Metabolic Dysfunction and Oxidative Stress in Epilepsy

Department of Pharmaceutical Sciences, University of Colorado, Anschutz Medical Campus, Aurora, CO 80045, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2017, 18(11), 2365; https://doi.org/10.3390/ijms18112365
Submission received: 27 September 2017
/
Revised: 27 October 2017
/
Accepted: 30 October 2017
/
Published: 8 November 2017
(This article belongs to the Special Issue Commemorative Issue in Honor of Professor Uwe Heinemann: Metabolic Epilepsies)
Abstract
:The epilepsies are a heterogeneous group of disorders characterized by the propensity to experience spontaneous recurrent seizures. Epilepsies can be genetic or acquired, and the underlying mechanisms of seizure initiation, seizure propagation, and comorbid conditions are incompletely understood. Metabolic changes including the production of reactive species are known to result from prolonged seizures and may also contribute to epilepsy development. In this review, we focus on the evidence that metabolic and redox disruption is both cause and consequence of epileptic seizures. Additionally, we discuss the promise of targeting redox processes as a therapeutic option in epilepsy.
1. Introduction
Epilepsy is the fourth most common neurological condition, affecting approximately 65 million people worldwide [1]. It is a complex spectrum of disorders defined by the tendency to experience abnormal, highly synchronous brain activity known as a seizure. Known risk factors for the development of epilepsy fall into two main categories, those that have genetic origins and those that are acquired; however, in some cases the cause is unknown [1]. Currently available therapeutics include those that target ion channels or neurotransmitter systems [2]. These types of medications are effective at controlling seizure activity in about 60% of people with epilepsy but provide merely symptomatic relief, are ineffective in roughly 40% of people, and have the potential to exacerbate comorbidities [2,3]. There is therefore a great need to identify mechanisms that cause epilepsy and thus generate new therapeutic targets.
