angiotensin II occurs via classical IL-6 signaling. These findings are important in that they demonstrate that IL-6 can have differential effects depending on which IL-6R signaling pathway is activated.
7. Effect of Interleukin-6 on Fibroblasts
Increased expression of IL-6 has been implicated as a contributing factor in vascular fibrosis [75,76,144]. Fibrosis of blood vessels is associated with reductions in vascular compliance and is associated with arterial stiffness and hypertension [161,162,163]. Both cardiac and vascular fibroblasts stimulated with angiotensin II are associated with increases in oxidative stress and IL-6 production [61,164,165]. For example, when cardiac fibroblasts, which normally express little to no IL-6, are treated with angiotensin II they produce IL-6 in both a concentration- and time-dependent manner [165]. Interestingly, in studies of vascular wall explants, adventitia produces much more IL-6 than either endothelium or the medial wall, suggesting that adventitia is a prominent source of IL-6 in the vascular wall [166]. Moreover, when cardiac fibroblasts are co-cultured with macrophages, IL-6 production in response to angiotensin II treatment increases more than 10-fold compared to IL-6 levels when either cell type is cultured alone and then treated with angiotensin II [165]. These findings suggest that fibroblasts in the presence of macrophages, which is most likely indicative of the situation that occurs in vivo in response to angiotensin II or hypertension, synergize to enhance IL-6 expression. Incubation of fibroblasts from IL-6-deficient mice with angiotensin II is associated with negligible IL-6 production [153]. Treatment of fibroblasts with IL-6 results in increased αSMA, a marker of fibroblast differentiation, and collagen expression in wild-type, but not IL-6-deficient, fibroblasts [153]. Inhibition of Jak or ERK signaling is associated with reduced expression of αSMA in response to IL-6 in cardiac fibroblasts [164]. Incubation of fibroblasts with IL-6 has similar effects as angiotensin II, including increases in TGF-β signaling and type I collagen expression [75].
Taking the cultured fibroblast studies back to the whole animal, angiotensin II infusion in wild-type mice was found to produce marked increases in cardiac fibrosis, which was not present in angiotensin II-infused IL-6-deficient mice [164]. IL-6 is also associated with increased expression of TGF-β, a pro-fibrotic cytokine and a key mediator of fibrogenesis, as TGF-β drives collagen production and extracellular matrix remodeling [167]. Co-culturing fibroblasts and macrophages and treating them with angiotensin II is associated with increased αSMA and type I collagen expression as well as TGF-β and Smad3 phosphorylation (markers of fibroblast activation), effects which are absent in fibroblasts from IL-6-deficient mice [168]. Interestingly, IL-6-deficient mice are characterized by impaired wound healing, a process that involves fibroblasts, TGF-β signaling, and extracellular matrix remodeling [169].
Fibroblasts exposed to physiological concentrations of hydrogen peroxide, a reactive oxygen species formed from the metabolism of superoxide, display transient increases in intracellular calcium that precede increases in fibroblast-derived IL-6 [116]. Inhibition of intracellular calcium release with zestospongin, an antagonist of the inositol 1,4,5-triphosphate receptor and reticular calcium, results in lowering of peroxide-induced IL-6 expression, suggesting fibroblast IL-6 expression in response to peroxide is calcium-dependent [116]. In addition to peroxide, angiotensin II-induced superoxide formation is associated with cardiac fibrosis. Both candesartan, an AT1R antagonist and DPI an inhibitor of flavin-containing enzymes, including NADPH oxidase is associated with reduction in angiotensin II-induced fibrosis [61].
Evidence suggests a correlation between IL-6 levels and increases in MMP-2 and TIMP-1 and -2 expression [153]. IL-6-deficiency is associated with alterations in MMP-2, TIMP-1 and TIMP-2 [153]. TIMPs are endogenous inhibitors of MMPs. IL-6 treatment reduces TIMP-2 expression in cultured fibroblasts. Interestingly, IL-6-induced inhibition of TIMP-2 expression may serve to promote angiogenesis, an effect that is absent in IL-6-deficient mice [153].
Dermal fibroblasts produce very little IL-6 when cultured as monolayers, but when seeded onto a collagenous matrix IL-6 expression increases more than 10-fold [144]. In addition to collagen, dermal fibroblasts respond to inflammatory cytokines such as IL-1 with increased production of IL-6. Incubation of primary human aortic adventitial fibroblasts with angiotensin II is associated with increased IL-6 and MCP-1 expression and monocyte recruitment, which activates fibroblast proliferation and adventitial thickening thereby creating a fibroblast-monocyte amplification loop [66,71]. Infusion of angiotensin II is similarly associated with marked increases in aortic IL-6, vascular superoxide, MCP-1 expression, and vascular monocyte/macrophage accumulation and activation in wild-type, but not IL-6-deficient, mice [71]. Interestingly, chronic infusion of supraphysiological concentrations of angiotensin II is associated with the development of aortic aneurysms and dissection, in essence a hyper-amplification of the fibroblast-monocyte/macrophage loop [71].
8. Effects of Interleukin-6 on Endothelial Function and Oxidative Stress
Although IL-6 is a pro-inflammatory cytokine, IL-6 has been found to have little to no effect on endothelial function [57,170]. For example, acute incubation (<22 h) of blood vessels with IL-6 has no apparent effect on responses to either acetylcholine or nitroprusside in carotid artery from healthy wild-type mice [57]. The lack of effect of IL-6 on endothelial function is consistent with IL-6 infusion having no effect on arterial pressure as discussed in the following section. The fact that IL-6 does not affect endothelial responses is somewhat surprising, as IL-6 has been shown to produce dose-dependent reductions in eNOS expression in cultured endothelial cells [150,157]. The reduction in eNOS produced by IL-6 reflects a decrease in steady-state levels of both eNOS message and protein [150]. The effect of IL-6 on eNOS expression appears to be mediated by increased STAT3-mediated inhibition of the eNOS promoter [150]. While there is some evidence to suggest that human endothelial cells express IL-6R, it is not clear to what level IL-6R is expressed in vivo under basal conditions. Low or negligible IL-6R expression may explain, in part, why IL-6 infusion has been found to have little effect on vascular responses in vivo in the absence of disease. Future studies are required to delineate levels of endogenous IL-6R expression and IL-6R expression in response to IL-6 infusion.
Despite the effects of IL-6 on endothelial function and eNOS expression and activity in wild-type mice, IL-6 was observed to significantly impair endothelial responses in vessels with inherent reductions in eNOS expression [152]. For example, despite a 60% reduction in eNOS expression endothelial responses to acetylcholine are normal in eNOS+/− mice. Incubation of vessels from eNOS+/− with IL-6 however produces marked impairment of endothelial responses to acetylcholine [152]. IL-6-mediated impairment of endothelial function in eNOS+/− mice is associated with increases in NADPH-stimulated superoxide levels, suggesting that IL-6-mediated increases in NADPH oxidase activity are greater in the absence of a single eNOS gene [152]. These data suggest that (1) despite marked reduction in eNOS expression, blood vessels such as carotid artery maintain their dependence on eNOS and NO in the absence a single eNOS gene, and (2) NADPH oxidase is an important source of vascular superoxide in response to IL-6 and that in the absence of a single eNOS gene the effect of IL-6 on NADPH oxidase activity is greatly enhanced. These findings may explain why IL-6 infusion is associated with increased arterial pressure and enhanced vascular contraction in pregnant but not non-pregnant rats, where eNOS expression may be altered in the former [171,172]. Taken together, such findings suggest that the effect of IL-6 on endothelial function only becomes apparent with marked reductions in eNOS expression (≥60%) such as that associated with heterozygous eNOS deficiency, eNOS polymorphisms, or cardiovascular risk factors associated with reduced eNOS expression.
Angiotensin II is elevated in several diseases, including atherosclerosis and hypertension [41,173,174]. Angiotensin II promotes increases in superoxide and a particularly important source of superoxide in response to angiotensin II involves NADPH oxidase [53,56,57]. NADPH oxidase expression increases in response to angiotensin II via an AT1 receptor dependent mechanism, as angiotensin II receptor blockers and AT1R deficiency are associated with reductions in angiotensin II-induced superoxide production [56]. IL-6 promotes increases in angiotensin II receptor expression further promoting increases in angiotensin II-induced superoxide. Angiotensin II stimulated increases in vascular superoxide is a key activator of NFκB-dependent IL-6 transcription. Similarly, IL-6-induced increases in vascular superoxide is dependent on Nox2 expression [53]. Thus, angiotensin II elevates superoxide levels, which leads to increases in IL-6 and IL-6-induced increases in Nox2-derived superoxide providing a vicious cycle promoting elevations in IL-6 and superoxide in the vascular wall. The increases in IL-6 and superoxide have been shown to impact both vascular function and vascular hypertrophy [53].
IL-6 has been shown to up-regulate AT1 receptor expression, leading to even greater levels of superoxide [56]. Angiotensin II impairs endothelial function via increases in vascular superoxide and reductions in nitric oxide bioavailability and deficiency of IL-6 protects against angiotensin II-induced endothelial dysfunction and increases in vascular superoxide [57]. In reconstitution studies, incubation of vessels from IL-6-deficient mice with recombinant IL-6 recapitulates the impaired vascular phenotypes produced by angiotensin II, providing strong and convincing evidence that IL-6 is a critical mediator of angiotensin II-induced endothelial dysfunction [57]. Moreover, NADPH oxidase appears to be a major source of vascular superoxide and deficiency of Nox2 (a major catalytic NADPH oxidase subunit) also protects against angiotensin II-induced impairment of endothelial function [57]. Incubation of vessels from Nox2-deficient mice with angiotensin II or recombinant IL-6 is not associated with endothelial dysfunction thereby strongly implicating Nox2-derived superoxide as an important vascular source of superoxide in response to angiotensin II and IL-6 [57]. Thus, angiotensin II is an important stimulus that increases vascular superoxide and vascular IL-6 expression, with increases in vascular superoxide being necessary for increased IL-6 expression presumably through activation of redox sensitive transcription factors such as NFκB [53].
9. Interleukin-6 and Blood Pressure
Early data from the literature suggested an association between blood pressure and circulating IL-6 levels in otherwise healthy men with IL-6 levels correlating in a linear fashion with every 10-mmHg increase in blood pressure [173]. Since this initial observation, several other studies provide additional support for increased IL-6 expression in hypertensive patients as well as several experimental models of hypertension [174,175,176]. Taken together, the clinical data links increased plasma IL-6 with disease progression. While such data provides evidence supporting an association between blood pressure and IL-6 it does not specifically demonstrate causality.
Several important questions regarding the role of IL-6 in blood pressure remain unanswered, such as what role does IL-6 levels play in maintenance of blood pressure in apparently healthy individuals. It is not clear whether the observed increase in IL-6 is due to increases in blood pressure or vice versa. Additionally, cardiovascular diseases associated with increases in IL-6, such as atherosclerosis and hypertension, are also associated with oxidative stress, particularly increases in vascular superoxide, suggesting a potential relationship between superoxide, IL-6 and arterial pressure.
One way to determine whether IL-6 affects blood pressure directly is to infuse IL-6. Interestingly, infusion of IL-6 at physiological concentrations associated with pathological conditions, but lower than that observed in sepsis, has little to no effect on arterial pressure when infused acutely or chronically [177,178,179]. (Table 2) The fact that IL-6 has little effect on blood pressure suggest IL-6 alone is not sufficient to affect blood pressure and that perhaps IL-6 requires activation of other inflammatory pathways in order to affect arterial pressure. Despite the lack of effect of IL-6 on arterial pressure, IL-6 is associated with a significant degree of cardiac fibrosis and hypertrophy, suggesting that IL-6 can produce cardiac alterations independent of changes in arterial pressure [179].
IL-6-transgenic mice are characterized by elevated plasma IL-6 levels as well as a number of outward phenotypes, including pulmonary hypertension [180,181]. Surprisingly, there have been no reported measurements of arterial pressure in IL-6-transgenic mice, perhaps due to the fact that transgenic expression of IL-6 is associated with enhanced mortality [180,182,183]. Generation of either vascular or immune cell specific IL-6-transgenic mice would be extremely helpful in determining the long term consequences, if any, associated with IL-6 overexpression on arterial pressure.
