Impact of Labile Zinc on Heart Function: From Physiology to Pathophysiology

Department of Biophysics, Ankara University, Faculty of Medicine, 06100 Ankara, Turkey
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2017, 18(11), 2395; https://doi.org/10.3390/ijms18112395
Submission received: 14 October 2017
/
Revised: 2 November 2017
/
Accepted: 8 November 2017
/
Published: 12 November 2017
(This article belongs to the Special Issue Zinc Signaling in Physiology and Pathogenesis)
Abstract
:Zinc plays an important role in biological systems as bound and histochemically reactive labile Zn2+. Although Zn2+ concentration is in the nM range in cardiomyocytes at rest and increases dramatically under stimulation, very little is known about precise mechanisms controlling the intracellular distribution of Zn2+ and its variations during cardiac function. Recent studies are focused on molecular and cellular aspects of labile Zn2+ and its homeostasis in mammalian cells and growing evidence clarified the molecular mechanisms underlying Zn2+-diverse functions in the heart, leading to the discovery of novel physiological functions of labile Zn2+ in parallel to the discovery of subcellular localization of Zn2+-transporters in cardiomyocytes. Additionally, important experimental data suggest a central role of intracellular labile Zn2+ in excitation-contraction coupling in cardiomyocytes by shaping Ca2+ dynamics. Cellular labile Zn2+ is tightly regulated against its adverse effects through either Zn2+-transporters, Zn2+-binding molecules or Zn2+-sensors, and, therefore plays a critical role in cellular signaling pathways. The present review summarizes the current understanding of the physiological role of cellular labile Zn2+ distribution in cardiomyocytes and how a remodeling of cellular Zn2+-homeostasis can be important in proper cell function with Zn2+-transporters under hyperglycemia. We also emphasize the recent investigations on Zn2+-transporter functions from the standpoint of human heart health to diseases together with their clinical interest as target proteins in the heart under pathological condition, such as diabetes.
1. Introduction
Zinc is a redox inactive element and presents in almost all biological tissues. Zinc is accepted to be a component of antioxidant defense system and contributes to maintain the cell redox balance through different mechanisms [1]. There is a close relation between cellular labile zinc (Zn2+), endogenous processes, and biological macromolecules under physiological condition, mainly due to its unique property being their structural component or major regulator of macromolecules [2]. Labile Zn2+ plays an important role as a signaling molecule in large number of cells and tissues being a constituent of many proteins and enzymes in human body [3,4,5]. For this reason, even mild zinc-deficiency may impact numerous aspects of human health, including heart function [6]. Although Zn2+ has traditionally been regarded as relatively nontoxic, recent studies have shown how high intracellular labile Zn2+ level ([Zn2+]i) can be a potent killer of numerous cell types [7] including cardiomyocytes [8,9,10]. In cardiomyocytes, [Zn2+]i is measured to be less than one nanomolar under physiological conditions being much less than the intracellular free Ca2+ ([Ca2+]i) [8,11]. Moreover, oxidants caused about 30-fold increase in [Zn2+]i but only 2-fold in [Ca2+]i in cardiomyocytes [8]. Therefore, any increase in [Zn2+]i may be much more toxic biologically than is generally realized. Furthermore, any evolution of biomolecules to scavenge [Zn2+]i is crucially important in ameliorating the cellular toxicity. Supporting these above statements, early studies showed that any disruption in [Zn2+]i homeostasis could be associated with severe disorders, including injuries to cardiac tissues [6,12]. Another interesting action of [Zn2+]i has been shown its insulino-mimetic activity in diabetic patients [13].
It has been shown that [Zn2+]i in mammalian cardiomyocytes plays an important role in excitation-contraction coupling [8,9] and in excitation-transcription coupling [14]. In mammalian cells, [Zn2+]i homeostasis is regulated with different ways. Mainly, cellular [Zn2+]i is controlled via its pools. It can be released from metalloproteins (a structural component) or metalloenzymes (a cofactor) under pathological conditions. As up to 15% of the total cellular zinc can be bound to metallothioneins (MT), they can represent a significant pool of Zn2+ [15,16]. Other type storage for Zn2+ is intracellular compartments such as organelles and vesicles. Zn2+ transport is mediated by Zn2+-dependent proteins, named Zn2+ transporters, which are localized to both sarcolemma and intracellular membranes [17,18]. MTs and two Zn2+ transporter families, solute carrier 39A [SLC39A] or Zrt-, Irt-like proteins 7ZIPs and solute carrier 30A (SLC30A) or ZnTs play crucial roles to maintain the cellular Zn2+ homeostasis [19,20,21,22,23]. Although there are many recent very good review articles focused on recent progress to describe the physiological and biological functions of ZIP and ZnT transporters and provided a better understanding of Zn2+ biology, in here, we aimed to review the recent data on Zn2+ signaling by Zn2+ transporters in the heart under physiological and pathological conditions, and therefore, to update our current understanding on the role of cellular [Zn2+]i regulation in heart function.
2. Role of Zinc in Human Health
Zinc, as one of important micronutrients in human nutrition, has very prominent role in the maintenance of tissue functions. As mentioned in a number of studies and review articles, its recognition for biological systems mostly depends on its being an essential component of many enzymes and its role in many physiological or metabolic processes in the mammalian body due to it being the most abundant intracellular metal ion found in cytosol, vesicles, organelles, and in the nucleus [2,3,4,7,24,25,26,27,28,29,30]. Therefore, trace element zinc has greatly attracted the attention of biological scientists for its importance in clinical medicine. Furthermore, its role in several nutritional disorders has been clearly established [2,31,32,33,34]. Although the demonstration of zinc essentiality for the growth of Aspergillus niger is very early in the literature [35], a recognition of its importance for the growth of plants and animals was not appreciated until the 1950–60s, what was considered improbable until 1960. It was believed that this element could be toxic if it was over its physiological level with high zinc intakes [36,37,38], although zinc was essential for human health and that its deficiency in humans would never occur [39].
Although Zn2+ is the most abundant intracellular metal ion found in cytosol, vesicles, organelles, and in the nucleus of mammalian cells including cardiomyocytes [11,40], even a small deficiency is a disaster to human health due to the number of biological dysfunctions [39]. In the literature, there are about 7000 articles searched using the key words “zinc deficiency and humans.” However, there is an early study on this topic published in Nature related with role of zinc deficiency in fruit trees in Britain by Bold C et al. [41]. Following this work, most studies were performed in animals, until the first observation in humans by Prasad et al. [39]. Important roles of Zn2+ associated with health implications and pharmacological targets have evoked further interest regarding its status in human health and nutrition.
Several review articles besides original works emphasized the central role of zinc content in human health starting from maternal period using documents from both underdeveloped and developing countries [27]. Zinc deficiency in humans seems to be widespread throughout the world. Generally, it is assumed that zinc deficiency in humans is due to inadequate dietary intake, gut malabsorption, or defective metabolism whereas at the cellular level, decrease zinc level can be caused a large series of morbid processes [5,8,29]. Recent clinical studies focusing on population-based groups demonstrated that there are conflicting results associated with zinc deficiency and related symptoms in different populations [42,43,44]. Moreover, some studies showed a positive correlation between serum zinc and type 2 diabetes risks in either middle-aged and older Finnish men or the Norwegian population as well as in development of liver fibrosis in the Miami adults [45,46,47,48]. Additionally, Kunutsor and Laukkanen [49] performed a population-based cohort study in the same country and, due to their data, they suggested that a higher serum zinc concentration is positively and independently associated with incident hypertension in men. However, clinical diagnosis of marginal zinc deficiency in humans remains problematic [50].
It is now well accepted that a well-controlled Zn2+ homeostasis via Zn2+ regulatory mechanisms can control influx/efflux of zinc to prevent toxic zinc accumulation into cells, particularly endogenous labile Zn2+ plays a significant role in cytotoxic events at single cell level, including cardiomyocytes [8,9,10,40,51]. As mentioned in many articles, Zn2+ is essential for growth and development for both plants and mammalians. At the cellular level, it is critically involved in proliferation, differentiation, and apoptosis [52,53,54,55,56]. Indeed, Zn2+ was known as relatively harmless comparison to several other metal ions with similar chemical properties, while there is not much interest to Zn2+ toxicity in biological systems although it has been known to be hazard in industrial aspects [36,57,58]. It is believed that overt symptoms of zinc toxicity require its large amount ingestion by the body. On the other hand, it has been known how zinc compounds as supplementary agents in daily diets are important for humans, particularly under some special conditions.
However, there are still widespread controversies and ambiguities with respect to the toxic effects and mechanisms of excess zinc in humans. Authors using different zinc-compounds, particularly metallic nanoparticles including zinc, demonstrated that the solubility and the size of zinc compounds have a major role in the induced toxic responses, whereas uptake of their large ones inside the cells was likely to play a key role in the detected cell cycle arrest [59]. In a recent interesting study demonstrated another important role of heavy metals including zinc in pathological biomineralization of cancer tissue due to their important roles in morphogenesis of tumors considering their ability to enter into covalent bonds with calcium salt molecules. Romanjuk et al. [60] examined the role of microelements in breast cancer calcifications. Their data demonstrated how excess heavy metals (such as iron, copper, chromium, and nickel) in the body could lead to pathologies in the tissues/organs, at most, via progressively increasing rate of degenerative/necrotic alterations. However, Hoang et al. [61] discussed in a widespread manner how zinc is an important metal as a possible preventive and therapeutic agent in pancreatic, prostate, and breast cancer in humans.
As summary of this part, the body zinc level in humans as well as zinc intake with nutrition, are receiving increasing attention, also due to its putative role in the development of different pathological conditions, there are serious conflicts between publications.
3. Labile Zn2+ in Cardiac Physiology and Pathology
As discussed in many review articles, zinc is a vital nutrient for human health via its incredibly important roles in physiology and pathology of many organ functions, at most, due to its important roles in proteins and enzymes [62,63,64]. As a consequence, zinc is required for the function of organs, including the heart. Furthermore, impairment of Zn2+ homeostasis is associated with a variety of health problems such as cardiovascular disorders [65,66,67,68]. Previously, several authors have documented the importance of zinc in cardiovascular function and diseases in many good review articles [1,66,69,70,71,72,73]. In these articles, it has been focused on the critical role of intracellular labile Zn2+ in the redox signaling pathway, where certain triggers lead to release of Zn2+ from proteins/intracellular pools and cause myocardial damage [8,14,40,51,74]. Thus, the area of Zn2+-homeostasis seems to be emerging in cardiovascular disease. However, it has been demonstrated that labile Zn2+ outside a narrow concentration range are toxic to a variety of cells [69], including cardiomyocytes [8,9,10,14,40,51,75].
It has been known, at cellular levels, that certain pathological conditions including hyperglycemia and/or changes in specific signaling pathways can stimulate the production of molecules related with the redox-state of the cells. Among these changes, the alterations in the voltage-dependent ionic channels, ion transporters and some ion exchanges [76]. Similar to [Ca2+]i homeostasis, any alteration in [Zn2+]i-homeostasis, including redox-status of the cells under hyperglycemia, can be involved in development of cardiac dysfunction [8,9,77,78,79,80,81]. In that regard, it is clear that a well-controlled redox-status in cardiomyocytes can be very beneficial, in part, via mediation of either [Ca2+]i homeostasis, [Zn2+]i homeostasis or both for a cardioprotective approach in diabetic cardiomyopathy as well as other pathologies in patients with heart disease [51,82]. Therefore, it can be suggested that a well-controlled regulation of [Zn2+]i homeostasis at cellular level, similar to [Ca2+]i homeostasis, can have important strategy to protect heart against redox-unbalanced pathologies [12]. In this regard, in an early study by Kamalov et al. [83], it has been mentioned that [Zn2+]i/[Ca2+]i ratio in cardiomyocytes and mitochondria have optimal ranges, having important roles to control the redox-status as well as oxidative stress status of cells. As shown in many articles, any enhancement of antioxidant defenses in cells are providing benefits against these pathological conditions, including the control of [Zn2+]i associated control of [Ca2+]i homeostasis, particularly, via RyR2 [40,51,82,84,85]. As summary, ours and others’ studies have documented that, at tissue level, [Zn2+]i-homeostasis in heart is impaired by different signaling mechanisms, including oxidative stress in the heart, and thereby, plays important role in the pathogenesis of myocardial injury. Our current understanding of the roles of [Zn2+]i homeostasis and [Zn2+]i signaling in human myocardial injury is yet limited under any pathological stimulus.
The importance and complexity of Zn2+ action has been presumed to parallel the degree of Ca2+’s participation in cellular processes [9,51,72,86]. At cellular level, Zn2+ homeostasis is regulated through Zn2+ transporters, Zn2+-binding molecules, and Zn2+ sensors. Interestingly, most of studies related with the role of intracellular labile Zn2+ in cell function are associated with its toxicity, most probably, due to its service as an important secondary messenger in various intracellular signal transduction pathways [79,87].
As mentioned, in previous paragraphs, the cellular toxicity of exogenous and a redox-inert labile Zn2+ is characterized generally by cellular responses such as mitochondrial dysfunction, elevated production of reactive oxygen species/reactive nitrogen species ROS/RNS, loss of signaling quiescence leading to apoptosis in cells, cell death and increased expression of adaptive and inflammatory genes [2,4,5,6,8,9,14,23,88]. Central to the molecular effects of Zn2+ are its interactions with cysteinyl-thiols of proteins, which alter their functionality by modulating their reactivity and participation in redox reactions, as well as its a cis-acting element that is the binding site for metal-responsive transcription factor-1 (MTF-1). When cytosolic labile Zn2+ is increased, it can lead to an increase in MTF-1 activity, which in turn leads to an increase in activation of MTF-1 target genes [8,9,51,78,89].
Ongoing studies together with early studies demonstrated that both Zn2+ deficiency and excess are detrimental to cells, causing growth retardation, important metabolic disorders and, particularly, inducing an impaired excitation-contraction cycling in cardiomyocytes [8,9,14,90]. Although total cellular Zn2+ is about 200 µM, the cytosolic labile [Zn2+]i in cardiomyocytes is less than 1 nM under physiological conditions [8,11], whereas it can increase either ~30-fold with acute oxidant exposure or ~2-fold under chronic hyperglycemia [8,9,51]. Therefore, in general, not only can Zn2+ deficiency be detrimental, causing depressed growth and serious metabolic disorders, but excess Zn2+ can also be toxic to many cells [8,70,91,92].
