Sphingosine 1-Phosphate (S1P) Signaling in Glioblastoma Multiforme—A Systematic Review


Abstract
:
1. Introduction
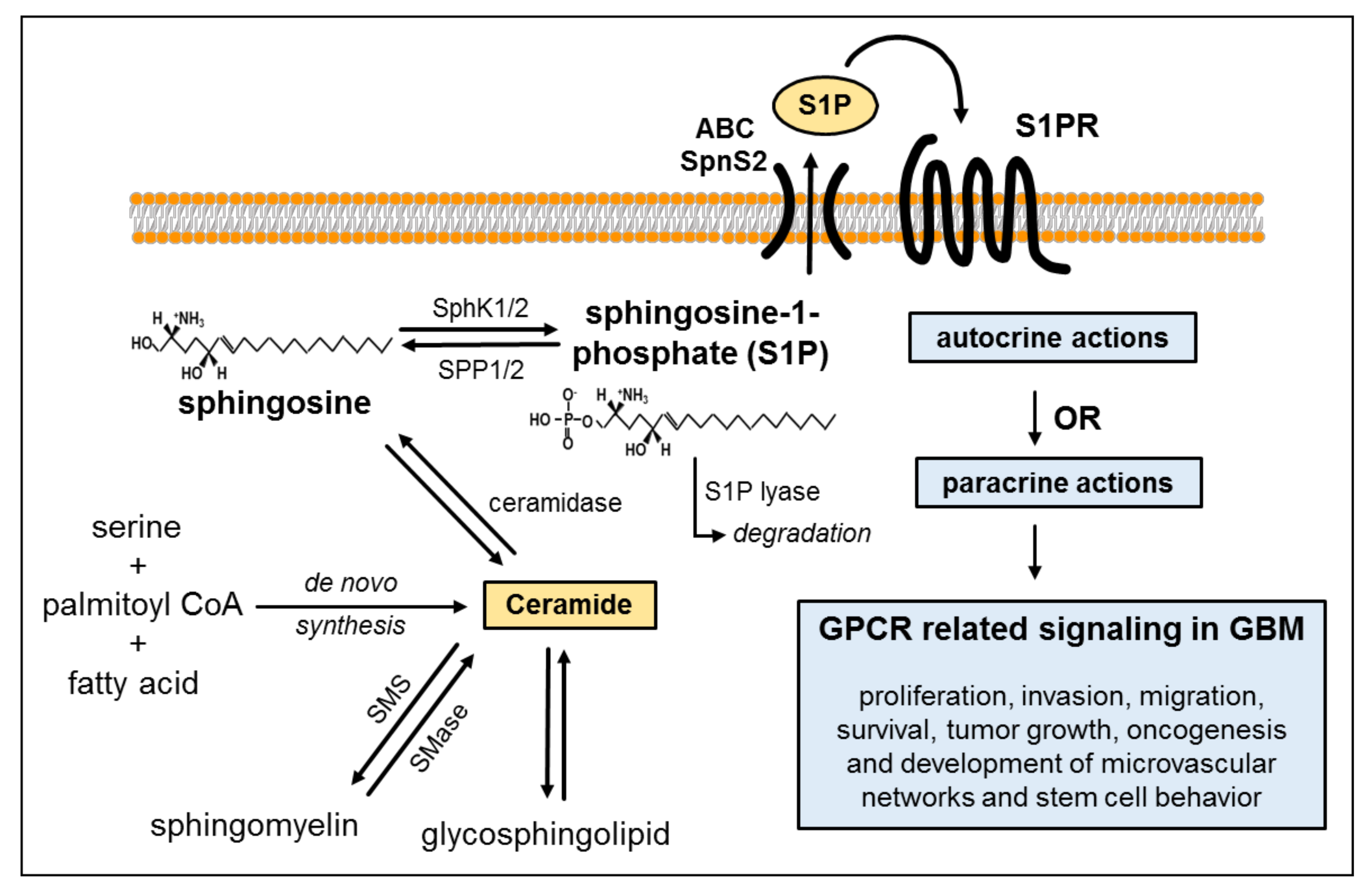
2. Biosynthesis of Ceramide and Sphingolipids
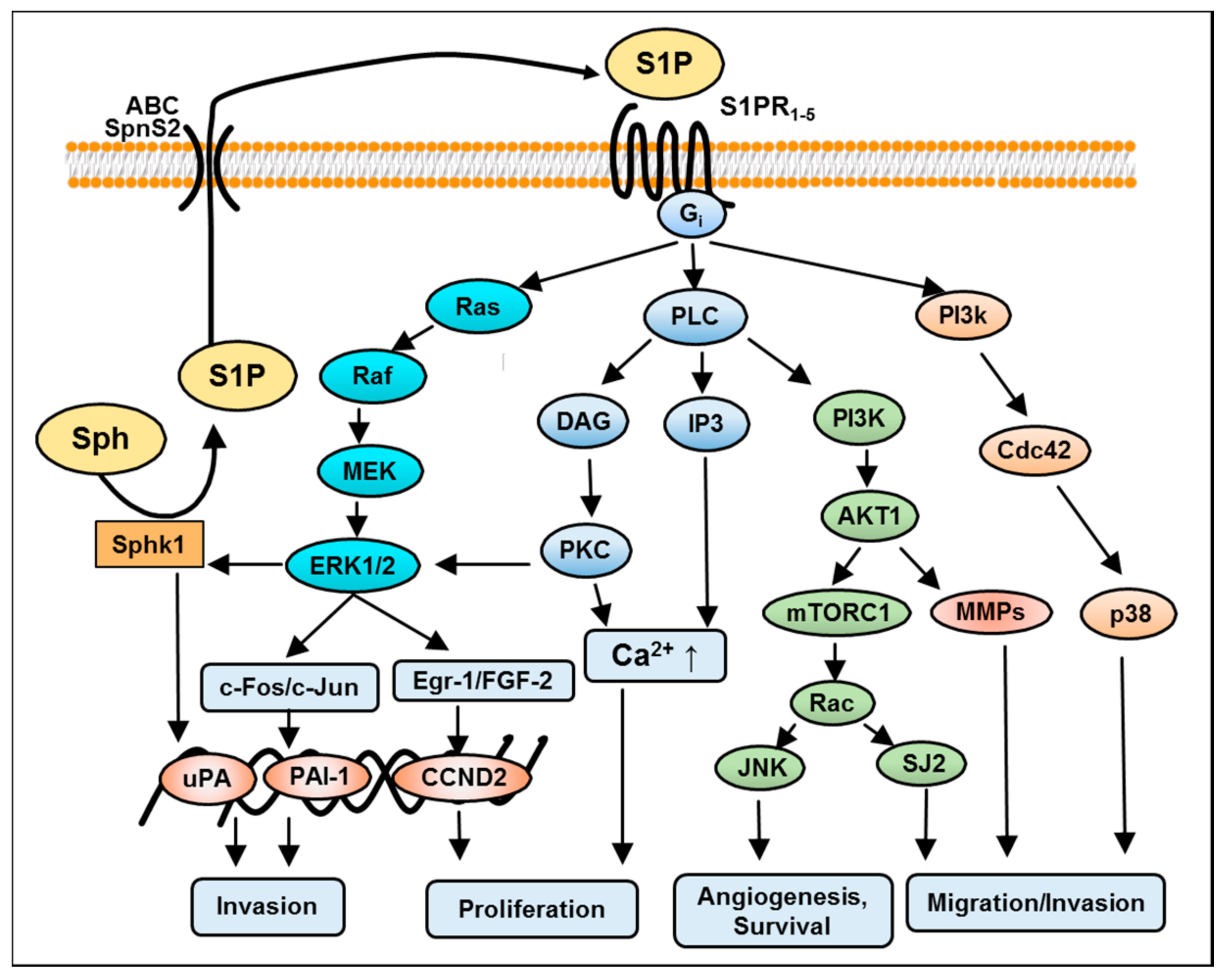
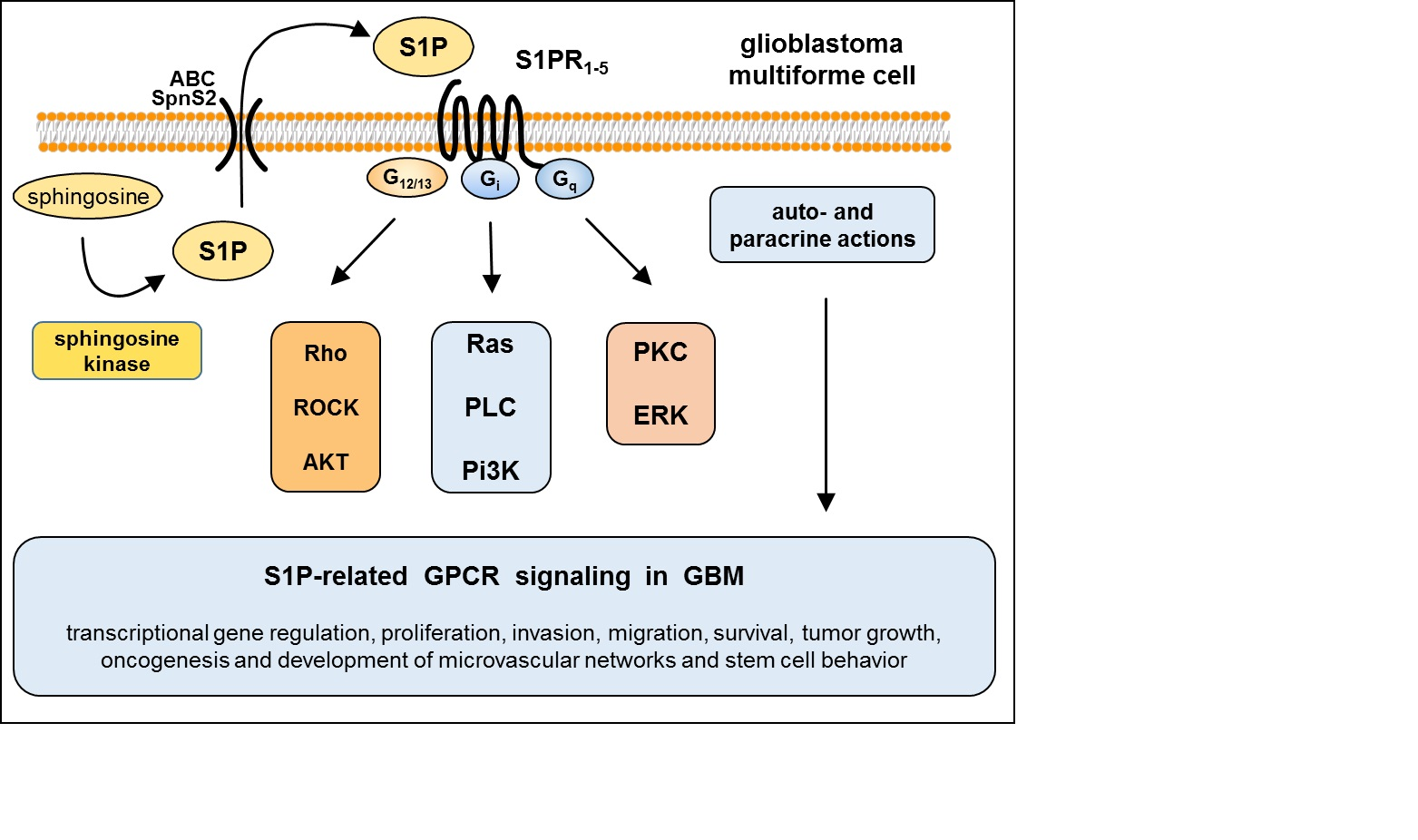
3. S1P Signaling in GBM
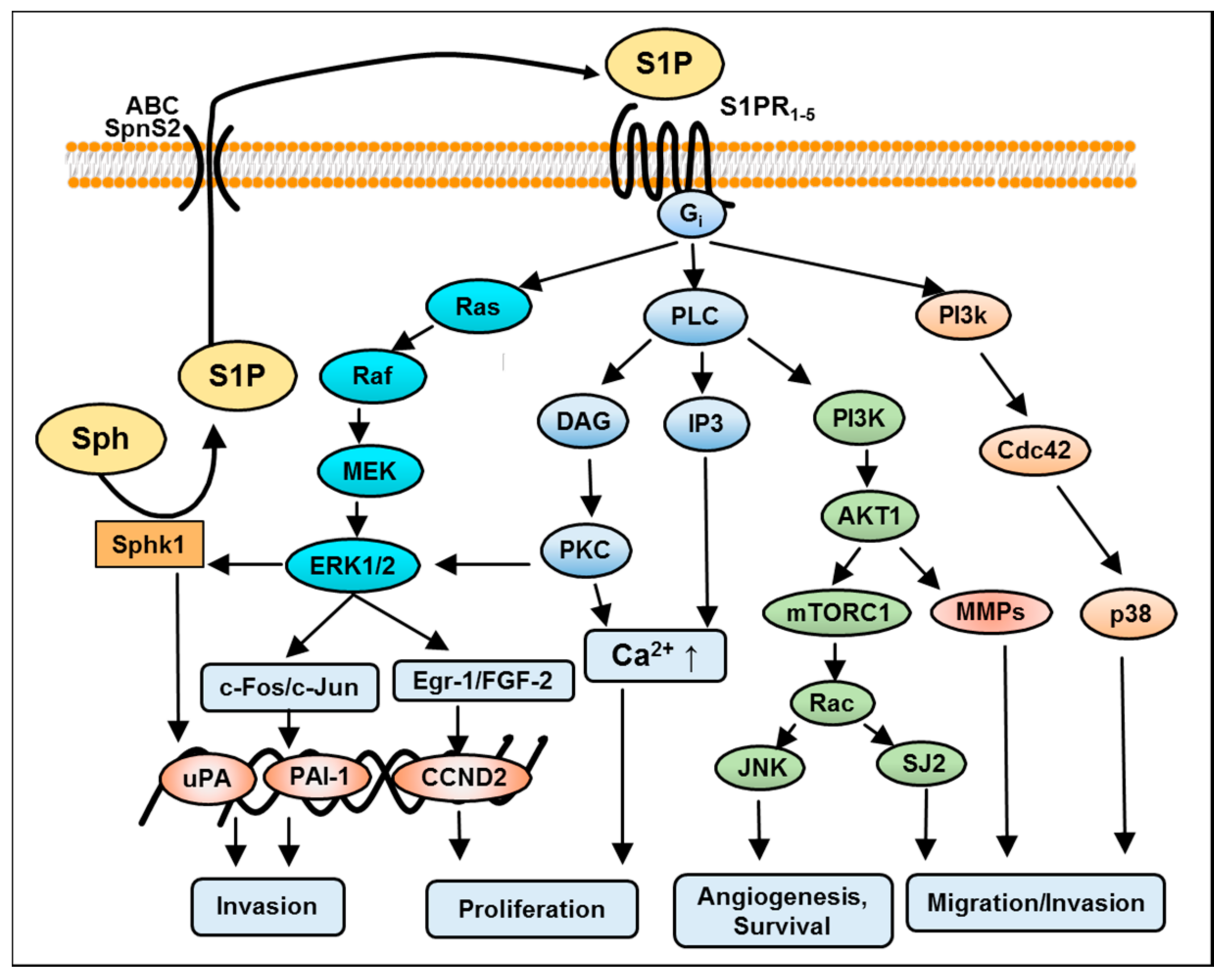
4. S1P-Induced Mitogen-Activated Protein Kinase (MAPK)/Extracellular Signal-Regulated Kinase (ERK) Kinase Signaling in GBM
5. S1P-Mediated Phosphoinositide 3-Kinase/AKT Pathway in GBM
6. S1P-Mediated Activation of Protein Kinase C in GBM
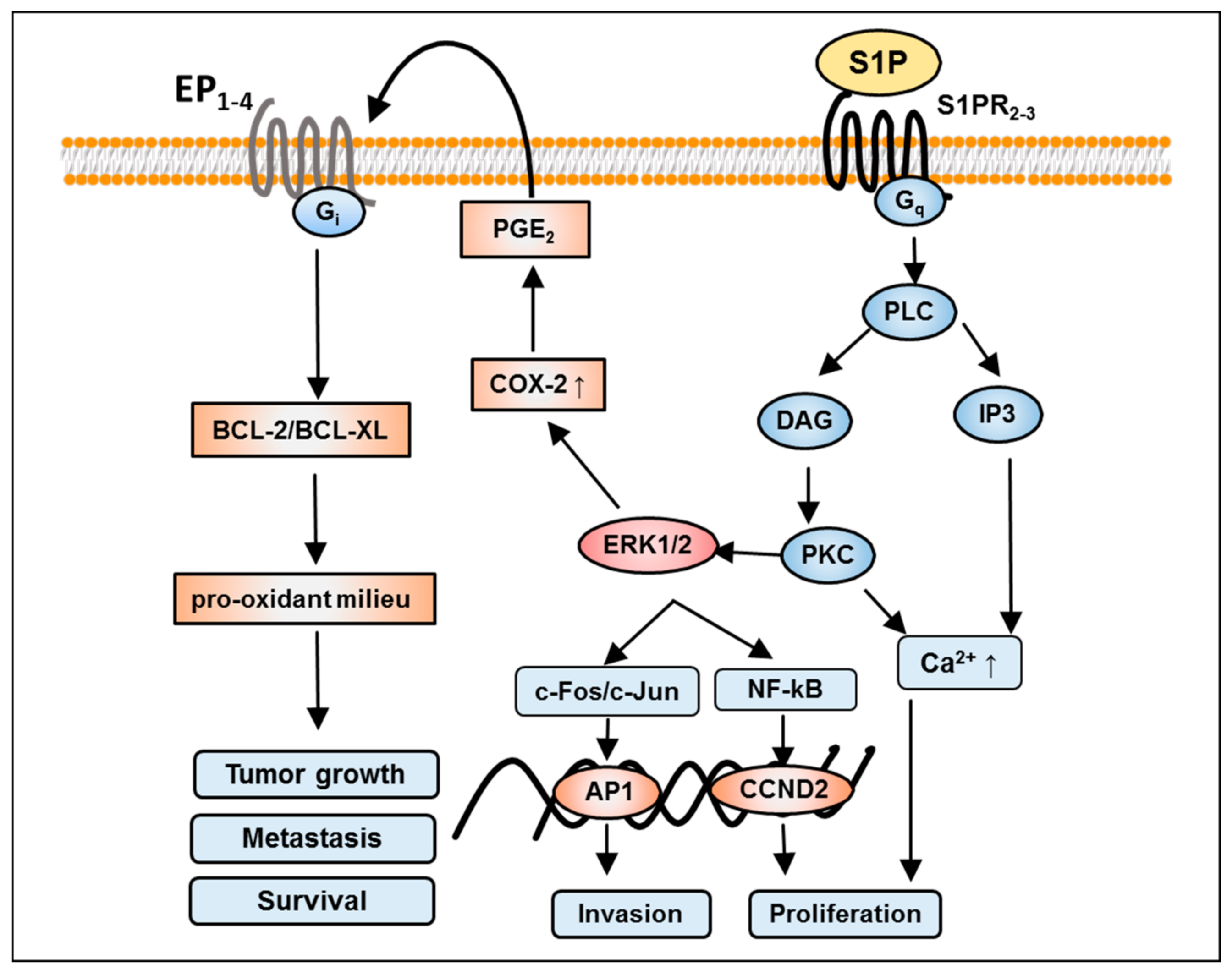
7. S1P-Mediated Activation of Phospholipase C and D in GBM
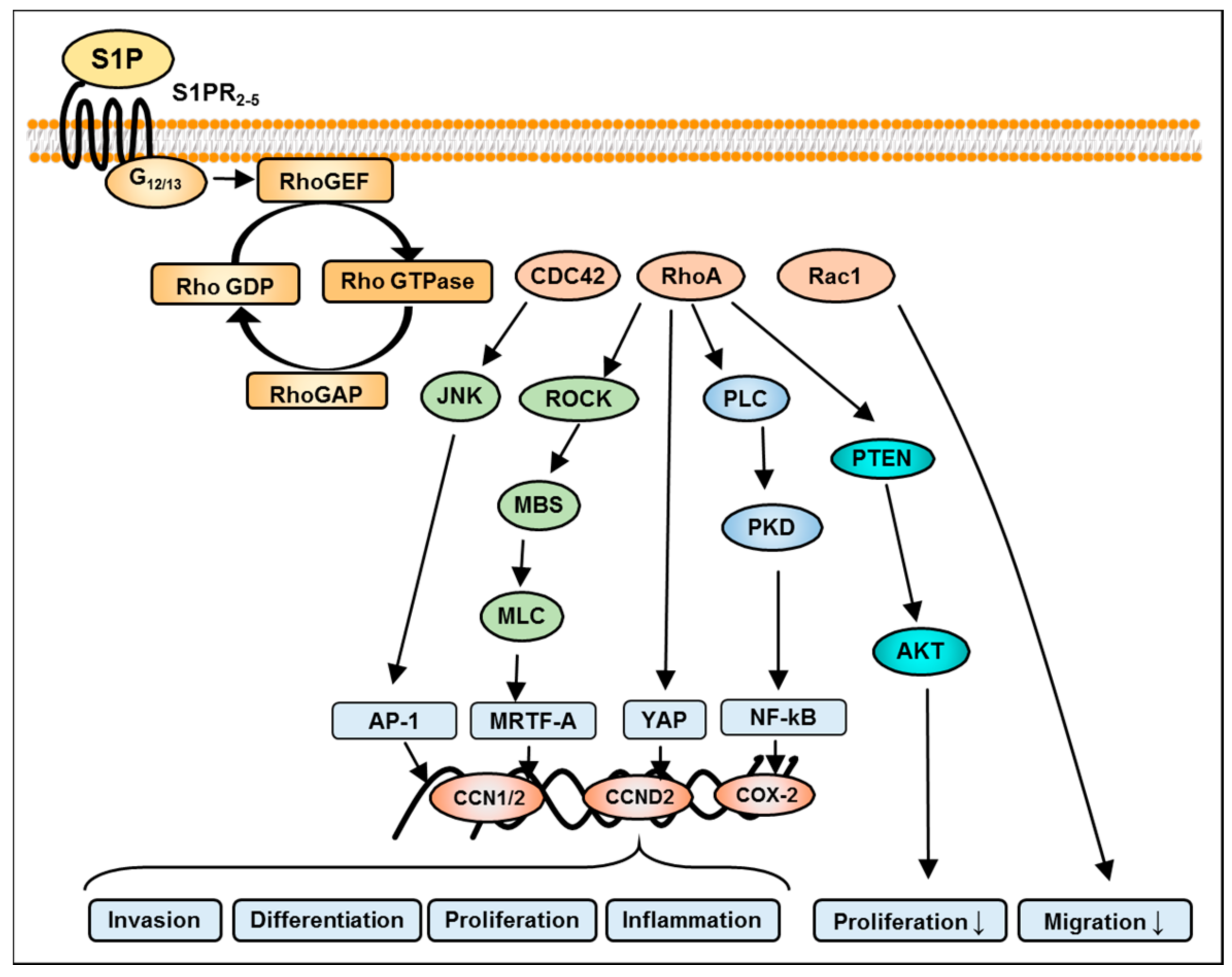
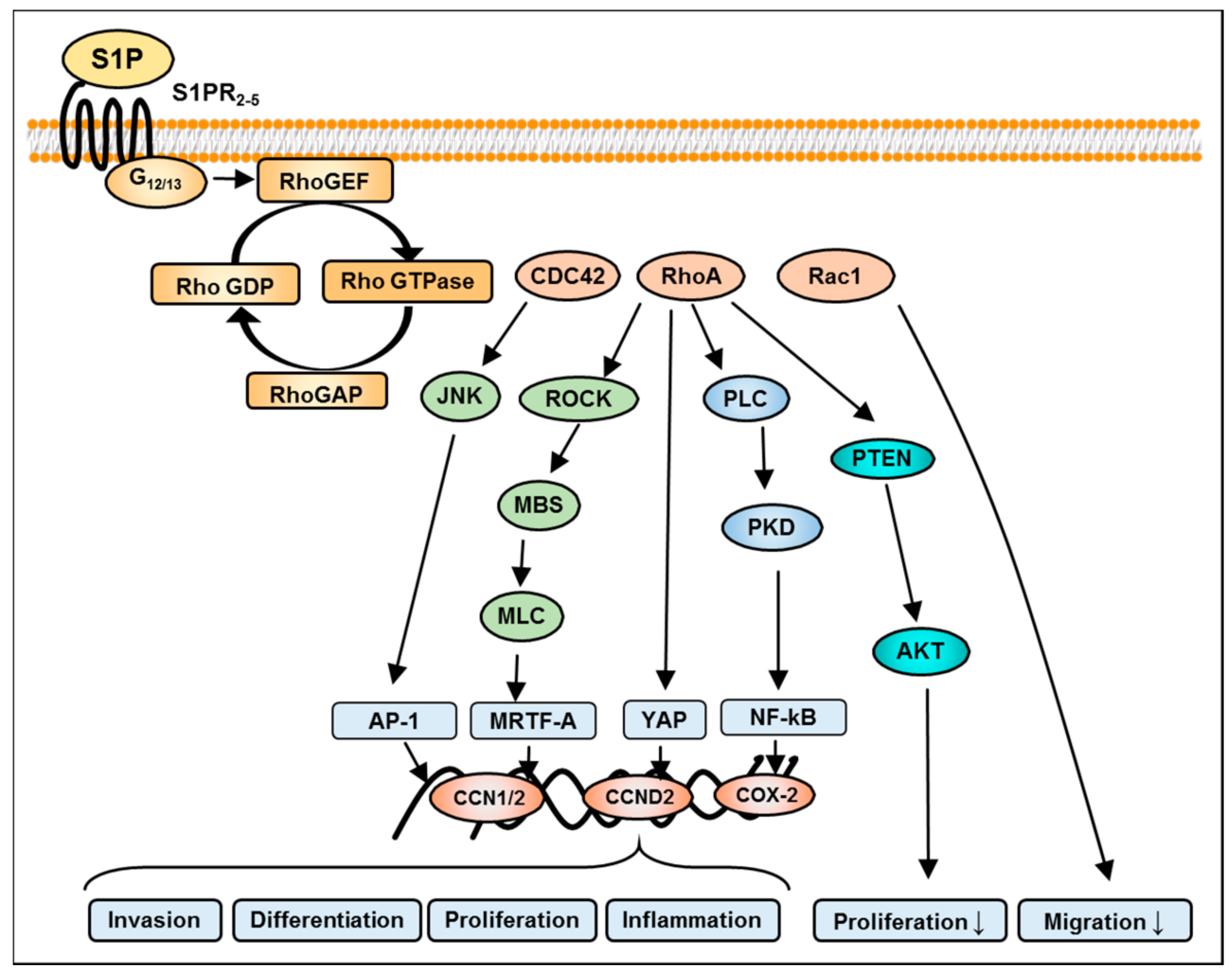
8. S1P-Mediated Activation of Rho Signaling in GBM
9. Summary
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pyne, N.J.; Pyne, S. Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 2010, 10, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Ponnusamy, S.; Meyers-Needham, M.; Senkal, C.E.; Saddoughi, S.A.; Sentelle, D.; Selvam, S.P.; Salas, A.; Ogretmen, B. Sphingolipids and cancer: Ceramide and sphingosine-1-phosphate in the regulation of cell death and drug resistance. Future Oncol. 2010, 6, 1603–1624. [Google Scholar] [CrossRef] [PubMed]
- Pyne, N.J.; Ohotski, J.; Bittman, R.; Pyne, S. The role of sphingosine 1-phosphate in inflammation and cancer. Adv. Biol. Regul. 2014, 54, 121–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oskouian, B.; Saba, J. Sphingosine-1-phosphate metabolism and intestinal tumorigenesis: Lipid signaling strikes again. Cell Cycle 2007, 6, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Tilly, J.L.; Kolesnick, R.N. Sphingolipids, apoptosis, cancer treatments and the ovary: Investigating a crime against female fertility. BBA-Mol. Cell. Biol. Lipids 2002, 1585, 135–138. [Google Scholar] [CrossRef]
- Sutphen, R.; Xu, Y.; Wilbanks, G.D.; Fiorica, J.; Grendys, E.C.; LaPolla, J.P.; Arango, H.; Hoffman, M.S.; Martino, M.; Wakeley, K. Lysophospholipids are potential biomarkers of ovarian cancer. Cancer Epidemiol Biomark. Prev. 2014, 13, 1185–1191. [Google Scholar]
- Watson, C.; Long, J.S.; Orange, C.; Tannahill, C.L.; Mallon, E.; McGlynn, L.M.; Pyne, S.; Pyne, N.J.; Edwards, J. High expression of sphingosine 1-phosphate receptors, S1P1 and S1P3, sphingosine kinase 1, and extracellular signal-regulated kinase-1/2 is associated with development of tamoxifen resistance in estrogen receptor-positive breast cancer patients. Am. J. Pathol. 2010, 177, 2205–2215. [Google Scholar] [CrossRef] [PubMed]
- Takabe, K.; Spiegel, S. Export of sphingosine-1-phosphate and cancer progression. J. Lipid Res. 2014, 55, 1839–1846. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Maceyka, M.; Hait, N.C.; Paugh, S.W.; Sankala, H.; Milstien, S.; Spiegel, S. Sphingosine kinase 1 is required for migration, proliferation and survival of MCF-7 human breast cancer cells. FEBS Lett. 2005, 579, 5313–5317. [Google Scholar] [CrossRef] [PubMed]
- Van Brocklyn, J.R.; Jackson, C.A.; Pearl, D.K; Kotur, M.S.; Snyder, P.J.; Prior, T.W. Sphingosine kinase-1 expression correlates with poor survival of patients with glioblastoma multiforme: Roles of sphingosine kinase isoforms in growth of glioblastoma cell lines. J. Neuropathol. Exp. Neurol. 2005, 64, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Bien-Möller, S.; Lange, S.; Holm, T.; Böhm, A.; Paland, H.; Küpper, J.; Herzog, S.; Weitmann, K.; Havemann, C.; Vogelgesang, S. Expression of S1P metabolizing enzymes and receptors correlate with survival time and regulate cell migration in glioblastoma multiforme. Oncotarget 2016, 7, 13031–13046. [Google Scholar] [CrossRef] [PubMed]
- Obinata, H.; Hla, T. Sphingosine 1-phosphate in coagulation and inflammation. Semin. Immunopathol. 2012, 34, 73–91. [Google Scholar] [CrossRef] [PubMed]