At the other end of the spectrum, IL-6-deficient mice have been used in a number of studies examining the effect of IL-6-deficiency on both baseline blood pressure and on the development of hypertension [57,73,81,163,184,185,186,187,188,189,190]. Baseline blood pressure in IL-6-deficient mice does not appear to be associated with alterations in arterial pressure, a consistent finding among several studies (Table 2). Such genetic findings provide strong evidence that IL-6 per se does not contribute to regulation of basal blood pressure. These findings are consistent with the concept that IL-6 expression would be negligible or minimal in the absence of disease.
While there is a clear consensus on the effect of IL-6 deficiency on baseline blood pressure, the effect of IL-6-deficiency on the development of hypertension is much more varied. For example, IL-6-deficiency has been associated with either no effect as well as partial inhibition of hypertension in various experimental models, such as that produced by angiotensin II-infusion alone or in combination with high-salt (Table 3). This is somewhat surprising as IL-6 levels are increased in response to angiotensin II infusion, a highly consistent finding among these same studies [186,188]. More recently, our laboratory found a direct correlation with plasma IL-6 levels and hypertension produced by angiotensin II infusion over a wide range of doses as well as with plasma angiotensin II levels [58].
Such observed differences in the effect of IL-6-deficiency on hypertension is most likely reflective of differences in the model of hypertension employed, such as angiotensin II-induced versus stress-induced hypertension, as well as differences in methods used to measure blood pressure (tail-cuff plethysmography versus radiotelemetry) [57,184,185,186,187,188,189,190]. Even when similar methods are used to produce hypertension and measure blood pressure, such as radiotelemetry, the gold standard for long-term and direct measurement of arterial pressure, major differences remain [184,186,190]. Thus, additional studies are needed to more clearly define the role of IL-6 in the development and maintenance of the hypertensive phenotype.
An important and most likely under reported event associated with angiotensin II infusion in C57Bl/6 mice, is the moderate incidence (≤30% incidence, depending on dose) of abdominal aortic aneurysm formation and rupture [191,192]. Some conflicting effects of IL-6 deficiency on blood pressure may be influenced by changes in aortic structure and death associated with aneurysm rupture such that a moderate degree of selection bias is introduced if the incidence of aneurysm is not considered or properly measured. Nonetheless, it is clear that IL-6 does not contribute to basal or resting blood pressure whereas IL-6 appears to contribute, in part, to the increase in blood pressure produced in experimental hypertension, including that produced by angiotensin II.
Examination of downstream mediators of IL-6 signaling, such as STAT3, has also revealed important insight into the role of IL-6 in experimental hypertension. For example, pharmacological inhibition of STAT3 with small-molecule inhibitors, such as S3I-201, which selectively inhibits STAT3 SH2-dependent complex formation, STAT3 dimerization and subsequent STAT3-dependent gene transcription, is associated with complete inhibition of angiotensin II-induced hypertension [160]. S31-201 also prevents angiotensin-II induced increases in vascular superoxide [160]. Interestingly, transgenic knockin mice expressing a functionally inactive STAT3 binding site on gp130, S737A mice, has been found to have no effect on baseline blood pressure and a normal pressor response to chronic angiotensin II infusion [193]. Whether endothelial responses and oxidative stress levels are altered in S737A mice, in general or in response to angiotensin II infusion, has not yet been determined. Taken together, these pharmacological and genetic data provide the first insights into downstream mechanisms of IL-6 signaling in angiotensin II-induced hypertension.
10. Summary
The relationship between angiotensin II, oxidative stress, and IL-6 is highly interdependent. Moreover, vascular cells can produce IL-6 in response to stimuli such as angiotensin II and IL-6 produced by vascular cells results in numerous and distinct effects on each vascular cell type (Figure 3). Angiotensin II increases IL-6 expression in the vascular wall through activation of the AT1 receptor and downstream activation of NADPH oxidase resulting in elevated oxidative stress, particularly superoxide that activates transcriptional mechanisms that directly promote IL-6 gene expression. Reciprocally, IL-6 promotes AT1 receptor expression further sensitizing the vascular wall to angiotensin II-dependent signaling mechanisms, which further promotes increases in oxidative stress and IL-6 expression. Vascular derived IL-6 plays an important role in the initiation of immune cell recruitment to the vascular wall. The increase in immune cells to the vascular wall serves to further promote endothelial dysfunction and recruitment of IL-6 and superoxide producing cells, such as neutrophils and macrophages. IL-6 is also associated with reductions in NO bioavailability through alterations in eNOS expression and activity. While IL-6 has many effects on vascular function and fibrosis, IL-6 does not appear to play a significant role in regulation of basal blood pressure. In contrast, IL-6 appears to contribute, in part, to the maintenance of the hypertensive phenotype as IL-6-deficiency is limits the rise in arterial pressure associated with some, but not all, forms of experimental hypertension. The role of IL-6 in hypertension development requires additional study in order define the specific contribution of IL-6 in the initiation and maintenance of the hypertensive phenotype. Nonetheless, IL-6 mediates many of the negative effects associated with hypertension and angiotensin II on vascular structure and function independent of blood pressure. While recent studies have begun to examine the role of downstream molecules, such as STAT3, in vascular function and oxidative stress, additional studies are required in order to provide a better understanding of the impact of IL-6 on both the cellular and oxidative mechanisms associated with IL-6 signaling in the vasculature.
Acknowledgments
This work was supported by a grant from the National Institutes of Health (HL-107632).
Conflicts of Interest
The author declares no conflict of interest.
References
- Handy, D.E.; Loscalzo, J. Responses to reductive stress in the cardiovascular system. Free Radic. Biol. Med. 2017, 109, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, P.M.; Zhao, Y.; Xu, A.; Leung, S.W. Thirty years of saying NO: Sources, fate, actions, and misfortunes of the endothelium-derived vasodilator mediator. Circ. Res. 2016, 119, 375–396. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Touyz, R.M. Oxidative stress, inflammation, and vascular aging in hypertension. Hypertension 2017, 70, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Félétou, M.; Köhler, R.; Vanhoutte, P.M. Endothelium-derived vasoactive factors and hypertension: Possible roles in pathogenesis and as treatment targets. Curr. Hypertens. Rep. 2010, 12, 267–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Caterina, R.; Libby, P.; Peng, H.B.; Thannickal, V.J.; Rajavashisth, T.B.; Gimbrone, M.A., Jr.; Shin, W.S.; Liao, J.K. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J. Clin. Investig. 1995, 96, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Devlin, A.M.; Brosnan, M.J.; Graham, D.; Morton, J.J.; McPhaden, A.R.; McIntyre, M.; Hamilton, C.A.; Reid, J.L.; Dominiczak, A.F. Vascular smooth muscle cell polyploidy and cardiomyocyte hypertrophy due to chronic NOS inhibition in vivo. Am. J. Physiol. 1998, 274, H52–H59. [Google Scholar] [PubMed]
- Rudic, R.D.; Shesely, E.G.; Maeda, N.; Smithies, O.; Segal, S.S.; Sessa, W.C. Direct evidence for the importance of endothelium-derived nitric oxide in vascular remodeling. J. Clin. Investig. 1998, 101, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Baumbach, G.L.; Sigmund, C.D.; Faraci, F.M. Structure of cerebral arterioles in mice deficient in expression of the gene for endothelial nitric oxide synthase. Circ. Res. 2004, 95, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, T.; Pappas, P.J.; Hobson, R.W., 2nd; Boric, M.P.; Sessa, W.C.; Durán, W.N. Endothelial nitric oxide synthase regulates microvascular hyperpermeability in vivo. J. Physiol. 2006, 574, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.L.; Huang, Z.; Mashimo, H.; Bloch, K.D.; Moskowitz, M.A.; Bevan, J.A.; Fishman, M.C. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 1995, 377, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Shesely, E.G.; Maeda, N.; Kim, H.S.; Desai, K.M.; Krege, J.H.; Laubach, V.E.; Sherman, P.A.; Sessa, W.C.; Smithies, O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 1996, 93, 13176–13181. [Google Scholar] [CrossRef] [PubMed]
- Faraci, F.M.; Sigmund, C.D.; Shesely, E.G.; Maeda, N.; Heistad, D.D. Responses of carotid artery in mice deficient in expression of the gene for endothelial NO synthase. Am. J. Physiol. 1998, 274, H564–H570. [Google Scholar] [PubMed]
- Mashimo, H.; Goyal, R.K. Lessons from genetically engineered animal models. IV. Nitric oxide synthase gene knockout mice. Am. J. Physiol. 1999, 277, G745–G750. [Google Scholar] [PubMed]
- Fleming, I.; Busse, R. Molecular mechanisms involved in the regulation of the endothelial nitric oxide synthase. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 284, R1–R12. [Google Scholar] [CrossRef] [PubMed]
- Searles, C.D. Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression. Am. J. Physiol. Cell Physiol. 2006, 291, C803–C816. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Fulton, D. Post-translational regulation of endothelial nitric oxide synthase in vascular endothelium. Front. Physiol. 2013, 4, 347. [Google Scholar] [CrossRef] [PubMed]
- Wei, E.P.; Kontos, H.A.; Christman, C.W.; DeWitt, D.S.; Povlishock, J.T. Superoxide generation and reversal of acetylcholine-induced cerebral arteriolar dilation after acute hypertension. Circ. Res. 1985, 57, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Giugliano, D.; Quatraro, A.; Lefebvre, P.J. Antioxidants shown an antihypertensive effect in diabetic and hypertensive subjects. Clin. Sci. 1991, 81, 739–742. [Google Scholar] [CrossRef] [PubMed]
- Nakazono, K.; Watanabe, N.; Matsuno, J.; Saski, J.; Sato, T.; Inoue, M. Does superoxide underlie the pathogenesis of hypertension? Proc. Natl. Acad. Sci. USA 1991, 88, 10045–10048. [Google Scholar] [CrossRef] [PubMed]
- Pagano, P.J.; Chanock, S.J.; Siwik, D.A.; Colucci, W.S.; Clark, J.K. Angiotensin II induces p67phox mRNA expression and NADPH oxidase superoxide generation in rabbit aortic adventitial fibroblasts. Hypertension 1998, 32, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes, M.E.; Rey, F.E.; Carretero, O.A.; Pagano, P.J. Upregulation of p67(phox) and gp91(phox) in aortas from angiotensin II-infused mice. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H2234–H2240. [Google Scholar] [PubMed]