4. Role of Cellular Labile Zn2+ in Electrical Properties of Cardiomyocytes
It is well accepted that a controlled Ca2+ signaling is key essential mechanism for regular function of cardiomyocytes. As mentioned in Section 3, the intracellular Ca2+ accumulation induced by increased production of ROS can cause injury and dysfunction of cardiomyocytes [93,94,95].
The sarcoplasmic reticulum (SR) is a main intracellular Ca2+ store in cardiomyocytes and ryanodine receptors (RyR2s) on the membrane of the SR play a central role in modulating Ca2+ signaling. These SR Ca2+ release channels contain many cysteine residues in their regulatory domain and putative Ca2+ pore region. These residues are susceptible to modification by oxidants [94,96,97].
Supporting the previous data, Woodier and co-workers [98] demonstrated nicely how cytosolic Zn2+ can act as a high affinity activator of RyR2 while their experimental approach enabled the study of RyR2 function under tight control of the chemical environment. Importantly, it has been widely discussed later how Zn2+ at 1 nM concentration has an ability to directly activate RyR2, which have a much higher affinity for Zn2+ than Ca2+ (about three-fold) [99]. Moreover, their data provided important information on the role of Zn2+ on RyR2 modulatory function in the absence of Ca2+ and presented a paradigm shift in our general understanding RyR2 activation during excitation-contraction coupling. Therefore, the already known data together with these new data provided an important mechanistic explanation associated with [Zn2+]i dishomeostasis and certain cardiomyopathies, including diabetic cardiomyopathy [51,82,98,99].
In our early study, we, for the time, have shown that the intracellular labile Zn2+ level, [Zn2+]i in rabbit cardiomyocytes, is less than 1 nM in physiological conditions [8], about 100-fold less than [Ca2+]i. At various concentrations, labile Zn2+ leads to the release of toxic ROS [79]. More importantly, our data demonstrated that [Zn2+]i was increased markedly (about 30-fold but only doubled [Ca2+]i) with thiol-reactive oxidants and contributed to oxidant-induced alterations of excitation-contraction coupling ECC under in vitro conditions. Therefore, that information emphasized the importance of [Zn2+]i measurement such as, how it could lead to significant overestimation of [Ca2+]i, if it was overlooked [8]. In later studies, under in vivo conditions, [Zn2+]i was increased by 70% in diabetes [51,78] and over 200% in aldosteronism [100]. Interestingly, Xie and Zhu [101] aimed to understand better the modulation of RyR2s during oxidative stress and showed how RyR2 in rat ventricular myocytes was modulated biphasically by sulfhydryl oxidation, contribution of increased [Zn2+]i besides increased [Ca2+]i.
Although it has been shown the resting level of [Zn2+]i in cardiomyocytes, there was very little information about precise mechanisms controlling intracellular distribution of Zn2+ and its variations during cardiac function. Therefore, we aimed to investigate the rapid changes in Zn2+ homeostasis in detailed and using the Zn2+-specific fluorescent dye, FluoZin-3, in comparison to Ca2+-dependent Fluo-3 fluorescence, we, for the first time, vizualised the existence of Zn2+ sparks and Zn2+ transients, in quiescent and electrically-stimulated cardiomyocytes, similarly to known rapid Ca2+ changes, while both Zn2+ sparks and Zn2+ transients required Ca2+ entry [9]. Inhibiting the SR-Ca2+ release, or increasing the Ca2+ load in a low-Na+ solution, suppressed or increased Zn2+ movements, respectively. Moreover, oxidation by H2O2 facilitated, and acidic pH inhibited the Ca2+-dependent Zn2+ release in freshy isolated rat ventricular cardiomyocytes.
In historical background, studies show that most experimental studies on the role of [Zn2+]i were performed in nervous system, at most, due to the localization of Zn2+ in nerve terminals and synaptic vesicles of excitatory neurons in the central nervous system [102,103]. The [Zn2+]i homeostasis in mammalian cells results from a coordinated regulation by different proteins involved in the uptake, excretion, and intracellular storage/trafficking of Zn2+ [104]. It has been shown the Zn2+ influx via the L-type Ca2+ channels in heart cells [8,14,75] while it was via L-type and N-type Ca2+ channels in neurons [105]. As can be seen in Figure 1, here, we reinvestigated the effects of extracellular and intracellular Zn2+ on the L-type Ca2+ current. In the presence of external Ca2+, the L-type Ca2+ current is inhibited by external Zn2+ (ZnCl2) and intracellular Zn2+ loading (with Zn2+-pyrithione, ZnPT) also reduces the L-type Ca2+ current as a concentration- and time-dependent manner (Figure 1A). Although both effects are washable, the intracellular Zn2+ loading induced a marked leftward shift in inactivation of the channels (Figure 1C) without any effect under external Zn2+ exposure (Figure 1B). Similar to ours, Alvarez-Collazo et al. [106] demonstrated the modulation of transmembrane Ca2+ movements and their regulation by β-adrenergic stimulation with both basal intracellular and extracellular Zn2+, emphasizing the importance of well-controlled cellular [Zn2+]i-homeostasis for prevention/treatment of cardiac dysfunction, whereas Traynelis et al. [107] showed that both T-type Ca2+ current and L-type Ca2+ current are inhibited by excess external Zn2+ via inhibition of N-methyl-D-aspartate (NMDA) receptors. Although the exact underlying mechanisms of Zn2+ effects are not clear yet, surface charge effects could be invoked to explain some of the Zn2+ actions. However, as for other divalent metal cations, most of the effects of Zn2+ could be well explained by changes in the gating of ion channels [108,109,110] that we have shown in Figure 1 similar to our previous data [75]. Furthermore, squid K+ channels are far more sensitive to Zn2+ than Na+ channels but the interactions of Zn2+ with gating charges appear similar in both cases [109]. In that regard, Aras [111] reported that, during sublethal ischemia, the early rise in neuronal Zn2+, preceding the rise in intracellular Ca2+, was responsible for the hyperpolarizing shift in the voltage dependency of the delayed rectifier Kv2.1 channels.
Labile Zn2+, with even picomolar concentrations, modulates many cellular processes via either inhibiting or activating many proteins, enzymes, kinases and phosphatases, particularly, at Ser/Thr or Tyr sites [2,29,112,113,114]. Therefore, these findings indicate clearly that cellular labile Zn2+ level has critical importance for cellular physiological function besides its physiopathological or toxicological role, and strongly suggest its modulatory activity of signal transduction processes [106,115]. Furthermore, it is known that fluctuations of [Zn2+]i participate in important cellular functions of not only breast cancer cells [116] but also mammalian cardiomyocyte [51,82,89]. In diabetic rat heart, an important actor in contractile machinery family, RyR2 and its accessory kinases protein kinase A (PKA) and calmodulin-dependent protein kinase II (CaMKII) are phosphorylated, significantly, at most, due to increased oxidative stress via increased [Zn2+]i. Interestingly, these changes could be prevented with antioxidant treatment under either in vivo or in vitro conditions. Indeed, either excess Zn2+ exposure to cardiomyocytes or labile Zn2+ loading of cardiomyocytes with zinc-ionophore could induce marked phoshorylation in RyR2, PKA and CaMKII as well as transcription factors such as nuclear factor κB (NFκB) and glycogen synthase kinase-3 (GSK) and other endogenous actors such as protein kinase B also known as Akt [89]. Parallel to our data, it has been shown that labile Zn2+ inhibits the activity of adenylyl cyclases as well as the hormone and forskolin stimulation of cAMP synthesis in N18TG2 cells [117], and, by preventing guanosine-5'-triphosphate (GTP) binding to the GTPase [118]. Also, in the presence of Ca2+ and calmodulin, increasing concentrations (in micromolars) of Zn2+ caused a progressive inhibition of substrate phosphorylation by CaMKII such as to produce a concentration-dependent inhibition of phospholamban phosphorylation [119]. However, Yi and coworkers [120] examined the role of extracellular Zn2+ exposure on cardiomyocyte contraction-relaxation function by using molecular and cellular techniques and showed that RyR2 and phospholamban were markedly dephosphorylated after permeating the hearts with 50 μM Zn2+. The different results associated with RyR2 phosphorylation level with Zn2+-exposures, most probably depending on the differences between Zn2+-exposure periods. One group experiments were performed in hours, while others in minutes.
Recently, since M-type (Kv7, KCNQ) potassium channels are important for the control of the excitability of neurons and muscle cells, Gao et al. [121] studied the effect of intracellular labile Zn2+ on M-type (Kv7, KCNQ) K+-channels. Their results reported that [Zn2+]i directly and reversibly augments the activity of recombinant and native M channels, being mechanistically distinct from the known redox-dependent KCNQ channel potentiation.
Taken into consideration the facts of [Zn2+]i in many cell types such as operation of many physiological and pathological mechanisms of cell excitation via the suppression of activity or expression of ion-channels, transporters, pumps, and receptors or pharmacological augmentation of their activities as a recognized strategy for the treatment of hyper-excitability disorders, it would be very helpful to understand well the action of [Zn2+]i in cardiomyocytes. However, physiological mechanisms resulting in ionic channel potentiation are rare. As short due to already known data, a large amount of Zn2+-proteins that are modulated by or contain Zn2+ can directly or indirectly affect the many cellular processes, at most, due to labile Zn2+ action on cell redox-balance [9,76]. At the cellular level, the effects of labile Zn2+ in cardiomyocytes via its action in membrane receptors, transporters and ionic channels as well as endogenous accessory proteins of contractile machinery and some transcription factors are summarized in Table 1 and Table 2, respectively.
5. Labile Zn2+ Pools in Cardiomyocytes
As described in previous sections, labile Zn2+ regulates the expression and activation of biological molecules such as ion channels, transcription factors, enzymes, adapters, and growth factors, along with their receptors in many cell types, including cardiomyocytes. Excess Zn2+ can be detrimental to cells, particularly that of cardiomyocytes [8,9,10,14]. As an intracellular signal transducer in multiple cellular functions, it has been shown that intracellular labile Zn2+ has an important role in ECC in cardiomyocytes by shaping Ca2+ dynamics [9,51,98], while it acts as a neuromodulator in synaptic transmissions [122,123]. In the regulation of cellular labile Zn2+, subcellular pools as well as metalloproteins are important actors, which also include manyZn2+-transporters [29,87,124].
In our previous study performed with freshly isolated ventricular cardiomyocytes, we demonstrated that rapid changes in labile Zn2+ mostly resulted from Zn2+ displacement by Ca2+ ions from intracellular binding sites that were highly sensitive to the redox status of the cardiomyocytes by using Zn2+-specific fluorescence dye, FluoZin-3 [9]. In order to examine the physiological importance of the protein-bound Zn2+ pools, similar to other studies [125] by applying acidic pH to the cardiomyocytes or by causing an oxidative stress with H2O2, we induced chemical modifications of the thiol groups in the proteins and demonstrated that Zn2+ binding to metallothioneins decreased at acid pH and significantly reduced contraction without altering [Ca2+]i.
Our testing S(E)R as an possible intracellular Zn2+ pool by using ryanodine application, we observed significantly and simultaneously decreases in the intensities of both Zn2+ transients and Ca2+ transients in field-stimulated cardiomyocytes without affecting their basal levels [9]. Furthermore, we performed additional experiments with caffeine. As can be seen in Figure 2, we observed two different responses in Fluo-3 or FluoZin-3 loaded cells. In Fluo-3 loaded cells, there was fast transitory and short-lived large increase as response to caffeine application, whereas there was a slow initial large increase followed with a stable long-lived plateau in FluoZin-3 loaded cells. These data support the hypothesis of sarco(endo)plasmic reticulum, S(E)R could be a Zn2+ pool similar to Ca2+ in cardiomyocytes. Furthermore, recently by using Förster resonance energy transfer (FRET)-based recombinant-targeted Zn2+-probes [11], we have shown that [Zn2+]i in cardiomyocytes is calculated less than 1 nM, while ~5-fold higher in S(E)R, and less than cytosolic-level in mitochondria. Elevated cytosolic Zn2+ appears to contribute to deleterious changes in many cellular signaling-pathways including hyperglycemia-challenged cardiomyocytes [8,9,10,19,51]. Moreover, we also demonstrated that elevated cytosolic Zn2+ appears to be associated with loss of S(E)R Zn2+ via hyperphosphorylation of Zn2+-transporter ZIP7, which further induces endoplasmic reticulum (ER) stress in cardiomyocytes in the diabetic rat heart [40]. Of note, in eukaryotes, Ellis et al. [126] demonstrated the Zn2+-deficiency associated disruption in ER such as alteration in its function and induction of ER stress.
Additional studies also pointed out mitochondria to be another intracellular Zn2+-pool in cardiomyocytes [9]. Expose of FluoZin-3 loaded cardiomyocytes either to a mitochondrial complex I inhibitor or to carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) a mitochondrial protonophore induced rapid and significant inhibitory effects on the Zn2+-changes with only mild initial effects on the Fluo-3 loaded cells. In this regard, in cortical neurons, it was proposed that the source of Ca2+-dependent Zn2+ release appears largely to be mitochondria [127]. In cardiomyocytes, since mitochondria constitute the major source of intracellular ROS production [128], mitochondria-related excessive ROS production has been implicated in the pathogenesis of many cardiovascular diseases. Indeed, the Ca2+-dependency of the glutamate mobilization of intracellular Zn2+ in neurons is attributed to the generation of ROS arising from both cytosolic and mitochondrial sources [129]. In cardiomyocytes, Ca2+ influx might trigger transitory change in ROS production leading to Zn2+ transients even on this time scale. Such a hypothesis could account for our recent observations [40].