- Heffernan-Stroud, L.A.; Obeid, L.M. Sphingosine kinase 1 in cancer. Adv. Cancer Res. 2013, 117, 201–235. [Google Scholar] [PubMed]
- Le Stunff, H.; Milstien, S.; Spiegel, S. Generation and metabolism of bioactive sphingosine-1-phosphate. J. Cell. Biochem. 2004, 92, 882–899. [Google Scholar] [CrossRef] [PubMed]
- Mora, R.; Dokic, I.; Kees, T.; Hüber, C.M.; Keitel, D.; Geibig, R.; Brügge, B.; Zentgraf, H.; Brady, N.R; Régnier-Vigouroux, A. Sphingolipid rheostat alterations related to transformation can be exploited for specific induction of lysosomal cell death in murine and human glioma. Glia 2010, 58, 1364–1383. [Google Scholar] [PubMed]
- Ohgaki, H.; Kleihues, P. Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 2007, 170, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- Rulseh, A.M.; Keller, J.; Klener, J.; Šroubek, J.; Dbalý, V.; Syrůček, M.; Tovaryš, F.; Vymazal, J. Long-term survival of patients suffering from glioblastoma multiforme treated with tumor-treating fields. World J. Surg. Oncol. 2012, 10, 220. [Google Scholar] [CrossRef] [PubMed]
- Sordillo, L.A.; Sordillo, P.P.; Helson, L. Sphingosine kinase inhibitors as maintenance therapy of glioblastoma after ceramide-induced response. Anticancer Res. 2016, 36, 2085–2095. [Google Scholar] [PubMed]
- Bassi, R.; Anelli, V.; Giussani, P.; Tettamanti, G.; Viani, P.; Riboni, L. Sphingosine-1-phosphate is released by cerebellar astrocytes in response to bFGF and induces astrocyte proliferation through G1-protein-coupled receptors. Glia 2006, 53, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Anelli, V.; Gault, C.R.; Cheng, A.B.; Obeid, L.M. Sphingosine kinase 1 is up-regulated during hypoxia in U87-MG glioma cells role of hypoxia-inducible factors 1 and 2. J. Biol. Chem. 2008, 283, 3365–3375. [Google Scholar] [CrossRef] [PubMed]
- Strub, G.M.; Maceyka, M.; Hait, N.C.; Milstien, S.; Spiegel, S. Extracellular and Intracellular Actions of Sphingosine-1-Phosphate. Adv. Exp. Med. Biol. 2010, 688, 141–155. [Google Scholar] [PubMed]
- Marfia, G.; Campanella, R.; Navone, S.E.; Di Vito, C.; Riccitelli, E.; Hadi, L.A.; Bornati, A.; de Rezende, G.; Giussani, P.; Tringali, C.; et al. Autocrine/paracrine sphingosine-1-phosphate fuels proliferative and stemness qualities of glioblastoma stem cells. Glia 2014, 62, 1968–1981. [Google Scholar] [CrossRef] [PubMed]
- Abuhusain, H.J.; Matin, A.; Qiao, Q.; Shen, H.; Kain, N.; Day, B.W.; Stringer, B.W.; Daniels, B.; Laaksonen, M.A.; Teo, C.; et al. Metabolic Shift Favoring Sphingosine 1-Phosphate at the Expense of Ceramide Controls Glioblastoma Angiogenesis. J. Biol. Chem. 2013, 288, 37355–37364. [Google Scholar] [CrossRef] [PubMed]
- Böhm, A.; Flößer, A.; Ermler, S.; Fender, A.C.; Lüth, A.; Kleuser, B.; Schrör, K.; Rauch, B.H. Factor-Xa-induced mitogenesis and migration require sphingosine kinase activity and S1P formation in human vascular smooth muscle cells. Cardiovasc. Res. 2013, 99, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Mahajan-Thakur, S.; Sostmann, B.D.; Fender, A.C.; Behrendt, D.; Felix, S.B.; Schrör, K.; Rauch, B.H. Sphingosine-1-phosphate induces thrombin receptor PAR-4 expression to enhance cell migration and COX-2 formation in human monocytes. J. Leukocyte Biol. 2014, 96, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Van Brocklyn, J.R. Sphingolipid signaling pathways as potential therapeutic targets in gliomas. Mini Rev. Med. Chem. 2007, 7, 984–990. [Google Scholar] [CrossRef] [PubMed]
- Hla, T. Signaling and biological actions of sphingosine 1-phosphate. Pharmacol. Res. 2003, 47, 401–407. [Google Scholar] [CrossRef]
- Rosen, H.; Goetzl, E.J. Sphingosine 1-phosphate and its receptors: An autocrine and paracrine network. Nat. Rev. Immunol. 2005, 5, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Nakada, M.; Sugimoto, N.; Harada, T.; Hayashi, Y.; Kita, D.; Uchiyama, N.; Hayashi, Y.; Yachie, A.; Takuwa, Y. Sphingosine-1-phosphate receptor type 1 regulates glioma cell proliferation and correlates with patient survival. Int. J. Cancer 2010, 126, 2341–2352. [Google Scholar] [CrossRef] [PubMed]