- Hsich, E.; Segal, B.H.; Pagano, P.J.; Rey, F.E.; Paigen, B.; Deleonardis, J.; Hoyt, R.F.; Holland, S.M.; Finkel, T. Vascular effects following homozygous disruption of p47(phox): An essential component of NADPH oxidase. Circulation 2000, 101, 1234–1236. [Google Scholar] [CrossRef] [PubMed]
- Rey, F.E.; Cifuentes, M.E.; Kiarash, A.; Quinn, M.T.; Pagano, P.J. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(-) and systolic blood pressure in mice. Circ. Res. 2001, 89, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Didion, S.P.; Faraci, F.M. Effects of NADH and NADPH on superoxide levels and cerebral vascular tone. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H688–H695. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, F.; Yang, X.P.; Jankowski, M.; Pagano, P.J. NAD(P)H oxidase mediates angiotensin II-induced vascular macrophage infiltration and medial hypertrophy. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 776–782. [Google Scholar] [CrossRef] [PubMed]
- Didion, S.P.; Faraci, F.M. Angiotensin II produces superoxide-mediated impairment of endothelial function in cerebral arterioles. Stroke 2003, 34, 2038–2042. [Google Scholar] [CrossRef] [PubMed]
- Byrne, J.A.; Grieve, D.J.; Bendall, J.K.; Li, J.M.; Gove, C.; Lambeth, J.D.; Cave, A.C.; Shah, A.M. Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ. Res. 2003, 93, 802–805. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, K.; Yamada, H.; Iwata, K.; Jin, D.; Katsuyama, M.; Matsuki, M.; Takai, S.; Yamanishi, K.; Miyazaki, M.; Matsubara, H.; et al. Nox1 is involved in angiotensin II-mediated hypertension: A study in Nox1-deficient mice. Circulation 2005, 112, 2677–2685. [Google Scholar] [CrossRef] [PubMed]
- Grote, K.; Ortmann, M.; Salguero, G.; Doerries, C.; Landmesser, U.; Luchtefeld, M.; Brandes, R.P.; Gwinner, W.; Tschernig, T.; Brabant, E.G.; et al. Critical role for p47phox in renin-angiotensin system activation and blood pressure regulation. Cardiovasc. Res. 2006, 71, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Gavazzi, G.; Banfi, B.; Deffert, C.; Fiette, L.; Schappi, M.; Herrmann, F.; Krause, K.H. Decreased blood pressure in NOX1-deficient mice. FEBS Lett. 2006, 580, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.I.; Nazarewicz, R.R.; Bikineyeva, A.; Hilenski, L.; Lassègue, B.; Griendling, K.K.; Harrison, D.G.; Dikalova, A.E. Nox2-induced production of mitochondrial superoxide in angiotensin II-mediated endothelial oxidative stress and hypertension. Antioxid. Redox Signal. 2014, 20, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Kofler, S.; Nickel, T.; Weis, M. Role of cytokines in cardiovascular diseases: A focus on endothelial responses to inflammation. Clin. Sci. 2005, 108, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Dworakowski, R.; Alom-Ruiz, S.P.; Shah, A.M. NADPH oxidase-derived reactive oxygen species in the regulation of endothelial phenotype. Pharmacol. Rep. 2008, 60, 21–28. [Google Scholar] [PubMed]
- Zhang, C. The role of inflammatory cytokines in endothelial dysfunction. Basic Res. Cardiol. 2008, 103, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Sprague, A.H.; Khalil, R.A. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem. Pharmacol. 2009, 78, 539–552. [Google Scholar] [CrossRef] [PubMed]
- McMaster, W.G.; Kirabo, A.; Madhur, M.S.; Harrison, D.G. Inflammation, immunity, and hypertensive end-organ damage. Circ. Res. 2015, 116, 1022–1033. [Google Scholar] [CrossRef] [PubMed]
- Karbach, S.; Wenzel, P.; Waisman, A.; Munzel, T.; Daiber, A. eNOS uncoupling in cardiovascular diseases—The role of oxidative stress and inflammation. Curr. Pharm. Des. 2014, 20, 3579–3594. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Minieri, C.A.; Ollerenshaw, J.D.; Alexander, R.W. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 1994, 74, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Mohazzab, K.M.; Kaminski, P.M.; Wolin, M.S. NADH oxidoreductase is a major source of superoxide anion in bovine coronary artery endothelium. Am. J. Physiol. 1994, 266, H2568–H2572. [Google Scholar] [PubMed]
- Nguyen Dinh Cat, A.; Montezano, A.C.; Burger, D.; Touyz, R.M. Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid. Redox Signal. 2013, 19, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Lassègue, B.; San Martín, A.; Griendling, K.K. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ. Res. 2012, 110, 1364–1390. [Google Scholar] [CrossRef] [PubMed]
- Drummond, G.R.; Selemidis, S.; Griendling, K.K.; Sobey, C.G. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH oxidases in vascular pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef] [PubMed]
- Vara, D.; Pula, G. Reactive oxygen species: Physiological roles in the regulation of vascular cells. Curr. Mol. Med. 2014, 14, 1103–1125. [Google Scholar] [CrossRef] [PubMed]
- Schnackenberg, C.G.; Welch, W.J.; Wilcox, C.S. Normalization of blood pressure and renal vascular resistance in SHR with a membrane-permeable superoxide dismutase mimetic: Role of nitric oxide. Hypertension 1998, 32, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Di Wang, H.; Hope, S.; Du, Y.; Quinn, M.T.; Cayatte, A.; Pagano, P.J.; Cohen, R.A. Paracrine role of adventitial superoxide anion in mediating spontaneous tone of the isolated rat aorta in angiotensin II-induced hypertension. Hypertension 1999, 33, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Didion, S.P.; Ryan, M.J.; Baumbach, G.L.; Sigmund, C.D.; Faraci, F.M. Superoxide contributes to vascular dysfunction in mice that express human renin and angiotensinogen. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1569–H1576. [Google Scholar] [CrossRef] [PubMed]
- Makino, A.; Skelton, M.M.; Zou, A.P.; Roman, R.J.; Cowley, A.W., Jr. Increased renalmedullary oxidative stress produces hypertension. Hypertension 2002, 39, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.C.; Lazartigues, E.; Lang, J.A.; Sinnayah, P.; Ahmad, I.M.; Spitz, D.R.; Davisson, R.L. Superoxide mediates the actions of angiotensin II in the central nervous system. Circ. Res. 2002, 91, 1038–1045. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.C.; Lazartigues, E.; Sharma, R.V.; Davisson, R.L. Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system. Circ. Res. 2004, 95, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Didion, S.P.; Kinzenbaw, D.A.; Faraci, F.M. Critical role for CuZn-superoxide dismutase in preventing angiotensin II-induced endothelial dysfunction. Hypertension 2005, 46, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi, Y.; Ichiki, T.; Ito, K.; Takeshita, A. Induction of interleukin-6 expression by angiotensin II in rat vascular smooth muscle cells. Hypertension 1999, 34, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Runge, M.S.; Brasier, A.R. Angiotensin II induces interleukin-6 transcription in vascular smooth muscle cells through pleiotropic activation of nuclear factor-kappaB transcription factors. Circ. Res. 1999, 84, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Kranzhöfer, R.; Schmidt, J.; Pfeiffer, C.A.; Hagl, S.; Libby, P.; Kübler, W. Angiotensin induces inflammatory activation of human vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1623–1629. [Google Scholar] [CrossRef] [PubMed]
- Schieffer, B.; Schieffer, E.; Hilfiker-Kleiner, D.; Hilfiker, A.; Kovanen, P.T.; Kaartinen, M.; Nussberger, J.; Harringer, W.; Drexler, H. Expression of angiotensin II and interleukin 6 in human coronary artery atherosclerotic plaques: Potential implications for inflammation and plaque instability. Circulation 2000, 101, 1372–1378. [Google Scholar] [CrossRef] [PubMed]
- Wassmann, S.; Stumpf, M.; Strehlow, K.; Schmid, A.; Schieffer, B.; Böhm, M.; Nickenig, G. Interleukin-6 induces oxidative stress and endothelial dysfunction by overexpression of the angiotensin II type 1 receptor. Circ. Res. 2004, 94, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Schrader, L.I.; Kinzenbaw, D.A.; Johnson, A.W.; Faraci, F.M.; Didion, S.P. IL-6 deficiency protects against angiotensin II induced endothelial dysfunction and hypertrophy. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2576–2581. [Google Scholar] [CrossRef] [PubMed]
- Gomolak, J.R.; Didion, S.P. Angiotensin II-induced endothelial dysfunction is temporally linked with increases in interleukin-6 and vascular macrophage accumulation. Front. Physiol. 2014, 5, 396. [Google Scholar] [CrossRef] [PubMed]
- Ala, Y.; Palluy, O.; Favero, J.; Bonne, C.; Modat, G.; Dornand, J. Hypoxia/reoxygenation stimulates endothelial cells to promote interleukin-1 and interleukin-6 production. Effects of free radical scavengers. Agents Actions 1992, 37, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Maruyama, M.; Fujita, T.; Arai, N.; Hayashi, R.; Araya, J.; Matsui, S.; Yamashita, N.; Sugiyama, E.; Kobayashi, M. Reactive oxygen intermediates stimulate interleukin-6 production in human bronchial epithelial cells. Am. J. Physiol. 1999, 276, L900–L908. [Google Scholar] [PubMed]
- Sano, M.; Fukuda, K.; Sato, T.; Kawaguchi, H.; Suematsu, M.; Matsuda, S.; Koyasu, S.; Matsui, H.; Yamauchi-Takihara, K.; Harada, M.; et al. ERK and p38 MAPK, but not NF-kappaB, are critically involved in reactive oxygen species-mediated induction of IL-6 by angiotensin II in cardiac fibroblasts. Circ. Res. 2001, 89, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Hirohata, S.; Kashiwado, T.; Itoh, K.; Ishii, H. Cytokine regulation of hemostatic property and IL-6 production of human endothelial cells. Inflammation 1992, 16, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Hooper, W.C.; Phillips, D.J.; Renshaw, M.A.; Evatt, B.L.; Benson, J.M. The up-regulation of IL-6 and IL-8 in human endothelial cells by activated protein C. J. Immunol. 1998, 161, 2567–2573. [Google Scholar] [PubMed]
- Marin, V.; Montero-Julian, F.A.; Grès, S.; Boulay, V.; Bongrand, P.; Farnarier, C.; Kaplanski, G. The IL-6-soluble IL-6Ralpha autocrine loop of endothelial activation as an intermediate between acute and chronic inflammation: An experimental model involving thrombin. J. Immunol. 2001, 167, 3435–3442. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.; Whittaker, S.; Smith, N.; Vora, A.J.; Dumonde, D.C.; Brown, K.A. IL-6 acts on endothelial cells to preferentially increase their adherence for lymphocytes. Clin. Exp. Immunol. 1996, 105, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Sironi, M.; Toniatti, C.; Polentartutti, N.; Fruscella, P.; Ghezzi, P.; Faggioni, R.; Luini, W.; van Hinsbergh, V.; Sozzani, S.; et al. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity 1997, 6, 315–325. [Google Scholar] [CrossRef]
- Hurst, S.M.; Wilkinson, T.S.; McLoughlin, R.M.; Jones, S.; Horiuchi, S.; Yamamoto, N.; Rose-John, S.; Fuller, G.M.; Topley, N.; Jones, S.A. IL-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity 2001, 14, 705–714. [Google Scholar] [CrossRef]