Besides voltage-dependent ionic channels, transient receptor potential (TRP) channels are a large family associated with multi-signal transducers and play important roles in different organ function, including heart function. The functional and structural control of TRP channels by trace metal ions, including Zn2+, has been demonstrated in different cell types [130]. For example, the high expression levels of both transient receptor potential cation channel3(TRPC3) and TRPC6 have been shown in the heart and could participate in the pathogenesis of cardiac hypertrophy and heart failure as a pathological response to chronic mechanical stress [131]. Additionally, the activation of transient receptor potential cation channel subfamily M member 4 (TRPM4), a Ca2+-activated, but Ca2+-impermeable non-selective cation channel, has been also demonstrated to have role in conduction block and other arrhythmic propensities associated with cardiac remodeling and injury [132]. Furthermore, Uchida and coworkers [133] have documented that extracellular Zn2+ regulates TRPM5 channel activation [133]. On the other hand, Lambert and coworkers demonstrated that extracellular Zn2+ exposure to HEK293 cells did inhibit TRPM5 and TRPM1 activity [134]. Moreover, Abiria and coworkers [135] recently showed that the majority of TRPM7 is localized in abundant intracellular vesicles in HEK293 cells and ROS-mediated TRPM7 activation releases Zn2+ from these vesicles following Zn2+ overload. They emphasized the important role of TRPM7-mediated Zn2+ release and the regulation of ROS signaling processes during postnatal stress/injury. Therefore, one can emphasized how will be very important to understand the roles of TRP channels as detailed in the regard of their contribution to the key procedures for the development of cardiovascular disorders. Furthermore, it seems that they may provide basic scientific knowledge for the development of new preventive and therapeutic approaches to manage patients with cardiovascular diseases [136].
On this basis, the effects of increased intracellular labile Zn2+ on the structure of cardiomyocytes particularly focused on ultrastructure of mitochondria were examined by electron microscopy by using short-term ZnPT incubation (0.1 or 1 μM for 15–20 min) in freshly isolated ventricular cells. As can be seen in Figure 3A–C, marked irregular mitochondrial cristae and significantly clustered and degenerated mitochondria between the myofibrils together with electron-dense matrix were observed in labile Zn2+ loaded cells. Additionally, there were an important amount of fragmented mitochondria and rounding and swelling in mitochondrion. Although the sarcomere showed normal structural appearance with regular myofibrils and mitochondrial structure in the control group cells (Figure 3A), there were dramatic signs of injury in the form of condensation, increased matrix density, and deposits of electron-dense material in the loaded cardiomyocytes. These Zn2+ loading effects in mitochondria can support its Zn2+ sensing pool in cardiomyocytes. However, the early data by Jang et al. [137] showed that NO mobilizes intracellular Zn2+ via Cyclic guanosine monophosphate/Protein Kinase G (cGMP/PKG) signaling pathway and prevents mitochondrial oxidant damage in cardiomyocytes.
6. Zn2+ Transporters in Cardiomyocytes
Recent review articles well summarized the already published data, performed in different mammalian cells except cardiomyocytes, which showed the regulation of Zn2+ homeostasis via a number of Zn2+-transporters and how they are crucial for proper cellular functions [22,23,29,138,139,140,141,142]. In the review by Kambe [143], it has been pointed out how the impaired Zn2+ transporter functions into and out of cells strongly linked to clinical human diseases [144]. Since the membrane transporters having the great potential for drug targets [145,146], hence, cytosolic labile Zn2+ and Zn2+ transporters should be considered as novel therapeutic targets for diseases, including heart diseases. In this section, we aimed to describe the physiological and molecular functions of Zn2+-transporters, which regulate [Zn2+]i homeostasis and are involved in signal transduction and heart diseases, particularly such as diabetes.
Even in early reviews, tightly control of [Zn2+]i homeostasis is defined due to existence of specific Zn2+-transporters, including the coordinated regulation of [Zn2+]i homeostasis in terms of its uptake, efflux, distribution, and storage, which are documented in several review articles, nicely [20,147]. In general, Zn2+ transporters belong to a family of transmembrane proteins that control the flux of Zn2+ across cellular membranes into cytosol (ZIPs) and out of cytosol (ZnTs) in many types of cells, therefore, contribute to the distribution, storage, and compartmentalization of Zn2+. Additionally, these all predict proteins with multiple membrane spanning regions, and most have a histidine-rich intracellular loop. The first described Zn2+ transporter in mammalian cells is ZnT-1 in kidney cells [148] while the second one is ZnT3 in regions of the brain [149]. The ZnT proteins (solute-linked carrier 30, SLC30) and the ZIP (zinc-regulated trans- porte /iron-regulated transporter Zrt/Irt)-like, solute-linked carrier, 39, SLC39) have been identified in mammalian tissues [22,23,150]. In mammals, there are 10 members of the Zn2+ efflux transporters (ZnT1–10) and 14 members of the zinc influx transporters (ZIP1–14).
ZIP proteins are thought to form homodimers to transport Zn2+ across the cellular membrane [151], while the conserved hydrophilic residue seems to sense metal specificity [152]. Supporting this statement, it has been demonstrated that ZIP8 and ZIP14, possessing glutamic acid instead of the conserved histidine, can efficiently transport Fe2+, Mn2+, and Cd2+ in addition to Zn2+. Human genetic disorders caused by mutations and single-nucleotide polymorphisms in Zn2+ transporter genes were summarized by Kambe et al. [144,153]. They documented that a number of genetic disorders are caused by mutations in the genes encoding ZIPs and ZnTs, such as ZIP4 in acrodermatitis enteropathica, ZIP13 in the spondylocheiro dysplastic form of Ehlers-Danlos syndrome, ZnT2 in transient neonatal zinc-deficiency, ZnT8 in type 1 and 2 diabetes mellitus, and ZnT10 in Parkinsonism and dystonia [22,23,141,142].
Furthermore, in the recent review article by Hara et al. [29], mechanisms of Zn2+-transporter expression and modification have been documented, very widely in different mammalian cells/tissues, focusing on their physiological roles from their molecular basis to genetic importance. In that review, they presented the role of ZnTs such as ZnT2, ZnT3, ZnT4, and ZnT8, which localize to acidic compartments and to vesicles such as endosomes/lysosomes, synaptic vesicles, and insulin granules as well as the ZIPs such as ZIP4, ZIP5, ZIP6, ZIP7, ZIP8, ZIP10, ZIP12, ZIP13, and ZIP14 in different cells. A number of cellular proteins, enzymes, kinases and phosphatases interact with Zn2+ for their biological functions. Studies have revealed that Zn2+ acts not only as an accessory molecule for proteins but also as a signaling molecule, much like Ca2+ [15,29].
Among others, ZnT7 has been demonstrated to play an important role in both growth and the accumulation of body fat in mice [154] as well as the association between its deficiency and metabolic disorders such as insulin and glucose intolerance and hyperglycemia [155]. Furthermore, studies on ZnT8 showed that the ZnT8 is strongly related to type 1 and 2 diabetes [156,157]. In this regard, later studies emphasized that ZnT8 expressed in pancreatic β-cells is involved in secreting insulin, forming crystals [158,159,160], and eliminating insulin by the liver [161]. In this field, the recent review articles by Chabosseau and Rutter [162] and by Rutter et al. [163] reviewed the regulation and roles of Zn2+ in islet cells and the mechanisms by which ZnT8 variants might affect glucose homeostasis and diabetes risk. Correspondingly, they presented that genetic variants in the ZnT8 gene, which encodes the diabetes-associated granule-resident ZnT8, are associated with an altered risk of type 2 diabetes. Additionally, they discussed the effects on insulin secretion and action of deleting or over-expressing ZnT8 highly selectively in the pancreatic β-cell, and the role of Zn2+ in insulin signaling. Due to their own data together with the others’ data, it has been concluded that maintenance of glucose homeostasis, and therefore lower diabetes risk, due to a proper intake level of dietary zinc at systemic level and a well-controlled [Zn2+]i homeostasis at cellular level is provided with both insulin release and insulin action at physiological levels.
In the content of ZIPs’ roles, there are a limited number of studies in mammalian cells in the literature without any in cardiomyocytes except our study [40]. However, Ellis et al. [126] demonstrated that the zinc deficiency in ER leads to an unfolded protein response (UPR) in human cells. In a later study, Huang et al. [155] proposed that ZIP7 is localized to Golgi apparatus in Chinese-Hamster Ovary-cells, allowing Zn2+-release from Golgi lumen into cytosol. It has been also suggested that ZIP7 facilitates release of Zn2+ from ER [164] and behaves as a critical component in sub-cellular re-distribution of Zn2+ in other systems [165]. Additionally, it has been hypothesized that protein kinase-2 (CK2) triggers cytosolic Zn2+-signaling-pathways by phosphorylating ZIP7 [116], while some studies have also highlighted its important contribution to Zn2+-homeostasis under pathological conditions [166,167,168]. In addition, in a recent study, it was demonstrated that ZIP7, which predominantly localizes to the ER membrane, promotes rapid cell proliferation in intestinal crypts by maintaining ER function. They also found that mice with an intestinalepithelium-specific ZIP7 deletion exhibited extensive apoptosis in the stem-cell-derived transit-amplifying cells due to increased ER stress. They further showed that UPR signaling upregulates ZIP7, which maintains [Zn2+]i homeostasis under ER stress and facilitates epithelial proliferation. Therefore, ZIP7 is considered as a novel regulator of [Zn2+]i homeostasis of the intestinal epithelium [169].
Although studies have shown the presence of weakly expressed ZIP7 and ZnT7 in mammalian heart [170,171], their subcellular localizations and functional roles in cardiomyocytes were not yet known well. In that regard, we hypothesized that disruption of Zn2+-transporters and Zn2+-axis such as ZIP7 and ZnT7 might contribute to deleterious changes in diabetic cardiomyocytes. Therefore, we first clarified their subcellular localizations into S(E)R and then explored their functional roles in Zn2+ homeostasis, particularly under hyperglycemia. Additionally, we tested their roles in cytosolic Zn2+ re-distribution and development of ER-stress in hyperglycemic conditions, at most due to activation of casein kinase 2 alpha (CK2α) [40]. We observed markedly increased mRNA and protein levels of ZIP7 in ventricular cardiomyocytes from diabetic rats or high glucose-treated H9c2 cells whilst ZnT7 expression was low comparison to those of controls. Additionally, we observed increased ZIP7 phosphorylation in response to high glucose in vivo and in vitro in ventricular cardiomyocytes. Using recombinant targeted FRET-based sensors, we showed that hyperglycemia induced a marked redistribution of cellular labile Zn2+, increasing cytosolic labile Zn2+ and lowering labile Zn2+ in the S(E)R. These changes involve alterations in ZIP7-phosphorylation and were suppressed by siRNA-mediated silencing of CK2α. Due to our whole data, we, for the first time, demonstrated that opposing changes in the expression of ZIP7 and ZnT7 observed in hyperglycemia is very important for development of ER stress in the heart. In addition, we also pointed out an importance of sub-cellular labile Zn2+ re-distribution in the hyperglycemic heart, which is resulting from altered ZIP7 and ZnT7 activity and contributing to cardiac dysfunction in diabetes [40]. Furthermore, Myers [140] previously discussed very widely the roles of Zn2+ transporters and Zn2+ signaling by using recent new roles of Zn2+ and its transporters in the synthesis, secretion, and action of insulin are dependent on zinc and the transporters in type 2 diabetes. Author, particularly, emphasized the role of cellular Zn2+’s dynamic as a “intracellular second messenger” to control insulin signaling and glucose homeostasis. Therefore, it was raised extensively a new research field into the pathophysiology of insulin resistance and possibility of new this-field related drug targets in diabetes [13,172,173,174,175].
In conclusion, the already known data associated with the role of ZIPs and ZnTs provide novel insights into regulation of cellular-Zn2+ and its role in the heart under pathological conditions, including hyperglycemia/diabetes-associated cardiac dysfunction. Additionally, all findings can provide new targets such as cellular [Zn2+]i-regulation via mediation of Zn2+-transporters and suggest that modulation of some endogenous kinases such as CK2α may provide a novel means to correct cardiac dysfunction under any pathological condition.
7. Concluding Remarks
Both early and recent studies strongly emphasized how [Zn2+]i homeostasis is tightly controlled by the coordinated regulation of its uptake, efflux, distribution, and storage in mammalian cells. A number of proteins involved in different signaling pathways, mitochondrial metabolism, and ion channels, which are also common targets of labile Zn2+, play pivotal roles in controlling cardiac contractility. The already known documents associated with the role of zinc in cardiac function are summarized in Figure 4. However, these regulatory actions of Zn2+ are not limited to the function of the heart, but also extend to numerous other organ systems in mammalians. In this review, the regulation of cellular labile Zn2+ levels, Zn2+-mediated signal transduction, impacts of Zn2+ on ionic channels and S(E)R, and finally, the roles of Zn2+ transporters in healthy and diseased heart, including diabetic heart, were outlined to help widen the current understanding of the versatile and complex roles of Zn2+. Although much has been learned from recent studies, revealed important relationships between Zn2+ transporters and heart diseases and indicating the potential of Zn2+ transporters as therapeutic targets, their precise physiological functions are not clear. Given the multiple roles of Zn2+ in various cell types and the detailed research development of Zn2+-containing new markers/sensors will improve the ways to handle heart failure in humans.