- Bernhart, E.; Damm, S.; Wintersperger, A.; Nusshold, C.; Brunner, A.M.; Plastira, I.; Rechberger, G.; Reicher, H.; Wadsack, C.; Zimmer, A. Interference with distinct steps of sphingolipid synthesis and signaling attenuates proliferation of U87-MG glioma cells. Biochem. Pharmacol. 2015, 96, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Kim, Y.L.; Sacket, S.J.; Kim, H.L.; Han, M.; Park, D.S.; Lee, B.K.; Lee, W.K.; Ha, H.J.; Im, D.S. Sphingosine 1-phosphate (S1P) induces shape change in rat C6 glioma cells through the S1P2 receptor: Development of an agonist for S1P receptors. J. Pharm. Pharmacol. 2007, 59, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Quint, K.; Stiel, N.; Neureiter, D.; Schlicker, H.U.; Nimsky, C.; Ocker, M.; Strik, H.; Kolodziej, M.A. The role of sphingosine kinase isoforms and receptors S1P1, S1P2, S1P3, and S1P5 in primary, secondary, and recurrent glioblastomas. Tumour Biol. 2014, 35, 8979–8989. [Google Scholar] [CrossRef] [PubMed]
- Siehler, S.; Manning, D.R. Pathways of transduction engaged by sphingosine 1-phosphate through G protein-coupled receptors. BBA-Mol. Cell. Biol. Lipids 2002, 1582, 94–99. [Google Scholar] [CrossRef]
- Rekers, H.; Sminia, P.; Peters, G.J. Towards tailored therapy of glioblastoma multiforme. J. Chemother. 2011, 23, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Bryan, L.; Paugh, B.S.; Kapitonov, D.; Wilczynska, K.M.; Alvarez, S.M.; Singh, S.K.; Milstien, S.; Spiegel, S.; Kordula, T. Sphingosine-1-phosphate and interleukin-1 independently regulate plasminogen activator inhibitor-1 and urokinase-type plasminogen activator receptor expression in glioblastoma cells: Implications for invasiveness. Mol. Cancer Res. 2008, 6, 1469–1477. [Google Scholar] [CrossRef] [PubMed]
- Young, N.; Van Brocklyn, J.R. Roles of sphingosine-1-phosphate (S1P) receptors in malignant behavior of glioma cells. Differential effects of S1P2 on cell migration and invasiveness. Exp. Cell Res. 2007, 313, 1615–1627. [Google Scholar] [CrossRef] [PubMed]
- Fortier, S.; Labelle, D.; Sina, A.; Moreau, R.; Annabi, B. Silencing of the MT1-MMP/G6PT axis suppresses calcium mobilization by sphingosine-1-phosphate in glioblastoma cells. FEBS Lett. 2008, 582, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Kapitonov, D.; Allegood, J.C.; Mitchell, C.; Hait, N.C.; Almenara, J.A.; Adams, J.K.; Zipkin, R.E.; Dent, P.; Kordula, T.; Milstien, S.; Spiegel, S. Targeting sphingosine kinase 1 inhibits Akt signaling, induces apoptosis, and suppresses growth of human glioblastoma cells and xenografts. Cancer Res. 2009, 69, 6915–6923. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Tomura, H.; Igarashi, Y.; Ui, M.; Okajima, F. Possible involvement of cell surface receptors in sphingosine 1-phosphate-induced activation of extracellular signal-regulated kinase in C6 glioma cells. Mol. Pharmacol. 1999, 55, 126–133. [Google Scholar] [PubMed]
- Malchinkhuu, E.; Sato, K.; Horiuchi, Y.; Mogi, C.; Ohwada, S.; Ishiuchi, S.; Saito, N.; Kurose, H.; Tomura, H.; Okajima, F. Role of p38 mitogen-activated kinase and c-Jun terminal kinase in migration response to lysophosphatidic acid and sphingosine-1-phosphate in glioma cells. Oncogene 2005, 24, 6676–6688. [Google Scholar] [CrossRef] [PubMed]
- Yu, O.M.; Miyamoto, S.; Brown, J.H. Myocardin-Related transcription factor A and yes-associated protein exert dual control in G protein-coupled receptor- and RhoA-mediated transcriptional regulation and cell proliferation. Mol. Cell Biol. 2015, 36, 39–49. [Google Scholar] [PubMed]
- Van Brocklyn, J.R.; Young, N.; Roof, R. Sphingosine-1-phosphate stimulates motility and invasiveness of human glioblastoma multiforme cells. Cancer Lett. 2003, 199, 53–60. [Google Scholar] [CrossRef]