- Fielding, C.A.; McLoughlin, R.M.; McLeod, L.; Colmont, C.S.; Najdovska, M.; Grail, D.; Ernst, M.; Jones, S.A.; Topley, N.; Jenkins, B.J. IL-6 regulates neutrophil trafficking during acute inflammation via STAT3. J. Immunol. 2008, 181, 2189–2195. [Google Scholar] [CrossRef] [PubMed]
- Tieu, B.C.; Lee, C.; Sun, H.; LeJeune, W.; Recinos, A., 3rd; Ju, X.; Spratt, H.; Guo, D.C.; Milewicz, D.; Tilton, R.G.; et al. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J. Clin. Investig. 2009, 119, 3637–3651. [Google Scholar] [CrossRef] [PubMed]
- Shoji, M.; Furuyama, F.; Yokota, Y.; Omori, Y.; Sato, T.; Tsunoda, F.; Iso, Y.; Koba, S.; Geshi, E.; Katagiri, T.; et al. IL-6 mobilizes bone marrow-derived cells to the vascular wall, resulting in neointima formation via inflammatory effects. J. Atheroscler. Thromb. 2014, 21, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.H.; Schlidt, S.A.; Chandel, N.S.; Hynes, K.L.; Schumacker, P.T.; Gewertz, B.L. Endothelial permeability and IL-6 production during hypoxia: Role of ROS in signal transduction. Am. J. Physiol. 1999, 277, L1057–L1065. [Google Scholar] [PubMed]
- Desai, T.R.; Leeper, N.J.; Hynes, K.L.; Gewertz, B.L. Interleukin-6 causes endothelial barrier dysfunction via the protein kinase C pathway. J. Surg. Res. 2002, 104, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Coles, B.; Fielding, C.A.; Rose-John, S.; Scheller, J.; Jones, S.A.; O’Donnell, V.B. Classic interleukin-6 receptor signaling and interleukin-6 trans-signaling differentially control angiotensin II-dependent hypertension, cardiac signal transducer and activator of transcription-3 activation, and vascular hypertrophy in vivo. Am. J. Pathol. 2007, 171, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Tieu, B.C.; Ju, X.; Lee, C.; Sun, H.; Lejeune, W.; Recinos, A., 3rd; Brasier, A.R.; Tilton, R.G. Aortic adventitial fibroblasts participate in angiotensin-induced vascular wall inflammation and remodeling. J. Vasc. Res. 2011, 48, 261–272. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, S.; Ciechomska, M.; Cant, R.; van Laar, J.M. Interleukin-6 (IL-6) trans signaling drives a STAT3-dependent pathway that leads to hyperactive transforming growth factor-β (TGF-β) signaling promoting SMAD3 activation and fibrosis via Gremlin protein. J. Biol. Chem. 2014, 289, 9952–9960. [Google Scholar] [CrossRef] [PubMed]
- Fielding, C.A.; Jones, G.W.; McLoughlin, R.M.; McLeod, L.; Hammond, V.J.; Uceda, J.; Williams, A.S.; Lambie, M.; Foster, T.L.; Liao, C.T.; et al. Interleukin-6 signaling drives fibrosis in unresolved inflammation. Immunity 2014, 40, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Castell, J.V.; Andus, T. Interleukin-6 and the acute phase response. Biochem. J. 1990, 265, 621–636. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A.; Jones, S.A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015, 16, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. The soluble interleukin-6 receptor and related proteins. Best Prac. Res. Clin. Endocrinol. Metab. 2015, 29, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Chava, K.R.; Karpurapu, M.; Wang, D.; Bhanoori, M.; Kundumani-Sridharan, V.; Zhang, Q.; Ichiki, T.; Glasgow, W.C.; Roa, G.N. CREB-mediated IL-6 expression is required for 15(S)-hydroxyeicosatetraenoic acid-induced vascular smooth muscle cell migration. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Sturgis, L.C.; Cannon, J.G.; Schreihofer, D.A.; Brands, M.W. The role of aldosterone in mediating the dependence of angiotensin hypertension on IL-6. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1742–R1748. [Google Scholar] [CrossRef] [PubMed]
- Luther, J.M.; Gainer, J.V.; Murphey, L.J.; Yu, C.; Vaughan, D.E.; Morrow, J.D.; Brown, N.J. Angiotensin II induces interleukin-6 in humans through a mineralocorticoid receptor-dependent mechanism. Hypertension 2006, 48, 1050–1057. [Google Scholar] [CrossRef] [PubMed]
- Inanaga, K.; Ichiki, T.; Matsuura, H.; Miyazaki, R.; Hashimoto, T.; Takeda, K.; Sunagawa, K. Resveratrol attenuates angiotensin II-induced interleukin-6 expression and perivascular fibrosis. Hypertens. Res. 2009, 32, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Browatzki, M.; Schmidt, J.; Kübler, W.; Kranzhöfer, R. Endothelin-1 induces interleukin-6 release via activation of the transcription factor NF-kappaB in human vascular smooth muscle cells. Basic Res. Cardiol. 2000, 95, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Stankova, J.; Rola-Pleszczynski, M.; D’Orleans-Just, P. Endothelin 1 and thrombin synergistically stimulate IL-6 mRNA expression and protein production in human umbilical vein endothelial cells. J. Cardiovasc. Pharmacol. 1995, 26, S505–S507. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.P.; Liu, J.T.; Liu, N.; Guo, F.; Ji, Y.Y.; Pang, X. Pro-inflammatory effect of fibrinogen and FDP on vascular smooth muscle cells by IL-6, TNF-α and iNOS. Life Sci. 2011, 88, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Delneste, Y.; Lassalle, P.; Jeannin, P.; Joseph, M.; Tonnel, A.B.; Gosset, P. Histamine induces IL-6 production by human endothelial cells. Clin. Exp. Immunol. 1994, 98, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chi, L.; Stechschulte, D.J.; Dileepan, K.N. Histamine-induced production of interleukin-6 and interleukin-8 by human coronary artery endothelial cells is enhanced by endotoxin and tumor necrosis factor-alpha. Microvasc. Res. 2001, 61, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jin, M.; Hu, X.S.; Zhu, J.H. Homocysteine stimulates nuclear factor kappaB activity and interleukin-6 expression in rat vascular smooth muscle cells. Cell Biol. Int. 2006, 30, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.F.; Tritto, I.; Pinsky, D.; Liao, H.; Huang, J.; Fuller, G.; Brett, J.; May, L.; Stern, D. Induction of interleukin-6 (IL-6) by hypoxia in vascular cells. J. Biol. Chem. 1995, 270, 11463–11471. [Google Scholar] [CrossRef] [PubMed]
- May, L.T.; Torcia, G.; Cozzolino, F.; Ray, A.; Tatter, S.B.; Santhanam, U.; Sehgal, P.B.; Stern, D. Interleukin-6 gene expression in human endothelial cells: RNA start sites, multiple IL-6 proteins and inhibition of proliferation. Biochem. Biophys. Res. Commun. 1989, 159, 991–998. [Google Scholar] [CrossRef]
- Jirik, F.R.; Podor, T.J.; Hirano, T.; Kishimoto, T.; Loskutoff, D.J.; Carson, D.A.; Lotz, M. Bacterial lipopolysaccharide and inflammatory mediators augment IL-6 secretion by human endothelial cells. J. Immunol. 1989, 142, 144–147. [Google Scholar] [PubMed]
- Podor, T.J.; Jirik, F.R.; Loskutoff, D.J.; Carson, D.A.; Lotz, M. Human endothelial cells produce IL-6. Lack of response to exogenous IL-6. Ann. N. Y. Acad. Sci. 1989, 557, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Sironi, M.; Breviario, F.; Propserpio, P.; Biondi, A.; Vecchi, A.; van Damme, J.; Dejana, E.; Mantovani, A. IL-1 stimulates IL-6 production in endothelial cells. J. Immunol. 1989, 142, 549–553. [Google Scholar] [PubMed]
- Loppnow, H.; Libby, P. Adult human vascular endothelial cells express the IL6 gene differentially in response to LPS or IL1. Cell. Immunol. 1989, 122, 493–503. [Google Scholar] [CrossRef]
- Loppnow, H.; Libby, P. Comparative analysis of cytokine induction in human vascular endothelial and smooth muscle cells. Lymphokine Res. 1989, 8, 293–299. [Google Scholar] [PubMed]
- Loppnow, H.; Libby, P. Proliferating or interleukin 1-activated human vascular smooth muscle cells secrete copious interleukin 6. J. Clin. Investig. 1990, 85, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Norioka, K.; Hara, M.; Harigai, M.; Kitani, A.; Hirose, T.; Hirose, W.; Suzuki, K.; Kawakami, M.; Kawagoe, M.; Nakamura, H. Pretreatment of human vascular smooth muscle cells with interleukin-1 enhances interleukin-6 production and cell proliferation (action of IL-1 on vascular smooth muscle cells). Autoimmunity 1990, 7, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Howells, G.; Pham, P.; Taylor, D.; Foxwell, B.; Feldmann, M. Interleukin 4 induces interleukin 6 production by endothelial cells: Synergy with interferon-gamma. Eur. J. Immunol. 1991, 21, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.W.; Lee, W.H.; Kim, P.H. Oxidative mechanisms of IL-4-induced IL-6 expression in vascular endothelium. Cytokine 2010, 49, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Sironi, M.; Borrè, A.; Luini, W.; Maddalena, F.; Mantovani, A. Interleukin 4 amplifies monocyte chemotactic protein and interleukin 6 production by endothelial cells. Cytokine 1992, 4, 24–28. [Google Scholar] [CrossRef]
- Chen, C.C.; Manning, A.M. TGF-beta 1, IL-10 and IL-4 differentially modulate the cytokine-induced expression of IL-6 and IL-8 in human endothelial cells. Cytokine 1996, 8, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Leeuwenberg, J.F.; von Asmuth, E.J.; Jeunhomme, T.M.; Buurman, W.A. IFN-gamma regulates the expression of the adhesion molecule ELAM-1 and IL-6 production by human endothelial cells in vitro. J. Immunol. 1990, 145, 2110–2114. [Google Scholar] [PubMed]
- Chi, L.; Li, Y.; Stehno-Bittel, L.; Gao, J.; Morrison, D.C.; Stechschulte, D.J.; Dileepan, K.N. Interleukin-6 production by endothelial cells via stimulation of protease-activated receptors is amplified by endotoxin and tumor necrosis factor-alpha. J. Interferon Cytokine Res. 2001, 21, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Jehle, A.B.; Li, Y.; Stechschulte, A.C.; Stechschulte, D.J.; Dileepan, K.N. Endotoxin and mast cell granule proteases synergistically activate human coronary artery endothelial cells to generate interleukin-6 and interleukin-8. J. Interferon Cytokine Res. 2000, 20, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Von Asmuth, E.J.; Leeuwenberg, J.F.; Ceska, M.; Buurman, W.A. LPS and cytokine-induced endothelial cell IL-6 release and ELAM-1 expression; involvement of serum. Eur. Cytokine Netw. 1991, 2, 291–297. [Google Scholar] [PubMed]
- Sasamoto, A.; Nagino, M.; Kobayashi, S.; Naruse, K.; Nimura, Y.; Sokabe, M. Mechanotransduction by integrin is essential for IL-6 secretion from endothelial cells in response to uniaxial continuous stretch. Am. J. Physiol. Cell Physiol. 2005, 288, C1012–C1022. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Nagino, M.; Komatsu, S.; Naruse, K.; Nimura, Y.; Nakanishi, M.; Sokabe, M. Stretch-induced IL-6 secretion from endothelial cells requires NF-kappaB activation. Biochem. Biophys. Res. Commun. 2003, 308, 306–312. [Google Scholar] [CrossRef]
- Kawai, M.; Naruse, K.; Komatsu, S.; Kobayashi, S.; Nagino, M.; Nimura, Y.; Sokabe, M. Mechanical stress-dependent secretion of interleukin 6 by endothelial cells after portal vein embolization: Clinical and experimental studies. J. Hepatol. 2002, 37, 240–246. [Google Scholar] [CrossRef]
- Zampetaki, A.; Zhang, Z.; Hu, Y.; Xu, Q. Biomechanical stress induces IL-6 expression in smooth muscle cells via Ras/Rac1-p38 MAPK-NF-kappaB signaling pathways. Am. J. Heart Circ. Physiol. 2005, 288, H2946–H2954. [Google Scholar] [CrossRef] [PubMed]