Acknowledgments
We thank our many colleagues for their excellent works. This work is supported through grant TUBITAK SBAG-113S466.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Oteiza, P.I. Zinc and the modulation of redox homeostasis. Free Radic. Biol. Med. 2012, 53, 1748–1759. [Google Scholar] [CrossRef] [PubMed]
- Vallee, B.L.; Falchuk, K.H. The biochemical basis of Zinc physiology. Physiol. Rev. 1993, 73, 79–118. [Google Scholar] [PubMed]
- Drinker, H.S. Concerning Modern Corporate Mortgages. Univ. Pa. Law Rev. Am. Law 1926, 74, 360–366. [Google Scholar] [CrossRef]
- Murakami, M.; Hirano, T. Intracellular Zinc homeostasis and Zinc signaling. Cancer Sci. 2008, 99, 1515–1522. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Zinc and human disease. Met. Ions Life Sci. 2013, 13, 389–414. [Google Scholar] [PubMed]
- Coudray, C.; Charlon, V.; de Leiris, J.; Favier, A. Effect of Zinc deficiency on lipid peroxidation status and infarct size in rat hearts. Int. J. Cardiol. 1993, 41, 109–113. [Google Scholar] [CrossRef]
- Maret, W. Zinc biochemistry: From a single Zinc enzyme to a key element of life. Adv. Nutr. (Bethesda, Md.) 2013, 4, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Turan, B.; Fliss, H.; Desilets, M. Oxidants increase intracellular free Zn2+ concentration in rabbit ventricular myocytes. Am. J. Physiol. 1997, 272, H2095–H2106. [Google Scholar] [PubMed]
- Tuncay, E.; Bilginoglu, A.; Sozmen, N.N.; Zeydanli, E.N.; Ugur, M.; Vassort, G.; Turan, B. Intracellular free Zinc during cardiac excitation-contraction cycle: Calcium and redox dependencies. Cardiovasc. Res. 2011, 89, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Tuncay, E.; Turan, B. Intracellular Zn2+ increase in cardiomyocytes induces both electrical and mechanical dysfunction in heart via endogenous generation of reactive nitrogen species. Biol. Trace Element Res. 2016, 169, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Chabosseau, P.; Tuncay, E.; Meur, G.; Bellomo, E.A.; Hessels, A.; Hughes, S.; Johnson, P.R.; Bugliani, M.; Marchetti, P.; Turan, B.; et al. Mitochondrial and ER-targeted eCALWY probes reveal high levels of free Zn2+. ACS Chem. Biol. 2014, 9, 2111–2120. [Google Scholar] [CrossRef] [PubMed]
- Crawford, A.J.; Bhattacharya, S.K. Excessive intracellular Zinc accumulation in cardiac and skeletal muscles of dystrophic hamsters. Exp. Neurol. 1987, 95, 265–276. [Google Scholar] [CrossRef]
- Jansen, J.; Rosenkranz, E.; Overbeck, S.; Warmuth, S.; Mocchegiani, E.; Giacconi, R.; Weiskirchen, R.; Karges, W.; Rink, L. Disturbed Zinc homeostasis in diabetic patients by in vitro and in vivo analysis of insulinomimetic activity of Zinc. J. Nutr. Biochem. 2012, 23, 1458–1466. [Google Scholar] [CrossRef] [PubMed]
- Atar, D.; Backx, P.H.; Appel, M.M.; Gao, W.D.; Marban, E. Excitation-transcription coupling mediated by Zinc influx through voltage-dependent Calcium channels. J. Biol. Chem. 1995, 270, 2473–2477. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Metals on the move: Zinc ions in cellular regulation and in the coordination dynamics of Zinc proteins. Biometals 2011, 24, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Coyle, P.; Philcox, J.C.; Carey, L.C.; Rofe, A.M. Metallothionein: The multipurpose protein. Cell. Mol. Life Sci. 2002, 59, 627–647. [Google Scholar] [CrossRef] [PubMed]
- Hennigar, S.R.; Kelleher, S.L. Zinc networks: The cell-specific compartmentalization of Zinc for specialized functions. Biol. Chem. 2012, 393, 565–578. [Google Scholar] [CrossRef] [PubMed]
- Kambe, T. An overview of a wide range of functions of ZnT and ZIP Zinc transporters in the secretory pathway. Biosci. Biotechnol. Biochem. 2011, 75, 1036–1043. [Google Scholar] [CrossRef] [PubMed]
- Kambe, T.; Tsuji, T.; Hashimoto, A.; Itsumura, N. The physiological, biochemical, and molecular roles of Zinc transporters in Zinc homeostasis and metabolism. Physiol. Rev. 2015, 95, 749–784. [Google Scholar] [CrossRef] [PubMed]
- Kambe, T.; Yamaguchi-Iwai, Y.; Sasaki, R.; Nagao, M. Overview of mammalian Zinc transporters. Cell. Mol. Life Sci. 2004, 61, 49–68. [Google Scholar] [CrossRef] [PubMed]
- Eide, D.J. The SLC39 family of metal ion transporters. Pflugers Arch. Eur. J. Physiol. 2004, 447, 796–800. [Google Scholar] [CrossRef] [PubMed]
- Lichten, L.A.; Cousins, R.J. Mammalian Zinc transporters: Nutritional and physiologic regulation. Ann. Rev. Nutr. 2009, 29, 153–176. [Google Scholar] [CrossRef] [PubMed]
- Fukada, T.; Kambe, T. Molecular and genetic features of zinc transporters in physiology and pathogenesis. Met. Integr. Biomet. Sci. 2011, 3, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Todd, W.R.; Elvehjem, C.A.; Hart, E.B. Zinc in the nutrition of the rat. Am. J. Physiol. 1934, 107, 146–156. [Google Scholar] [CrossRef]
- Prasad, A.S.; Miale, A., Jr.; Farid, Z.; Sandstead, H.H.; Schulert, A.R. Zinc metabolism in patients with the syndrome of iron deficiency anemia, hepatosplenomegaly, dwarfism, and hypognadism. J. Lab. Clin. Med. 1963, 61, 537–549. [Google Scholar] [PubMed]
- Buamah, P.K.; Russell, M.; Bates, G.; Ward, A.M.; Skillen, A.W. Maternal zinc status: A determination of central nervous system malformation. Br. J. Obstet. Gynaecol. 1984, 91, 788–790. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.S. Impact of the discovery of human Zinc deficiency on health. J. Trace Elements Med. Biol. Organ Soc. Miner. Trace Elements (GMS) 2014, 28, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.P.; Zhang, Y.Q.; Tong, Y.P.; Xue, Y.F.; Liu, D.Y.; Zhang, W.; Deng, Y.; Meng, Q.F.; Yue, S.C.; Yan, P.; et al. Harvesting more grain Zinc of wheat for human health. Sci. Rep. 2017, 7, 7016. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Takeda, T.A.; Takagishi, T.; Fukue, K.; Kambe, T.; Fukada, T. Physiological roles of Zinc transporters: Molecular and genetic importance in Zinc homeostasis. J. Physiol. Sci. 2017, 67, 283–301. [Google Scholar] [CrossRef] [PubMed]
- Arquilla, E.R.; Packer, S.; Tarmas, W.; Miyamoto, S. The effect of Zinc on insulin metabolism. Endocrinology 1978, 103, 1440–1449. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.S. Zinc deficiency and effects of Zinc supplementation on sickle cell anemia subjects. Progress Clin. Biol. Res. 1981, 55, 99–122. [Google Scholar]
- Savin, J.A. Skin disease: The link with Zinc. Br. Med. J. (Clin. Res. Ed.) 1984, 289, 1476–1477. [Google Scholar] [CrossRef]
- Simmer, K.; Thompson, R.P. Maternal Zinc and intrauterine growth retardation. Clin. Sci. 1985, 68, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.H. Zinc and micronutrient supplements for children. Am. J. Clin. Nutr. 1998, 68, 495s–498s. [Google Scholar] [PubMed]
- Raulin, J. Chemical studies on vegetation. J. Ann. Sci. Nat. 1869, 11, 93–99. (In French) [Google Scholar]
- Fosmire, G.J. Zinc toxicity. Am. J. Clin. Nutr. 1990, 51, 225–227. [Google Scholar] [PubMed]
- Jones, M.M.; Schoenheit, J.E.; Weaver, A.D. Pretreatment and heavy metal LD50 values. Toxicol. Appl. Pharmacol. 1979, 49, 41–44. [Google Scholar] [CrossRef]
- Bentley, P.J.; Grubb, B.R. Effects of a Zinc-deficient diet on tissue Zinc concentrations in rabbits. J. Anim. Sci. 1991, 69, 4876–4882. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.S.; Walker, D.G.; Dehgani, A.; Halsted, J.A. Cirrhosis of the liver in Iran. Arch. Intern. Med. 1961, 108, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Tuncay, E.; Bitirim, V.C.; Durak, A.; Carrat, G.R.J.; Taylor, K.M.; Rutter, G.A.; Turan, B. Hyperglycemia-induced changes in ZIP7 and ZnT7 expression cause Zn2+ Release From the sarco(endo)plasmic reticulum and mediate ER stress in the heart. Diabetes 2017, 66, 1346–1358. [Google Scholar] [CrossRef] [PubMed]
- Bould, C.; Nicholas, D.J.; Tolhurst, J.A.H.; Wallace, T.; Potter, J.M.S. Zinc deficiency of fruit trees in Britain. Nature 1949, 164, 801. [Google Scholar] [CrossRef] [PubMed]
- Stelmach, I.; Grzelewski, T.; Bobrowska-Korzeniowska, M.; Kopka, M.; Majak, P.; Jerzynska, J.; Stelmach, W.; Polanska, K.; Sobala, W.; Gromadzinska, J.; et al. The role of Zinc, Copper, plasma glutathione peroxidase enzyme, and vitamins in the development of allergic diseases in early childhood: The Polish mother and child cohort study. Allergy Asthma Proc. 2014, 35, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Markiewicz-Zukowska, R.; Gutowska, A.; Borawska, M.H. Serum Zinc concentrations correlate with mental and physical status of nursing home residents. PLoS ONE 2015, 10, e0117257. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.K.; Lee, S.H.; Han, K.; Kang, B.; Lee, S.Y.; Yoon, K.H.; Kwon, H.S.; Park, Y.M. Lower serum Zinc levels are associated with unhealthy metabolic status in normal-weight adults: The 2010 korea national health and nutrition examination survey. Diabetes Metab. 2015, 41, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Yary, T.; Virtanen, J.K.; Ruusunen, A.; Tuomainen, T.P.; Voutilainen, S. Serum Zinc and risk of type 2 diabetes incidence in men: The kuopio ischaemic heart disease risk factor study. J. Trace Elements Med. Biol. Organ Soc. Miner. Trace Elements (GMS) 2016, 33, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.F.; Simic, A.; Asvold, B.O.; Romundstad, P.R.; Midthjell, K.; Syversen, T.; Flaten, T.P. Trace elements in early phase type 2 diabetes mellitus-A population-based study. The HUNT study in Norway. J. Trace Elements Med. Biol. Organ Soc. Miner. Trace Elements (GMS) 2017, 40, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Simic, A.; Hansen, A.F.; Asvold, B.O.; Romundstad, P.R.; Midthjell, K.; Syversen, T.; Flaten, T.P. Trace element status in patients with type 2 diabetes in Norway: The HUNT3 Survey. J. Trace Elements Med. Biol. Organ Soc. Miner. Trace Elements (GMS) 2017, 41, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Martinez, S.S.; Campa, A.; Li, Y.; Fleetwood, C.; Stewart, T.; Ramamoorthy, V.; Baum, M.K. Low plasma Zinc is associated with higher mitochondrial oxidative stress and faster liver fibrosis development in the miami adult studies in HIV cohort. J. Nutr. 2017, 147, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Kunutsor, S.K.; Laukkanen, J.A. Serum Zinc concentrations and incident hypertension: New findings from a population-based cohort study. J. Hypertens. 2016, 34, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.M.; Wolf, P.; Hauner, H.; Skurk, T. Effect of a fermented dietary supplement containing chromium and Zinc on metabolic control in patients with type 2 diabetes: A randomized, placebo-controlled, double-blind cross-over study. Food Nutr. Res. 2016, 60, 30298. [Google Scholar] [CrossRef] [PubMed]
- Tuncay, E.; Okatan, E.N.; Vassort, G.; Turan, B. β-blocker timolol prevents arrhythmogenic Ca2+ release and normalizes Ca2+ and Zn2+ dyshomeostasis in hyperglycemic rat heart. PLoS ONE 2013, 8, e71014. [Google Scholar] [CrossRef] [PubMed]
- Fraker, P.J.; Telford, W.G. A reappraisal of the role of zinc in life and death decisions of cells. Proc. Soc. Exp. Biol. Med. Soc. Exp. Biol. Med. (New York, NY) 1997, 215, 229–236. [Google Scholar] [CrossRef]
- Truong-Tran, A.Q.; Carter, J.; Ruffin, R.E.; Zalewski, P.D. The role of Zinc in caspase activation and apoptotic cell death. Biometals 2001, 14, 315–330. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, B.; Pal, S.; Tran, M.P.; Parsons, A.A.; Barone, F.C.; Erhardt, J.A.; Aizenman, E. p38 activation is required upstream of potassium current enhancement and caspase cleavage in thiol oxidant-induced neuronal apoptosis. J. Neurosci. 2001, 21, 3303–3311. [Google Scholar] [PubMed]
- Wiseman, D.A.; Wells, S.M.; Wilham, J.; Hubbard, M.; Welker, J.E.; Black, S.M. Endothelial response to stress from exogenous Zn2+ resembles that of NO-mediated nitrosative stress, and is protected by MT-1 overexpression. Am. J. Physiol. Cell Physiol. 2006, 291, C555–C568. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.E.; Kovacic, J.P. The ubiquitous role of Zinc in health and disease. J. Vet. Emerg. Crit. Care (San Antonio, Tex. 2001) 2009, 19, 215–240. [Google Scholar] [CrossRef] [PubMed]
- Hambidge, K.M.; Olivarasbach, J.; Jacobs, M.; Purcell, S.; Statland, C.; Poirier, J. Randomized study of Zinc supplementation during pregnancy. Fed. Proc. 1986, 45, 974. [Google Scholar]
- Brown, M.A.; Thom, J.V.; Orth, G.L.; Cova, P.; Juarez, J. Food poisoning involving zinc contamination. Arch. Environ. Health 1964, 8, 657–660. [Google Scholar] [CrossRef] [PubMed]
- Uski, O.; Torvela, T.; Sippula, O.; Karhunen, T.; Koponen, H.; Peraniemi, S.; Jalava, P.; Happo, M.; Jokiniemi, J.; Hirvonen, M.R.; et al. In vitro toxicological effects of Zinc containing nanoparticles with different physico-chemical properties. Toxicol. In Vitro 2017, 42, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Romanjuk, A.; Lyndin, M.; Moskalenko, R.; Gortinskaya, O.; Lyndina, Y. The role of heavy metal salts in pathological biomineralization of breast cancer tissue. Adv. Clin. Exp. Med. 2016, 25, 907–910. [Google Scholar] [CrossRef] [PubMed]
- Hoang, B.X.; Han, B.; Shaw, D.G.; Nimni, M. Zinc as a possible preventive and therapeutic agent in pancreatic, prostate, and breast cancer. Eur. J. Cancer Prev. 2016, 25, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Livingstone, C. Zinc: Physiology, deficiency, and parenteral nutrition. Nutr. Clin. Pract. 2015, 30, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, A.J. Zinc, aging, and immunosenescence: An overview. Pathobiol. Aging Age Relat. Dis. 2015, 5, 25592. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, T.; Kurokawa, Y.; Chiche, J.; Pouyssegur, J.; Sato, H.; Fukuzawa, H.; Nagao, M.; Kambe, T. Dissecting the process of activation of cancer-promoting Zinc-requiring ectoenzymes by Zinc metalation mediated by ZNT transporters. J. Biol. Chem. 2017, 292, 2159–2173. [Google Scholar] [CrossRef] [PubMed]