- Yoshida, Y.; Nakada, M.; Harada, T.; Tanaka, S.; Furuta, T.; Hayashi, Y.; Kita, D.; Uchiyama, N.; Hayashi, Y.; Hamada, J. The expression level of sphingosine-1-phosphate receptor type 1 is related to MIB-1 labeling index and predicts survival of glioblastoma patients. J. Neurooncol. 2010, 98, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Van Brocklyn, J.; Letterle, C.; Snyder, P.; Prior, T. Sphingosine-1-phosphate stimulates human glioma cell proliferation through Gi-coupled receptors: Role of ERK MAP kinase and phosphatidylinositol 3-kinase beta. Cancer Lett. 2002, 181, 195–204. [Google Scholar] [CrossRef]
- Hu, W.M.; Li, L.; Jing, B.Q.; Zhao, Y.S.; Wang, C.L.; Feng, L.; Feng, L.; Xie, Y.E. Effect of S1P5 on proliferation and migration of human esophageal cancer cells. World J. Gastroenterol. 2010, 15, 1859–1866. [Google Scholar] [CrossRef]
- Sato, K.; Ui, M.; Okajima, F. Differential roles of Edg-1 and Edg-5, sphingosine 1-phosphate receptors, in the signaling pathways in C6 glioma cells. Brain Res. Mol. Brain Res. 2000, 85, 151–160. [Google Scholar] [CrossRef]
- Beretta, F.; Bassani, S.; Binda, E.; Verpelli, C.; Bello, L.; Galli, R.; Passafaro, M. The GluR2 subunit inhibits proliferation by inactivating Src-MAPK signalling and induces apoptosis by means of caspase 3/6-dependent activation in glioma cells. Eur. J. Neurosci. 2009, 30, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Bunda, S.; Burrell, K.; Heir, P.; Zeng, L.; Alamsahebpour, A.; Kano, Y.; Raught, B.; Zhang, Z.Y.; Zadeh, G.; Ohh, M. Inhibition of SHP2-mediated dephosphorylation of Ras suppresses oncogenesis. Nat. Commun. 2015, 6, 8859. [Google Scholar] [CrossRef] [PubMed]
- Gault, C.R.; Eblen, S.T.; Neumann, C.A.; Hannun, Y.A.; Obeid, L.M. Oncogenic K-Ras regulates bioactive sphingolipids in a sphingosine kinase 1-dependent manner. J. Biol. Chem. 2012, 287, 31794–31803. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef] [PubMed]
- Westhoff, M.A.; Karpel-Massler, G.; Brühl, O.; Enzenmüller, S.; La Ferla-Brühl, K.; Siegelin, M.D.; Nonnenmacher, L.; Debatin, K.M. A critical evaluation of PI3K inhibition in Glioblastoma and Neuroblastoma therapy. Mol. Cell. Ther. 2014, 2, 32. [Google Scholar] [CrossRef] [PubMed]
- Takuwa, N.; Du, W.; Kaneko, E.; Okamoto, Y.; Yoshioka, K.; Takuwa, Y. Tumor-suppressive sphingosine-1-phosphate receptor-2 counteracting tumor-promoting sphingosine-1-phosphate receptor-1 and sphingosine kinase 1-Jekyll Hidden behind Hyde. Am. J. Cancer Res. 2011, 1, 460–481. [Google Scholar] [PubMed]
- Lepley, D.; Paik, J.H.; Hla, T.; Ferrer, F. The G protein-coupled receptor S1P2 regulates Rho/Rho kinase pathway to inhibit tumor cell migration. Cancer Res. 2005, 65, 3788–3795. [Google Scholar] [CrossRef] [PubMed]
- Malchinkhuu, E.; Sato, K.; Maehama, T.; Mogi, C.; Tomura, H.; Ishiuchi, S.; Yoshimoto, Y.; Kurose, H.; Okajima, F. S1P(2) receptors mediate inhibition of glioma cell migration through Rho signaling pathways independent of PTEN. Biochem. Biophys. Res. Commun. 2008, 366, 963–968. [Google Scholar] [CrossRef] [PubMed]
- McDowell, K.A.; Riggins, G.J.; Gallia, G.L. Targeting the AKT pathway in glioblastoma. Curr. Pharm. Des. 2011, 17, 2411–2420. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.; Meyerson, M.; Aldape, K.; Bigner, D.; Mikkelsen, T.; VandenBerg, S.; Kahn, A.; Penny, R.; Ferguson, M.L.; Gerhard, D.S.; et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar]
- Chen, H.C.; Appeddu, P.A.; Isoda, H.; Guan, J.L. Phosphorylation of tyrosine 397 in focal adhesion kinase is required for binding phosphatidylinositol 3-kinase. J. Biol. Chem. 1996, 271, 26329–26334. [Google Scholar] [CrossRef] [PubMed]
- Westhoff, M.A.; Bruhl, O.; Nonnenmacher, L.; Karpel-Massler, G.; Debatin, K.M. Killing me softly-future challenges in apoptosis research. Int. J. Mol. Sci. 2014, 15, 3746–3767. [Google Scholar] [CrossRef] [PubMed]