- Lubrano, V.; Baldi, S.; Ferrannini, E.; L’Abbate, A.; Natali, A. Role of thromboxane A2 receptor on the effects of oxidized LDL on microvascular endothelium nitric oxide, endothelin-1, and IL-6 production. Microcirculation 2008, 15, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, U.; Ikeda, M.; Oohara, T.; Oguchi, A.; Kamitani, K.; Tsuruya, Y.; Kano, S. Interleukin-6 stimulates the growth of vascular cells in a PDGF-dependent manner. Am. J. Physiol. 1991, 260, H1713–H1717. [Google Scholar] [PubMed]
- Gaumond, F.; Fortin, D.; Stankova, J.; Rola-Pleszczynski, M. Differential signaling pathways in platelet-activating factor-induced proliferation and interleukin-6 production by rat vascular smooth muscle cells. J. Cardiovasc. Pharmacol. 1997, 30, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Ikeda, U.; Shimpo, M.; Yamamoto, K.; Shimada, K. Serotonin increases interleukin-6 synthesis in human vascular smooth muscle cells. Circulation 2000, 102, 2522–2527. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Inagaki, Y.; Nakamura, K.; Abe, R.; Shimizu, T.; Yoshimura, A.; Imaizumi, T. Pigment epithelium-derived factor inhibits TNF-alpha-induced interleukin-6 expression in endothelial cells by suppressing NADPH oxidase-mediated reactive oxygen species generation. J. Mol. Cell. Cardiol. 2004, 37, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Colston, J.T.; Chandrasekar, B.; Freeman, G.L. A novel peroxide-induced calcium transient regulates interleukin-6 expression in cardiac-derived fibroblasts. J. Biol. Chem. 2002, 277, 23477–23483. [Google Scholar] [CrossRef] [PubMed]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochem. Biophys. Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Chalaris, A.; Garbers, C.; Rabe, B.; Rose-John, S.; Scheller, J. The soluble Interleukin 6 receptor: Generation and role in inflammation and cancer. Eur. J. Cell Biol. 2011, 90, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Mihara, M.; Hashizume, M.; Yoshida, H.; Suzuki, M.; Shiina, M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin. Sci. 2012, 122, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Yoshida, K.; Hibi, M.; Taga, T.; Kishimoto, T. Molecular cloning of a murine IL-6 receptor-associated signal transducer, gp130, and its regulated expression in vivo. J. Immunol. 1992, 148, 4066–4071. [Google Scholar] [PubMed]
- Demyanets, S.; Huber, K.; Wojta, J. Vascular effects of glycoprotein130 ligands- part II: Biomarkers and therapeutic targets. Vascul. Pharmacol. 2012, 57, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Klouche, M.; Bhakdi, S.; Hemme, M.; Rose-John, S. Novel path to activation of vascular smooth muscle cells: Up-regulation of gp130 creates an autocrine activation loop by IL-6 and its soluble receptor. J. Immunol. 1999, 163, 4583–4589. [Google Scholar] [PubMed]
- Boulanger, M.J.; Chow, D.C.; Brevnova, E.E.; Garcia, K.C. Hexameric structure and assembly of the interleukin-6/IL-6 α-receptor/gp130 complex. Science 2003, 300, 2101–2104. [Google Scholar] [CrossRef] [PubMed]
- Ernst, M.; Jenkins, B.J. Acquiring signalling specificity from the cytokine receptor gp130. Trends Genet. 2004, 20, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Schöbitz, B.; Pezeshki, G.; Pohl, T.; Hemmann, U.; Heinrich, P.C.; Holsboer, F.; Reul, J.M. Soluble interleukin-6 (IL-6) receptor augments central effects of IL-6 in vivo. FASEB J. 1995, 9, 659–664. [Google Scholar] [PubMed]
- Garbers, C.; Aparicio-Siegmund, S.; Rose-John, S. The IL-6/gp130/STAT3 signaling axis: Recent advances towards specific inhibition. Curr. Opin. Immunol. 2015, 34, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, M.; Rousseau, F.; Guilhot, F.; Malinge, P.; Magistrelli, G.; Herren, S.; Jones, S.A.; Jones, G.W.; Scheller, J.; Lissilaa, R.; et al. Novel Insights into Interleukin 6 (IL-6) Cis- and trans-signaling pathways by differentially manipulating the assembly of the IL-6 signaling complex. J. Biol. Chem. 2015, 290, 26943–26953. [Google Scholar] [CrossRef] [PubMed]
- Garbers, C.; Jänner, N.; Chalaris, A.; Moss, M.L.; Floss, D.M.; Meyer, D.; Koch-Nolte, F.; Rose-John, S.; Scheller, J. Species specificity of ADAM10 and ADAM17 proteins in interleukin-6 (IL-6) trans-signaling and novel role of ADAM10 in inducible IL-6 receptor shedding. J. Biol. Chem. 2011, 286, 14804–14811. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, N.; Meyer, D.; Mauermann, A.; von der Heyde, J.; Wolf, J.; Schwarz, J.; Knittler, K.; Murphy, G.; Michalek, M.; Garbers, C.; et al. Shedding of Endogenous Interleukin-6 Receptor (IL-6R) Is Governed by A Disintegrin and Metalloproteinase (ADAM) Proteases while a Full-length IL-6R Isoform Localizes to Circulating Microvesicles. J. Biol. Chem. 2015, 290, 26059–26071. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.; Jacobs, S.; Ehlers, M.; Vollmer, P.; Mullberg, J.; Wolf, E.; Brem, G.; Meyer zum Buschenfelde, K.H.; Rose-John, S. The function of the soluble interleukin 6 (IL-6) receptor in vivo: Sensitization of human soluble IL-6 receptor transgenic mice towards IL-6 and prolongation of the plasma half-life of IL-6. J. Exp. Med. 1996, 183, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Croker, B.A.; Krebs, D.L.; Zhang, J.G.; Wormald, S.; Willson, T.A.; Stanley, E.G.; Robb, L.; Greenhalgh, C.J.; Förster, I.; Clausen, B.E.; et al. SOCS3 negatively regulates IL-6 signaling in vivo. Nat. Immunol. 2003, 4, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Babon, J.J.; Varghese, L.N.; Nicola, N.A. Inhibition of IL-6 family cytokines by SOCS3. Semin. Immunol. 2014, 26, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Weissenbach, M.; Haan, S.; Heinrich, P.C.; Schaper, F. SOCS3 exerts its inhibitory function on interleukin-6 signal transduction through the SHP2 recruitment site of gp130. J. Biol. Chem. 2000, 275, 12848–12856. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Badgwell, D.B.; Bevers, J.J., 3rd; Schlessinger, K.; Murray, P.J.; Levy, D.E.; Watowich, S.S. IL-6 signaling via the STAT3/SOCS3 pathway: Functional analysis of the conserved STAT3 N-domain. Mol. Cell. Biochem. 2006, 288, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Narazaki, M.; Yasukawa, K.; Saito, T.; Ohsugi, Y.; Fukui, H.; Koishihara, Y.; Yancopoulos, G.D.; Taga, T.; Kishimoto, T. Soluble forms of the interleukin-6 signal-transducing receptor component gp130 in human serum possessing a potential to inhibit signals through membrane-anchored gp130. Blood 1993, 82, 1120–1126. [Google Scholar] [PubMed]
- Yasukawa, K.; Futatsugi, K.; Saito, T.; Yawata, H.; Narazaki, M.; Suzuki, H.; Taga, T.; Kishimoto, T. Association of recombinant soluble IL-6-signal transducer, gp130, with a complex of IL 6 and soluble IL-6 receptor, and establishment of an ELISA for soluble gp130. Immunol. Lett. 1992, 31, 123–130. [Google Scholar] [CrossRef]
- Wolf, J.; Waetzig, G.H.; Chalaris, A.; Reinheimer, T.M.; Wege, H.; Rose-John, S.; Garbers, C. Different Soluble Forms of the Interleukin-6 Family Signal Transducer gp130 Fine-tune the Blockade of Interleukin-6 Trans-signaling. J. Biol. Chem. 2016, 291, 16186–16196. [Google Scholar] [CrossRef] [PubMed]
- Garbers, C.; Thaiss, W.; Jones, G.W.; Waetzig, G.H.; Lorenzen, I.; Guilhot, F.; Lissilaa, R.; Ferlin, W.G.; Grötzinger, J.; Jones, S.A.; et al. Inhibition of classic signaling is a novel function of soluble glycoprotein 130 (sgp130), which is controlled by the ratio of interleukin 6 and soluble interleukin 6 receptor. J. Biol. Chem. 2011, 286, 42959–42970. [Google Scholar] [CrossRef] [PubMed]
- Müller-Newen, G.; Küster, A.; Hemmann, U.; Keul, R.; Horsten, U.; Martens, A.; Graeve, L.; Wijdenes, J.; Heinrich, P.C. Soluble IL-6 receptor potentiates the antagonistic activity of soluble gp130 on IL-6 responses. J. Immunol. 1998, 161, 6347–6355. [Google Scholar] [PubMed]
- Jostock, T.; Müllberg, J.; Özbek, S.; Atreya, R.; Blinn, G.; Voltz, N.; Fischer, M.; Neurath, M.F.; Rose-John, S. Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur. J. Biochem. 2001, 268, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Scheller, J.; Kovaleva, M.; Rabe, B.; Eichler, J.; Kallen, K.J.; Rose-John, S. Development of a monoclonal antibody-based enzyme-linked immunoabsorbent assay for the binding of gp130 to the IL-6/IL-6R complex and its competitive inhibition. J. Immunol. Methods 2004, 291, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Müller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Naka, T.; Nishimoto, N.; Kishimoto, T. The paradigm of IL-6: From basic science to medicine. Arthritis Res. 2002, 4, S233–S242. [Google Scholar] [CrossRef] [PubMed]
- Eckes, B.; Hunzelmann, N.; Ziegler-Heitbrock, L.Z.; Urbanski, A.; Luger, T.; Krieg, T.; Mauch, C. Interleukin-6 expression by fibroblasts grown in three-dimensional gel culture. FEBS Lett. 1992, 298, 229–232. [Google Scholar] [CrossRef]
- Song, Y.; Shen, H.; Schenten, D.; Shan, P.; Lee, P.J.; Goldstein, D.R. Aging enhances the basal production of IL-6 and CCL2 in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.; Rose-John, S.; Garbers, C. Interleukin-6 and its receptors: A highly regulated and dynamic system. Cytokine 2014, 70, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Wung, B.S.; Hsu, M.C.; Wu, C.C.; Hsieh, C.W. Resveratrol suppresses IL-6-induced ICAM-1 gene expression in endothelial cells: Effects on the inhibition of STAT3 phosphorylation. Life Sci. 2005, 78, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Wung, B.S.; Ni, C.W.; Wang, D.L. ICAM-1 induction by TNFalpha and IL-6 is mediated by distinct pathways via Rac in endothelial cells. J. Biomed. Sci. 2005, 12, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Kaplanski, G.; Marin, V.; Montero-Julian, F.; Mantovani, A.; Farnarier, C. IL-6: A regulator of the transition from neutrophil to monocyte recruitment during inflammation. Trends Immunol. 2003, 24, 25–29. [Google Scholar] [CrossRef]
- Saura, M.; Zaragoza, C.; Bao, C.; Herranz, B.; Rodriguez-Puyol, M.; Lowenstein, C.J. Stat3 mediates interleukin-6 inhibition of human endothelial nitric-oxide synthase expression. J. Biol. Chem. 2006, 281, 30057–30062. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.J.; Cherng, W.J.; Hung, M.Y.; Wu, H.T.; Pang, J.H. Interleukin-6 inhibits endothelial nitric oxide synthase activation and increases endothelial nitric oxide synthase binding to stabilized caveolin-1 in human vascular endothelial cells. J. Hypertens. 2010, 28, 940–951. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.I.; Chen, X.; Didion, S.P. Heterozygous eNOS deficiency is associated with oxidative stress and endothelial dysfunction in diet-induced obesity. Physiol. Rep. 2015, 3, e12630. [Google Scholar] [CrossRef] [PubMed]
- Luckett, L.R.; Gallucci, R.M. Interleukin-6 (IL-6) modulates migration and matrix metalloproteinase function in dermal fibroblasts from IL-6KO mice. Br. J. Dermatol. 2007, 156, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Schieffer, B.; Luchtefeld, M.; Braun, S.; Hilfiker, A.; Hilfiker-Kleiner, D.; Drexler, H. Role of NAD(P)H oxidase in angiotensin II-induced JAK/STAT signaling and cytokine induction. Circ. Res. 2000, 87, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, T.; Fujibayashi, M.; Fujiwara, Y.; Kaneko, T.; Xia, C.; Imai, E.; Kamada, T.; Ando, A.; Ueda, N. Angiotensin II stimulates interleukin-6 release from cultured mouse mesangial cells. J. Am. Soc. Nephrol. 1995, 6, 95–101. [Google Scholar] [PubMed]