One promising avenue of research is the study of how metabolic dysfunction can contribute to seizures and exacerbate related sequalae such as neuronal loss and cognitive impairment. In this review, we will discuss the evidence suggesting the role of metabolic and redox alterations in genetic and acquired epilepsies.
2. Sources of Reactive Species and Oxidative Stress
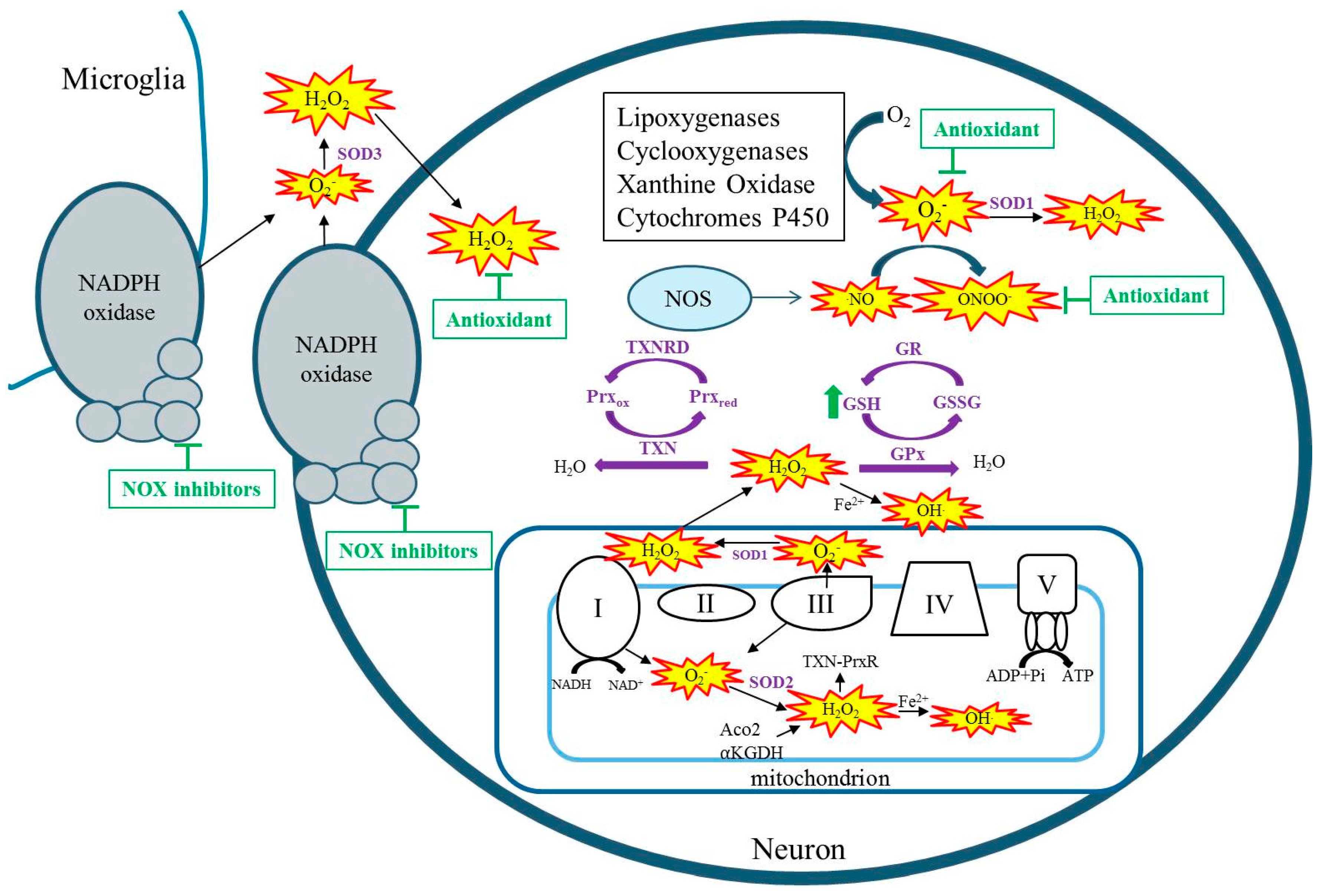
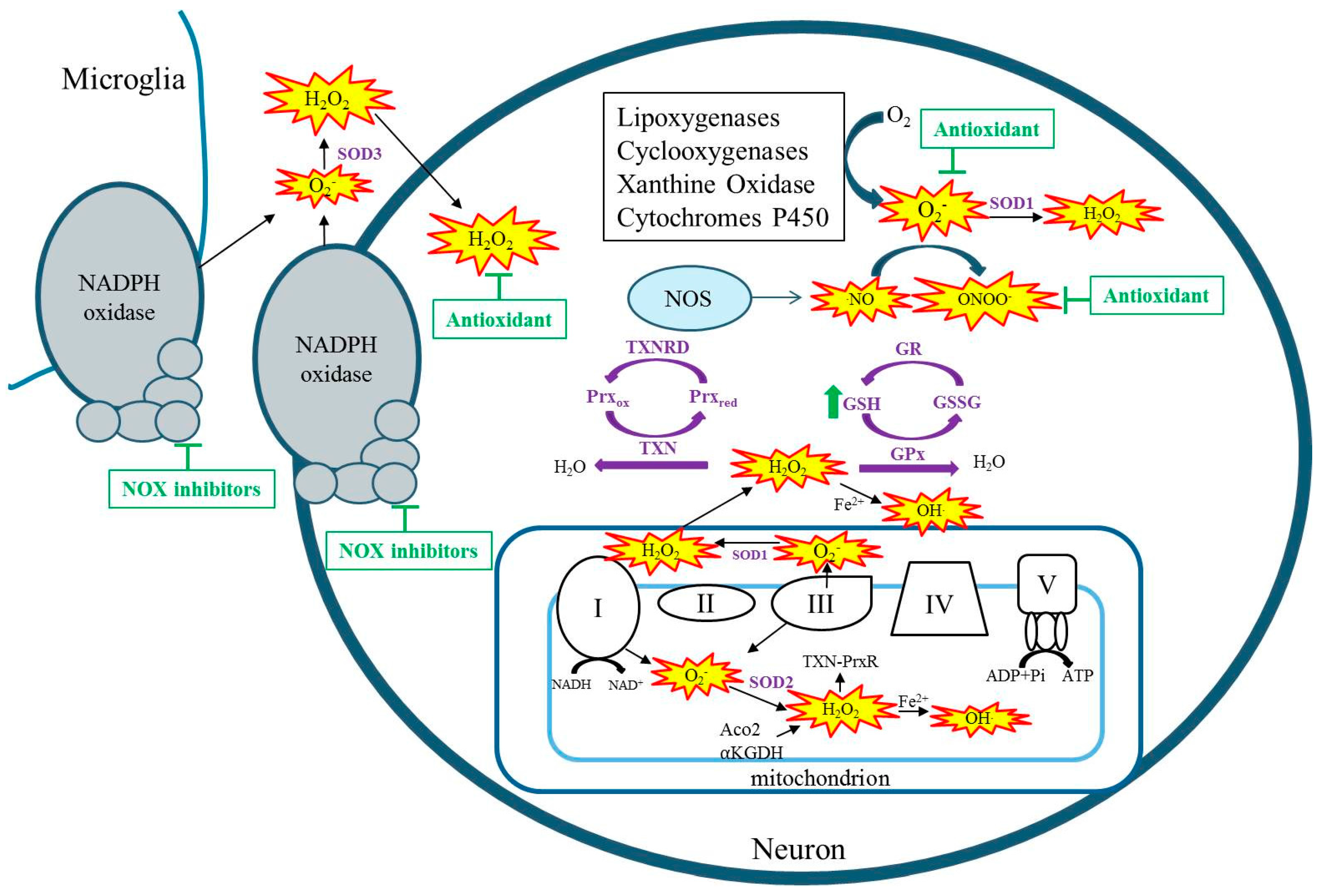
Free radicals are any chemical species with one or more unpaired electrons in the external orbit. Reactive species (RSs) i.e., reactive oxygen species (ROS) and reactive nitrogen species (RNS) collectively include singlet oxygen, superoxide (O2−), hydrogen peroxide (H2O2), nitric oxide (NO), hydroxyl radical (HO), and peroxynitrite (ONOO−). These RSs are produced by both enzymatic and non-enzymatic reactions and as a natural consequence of aerobic metabolism [4]. During aerobic metabolism, an infinitesimal percentage of oxygen consumed by mitochondria leaks from electron transport chain complexes I and III to form O2−, which can go on to form additional RSs such as H2O2 and OH (Figure 1) [5,6,7]. Whereas O2− is not a strong oxidant itself, it can oxidize certain susceptible targets such as the labile iron–sulfur center of aconitase(s) [8,9]. In the kainate model of acquired epilepsy, the production of mitochondrial O2 has been demonstrated by the inactivation of mitochondrial, not cytosolic aconitase, and the production of H2O2 in isolated respiring mitochondria from the hippocampus [10,11]. Additionally, studies have demonstrated inhibition of α ketoglutarate dehydrogenase (α-KGDH) in a model of acquired epilepsy, inhibition of complex I in human temporal lobe epilepsy tissue as well as in immature and adult models of acquired epilepsy [12,13,14,15,16]. Regardless of the original source of RSs, when mitochondrial aconitase, α-KGDH, or complex I are inhibited, the result is additional RS production capable of creating vicious cycles of RS production in mitochondria [17,18,19,20]. Reactive species are also