- Gulati, N.; Karsy, M.; Albert, L.; Murali, R.; Jhanwar-Uniyal, M. Involvement of mTORC1 and mTORC2 in regulation of glioblastoma multiforme growth and motility. Int. J. Oncol. 2009, 35, 731–740. [Google Scholar] [PubMed]
- Do Carmo, A.; Patricio, I.; Cruz, M.T.; Carvalheiro, H.; Oliveira, C.R.; Lopes, M.C. CXCL12/CXCR4 promotes motility and proliferation of glioma cells. Cancer Biol. Ther. 2010, 9, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Benedetti, L.G.; Abera, M.B.; Wang, H.; Abba, M.; Kazanietz, M.G. Protein kinase C and cancer: What we know and what we do not. Oncogene 2014, 33, 5225–5237. [Google Scholar] [CrossRef] [PubMed]
- Bazzi, M.D.; Nelsestuen, G.L. Mechanism of protein kinase C inhibition by sphingosine. Biochem. Biophys. Res. Commun. 1987, 146, 203–207. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Bell, R.M. Regulation of protein kinase C by sphingosine and lysosphingolipids. Clin. Chim. Acta 1989, 185, 333–345. [Google Scholar] [CrossRef]
- Bredel, M.; Pollack, I.F. The role of protein kinase C (PKC) in the evolution and proliferation of malignant gliomas, and the application of PKC inhibition as a novel approach to anti-glioma therapy. Acta Neurochir. 1997, 139, 1000–1013. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.J.; Procyk, K.J.; Leitges, M.; Parker, P.J. PKC alpha protein but not kinase activity is critical for glioma cell proliferation and survival. Int. J. Cancer 2008, 123, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Menon, K.; Alvarez, E.; Galbreath, E.; Shih, C.; Faul, M. Antiangiogenic and antitumor effects of a protein kinase Cbeta inhibitor in human T98G glioblastoma multiforme xenografts. Clin. Cancer Res. 2001, 7, 634–640. [Google Scholar] [PubMed]
- Mishima, K.; Ohno, S.; Shitara, N.; Yamaoka, K.; Suzuki, K. Opposite effects of the overexpression of protein kinase Cγ and δ on the growth properties of human glioma cell line U251 MG. Biochem. Biophys. Res. Commun. 1994, 201, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Uht, R.M.; Amos, S.; Martin, P.M.; Riggan, A.E.; Hussaini, I.M. The protein kinase C-η isoform induces proliferation in glioblastoma cell lines through an ERK/Elk-1 pathway. Oncogene 2007, 26, 2885–2893. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Win, H.; Desai, S.; Patel, K.; Matthews, J.A.; Acevedo-Duncan, M. Involvement of PKC-iota in glioma proliferation. Cell Prolif. 2008, 41, 122–135. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Goldthwait, D.A.; Mapstone, T. A search for glial expression in tumors of the central nervous system. Pediatr. Neurosurg. 1994, 20, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Sharif, T.R.; Sharif, M. Overexpression of protein kinase C epsilon in astroglial brain tumor derived cell lines and primary tumor samples. Int. J. Oncol. 1999, 15, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Goldthwait, D.A.; Mapstone, T. The identification of four protein kinase C isoforms in human glioblastoma cell lines: PKC alpha, gamma, epsilon, and zeta. J. Neurosurg. 1994, 81, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.R.; Becker, K.P.; Facchinetti, M.M.; Hannun, Y.A.; Obeid, L.M. PKC-dependent activation of sphingosine kinase 1 and translocation to the plasma membrane extracellular release of sphingosine-1-phosphate induced by phorbol 12-myristate 13-acetate(PMA). J. Biol. Chem. 2002, 277, 35257–35262. [Google Scholar] [CrossRef] [PubMed]
- Kotelevets, N.; Fabbro, D.; Huwiler, A.; Zangemeister-Wittke, U. Targeting sphingosine kinase 1 in carcinoma cells decreases proliferation and survival by compromising PKC activity and cytokinesis. PLoS ONE 2012, 7, e39209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, N.; Pearl, D.K.; Van Brocklyn, J.R. Sphingosine-1-phosphate regulates glioblastoma cell invasiveness through the urokinase plasminogen activator system and CCN1/Cyr61. Mol. Cancer Res. 2009, 7, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.G.; Wang, X.F.; Zhou, J.S.; Wang, F.C.; Li, X.W.; Lü, H.Z. Activation of PKCα is required for migration of C6 glioma cells. Acta Neurobiol. Exp. 2010, 70, 239–245. [Google Scholar]