- Satou, R.; Gonzalez-Villalobos, R.A.; Miyata, K.; Ohashi, N.; Katsurada, A.; Navar, L.G.; Kobori, H. Costimulation with angiotensin II and interleukin 6 augments angiotensinogen expression in cultured human renal proximal tubular cells. Am. J. Physiol. Ren. Physiol. 2008, 295, F283–F289. [Google Scholar] [CrossRef] [PubMed]
- Satou, R.; Gonzalez-Villalobos, R.A.; Miyata, K.; Ohashi, N.; Urushihara, M.; Acres, O.W.; Navar, L.G.; Kobori, H. IL-6 augments angiotensinogen in primary cultured renal proximal tubular cells. Mol. Cell. Endocrinol. 2009, 311, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Nabata, T.; Morimono, S.; Koh, E.; Shiraishi, T.; Ogihara, T. Interleukin-6 stimulates c-myc expression and proliferation of cultured vascular smooth muscle cells. Biochem. Int. 1990, 20, 445–453. [Google Scholar] [PubMed]
- Wang, Z.; Newman, W.H. Smooth muscle cell migration stimulated by interleukin 6 is associated with cytoskeletal reorganization. J. Surg. Res. 2003, 111, 261–266. [Google Scholar] [CrossRef]
- Johnson, A.W.; Kinzenbaw, D.A.; Modrick, M.L.; Faraci, F.M. Small-molecule inhibitors of signal transducer and activator of transcription 3 protect against angiotensin II-induced vascular dysfunction and hypertension. Hypertension 2013, 61, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Birukov, K.G. Cyclic stretch, reactive oxygen species, and vascular remodeling. Antioxid. Redox Signal. 2009, 11, 1651–1667. [Google Scholar] [CrossRef] [PubMed]
- Lemarié, C.A.; Tharaux, P.L.; Lehoux, S. Extracellular matrix alterations in hypertensive vascular remodeling. J. Mol. Cell. Cardiol. 2010, 48, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Briones, A.M.; Arribas, S.M.; Salaices, M. Role of extracellular matrix in vascular remodeling of hypertension. Curr. Opin. Nephrol. Hypertens. 2010, 19, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Mir, S.A.; Chatterjee, A.; Mitra, A.; Pathak, K.; Mahata, S.K.; Sarkar, S. Inhibition of signal transducer and activator of transcription 3 (STAT3) attenuates interleukin-6 (IL-6)-induced collagen synthesis and resultant hypertrophy in rat heart. J. Biol. Chem. 2012, 287, 2666–2677. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Li, Y.; Jia, L.; Han, Y.; Cheng, J.; Li, H.; Qi, Y.; Du, J. Macrophage-stimulated cardiac fibroblast production of IL-6 is essential for TGF β/Smad activation and cardiac fibrosis induced by angiotensin II. PLoS ONE 2012, 7, e35144. [Google Scholar] [CrossRef] [PubMed]
- Recinos, A., 3rd; LeJeune, W.S.; Sun, H.; Lee, C.Y.; Tieu, B.C.; Lu, M.; Hou, T.; Boldogh, I.; Tilton, R.G.; Brasier, A.R. Angiotensin II induces IL-6 expression and the Jak-STAT3 pathway in aortic adventitia of LDL receptor-deficient mice. Atherosclerosis 2007, 194, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-β signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Gallucci, R.M.; Lee, E.G.; Tomasek, J.J. IL-6 modulates alpha-smooth muscle actin in dermal fibroblasts from IL-6-deficient mice. J. Investig. Dermatol. 2006, 126, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.Q.; Kondo, T.; Ishida, Y.; Takayasu, T.; Mukaida, N. Essential involvement of IL-6 in the skin wound-healing process as evidenced by delayed wound healing in IL-6-deficient mice. J. Leukoc. Biol. 2003, 73, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Bhagat, K.; Vallance, P. Inflammatory cytokines impair endothelium-dependent dilatation in human veins in vivo. Circulation 1997, 96, 3042–3047. [Google Scholar] [CrossRef] [PubMed]
- Orshal, J.M.; Khalil, R.A. Interleukin-6 impairs endothelium-dependent NO-cGMP-mediated relaxation and enhances contraction in systemic vessels of pregnant rats. Am. J. Regul. Integr. Comp. Physiol. 2004, 286, R1013–R1023. [Google Scholar] [CrossRef] [PubMed]
- Orshal, J.M.; Khalil, R.A. Reduced endothelial NO-cGMP-mediated vascular relaxation and hypertension in IL-6-infused pregnant rats. Hypertension 2004, 43, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Chae, C.U.; Lee, R.T.; Rifai, N.; Ridker, P.M. Blood pressure and inflammation in apparently healthy men. Hypertension 2001, 38, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Gibas-Dorna, M.; Nowak, D.; Piatek, J.; Pupek-Musialik, D.; Krauss, H.; Kopczynski, P. Plasma ghrelin and interleukin-6 levels correlate with body mass index and arterial blood pressure in males with essential hypertension. J. Physiol. Pharmacol. 2015, 66, 367–372. [Google Scholar] [PubMed]
- Naya, M.; Tsukamoto, T.; Morita, K.; Katoh, C.; Furumoto, T.; Fujii, S.; Tamaki, N.; Tsutsui, H. Plasma interleukin-6 and tumor necrosis factor-alpha can predict coronary endothelial dysfunction in hypertensive patients. Hypertens. Res. 2007, 30, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Real, J.M.; Vayreda, M.; Richart, C.; Gutierrez, C.; Broch, M.; Vendrell, J.; Ricart, W. Circulating interleukin 6 levels, blood pressure, and insulin sensitivity in apparently healthy men and women. J. Clin. Endocrinol. Metab. 2001, 86, 1154–1159. [Google Scholar] [CrossRef] [PubMed]
- Preiser, J.C.; Schmartz, D.; Van der Linden, P.; Content, J.; Vanden Bussche, P.; Buurman, W.; Sebald, W.; Dupont, E.; Pinsky, M.R.; Vincent, J.L. Interleukin-6 administration has no acute hemodynamic or hematologic effect in the dog. Cytokine 1991, 3, 1–4. [Google Scholar] [CrossRef]
- Boesen, E.I.; Pollock, D.M. Effect of chronic IL-6 infusion on acute pressor responses to vasoconstrictors. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1745–H1749. [Google Scholar] [CrossRef] [PubMed]
- Melendez, G.C.; McLarty, J.L.; Levick, S.P.; Du, Y.; Janicki, J.S.; Brower, G.L. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension 2010, 56, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Suematsu, S.; Matsuda, T.; Aozasa, K.; Akira, S.; Nakano, N.; Ohno, S.; Miyazaki, J.I.; Yamamura, K.I.; Hirano, T.; Kishimoto, T. IgG1 plasmacytosis in interleukin 6 transgenic mice. Proc. Natl. Acad. Sci. USA 1989, 86, 7547–7551. [Google Scholar] [CrossRef] [PubMed]
- Steiner, M.K.; Syrkina, O.L.; Kolliputi, N.; Mark, E.J.; Hales, C.A.; Waxman, A.B. Interleukin-6 overexpression induces pulmonary hypertension. Circ. Res. 2009, 104, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Fattori, E.; Della Rocca, C.; Costa, P.; Giorgio, M.; Dente, B.; Pozzi, D.L.; Ciliberto, G. Development of progressive kidney damage and myeloma kidney in interleukin-6 transgenic mice. Blood 1994, 83, 2570–2579. [Google Scholar] [PubMed]
- Kovalchuk, A.L.; Kim, J.S.; Park, S.S.; Coleman, A.E.; Ward, J.M.; Morse, H.C., 3rd; Kishimoto, T.; Potter, M.; Janz, S. IL-6 transgenic mouse model for extraosseous plasmacytoma. Proc. Natl. Acad. Sci. USA 2002, 99, 1509–1514. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, G.E.; Rhaleb, N.E.; D’Ambrosio, M.A.; Nakagawa, P.; Liu, Y.; Leung, P.; Dai, X.; Yang, X.P.; Peterson, E.L.; Carretero, O.A. Deletion of interleukin-6 prevents cardiac inflammation, fibrosis and dysfunction without affecting blood pressure in angiotensin II-high salt-induced hypertension. J. Hypertens. 2015, 33, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.L.; Leite, R.; Fleming, C.; Pollock, J.S.; Webb, R.C.; Brands, M.W. Hypertensive response to acute stress is attenuated in interleukin-6 knockout mice. Hypertension 2004, 44, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.L.; Stergis, L.C.; Labazi, H.; Osborne, J.B., Jr.; Fleming, C.; Pollock, J.S.; Manhiani, M.; Imig, J.D.; Brands, M.W. Angiotensin II hypertension is attenuated in interleukin-6 knockout mice. Am. J. Heart Circ. Physiol. 2006, 290, H935–H940. [Google Scholar] [CrossRef] [PubMed]
- Brands, M.W.; Banes-Berceli, A.K.; Inscho, E.W.; Al-Azawi, H.; Allen, A.J.; Labazi, H. Interleukin 6 knockout prevents angiotensin II hypertension: Role of renal vasoconstriction and janus kinase 2/signal transducer and activator of transcription 3 activation. Hypertension 2010, 56, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, W.; Yu, H.; Zhang, Y.; Dai, Y.; Ning, C.; Tao, L.; Sun, H.; Kellems, R.E.; Blackburn, M.R.; et al. Interleukin 6 underlies angiotensin II-induced hypertension and chronic renal damage. Hypertension 2012, 59, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Manhiani, M.M.; Seth, D.M.; Banes-Berceli, A.K.; Satou, R.; Navar, L.G.; Brands, M.W. The role of IL-6 in the physiological versus hypertensive blood pressure actions of angiotensin II. Physiol. Rep. 2015, 3, e12595. [Google Scholar] [CrossRef] [PubMed]
- Manhiani, M.M.; Quigley, J.E.; Socha, M.J.; Motamed, K.; Imig, J.D. IL6 supression provides renal protection independent of blood pressure in a murine model of salt-sensitive hypertension. Kidney Blood Press. Res. 2007, 30, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Ju, X.; Ijaz, T.; Sun, H.; Ray, S.; Lejeune, W.; Lee, C.; Recinos, A., 3rd; Guo, D.C.; Milewicz, D.M.; Tilton, R.G.; et al. Interleukin-6-signal transducer and activator of transcription-3 signaling mediates aortic dissections induced by angiotensin II via the T-helper lymphocyte 17-interleukin 17 axis in C57BL/6 mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1612–1621. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Howatt, D.A.; Balakrishnan, A.; Moorleghen, J.J.; Rateri, D.L.; Cassis, L.A.; Daugherty, A. Subcutaneous angiotensin II infusion using osmotic pumps induces aortic aneurysms in mice. J. Vis. Exp. 2015. [Google Scholar] [CrossRef] [PubMed]
- Zouein, F.A.; Zgheib, C.; Hamza, S.; Fuseler, J.W.; Hall, J.E.; Soljancic, A.; Lopez-Ruiz, A.; Kurdi, M.; Booz, G.W. Role of STAT3 in angiotensin II-induced hypertension and cardiac remodeling revealed by mice lacking STAT3 serine 727 phosphorylation. Hypertens. Res. 2013, 36, 496–503. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Vascular expression of IL-6, such as that produced by angiotensin II, is associated with alterations in vascular function and structure. Increases in reactive oxygen species, such as superoxide, serve to increase IL-6 expression in vascular cells. Increases in vascular or immune cell-derived IL-6 serve as a key mediator of several vascular phenotypes that are produced in response to angiotensin II. IL-6, interleukin-6; ROS, reactive oxygen species.
Figure 1.
Vascular expression of IL-6, such as that produced by angiotensin II, is associated with alterations in vascular function and structure. Increases in reactive oxygen species, such as superoxide, serve to increase IL-6 expression in vascular cells. Increases in vascular or immune cell-derived IL-6 serve as a key mediator of several vascular phenotypes that are produced in response to angiotensin II. IL-6, interleukin-6; ROS, reactive oxygen species.
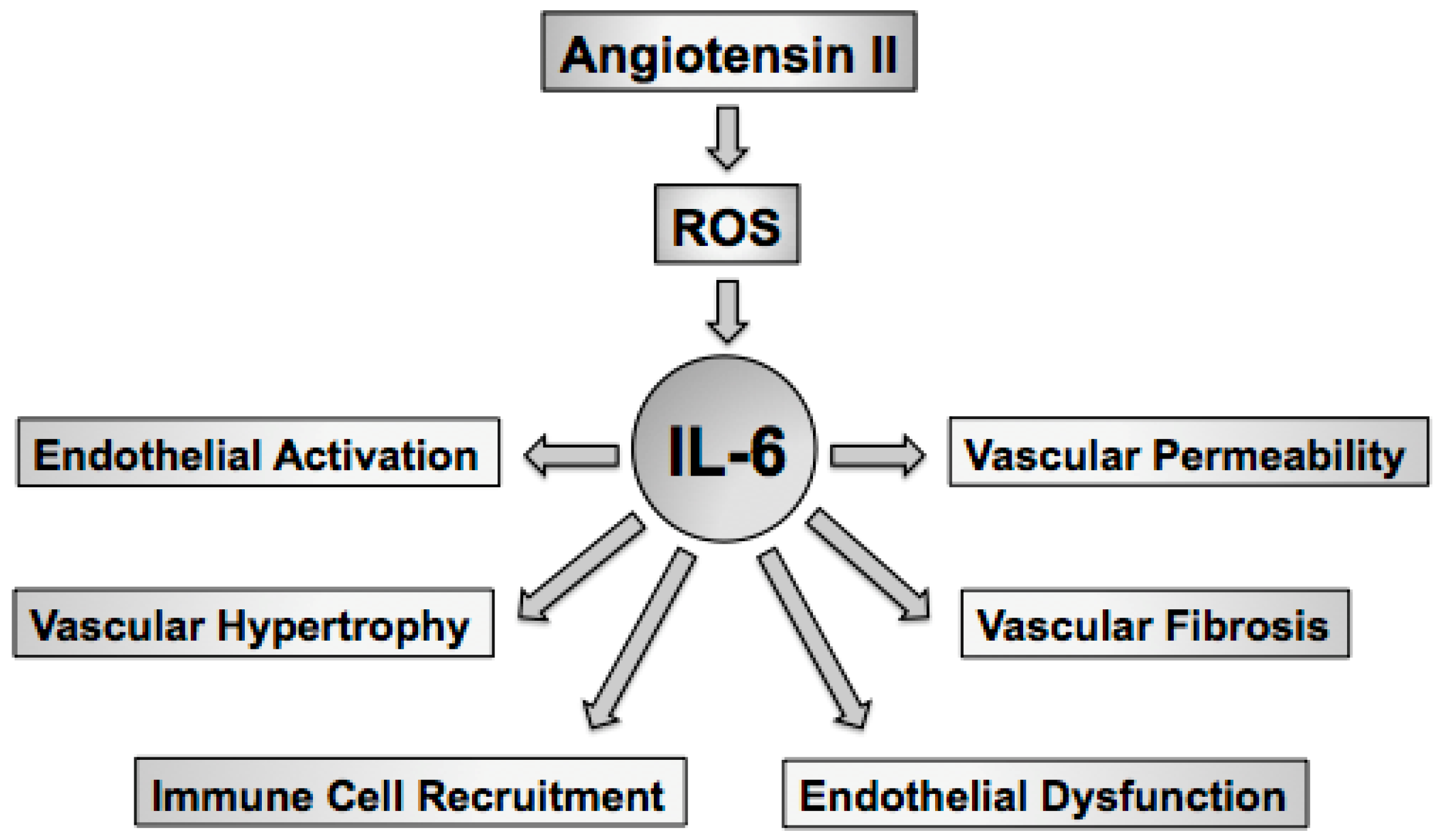
Figure 2.