- Aamodt, R.L.; Rumble, W.F.; Johnston, G.S.; Foster, D.; Henkin, R.I. Zinc metabolism in humans after oral and intravenous administration of Zn-69m. Am. J. Clin. Nutr. 1979, 32, 559–569. [Google Scholar] [PubMed]
- Little, P.J.; Bhattacharya, R.; Moreyra, A.E.; Korichneva, I.L. Zinc and cardiovascular disease. Nutrition 2010, 26, 1050–1057. [Google Scholar] [CrossRef] [PubMed]
- Gimelli, A.; Menichetti, F.; Soldati, E.; Liga, R.; Vannozzi, A.; Marzullo, P.; Bongiorni, M.G. Relationships between cardiac innervation/perfusion imbalance and ventricular arrhythmias: Impact on invasive electrophysiological parameters and ablation procedures. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 2383–2391. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, B.; Wang, Y.; Tong, Q.; Liu, Q.; Sun, J.; Zheng, Y.; Cai, L. Zinc prevents the development of diabetic cardiomyopathy in db/db mice. Int. J. Mol. Sci. 2017, 18, 580. [Google Scholar] [CrossRef] [PubMed]
- Bozym, R.A.; Chimienti, F.; Giblin, L.J.; Gross, G.W.; Korichneva, I.; Li, Y.; Libert, S.; Maret, W.; Parviz, M.; Frederickson, C.J.; et al. Free Zinc ions outside a narrow concentration range are toxic to a variety of cells in vitro. Exp. Biol. Med. (Maywood, NJ) 2010, 235, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Efeovbokhan, N.; Bhattacharya, S.K.; Ahokas, R.A.; Sun, Y.; Guntaka, R.V.; Gerling, I.C.; Weber, K.T. Zinc and the prooxidant heart failure phenotype. J. Cardiovasc. Pharmacol. 2014, 64, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Hashemian, M.; Poustchi, H.; Mohammadi-Nasrabadi, F.; Hekmatdoost, A. Systematic review of Zinc biochemical indicators and risk of coronary heart disease. ARYA Atheroscler. 2015, 11, 357–365. [Google Scholar] [PubMed]
- Lee, S.R.; Noh, S.J.; Pronto, J.R.; Jeong, Y.J.; Kim, H.K.; Song, I.S.; Xu, Z.; Kwon, H.Y.; Kang, S.C.; Sohn, E.H.; et al. The critical roles of Zinc: Beyond impact on myocardial signaling. Korean J. Physiol. Pharmacol. 2015, 19, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Chu, A.; Foster, M.; Samman, S. Zinc Status and risk of cardiovascular diseases and type 2 diabetes mellitus-a systematic review of prospective cohort studies. Nutrients 2016, 8, 707. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, T.; Fliss, H. Hypochlorous acid and chloramines increase endothelial permeability: Possible involvement of cellular zinc. Am. J. Physiol. 1994, 267 Pt 2, H1597–H1607. [Google Scholar]
- Turan, B. Zinc-induced changes in ionic currents of cardiomyocytes. Biol. Trace Element Res. 2003, 94, 49–60. [Google Scholar] [CrossRef]
- Zima, A.V.; Blatter, L.A. Redox regulation of cardiac calcium channels and transporters. Cardiovasc. Res. 2006, 71, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Dineley, K.E.; Richards, L.L.; Votyakova, T.V.; Reynolds, I.J. Zinc causes loss of membrane potential and elevates reactive oxygen species in rat brain mitochondria. Mitochondrion 2005, 5, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Ayaz, M.; Turan, B. Selenium prevents diabetes-induced alterations in [Zn2+]i and metallothionein level of rat heart via restoration of cell redox cycle. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1071–H1080. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Molecular aspects of human cellular zinc homeostasis: Redox control of zinc potentials and zinc signals. Biometals 2009, 22, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Kuster, G.M.; Lancel, S.; Zhang, J.; Communal, C.; Trucillo, M.P.; Lim, C.C.; Pfister, O.; Weinberg, E.O.; Cohen, R.A.; Liao, R.; et al. Redox-mediated reciprocal regulation of SERCA and Na+-Ca2+ exchanger contributes to sarcoplasmic reticulum Ca2+ depletion in cardiac myocytes. Free Radic. Biol. Med. 2010, 48, 1182–1187. [Google Scholar] [CrossRef] [PubMed]
- Ranasinghe, P.; Pigera, S.; Galappatthy, P.; Katulanda, P.; Constantine, G.R. Zinc and diabetes mellitus: Understanding molecular mechanisms and clinical implications. Daru 2015, 23, 44. [Google Scholar] [CrossRef] [PubMed]
- Tuncay, E.; Okatan, E.N.; Toy, A.; Turan, B. Enhancement of cellular antioxidant-defence preserves diastolic dysfunction via regulation of both diastolic Zn2+ and Ca2+ and prevention of RyR2-leak in hyperglycemic cardiomyocytes. Oxid. Med. Cell. Longev. 2014, 2014, 290381. [Google Scholar] [CrossRef] [PubMed]
- Kamalov, G.; Ahokas, R.A.; Zhao, W.; Zhao, T.; Shahbaz, A.U.; Johnson, P.L.; Bhattacharya, S.K.; Sun, Y.; Gerling, I.C.; Weber, K.T. Uncoupling the coupled calcium and zinc dyshomeostasis in cardiac myocytes and mitochondria seen in aldosteronism. J. Cardiovasc. Pharmacol. 2010, 55, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Reilly-O’Donnell, B.; Robertson, G.B.; Karumbi, A.; McIntyre, C.; Bal, W.; Nishi, M.; Takeshima, H.; Stewart, A.J.; Pitt, S.J. Dysregulated Zn2+ homeostasis impairs cardiac type-2 ryanodine receptor and mitsugumin 23 functions, leading to sarcoplasmic reticulum Ca2+ leakage. J. Biol. Chem. 2017, 292, 13361–13373. [Google Scholar] [CrossRef] [PubMed]
- Brugger, D.; Windisch, W.M. Short-term subclinical Zinc deficiency in weaned piglets affects cardiac redox metabolism and Zinc concentration. J. Nutr. 2017, 147, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Permyakov, E.A.; Kretsinger, R.H. Cell signaling, beyond cytosolic calcium in eukaryotes. J. Inorg. Biochem. 2009, 103, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Murakami, M.; Fukada, T.; Nishida, K.; Yamasaki, S.; Suzuki, T. Roles of Zinc and Zinc signaling in immunity: Zinc as an intracellular signaling molecule. Adv. Immunol. 2008, 97, 149–176. [Google Scholar] [PubMed]
- Cicek, F.A.; Tokcaer-Keskin, Z.; Ozcinar, E.; Bozkus, Y.; Akcali, K.C.; Turan, B. Di-peptidyl peptidase-4 inhibitor sitagliptin protects vascular function in metabolic syndrome: Possible role of epigenetic regulation. Mol. Biol. Rep. 2014, 41, 4853–4863. [Google Scholar] [CrossRef] [PubMed]
- Billur, D.; Tuncay, E.; Okatan, E.N.; Olgar, Y.; Durak, A.T.; Degirmenci, S.; Can, B.; Turan, B. Interplay between cytosolic free Zn2+ and mitochondrion morphological changes in rat ventricular cardiomyocytes. Biol. Trace Element Res. 2016, 174, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Luo, M.; Zhang, Z.; Gu, J.; Chen, J.; Payne, K.M.; Tan, Y.; Wang, Y.; Yin, X.; Zhang, X.; et al. Zinc deficiency exacerbates while Zinc supplement attenuates cardiac hypertrophy in high-fat diet-induced obese mice through modulating p38 MAPK-dependent signaling. Toxicol. Lett. 2016, 258, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Bromberg, P.A.; Samet, J.M. Zinc ions as effectors of environmental oxidative lung injury. Free Radic. Biol. Med. 2013, 65, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Plum, L.M.; Rink, L.; Haase, H. The essential toxin: Impact of Zinc on human health. Int. J. Environ. Res. Public Health 2010, 7, 1342–1365. [Google Scholar] [CrossRef] [PubMed]
- Yaras, N.; Ugur, M.; Ozdemir, S.; Gurdal, H.; Purali, N.; Lacampagne, A.; Vassort, G.; Turan, B. Effects of diabetes on ryanodine receptor Ca release channel (RyR2) and Ca2+ homeostasis in rat heart. Diabetes 2005, 54, 3082–3088. [Google Scholar] [CrossRef] [PubMed]
- Ermak, G.; Davies, K.J. Calcium and oxidative stress: From cell signaling to cell death. Mol. Immunol. 2002, 38, 713–721. [Google Scholar] [CrossRef]
- Chakraborti, T.; Ghosh, S.K.; Michael, J.R.; Batabyal, S.K.; Chakraborti, S. Targets of oxidative stress in cardiovascular system. Mol. Cell. Biochem. 1998, 187, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, M.; Okabe, E. Superoxide anion radical-triggered Ca2+ release from cardiac sarcoplasmic reticulum through ryanodine receptor Ca2+ channel. Mol. Pharmacol. 1998, 53, 497–503. [Google Scholar] [PubMed]
- Boraso, A.; Williams, A.J. Modification of the gating of the cardiac sarcoplasmic reticulum Ca2+-release channel by H2O2 and dithiothreitol. Am. J. Physiol. 1994, 267 Pt 2, H1010–H1016. [Google Scholar]
- Woodier, J.; Rainbow, R.D.; Stewart, A.J.; Pitt, S.J. Intracellular Zinc modulates cardiac ryanodine receptor-mediated Calcium release. J. Biol. Chem. 2015, 290, 17599–17610. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.J.; Pitt, S.J. Zinc controls RyR2 activity during excitation-contraction coupling. Channels 2015, 9, 227–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamalov, G.; Ahokas, R.A.; Zhao, W.; Shahbaz, A.U.; Bhattacharya, S.K.; Sun, Y.; Gerling, I.C.; Weber, K.T. Temporal responses to intrinsically coupled Calcium and Zinc dyshomeostasis in cardiac myocytes and mitochondria during aldosteronism. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H385–H394. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Zhu, P.H. Biphasic modulation of ryanodine receptors by sulfhydryl oxidation in rat ventricular myocytes. Biophys. J. 2006, 91, 2882–2891. [Google Scholar] [CrossRef] [PubMed]
- Perez-Clausell, J.; Danscher, G. Intravesicular localization of Zinc in rat telencephalic boutons. A histochemical study. Brain Res. 1985, 337, 91–98. [Google Scholar] [CrossRef]
- Slomianka, L. Neurons of origin of Zinc-containing pathways and the distribution of Zinc-containing boutons in the hippocampal region of the rat. Neuroscience 1992, 48, 325–352. [Google Scholar] [CrossRef]
- Sekler, I.; Sensi, S.L.; Hershfinkel, M.; Silverman, W.F. Mechanism and regulation of cellular Zinc transport. Mol. Med. 2007, 13, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Kerchner, G.A.; Canzoniero, L.M.; Yu, S.P.; Ling, C.; Choi, D.W. Zn2+ current is mediated by voltage-gated Ca2+ channels and enhanced by extracellular acidity in mouse cortical neurones. J. Physiol. 2000, 528 Pt 1, 39–52. [Google Scholar] [CrossRef]
- Alvarez-Collazo, J.; Diaz-Garcia, C.M.; Lopez-Medina, A.I.; Vassort, G.; Alvarez, J.L. Zinc modulation of basal and β-adrenergically stimulated l-type Ca2+ current in rat ventricular cardiomyocytes: Consequences in cardiac diseases. Pflugers Arch. 2012, 464, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Traynelis, S.F.; Burgess, M.F.; Zheng, F.; Lyuboslavsky, P.; Powers, J.L. Control of voltage-independent Zinc inhibition of NMDA receptors by the NR1 subunit. J. Neurosci. 1998, 18, 6163–6175. [Google Scholar] [PubMed]
- Zhang, S.; Kehl, S.J.; Fedida, D. Modulation of Kv1.5 potassium channel gating by extracellular Zinc. Biophys. J. 2001, 81, 125–136. [Google Scholar] [CrossRef]
- Gilly, W.F.; Armstrong, C.M. Slowing of sodium channel opening kinetics in squid axon by extracellular Zinc. J. Gen. Physiol. 1982, 79, 935–964. [Google Scholar] [CrossRef] [PubMed]
- Gilly, W.F.; Armstrong, C.M. Divalent cations and the activation kinetics of potassium channels in squid giant axons. J. Gen. Physiol. 1982, 79, 965–996. [Google Scholar] [CrossRef] [PubMed]
- Aras, M.A.; Saadi, R.A.; Aizenman, E. Zn2+ regulates Kv2.1 voltage-dependent gating and localization following ischemia. Eur. J. Neurosci. 2009, 30, 2250–2257. [Google Scholar] [CrossRef] [PubMed]
- Haase, H.; Maret, W. Intracellular Zinc fluctuations modulate protein tyrosine phosphatase activity in insulin/insulin-like growth factor-1 signaling. Exp. Cell Res. 2003, 291, 289–298. [Google Scholar] [CrossRef]
- von Bulow, V.; Rink, L.; Haase, H. Zinc-mediated inhibition of cyclic nucleotide phosphodiesterase activity and expression suppresses TNF-α and IL-1 β production in monocytes by elevation of guanosine 3′,5′-cyclic monophosphate. J. Immunol. 2005, 175, 4697–4705. [Google Scholar] [CrossRef] [PubMed]
- van der Heyden, M.A.; Wijnhoven, T.J.; Opthof, T. Molecular aspects of adrenergic modulation of cardiac L-type Ca2+ channels. Cardiovasc. Res. 2005, 65, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.; Hogstrand, C.; Maret, W. Picomolar concentrations of free zinc(II) ions regulate receptor protein-tyrosine phosphatase beta activity. J. Biol. Chem. 2012, 287, 9322–9326. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.M.; Hiscox, S.; Nicholson, R.I.; Hogstrand, C.; Kille, P. Protein kinase CK2 triggers cytosolic Zinc signaling pathways by phosphorylation of Zinc channel ZIP7. Sci. Signal. 2012, 5, ra11. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Sunahara, R.K.; Hudson, T.Y.; Heyduk, T.; Howlett, A.C. Zinc inhibition of cAMP signaling. J. Biol. Chem. 2002, 277, 11859–11865. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Du, Z.; Patel, T.B. Copper and Zinc inhibit Galphas function: A nucleotide-free state of Galphas induced by Cu2+ and Zn2+. J. Biol. Chem. 2005, 280, 2579–2586. [Google Scholar] [CrossRef] [PubMed]
- Baltas, L.G.; Karczewski, P.; Bartel, S.; Krause, E.G. The endogenous cardiac sarcoplasmic reticulum Ca2+/calmodulin-dependent kinase is activated in response to β-adrenergic stimulation and becomes Ca2+-independent in intact beating hearts. FEBS Lett. 1997, 409, 131–136. [Google Scholar] [CrossRef]
- Yi, T.; Vick, J.S.; Vecchio, M.J.; Begin, K.J.; Bell, S.P.; Delay, R.J.; Palmer, B.M. Identifying cellular mechanisms of Zinc-induced relaxation in isolated cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H706–H715. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Boillat, A.; Huang, D.; Liang, C.; Peers, C.; Gamper, N. Intracellular zinc activates KCNQ channels by reducing their dependence on phosphatidylinositol 4,5-bisphosphate. Proc. Natl. Acad. Sci. USA 2017, 114, E6410–E6419. [Google Scholar] [CrossRef] [PubMed]