- Graff, J.R.; McNulty, A.M.; Hanna, K.R.; Konicek, B.W.; Lynch, R.L.; Bailey, S.N.; Banks, C.; Capen, A.; Goode, R.; Lewis, J.E. The protein kinase Cβ-selective inhibitor, Enzastaurin (LY317615.HCl), suppresses signaling through the AKT pathway, induces apoptosis, and suppresses growth of human colon cancer and glioblastoma xenografts. Cancer Res. 2005, 65, 7462–7469. [Google Scholar] [CrossRef] [PubMed]
- Acevedo-Duncan, M.; Patel, R.; Whelan, S.; Bicaku, E. Human glioma PKC-iota and PKC-βII phosphorylate cyclin-dependent kinase activating kinase during the cell cycle. Cell Proliferat. 2002, 35, 23–36. [Google Scholar] [CrossRef]
- Paugh, B.S.; Paugh, S.W.; Bryan, L.; Kapitonov, D.; Wilczynska, K.M.; Gopalan, S.M.; Rokita, H.; Milstien, S.; Spiegel, S.; Kordula, T. EGF regulates plasminogen activator inhibitor-1 (PAI-1) by a pathway involving c-Src, PKCδ, and sphingosine kinase 1 in glioblastoma cells. FASEB J. 2008, 22, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Hwang, E.; Yoo, K.C.; Kang, S.G.; Kim, R.K.; Cui, Y.H.; Lee, H.J.; Kim, M.J.; Lee, J.S.; Kim, I.G.; Suh, Y.; et al. PKCδ activated by c-MET enhances infiltration of human glioblastoma cells through NOTCH2 signaling. Oncotarget 2016, 7, 4890–4902. [Google Scholar] [CrossRef] [PubMed]
- Alapati, K.; Gopinath, S.; Malla, R.R.; Dasari, V.R.; Rao, J.S. uPAR and cathepsin B knockdown inhibits radiation-induced PKC integrated integrin signaling to the cytoskeleton of glioma-initiating cells. Int. J. Oncol. 2012, 41, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Aeder, S.E.; Martin, P.M.; Soh, J.W.; Hussaini, I.M. PKC-η mediates glioblastoma cell proliferation through the Akt and mTOR signaling pathways. Oncogene 2004, 23, 9062–9069. [Google Scholar] [CrossRef] [PubMed]
- Okhrimenko, H.; Lu, W.; Xiang, C.; Hamburger, N.; Kazimirsky, G.; Brodie, C. Protein kinase C-epsilon regulates the apoptosis and survival of glioma cells. Cancer Res. 2005, 65, 7301–7309. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, R.M.; Parolin, D.A.; Lorimer, I.A. Regulation of glioblastoma cell invasion by PKCι and RhoB. Oncogene 2008, 27, 3587–3595. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Gu, F.; Li, W.; Zhang, B.; Niu, R.; Fu, L.; Zhang, N.; Ma, Y. Reduction of protein kinase C ζ inhibits migration and invasion of human glioblastoma cells. J. Neurochem. 2009, 109, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Mattie, M.; Brooker, G.; Spiegel, S. Sphingosine-1-phosphate, a putative second messenger, mobilizes calcium from internal storesvia an inositol trisphosphate-independent pathway. J. Biol. Chem. 1994, 269, 3181–3188. [Google Scholar] [PubMed]
- Mawrin, C.; Diete, S.; Treuheit, T.; Kropf, S.; Vorwerk, C.K.; Boltze, C.; Kirches, E.; Firsching, R.; Dietzmann, K. Prognostic relevance of MAPK expression in glioblastoma multiforme. Int. J. Oncol. 2003, 23, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Okajima, F.; Tomura, H.; Sho, K.; Nochi, H.; Tamoto, K.; Kondo, Y. Involvement of pertussis toxin-sensitive GTP-binding proteins in sphingosine 1-phosphate-induced activation of phospholipase C-Ca2+ system in HL60 leukemia cells. FEBS Lett. 1996, 379, 260–264. [Google Scholar] [CrossRef]
- Czajkowski, R.; Sabala, P.; Baranska, J. Sphingosine modulates Ca signals via phospholipase C dependent pathway in glioma C6 cells. Acta Neurobiol. Exp. 1997, 57, 353. [Google Scholar]