IL-6 signaling is a function of both classical IL-6 signaling and IL-6-trans-signaling. Classical IL-6 signaling involves IL-6 binding a membrane-bound IL-6 receptor (IL-6R), which facilitates dimerization of membrane-bound gp130. IL-6R lacks intrinsic signaling capability, however activated gp130 contains binding sites for recruitment and phosphorylation of signaling molecules such as Jak/STAT and SHP-2, which play important roles in transcription of IL-6 target genes, such as SOCS3. IL-6-trans-signaling occurs via IL-6 binding soluble IL-6 receptor (sIL-6R), which increases the half-life of IL-6 and serves as an important mechanism of IL-6 signaling in tissues that express gp130 but not IL-6R. Classical IL-6 signaling and IL-6-trans-signaling plays an important role in the diversity of IL-6 signaling. Expression of a soluble form of gp130 (sgp130) functions as an important endogenous inhibitor of IL-6 signaling by binding and preventing IL-6/sIL-6R complex binding and activation of membrane-bound gp130. Jak, Janus-associated Kinase; STAT, Signaling Transducer and Activator of Transcription; SOCS, Suppressor of Cytokine Signaling; PI3K, Phosphatidylinositol-4,5-bisphosphate 3-kinase; Akt, protein kinase B; ERK, Extracellular Signal-Regulated Kinase; MAPK, Mitogen-Activated Protein Kinase.
Figure 2.
IL-6 signaling is a function of both classical IL-6 signaling and IL-6-trans-signaling. Classical IL-6 signaling involves IL-6 binding a membrane-bound IL-6 receptor (IL-6R), which facilitates dimerization of membrane-bound gp130. IL-6R lacks intrinsic signaling capability, however activated gp130 contains binding sites for recruitment and phosphorylation of signaling molecules such as Jak/STAT and SHP-2, which play important roles in transcription of IL-6 target genes, such as SOCS3. IL-6-trans-signaling occurs via IL-6 binding soluble IL-6 receptor (sIL-6R), which increases the half-life of IL-6 and serves as an important mechanism of IL-6 signaling in tissues that express gp130 but not IL-6R. Classical IL-6 signaling and IL-6-trans-signaling plays an important role in the diversity of IL-6 signaling. Expression of a soluble form of gp130 (sgp130) functions as an important endogenous inhibitor of IL-6 signaling by binding and preventing IL-6/sIL-6R complex binding and activation of membrane-bound gp130. Jak, Janus-associated Kinase; STAT, Signaling Transducer and Activator of Transcription; SOCS, Suppressor of Cytokine Signaling; PI3K, Phosphatidylinositol-4,5-bisphosphate 3-kinase; Akt, protein kinase B; ERK, Extracellular Signal-Regulated Kinase; MAPK, Mitogen-Activated Protein Kinase.
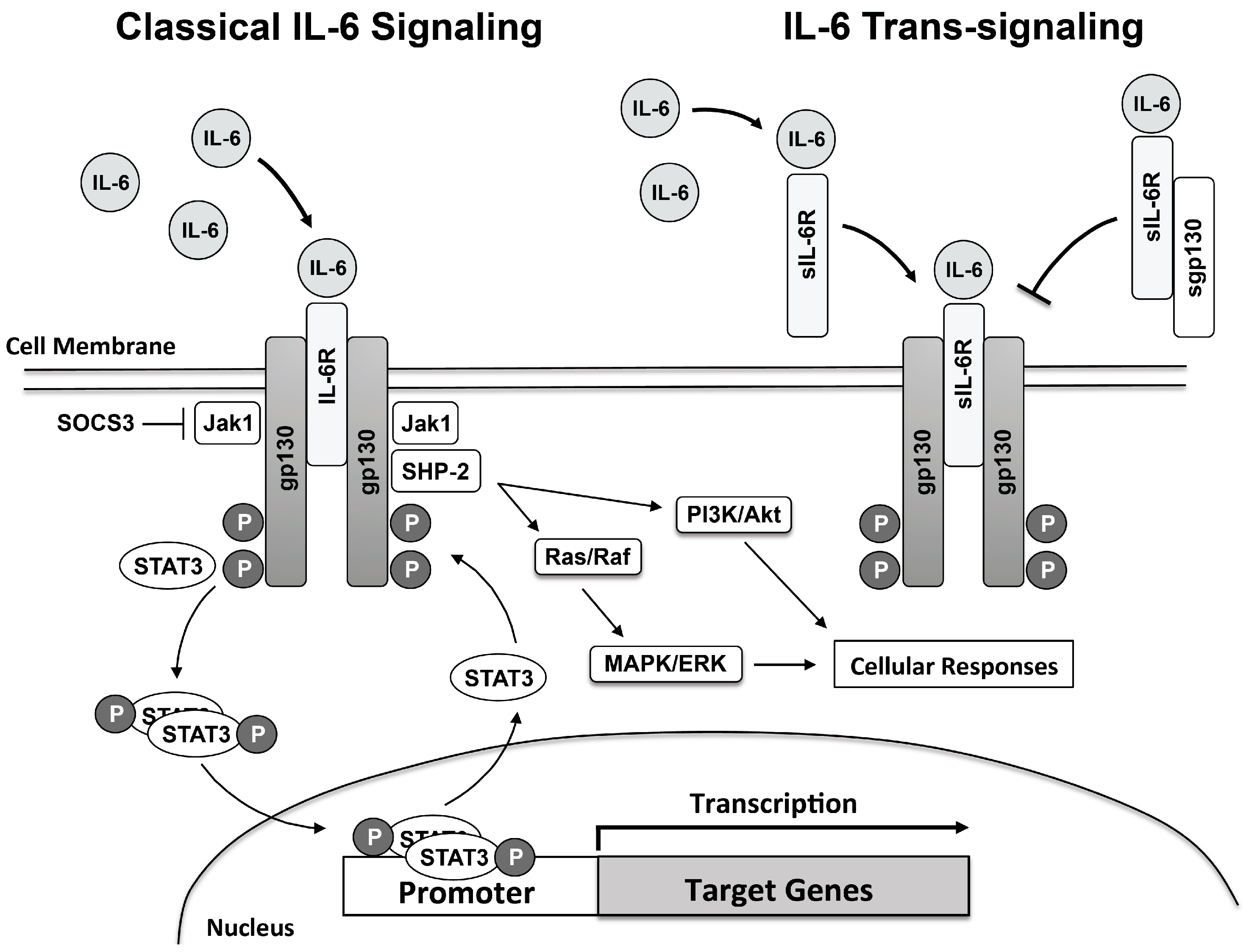
Figure 3.
IL-6 signaling is associated with reductions in NO bioavailability, due, in large part, to reductions in eNOS expression and activity as well as increases in NADPH oxidase-derived superoxide. This combination promotes oxidative stress, endothelial dysfunction as well as vascular hypertrophy and fibrosis. Many of the effects of angiotensin II, such as increased vascular superoxide, are enhanced by IL-6 mediated increases in AT1 receptor expression. Similarly, angiotensin II and IL-6 synergize to promote increases in fibroblast SMA-α, collagen, and MCP-1 (CCL2) expression which promote matrix expansion that ultimately contributes to increased arterial compliance and vascular stiffness. IL-6 also promotes immune cell infiltration of the vascular wall through increased expression of adhesion molecules, such as ICAM-1 and VCAM-1, and expression of MCP-1. IL-6, interleukin-6; eNOS, endothelial nitric oxide synthase; AT1R, angiotensin type 1 receptor; ICAM, intracellular adhesion molecule; VCAM, vascular cell adhesion molecule; MØ, macrophage; SMA, smooth muscle actin; MCP-1, monocyte chemoattractant protein-1.
Figure 3.
IL-6 signaling is associated with reductions in NO bioavailability, due, in large part, to reductions in eNOS expression and activity as well as increases in NADPH oxidase-derived superoxide. This combination promotes oxidative stress, endothelial dysfunction as well as vascular hypertrophy and fibrosis. Many of the effects of angiotensin II, such as increased vascular superoxide, are enhanced by IL-6 mediated increases in AT1 receptor expression. Similarly, angiotensin II and IL-6 synergize to promote increases in fibroblast SMA-α, collagen, and MCP-1 (CCL2) expression which promote matrix expansion that ultimately contributes to increased arterial compliance and vascular stiffness. IL-6 also promotes immune cell infiltration of the vascular wall through increased expression of adhesion molecules, such as ICAM-1 and VCAM-1, and expression of MCP-1. IL-6, interleukin-6; eNOS, endothelial nitric oxide synthase; AT1R, angiotensin type 1 receptor; ICAM, intracellular adhesion molecule; VCAM, vascular cell adhesion molecule; MØ, macrophage; SMA, smooth muscle actin; MCP-1, monocyte chemoattractant protein-1.