- Sensi, S.L.; Paoletti, P.; Bush, A.I.; Sekler, I. Zinc in the physiology and pathology of the CNS. Nat. Rev. Neurosci. 2009, 10, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J.; Koh, J.Y.; Bush, A.I. The neurobiology of Zinc in health and disease. Nat. Rev. Neurosci. 2005, 6, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Zinc coordination environments in proteins as redox sensors and signal transducers. Antioxid. Redox Signal. 2006, 8, 1419–1441. [Google Scholar] [CrossRef] [PubMed]
- Dineley, K.E.; Devinney, M.J., 2nd; Zeak, J.A.; Rintoul, G.L.; Reynolds, I.J. Glutamate mobilizes [Zn2+] through Ca2+ -dependent reactive oxygen species accumulation. J. Neurochem. 2008, 106, 2184–2193. [Google Scholar] [PubMed]
- Ellis, C.D.; Wang, F.; MacDiarmid, C.W.; Clark, S.; Lyons, T.; Eide, D.J. Zinc and the MSC2 Zinc transporter protein are required for endoplasmic reticulum function. J. Cell Biol. 2004, 166, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Sensi, S.L.; Ton-That, D.; Weiss, J.H. Mitochondrial sequestration and Ca2+-dependent release of cytosolic Zn2+ loads in cortical neurons. Neurobiol. Dis. 2002, 10, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [PubMed]
- Dedkova, E.N.; Blatter, L.A. Characteristics and function of cardiac mitochondrial nitric oxide synthase. J. Physiol. 2009, 587 Pt 4, 851–872. [Google Scholar] [CrossRef]
- Bouron, A.; Kiselyov, K.; Oberwinkler, J. Permeation, regulation and control of expression of TRP channels by trace metal ions. Pflugers Arch. 2015, 467, 1143–1164. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Iribe, G.; Nishida, M.; Naruse, K. Role of TRPC3 and TRPC6 channels in the myocardial response to stretch: Linking physiology and pathophysiology. Progress Biophys. Mol. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Duan, Y.; Takeuchi, A.; Hai-Kurahara, L.; Ichikawa, J.; Hiraishi, K.; Numata, T.; Ohara, H.; Iribe, G.; Nakaya, M.; et al. Uncovering the arrhythmogenic potential of TRPM4 activation in atrial-derived HL-1 cells using novel recording and numerical approaches. Cardiovasc. Res. 2017, 113, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K.; Tominaga, M. Extracellular Zinc ion regulates transient receptor potential melastatin 5 (TRPM5) channel activation through its interaction with a pore loop domain. J. Biol. Chem. 2013, 288, 25950–25955. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.; Drews, A.; Rizun, O.; Wagner, T.F.; Lis, A.; Mannebach, S.; Plant, S.; Portz, M.; Meissner, M.; Philipp, S.E.; et al. Transient receptor potential melastatin 1 (TRPM1) is an ion-conducting plasma membrane channel inhibited by Zinc ions. J. Biol. Chem. 2011, 286, 12221–12233. [Google Scholar] [CrossRef] [PubMed]
- Abiria, S.A.; Krapivinsky, G.; Sah, R.; Santa-Cruz, A.G.; Chaudhuri, D.; Zhang, J.; Adstamongkonkul, P.; DeCaen, P.G.; Clapham, D.E. TRPM7 senses oxidative stress to release Zn2+ from unique intracellular vesicles. Proc. Natl. Acad. Sci. USA 2017, 114, E6079–E6088. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Jiang, Y.; Huang, W.; Li, X.; Li, S. Role of transient receptor potential channels in heart transplantation: A potential novel therapeutic target for cardiac allograft vasculopathy. Med. Sci. Monit. 2017, 23, 2340–2347. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.; Wang, H.; Xi, J.; Mueller, R.A.; Norfleet, E.A.; Xu, Z. NO mobilizes intracellular Zn2+ via cGMP/PKG signaling pathway and prevents mitochondrial oxidant damage in cardiomyocytes. Cardiovasc. Res. 2007, 75, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Kambe, T.; Fukada, T.; Toyokuni, S. Editorial: The cutting edge of zinc biology. Arch. Biochem. Biophys. 2016, 611, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Kambe, T. The functions of metallothionein and ZIP and ZnT transporters: An overview and perspective. Int. J. Mol. Sci. 2016, 17, 336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, S.A. Zinc transporters and Zinc signaling: New insights into their role in type 2 diabetes. Int. J. Endocrinol. 2015, 2015, 167503. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Tepaamorndech, S. The SLC30 family of zinc transporters-a review of current understanding of their biological and pathophysiological roles. Mol. Aspects Med. 2013, 34, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Eide, D.J. The SLC39 family of Zinc transporters. Mol. Aspects Med. 2013, 34, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Kambe, T. Methods to evaluate Zinc transport into and out of the secretory and endosomal-lysosomal compartments in DT40 cells. Methods Enzymol. 2014, 534, 77–92. [Google Scholar] [PubMed]
- Kambe, T.; Hashimoto, A.; Fujimoto, S. Current understanding of ZIP and ZnT zinc transporters in human health and diseases. Cell. Mol. Life Sci. CMLS 2014, 71, 3281–3295. [Google Scholar] [CrossRef] [PubMed]
- Shima, T.; Jesmin, S.; Matsui, T.; Soya, M.; Soya, H. Differential effects of type 2 diabetes on brain glycometabolism in rats: Focus on glycogen and monocarboxylate transporter 2. J. Physiol. Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Bu, H.M.; Yang, C.Y.; Wang, M.L.; Ma, H.J.; Sun, H.; Zhang, Y. K(ATP) channels and MPTP are involved in the cardioprotection bestowed by chronic intermittent hypobaric hypoxia in the developing rat. J. Physiol. Sci. 2015, 65, 367–376. [Google Scholar] [CrossRef] [PubMed]
- King, J.C.; Shames, D.M.; Woodhouse, L.R. Zinc homeostasis in humans. J. Nutr. 2000, 130 (Suppl. S5), 1360s–1366s. [Google Scholar] [PubMed]
- Palmiter, R.D.; Findley, S.D. Cloning and functional characterization of a mammalian Zinc transporter that confers resistance to Zinc. EMBO J. 1995, 14, 639–649. [Google Scholar] [PubMed]
- Wenzel, H.J.; Cole, T.B.; Born, D.E.; Schwartzkroin, P.A.; Palmiter, R.D. Ultrastructural localization of Zinc transporter-3 (ZnT-3) to synaptic vesicle membranes within mossy fiber boutons in the hippocampus of mouse and monkey. Proc. Natl. Acad. Sci. USA 1997, 94, 12676–12681. [Google Scholar] [CrossRef] [PubMed]
- Kambe, T.; Weaver, B.P.; Andrews, G.K. The genetics of essential metal homeostasis during development. Genesis 2008, 46, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Bin, B.H.; Fukada, T.; Hosaka, T.; Yamasaki, S.; Ohashi, W.; Hojyo, S.; Miyai, T.; Nishida, K.; Yokoyama, S.; Hirano, T. Biochemical characterization of human ZIP13 protein: A homo-dimerized zinc transporter involved in the spondylocheiro dysplastic Ehlers-Danlos syndrome. J. Biol. Chem. 2011, 286, 40255–40265. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.M.; Morgan, H.E.; Smart, K.; Zahari, N.M.; Pumford, S.; Ellis, I.O.; Robertson, J.F.; Nicholson, R.I. The emerging role of the LIV-1 subfamily of Zinc transporters in breast cancer. Mol. Med. 2007, 13, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Kambe, T.; Geiser, J.; Lahner, B.; Salt, D.E.; Andrews, G.K. Slc39a1 to 3 (subfamily II) Zip genes in mice have unique cell-specific functions during adaptation to Zinc deficiency. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R1474–R1481. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Yu, Y.Y.; Kirschke, C.P.; Gertz, E.R.; Lloyd, K.K. Znt7 (Slc30a7)-deficient mice display reduced body Zinc status and body fat accumulation. J. Biol. Chem. 2007, 282, 37053–37063. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Kirschke, C.P.; Zhang, Y.; Yu, Y.Y. The ZIP7 gene (Slc39a7) encodes a Zinc transporter involved in Zinc homeostasis of the Golgi apparatus. J. Biol. Chem. 2005, 280, 15456–15463. [Google Scholar] [CrossRef] [PubMed]
- Sladek, R.; Rocheleau, G.; Rung, J.; Dina, C.; Shen, L.; Serre, D.; Boutin, P.; Vincent, D.; Belisle, A.; Hadjadj, S.; et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 2007, 445, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Wenzlau, J.M.; Juhl, K.; Yu, L.; Moua, O.; Sarkar, S.A.; Gottlieb, P.; Rewers, M.; Eisenbarth, G.S.; Jensen, J.; Davidson, H.W.; et al. The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc. Natl. Acad. Sci. USA 2007, 104, 17040–17045. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, K.; Ravier, M.A.; Schraenen, A.; Creemers, J.W.; Van de Plas, R.; Granvik, M.; Van Lommel, L.; Waelkens, E.; Chimienti, F.; Rutter, G.A.; et al. Insulin crystallization depends on Zinc transporter ZnT8 expression, but is not required for normal glucose homeostasis in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 14872–14877. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, T.J.; Bellomo, E.A.; Wijesekara, N.; Loder, M.K.; Baldwin, J.M.; Gyulkhandanyan, A.V.; Koshkin, V.; Tarasov, A.I.; Carzaniga, R.; Kronenberger, K.; et al. Insulin storage and glucose homeostasis in mice null for the granule zinc transporter ZnT8 and studies of the type 2 diabetes-associated variants. Diabetes 2009, 58, 2070–2083. [Google Scholar] [CrossRef] [PubMed]
- Wijesekara, N.; Dai, F.F.; Hardy, A.B.; Giglou, P.R.; Bhattacharjee, A.; Koshkin, V.; Chimienti, F.; Gaisano, H.Y.; Rutter, G.A.; Wheeler, M.B. β cell-specific ZnT8 deletion in mice causes marked defects in insulin processing, crystallisation and secretion. Diabetologia 2010, 53, 1656–1668. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, M.; Fujitani, Y.; Hara, A.; Uchida, T.; Tamura, Y.; Takeno, K.; Kawaguchi, M.; Watanabe, T.; Ogihara, T.; Fukunaka, A.; et al. The diabetes-susceptible gene Slc30A8/ZnT8 regulates hepatic insulin clearance. J. Clin. Investig. 2013, 123, 4513–4524. [Google Scholar] [CrossRef] [PubMed]
- Chabosseau, P.; Rutter, G.A. Zinc and diabetes. Arch. Biochem. Biophys. 2016, 611, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Rutter, G.A.; Chabosseau, P.; Bellomo, E.A.; Maret, W.; Mitchell, R.K.; Hodson, D.J.; Solomou, A.; Hu, M. Intracellular Zinc in insulin secretion and action: A determinant of diabetes risk? Proc. Nutr. Soc. 2016, 75, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Hogstrand, C.; Kille, P.; Nicholson, R.I.; Taylor, K.M. Zinc transporters and cancer: A potential role for ZIP7 as a hub for tyrosine kinase activation. Trends Mol. Med. 2009, 15, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.M.; Vichova, P.; Jordan, N.; Hiscox, S.; Hendley, R.; Nicholson, R.I. ZIP7-mediated intracellular Zinc transport contributes to aberrant growth factor signaling in antihormone-resistant breast cancer Cells. Endocrinology 2008, 149, 4912–4920. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Batchuluun, B.; Ho, L.; Zhu, D.; Prentice, K.J.; Bhattacharjee, A.; Zhang, M.; Pourasgari, F.; Hardy, A.B.; Taylor, K.M.; et al. Characterization of Zinc influx transporters (ZIPs) in pancreatic β cells: Roles in regulating cytosolic Zinc homeostasis and insulin secretion. J. Biol. Chem. 2015, 290, 18757–18769. [Google Scholar] [CrossRef] [PubMed]
- Grubman, A.; Lidgerwood, G.E.; Duncan, C.; Bica, L.; Tan, J.L.; Parker, S.J.; Caragounis, A.; Meyerowitz, J.; Volitakis, I.; Moujalled, D.; et al. Deregulation of subcellular biometal homeostasis through loss of the metal transporter, ZIP7, in a childhood neurodegenerative disorder. Acta Neuropathol. Commun. 2014, 2, 25. [Google Scholar] [CrossRef] [PubMed]
- Groth, C.; Sasamura, T.; Khanna, M.R.; Whitley, M.; Fortini, M.E. Protein trafficking abnormalities in Drosophila tissues with impaired activity of the ZIP7 Zinc transporter Catsup. Development 2013, 140, 3018–3027. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, W.; Kimura, S.; Iwanaga, T.; Furusawa, Y.; Irie, T.; Izumi, H.; Watanabe, T.; Hijikata, A.; Hara, T.; Ohara, O.; et al. Zinc Transporter Slc39A7/ZIP7 promotes intestinal epithelial self-renewal by resolving ER stress. PLoS Genet. 2016, 12, e1006349. [Google Scholar] [CrossRef] [PubMed]
- Kirschke, C.P.; Huang, L. ZnT7, a novel mammalian Zinc transporter, accumulates Zinc in the Golgi apparatus. J. Biol. Chem. 2003, 278, 4096–4102. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, Y.; Cui, X.; Yao, W.; Yu, X.; Cen, P.; Hodges, S.E.; Fisher, W.E.; Brunicardi, F.C.; Chen, C.; et al. Gene profile identifies Zinc transporters differentially expressed in normal human organs and human pancreatic cancer. Curr. Mol. Med. 2013, 13, 401–409. [Google Scholar] [PubMed]
- Haase, H.; Maret, W. Fluctuations of cellular, available zinc modulate insulin signaling via inhibition of protein tyrosine phosphatases. J. Trace Elements Med. Biol. Organ Soc. Miner. Trace Elements (GMS) 2005, 19, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, S.; Sakata-Sogawa, K.; Hasegawa, A.; Suzuki, T.; Kabu, K.; Sato, E.; Kurosaki, T.; Yamashita, S.; Tokunaga, M.; Nishida, K.; et al. Zinc is a novel intracellular second messenger. J. Cell Biol. 2007, 177, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Myers, S.A.; Nield, A.; Myers, M. Zinc transporters, mechanisms of action and therapeutic utility: Implications for type 2 diabetes mellitus. J. Nutr. Metab. 2012, 2012, 173712. [Google Scholar] [CrossRef] [PubMed]
- Jansen, J.; Karges, W.; Rink, L. Zinc and diabetes—Clinical links and molecular mechanisms. J. Nutr. Biochem. 2009, 20, 399–417. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A patch-clamp study on the time course of L-type Ca2+-current (ICa), depressed by Zn2+ exposed either extracellulary (ZnCl2) or intracellularly (i.e., loaded with Zn2+-ionophore pyrithione, ZnPT). (A) The ICa was evoked at 0 mV from a holding potential −80 mV and ICa was recorded in the presence of either ZnCl2 (10 and 100 µM) or ZnPT (1 and 10 µM) in whole-cell patch-clamped ventricular cardiomyocytes in the presence of 1.8 mM external Ca2+. WO represents the washout of applications. Corresponding availability curves of the ICa by either ZnCl2 (B) or ZnPT (C). Note about 10 mV left-shift by ZnPT application in availability curve of ICa.