- Malchinkhuu, E.; Sato, K.; Muraki, T.; Ishikawa, K.; Kuwabara, A.; Okajima, F. Assessment of the role of sphingosine 1-phosphate and its receptors in high-density lipoprotein-induced stimulation of astroglial cell function. Biochem. J. 2003, 370, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.A.; Thomas, P.G.; Lindsley, C.W. Targeting phospholipase D in cancer, infection and neurodegenerative disorders. Nat. Rev. Drug Discov. 2017, 16, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Kantonen, S.; Henkels, K.M.; Gomez-Cambronero, J. A new signaling pathway (JAK-Fes-phospholipase D) that is enhanced in highly proliferative breast cancer cells. J. Biol. Chem. 2013, 288, 9881–9891. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B.H.; Min, G.; Bae, Y.S.; Bae, Y.S.; Min, D.S. Phospholipase D is activated and phosphorylated by casein kinase-II in human U87 astroglioma cells. Exp. Mol. Med. 2006, 38, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Bruntz, R.C.; Taylor, H.E.; Lindsley, C.W.; Brown, H.A. Phospholipase D2 mediates survival signaling through direct regulation of Akt in glioblastoma cells. Pharmacol. Rev. 2014, 66, 1033–1079. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Ahn, B.H.; Hong, Y.K.; Min, S. Overexpression of phospholipase D enhances matrix metalloproteinase-2 expression and glioma cell invasion via protein kinase C and protein kinase A/NF-kappaB/Sp1-mediated signaling pathways. Carcinogenesis 2009, 30, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Etienne-Manneville, S.; Hall, A. Rho GTPases in cell biology. Nature 2002, 420, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Radeff-Huang, J.; Seasholtz, T.M.; Matteo, R.G.; Brown, J.H. G protein mediated signaling pathways in lysophospholipid induced cell proliferation and survival. J. Cell. Biochem. 2004, 92, 949–966. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, F.; Van Keymeulen, A.; Herzmark, P.; Straight, A.; Kelly, K.; Takuwa, Y.; Sugimoto, N.; Mitchison, T.; Bourne, H.R. Divergent signals and cytoskeletal assemblies regulate self-organizing polarity in neutrophils. Cell 2003, 114, 201–214. [Google Scholar] [CrossRef]
- Sanchez, T.; Thangada, S.; Wu, M.T; Kontos, C.D; Wu, D.; Wu, H.; Hla, T. PTEN as an effector in the signaling of antimigratory G protein-coupled receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 4312–4317. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, N.; Takuwa, N.; Okamoto, H.; Sakurada, S.; Takuwa, Y. Inhibitory and stimulatory regulation of Rac and cell motility by the G12/13-Rho and Gi pathways integrated downstream of a single G protein-coupled sphingosine-1-phosphate receptor isoform. Mol. Cell Biol. 2003, 23, 1534–1545. [Google Scholar] [CrossRef] [PubMed]
- Michaud, J.; Im, D.S.; Hla, T. Inhibitory role of sphingosine 1-phosphate receptor 2 in macrophage recruitment during inflammation. J. Immunol. 2010, 184, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Zohrabian, V.M.; Forzani, B.; Chau, Z.; Murali, R.; Jhanwar-Uniyal, M. Rho/ROCK and MAPK signaling pathways are involved in glioblastoma cell migration and proliferation. Anticancer Res. 2009, 29, 119–123. [Google Scholar] [PubMed]
- Khalil, B.D.; El-Sibai, M. Rho GTPases in primary brain tumor malignancy and invasion. J Neurooncol. 2012, 108, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models (in vitro/in vivo) | Involved Enzyme(s) or S1P Receptor(s) | Signaling Pathway(s) | Findings |
|---|---|---|---|
| LN-18 U87-MG | ↑ SphK1, ↑ S1PR1-3 | PI3K/AKT1 activation | poor prognosis and survival of GBM patients; inhibition of SphK1 reduces cell viability; inhibition of S1PR1/2 diminishes cell migration [11] |
| U373-MG | ↑ S1PR1-3 expression | MAPK/ERK and PI3Kβ pathway | S1P stimulates glioma cell proliferation [34] |
| U373-MG, GBM-6, and GBM-12 | ↑ SphK1 S1PR2 | MEK1/2 and Rho/ROCK | S1P induced ↑ mRNA and protein expression of PAI-1 and uPAR is correlated with invasion of glioblastoma cells [35] |
| U373-MG U118-MG | ↑ S1PR1, S1PR2, and S1PR3 | MAPK-ERK Rho/ROCK ↑ CCN1/Cyr61protein expression | S1P stimulates growth and invasiveness; S1PR2-mediates migration, and invasiveness; ↑ CCN1/Cyr61 linked to tumor cell adhesion and angiogenesis [36] |