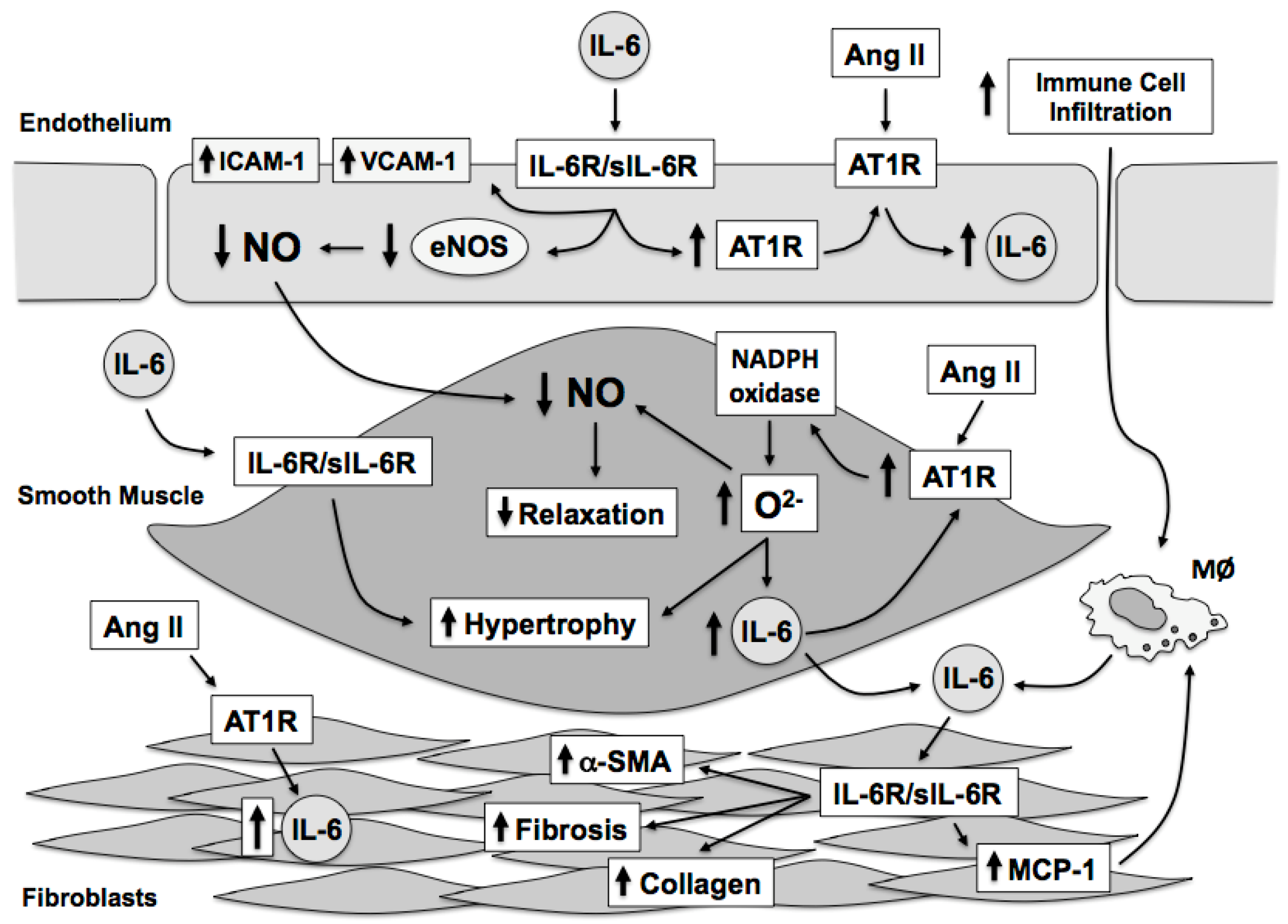

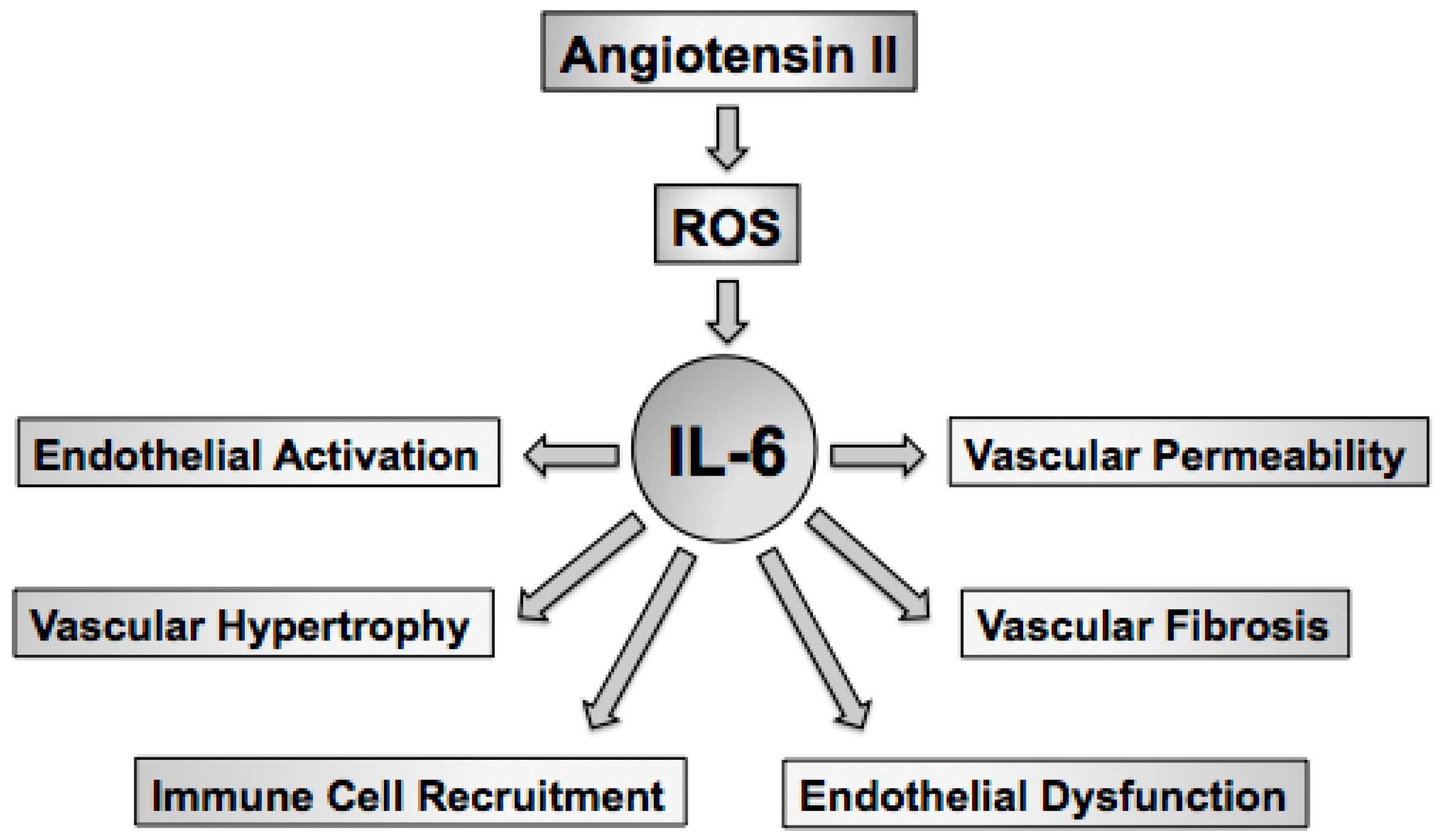{kind=link}
{kind=link}
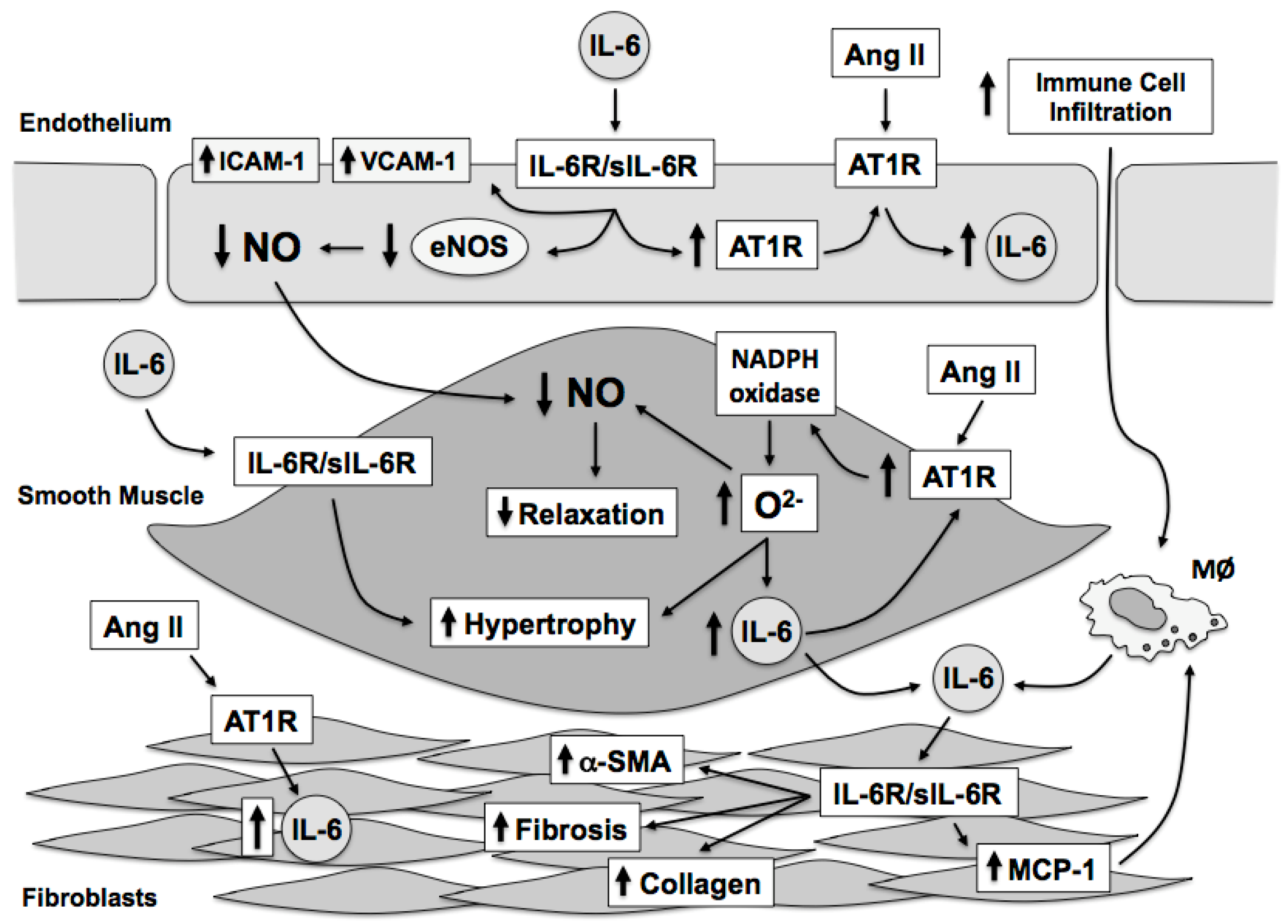{kind=link}
Table 1.
Stimuli that increase IL-6 expression in vascular cells.
| Stimulus | Reference |
|---|---|
| 15(S)-HETE | [80] |
| Activated Protein C | [63] |
| Aldosterone | [81] |
| Angiotensin II | [52,53,58,74,83] |
| Endothelin | [84,85] |
| Fibrinogen | [86] |
| Histamine | [87,88] |
| Homocysteine | [89] |
| Hydrogen Peroxide | [61,116] |
| Hypoxia | [59,71,90] |
| Interleukin-1α/β | [94,95,96,97,98] |
| Interleukin-4 | [99,100,101,102] |
| Interferon-γ | [98,103] |
| Lipopolysaccharide | [92,93,94,95,104,105,106] |
| Mechanical Stretch | [107,108,109,110] |
| Oxidized LDL | [111] |
| Platelet-derived Growth Factor | [112,113] |
| Serotonin | [114] |
| Thrombin | [86] |
| Tumor Necrosis Factor-α | [92,96,104,115] |
Table 2.
Effect of IL-6 infusion or IL-6-deficiency on Basal Blood Pressure.
| Basal Blood Pressure | Reference | |
|---|---|---|
| IL-6 Infusion | No effect | [177,178,179] |
| IL-6-Deficiency | No effect | [57,73,81,184,185,186,187,188,189] |
Table 3.
Effect of IL-6 Deficiency in Experimental Models of Hypertension.
| Model | Hypertension Development | Reference |
|---|---|---|
| Acute Stress | Partial Inhibition | [185] |
| Ang II | No Effect | [57,185] |
| Partial Inhibition | [73,187,188] | |
| Ang II + High-salt | No Effect | [184,190] |
| Partial Inhibition | [186] | |
| Ang II + RKM | No Effect | [189] |
| DOCA-salt | No Effect | [81] |
| High-salt | No Effect | [184,186] |
Ang II, angiotensin II; DOCA, deoxycorticosterone acetate-salt, RKM, reduced kidney mass.
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Didion, S.P. Cellular and Oxidative Mechanisms Associated with Interleukin-6 Signaling in the Vasculature. Int. J. Mol. Sci. 2017, 18, 2563. https://doi.org/10.3390/ijms18122563
AMA Style
Didion SP. Cellular and Oxidative Mechanisms Associated with Interleukin-6 Signaling in the Vasculature. International Journal of Molecular Sciences. 2017; 18(12):2563. https://doi.org/10.3390/ijms18122563
Chicago/Turabian StyleDidion, Sean P. 2017. "Cellular and Oxidative Mechanisms Associated with Interleukin-6 Signaling in the Vasculature" International Journal of Molecular Sciences 18, no. 12: 2563. https://doi.org/10.3390/ijms18122563
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.