Figure 1.
A patch-clamp study on the time course of L-type Ca2+-current (ICa), depressed by Zn2+ exposed either extracellulary (ZnCl2) or intracellularly (i.e., loaded with Zn2+-ionophore pyrithione, ZnPT). (A) The ICa was evoked at 0 mV from a holding potential −80 mV and ICa was recorded in the presence of either ZnCl2 (10 and 100 µM) or ZnPT (1 and 10 µM) in whole-cell patch-clamped ventricular cardiomyocytes in the presence of 1.8 mM external Ca2+. WO represents the washout of applications. Corresponding availability curves of the ICa by either ZnCl2 (B) or ZnPT (C). Note about 10 mV left-shift by ZnPT application in availability curve of ICa.
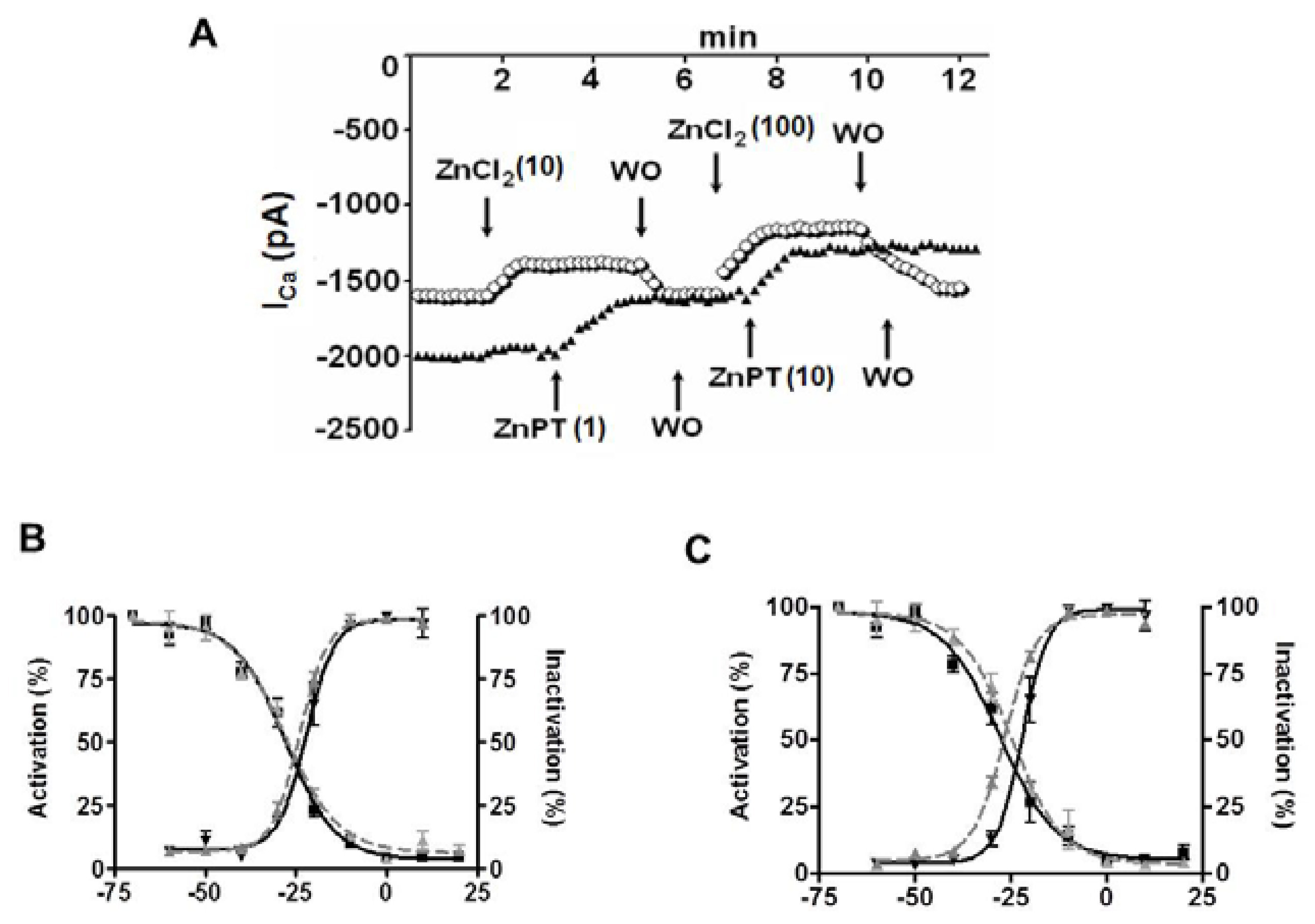
Figure 2.
FluoZin-3 and Fluo-3 responses to caffeine application demonstrate sarco(endo)plasmic reticulum as an intracellular labile Zn2+ pool in cardiomyocytes performed with confocal imaging. We used either a Zn2+-specific fluorescence dye, FluoZin-3 (A) or a Ca2+-specific fluorescence dye, Fluo-3 (B) loaded ventricular cardiomyocytes isolated from rat heart and stimulated them with 10 mM caffeine (Caff) in the either absence (A and B, respectively) or presence of a membrane-permeant Zn2+ chelator TPEN (N,N,N′,N′-tetrakis(2-pyridylmethyl) ethylenediamine, 30-µM) (C), respectively. Normalized caffeine responses are given as F/F0, where F is the fluorescence signal and F0 is the diastolic fluorescence. The mean (± standard error of mean (SEM)) values are given for the protocols in (D), n = 5–6 for hearts/protocol. * p < 0.05 vs. before application.
Figure 2.
FluoZin-3 and Fluo-3 responses to caffeine application demonstrate sarco(endo)plasmic reticulum as an intracellular labile Zn2+ pool in cardiomyocytes performed with confocal imaging. We used either a Zn2+-specific fluorescence dye, FluoZin-3 (A) or a Ca2+-specific fluorescence dye, Fluo-3 (B) loaded ventricular cardiomyocytes isolated from rat heart and stimulated them with 10 mM caffeine (Caff) in the either absence (A and B, respectively) or presence of a membrane-permeant Zn2+ chelator TPEN (N,N,N′,N′-tetrakis(2-pyridylmethyl) ethylenediamine, 30-µM) (C), respectively. Normalized caffeine responses are given as F/F0, where F is the fluorescence signal and F0 is the diastolic fluorescence. The mean (± standard error of mean (SEM)) values are given for the protocols in (D), n = 5–6 for hearts/protocol. * p < 0.05 vs. before application.

Figure 3.
An electron microscopy examination of the effect of increased intracellular labile Zn2+ on ultrastructure of cardiomyocytes. The microscopic examination of freshly isolated cardiomyocytes under physiological condition (A) showing regular myofibrils and mitochondrial (m) structure and under 0.1 μM ZnPT incubation for 15–20 min (B) showing irregular mitochondrial cristae (white arrows). Magnification is ×21, 560 and bars represent 500 nm. On the right, cardiomyocytes showing clustered and degenerated mitochondria (m) under 1 μM ZnPT incubation for 15–20 min(C). Black arrows are showing irregular Z-lines. Magnification is ×12,930 and bar represents 1000 nm.
Figure 3.
An electron microscopy examination of the effect of increased intracellular labile Zn2+ on ultrastructure of cardiomyocytes. The microscopic examination of freshly isolated cardiomyocytes under physiological condition (A) showing regular myofibrils and mitochondrial (m) structure and under 0.1 μM ZnPT incubation for 15–20 min (B) showing irregular mitochondrial cristae (white arrows). Magnification is ×21, 560 and bars represent 500 nm. On the right, cardiomyocytes showing clustered and degenerated mitochondria (m) under 1 μM ZnPT incubation for 15–20 min(C). Black arrows are showing irregular Z-lines. Magnification is ×12,930 and bar represents 1000 nm.

Figure 4.
A summary of zinc and its role in cardiac function. Basic mechanisms affected with either zinc-deficiency or zinc-excess and, the, in turn, underline the heart dysfunction, mainly as a zinc-concentration dependent manner in the heart.
Figure 4.
A summary of zinc and its role in cardiac function. Basic mechanisms affected with either zinc-deficiency or zinc-excess and, the, in turn, underline the heart dysfunction, mainly as a zinc-concentration dependent manner in the heart.