| U87-MG | S1P | MAPK-ERK Rho/ROCK; PLC; MT1-MMP/G6PT | S1P induces Ca2+ mobilization via MT1-MMP/ G6PT axis which is responsible for infiltrative and invasive properties of GBM [37] |
| LN-229 and U373-MG xenografts | SphK1 | AKT/JNK c-jun/ATF2 transcription | ↓ SphK1 expression or its inhibition by SK1-I reduces growth, migration and invasion of glioma cells in vitro and in vivo [38] |
| Rat C6 glioma | S1PR2 | MAPK/ERK, PKC and PLC/D Ca2+ signaling | S1P receptors are linked to at least two signaling pathways, i.e., the PTX-sensitive Gi /Go-protein pathway and the toxin-insensitive Gq/G11-phospholipase C-PKC pathway [39] |
| Rat C6 glioma, 1321-N1 astrocytoma cells | S1P, S1PR2 | PI3K/Cdc42/p38MAPK and PI3K/Rac1/JNK | S1P, through S1PR2, negatively regulated the migration [40] |
| 1321N1 | S1P levels | G12/13-mediated Rho/ROCK activation | S1P enhanced proliferation by activation of both MRTF-A and YAP [41] |
| U-373 MG, U87-MG, M059K, U-1242 and A172 | S1P levels ↑ S1PR1-3 | MAPK/ERK and PI3K | S1P enhances glioma cell motility and invasiveness via Gi-coupling [42] |
| T98G and G112 cells | S1P1, S1P2, S1P3 and S1P5 | PTEN/AKT/Egr | ↓ S1PR1 expression enhances the malignancy of glioblastoma; ↑ proliferation correlates with the shorter survival of patients with GBM [43] |
| human glioblastoma specimens | S1PR1 | NS | low expression of S1PR1 was significantly correlated with the high MIB-1 LI in glioblastomas from patients showing reduced survival [29] |
| U87-MG | S1P levels | NS | SphK1 inhibition reduced angiogenesis in a co-culture in vitro model [23] |
| glioblastoma primary tumors glioma specimens | ↑ S1P and SphK1 levels and ↓ SPP2 expression | NS | ↑ S1P and ↓ ceramide content; high SphK1 and low SPP2 observed [23] |
| Class | Type of Isoform | Model (in vitro/in vivo) | Signaling Pathway(s) | Findings |
|---|---|---|---|---|
| conventional isoforms | α | Rat C6 glioma, U87MG | ERK1/2 activation | cell proliferation, survival, invasion, migration [65,76] |
| βI | U87-MG, T98G xenografts | AKT/GSK3β and S6 kinase | regulation of cell growth [66,77] | |
| βII | U87-MG | CAK activation | cell cycle activation [78] | |
| γ | human glioblastoma cells | NS | expression of PKCγ in GBM cells [72] | |
| novel isoforms | δ | U373-MG, A172 U87-MG, T98G | EFG/Src and SphK1, elevated PAI-1 levels; c-MET/NOTCH2 | Cell motility, invasion and infiltration [79,80] |
| ε | GBM specimens and gliosarcoma samples | PI3K/AKT pathway | Overexpression in primary GBM tumors; regulates the apoptosis and survival [71] | |
| θ | U-251 and 5310 xenograft cell lines | MAPK/ERK signaling | Proapoptotic kinase; radioresistance [81] | |
| η | U-1242 and U-251 | AKT/mTOR and Erk/Elk-1 signaling | cell proliferation [68,82] | |
| μ | human glioblastoma cell lines | NS | proapoptotic kinase [83] | |
| atypical isoform | ι/λ | U87MG and gliomas | PI3K signaling | cell proliferation, motility and invasion [69,84] |
| ζ | U251 cell line and the 5310 xenograft glioma cell line | RhoA-dependent PKCζ/Raf1/MEK/ERK | cell proliferation, survival, invasion, and migration [81,85] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahajan-Thakur, S.; Bien-Möller, S.; Marx, S.; Schroeder, H.; Rauch, B.H. Sphingosine 1-Phosphate (S1P) Signaling in Glioblastoma Multiforme—A Systematic Review. Int. J. Mol. Sci. 2017, 18, 2448. https://doi.org/10.3390/ijms18112448
Mahajan-Thakur S, Bien-Möller S, Marx S, Schroeder H, Rauch BH. Sphingosine 1-Phosphate (S1P) Signaling in Glioblastoma Multiforme—A Systematic Review. International Journal of Molecular Sciences. 2017; 18(11):2448. https://doi.org/10.3390/ijms18112448
Chicago/Turabian StyleMahajan-Thakur, Shailaja, Sandra Bien-Möller, Sascha Marx, Henry Schroeder, and Bernhard H. Rauch. 2017. "Sphingosine 1-Phosphate (S1P) Signaling in Glioblastoma Multiforme—A Systematic Review" International Journal of Molecular Sciences 18, no. 11: 2448. https://doi.org/10.3390/ijms18112448