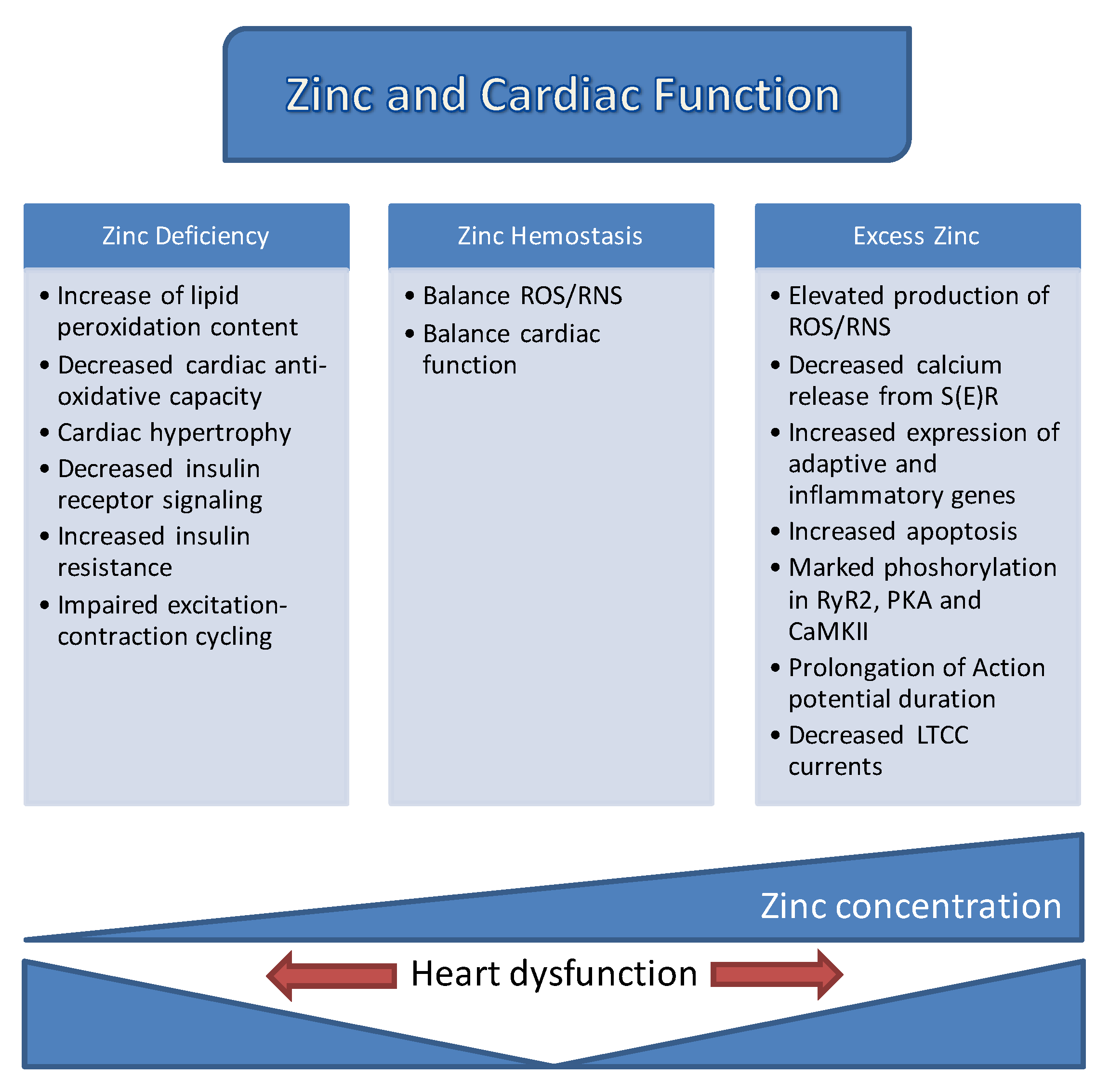

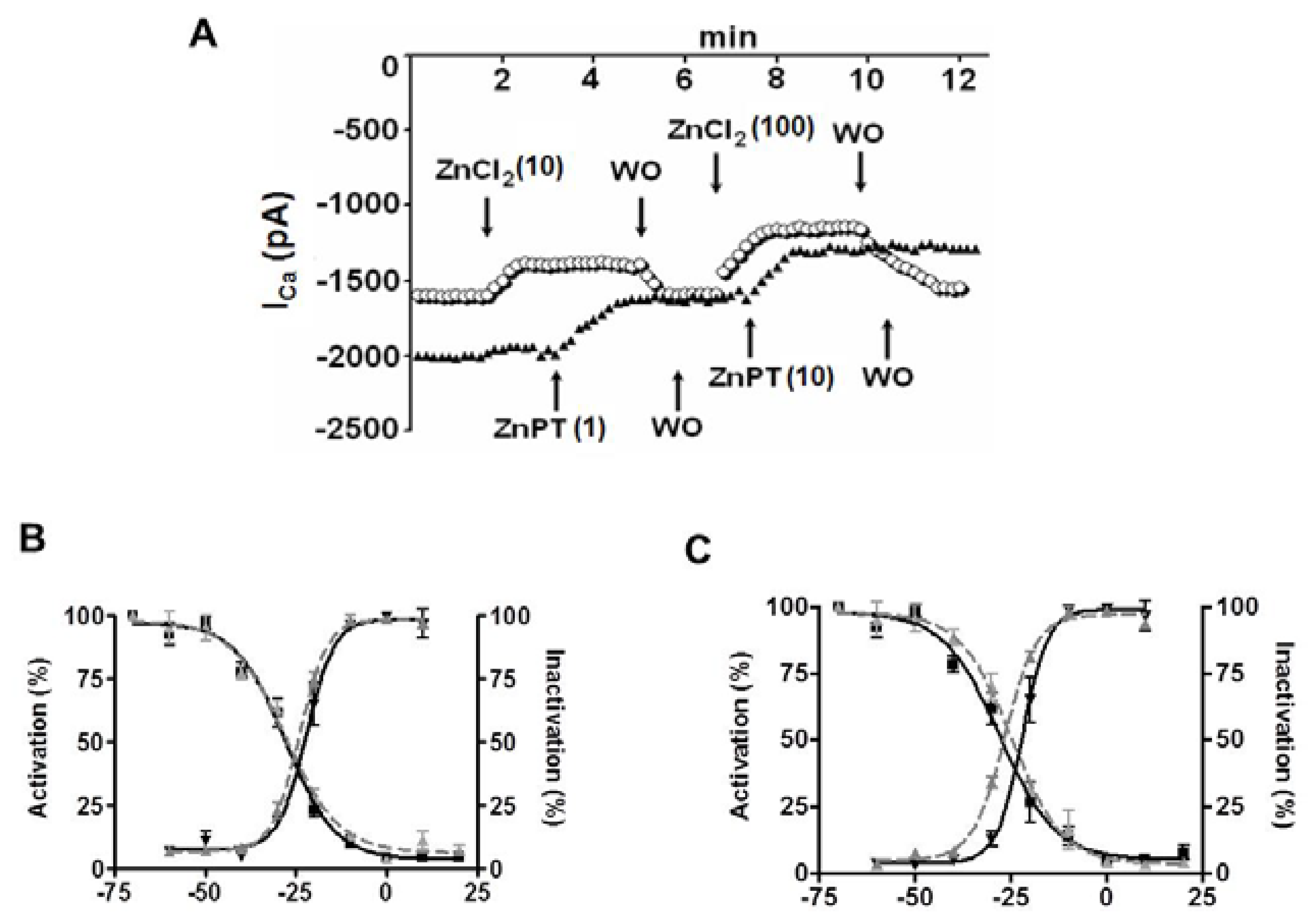{kind=link}
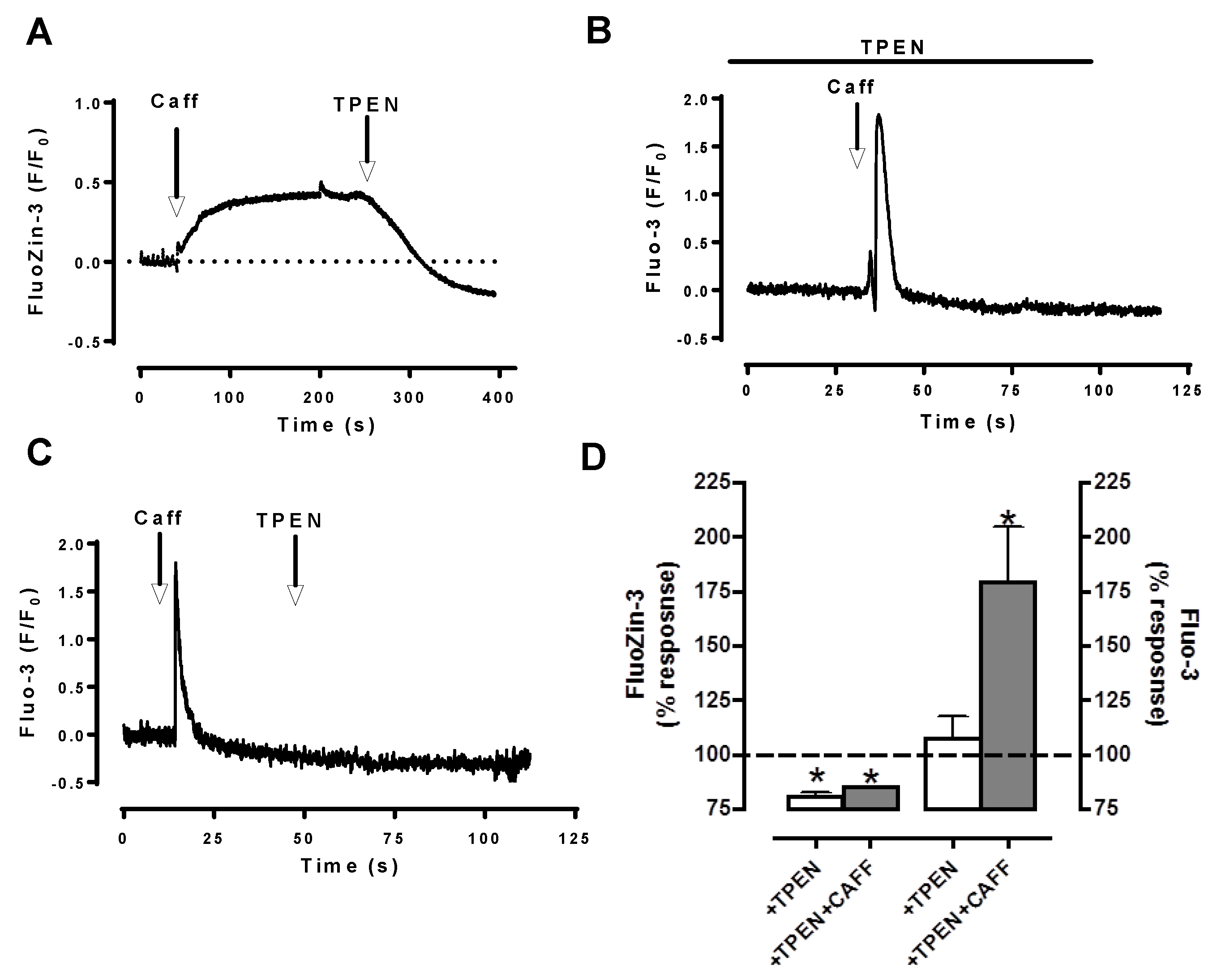{kind=link}
{kind=link}
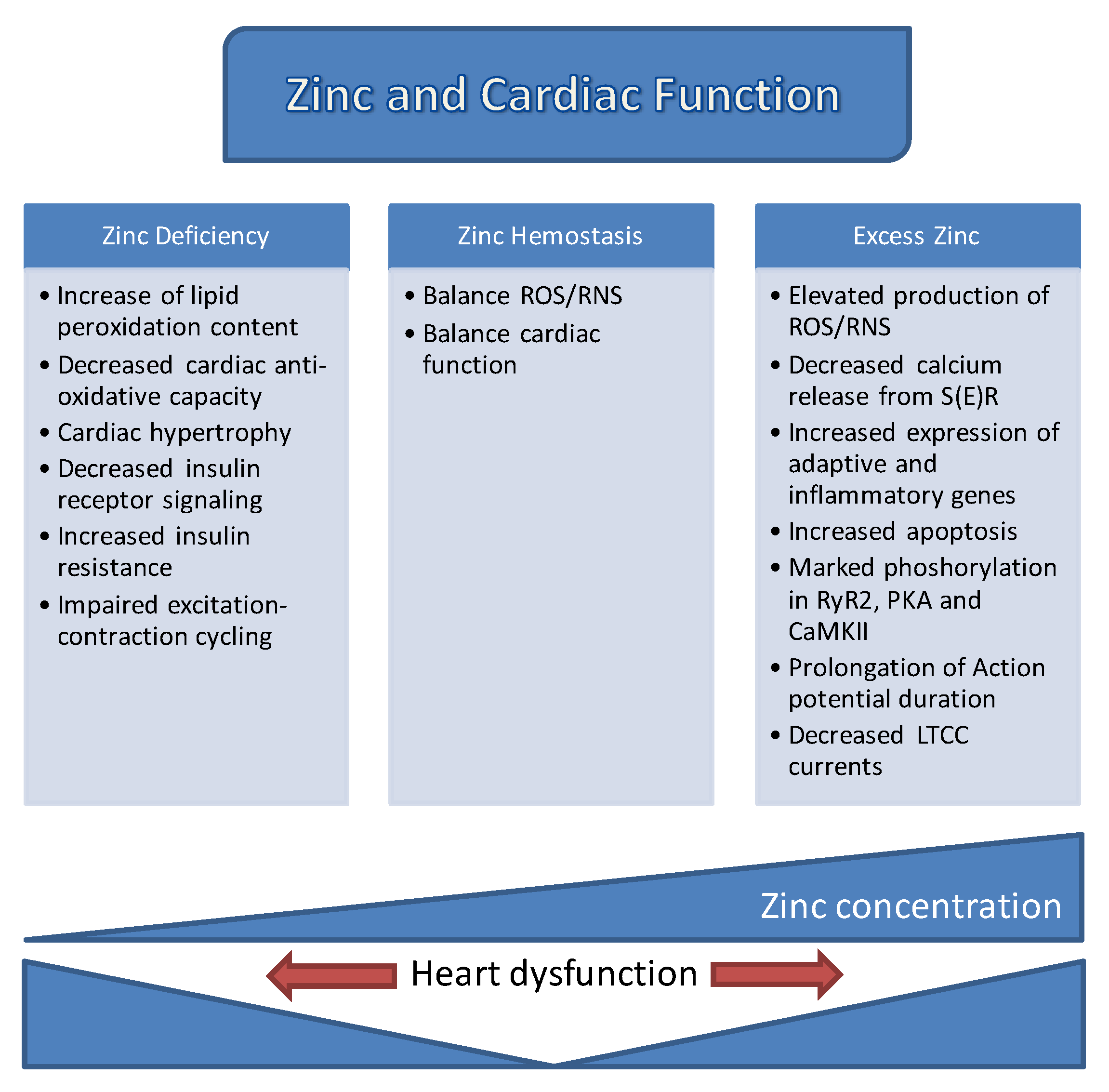{kind=link}
Table 1.
Effect of excess Zn2+ on cardiomyocyte electrical and mechanical activity.
| Parameters | Excess Zn2+ | Parameters | Excess Zn2+ |
|---|---|---|---|
| Electrical activity | Mechanical activity | ||
| Resting membrane potential | ↔ | Muscle Contraction | ↓ |
| Action potential duration (APD) Time to peak AP amplitude (TP) | ↔ ↑ | Contraction rate Relaxation rate | ↓ ↓ |
| Ca2+ transients | ↓ | Time to peak contraction | ↔ |
| L-type Ca2+ currents | ↓ | Time at 50% of relaxation | ↔ |
| Mitochondrial membrane potential | ↑ |
Arrows represent the increased (↑) decreased (↓) or unchanged (↔) electrical and mechanical activity as time or amplitude of the parameters.
Table 2.
Effects of excess Zn2+ on biochemical and ultrastructural parameters in cardiomycoytes.
| Parameters | Excess Zn2+ | Parameters | Excess Zn2+ | |
|---|---|---|---|---|
| Biochemical parameters | ||||
| pRyR2/RyR2 | ↑ | Promyelocytic leukemia(PML) | ↑ | |
| pPKA/PKA | ↑ | Bcl-2/BAX | ↓ | |
| FK506-binding protein(FKBP12.6) | ↔ | pAkt/Akt | ↑ | |
| pCaMKII/CaMKII | ↑ | pNFκB/NFκB | ↑ | |
| Calregulin | ↑ | pGSK/GSK | ↑ | |
| Glucose regulated protein (GRP78) | ↑ | |||
| Ultrastructure parameters | ||||
| Morphological changes in mitochondria | ↑ | Electron density of Z-lines | ↓ | |
| Number of lysosomes | ↑ | |||
Arrows represent the increased (↑), decreased (↓), or unchanged (↔) protein expression levels in biochemical parameters or number of lysosomes, Z-lines, and morphological changes in ultrastructure parameters.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Turan, B.; Tuncay, E. Impact of Labile Zinc on Heart Function: From Physiology to Pathophysiology. Int. J. Mol. Sci. 2017, 18, 2395. https://doi.org/10.3390/ijms18112395
AMA Style
Turan B, Tuncay E. Impact of Labile Zinc on Heart Function: From Physiology to Pathophysiology. International Journal of Molecular Sciences. 2017; 18(11):2395. https://doi.org/10.3390/ijms18112395
Chicago/Turabian StyleTuran, Belma, and Erkan Tuncay. 2017. "Impact of Labile Zinc on Heart Function: From Physiology to Pathophysiology" International Journal of Molecular Sciences 18, no. 11: 2395. https://doi.org/10.3390/ijms18112395
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.