produced from enzymatic processes outside of the mitochondria such as by xanthine oxidase, nitric oxide synthase, cyclooxygenase, lipoxygenase, cytochrome p450, and nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (Figure 1) [21,22]. Non-mitochondrial sources of ROS are activated by seizures, and targeting these enzymes has been shown to attenuate seizure-induced neurodegeneration [23,24,25,26]. When the production of RS exceeds the capacity of endogenous antioxidants to detoxify them, oxidative damage ensues.
The brain is particularly vulnerable to oxidant damage due to the abundance of mitochondria, high oxygen demand, poor repair capacity, and the presence of high concentrations of polyunsaturated fatty acids [27]. Oxidative stress can inflict damage to imperative cellular macromolecules including proteins, lipids, and DNA. Oxidation of proteins can change the structure, function, and activity of key enzymes [28]. Lipid peroxidation can compromise membrane structure leading to alterations in cell permeability and activity of membrane proteins, leading to hyperexcitability [29]. In the brain, oxidative damage to neural membranes can have profound effects on neurotransmitter uptake and release in addition to the maintenance of proper ionic gradients, again altering neuronal excitability [29]. Oxidative stress has been shown to contribute to the pathogenesis of a number of neurological conditions including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, stroke, and Amyotrophic Lateral Sclerosis [30,31]. In this review, we discuss the evidence which supports a key role of oxidative stress and mitochondrial function in the pathogenesis of the epilepsies. PubMed was searched for specific English terms including oxidative stress/damage in Alpers–Huttenlocher syndrome, oxidative stress, and Dravet syndrome. No restrictions regarding publication date, funder, or other article attributes were employed. The number of articles retrieved by such searches was relatively small (<100) and the relevance of the articles was assessed first by the abstract and second by the article text.
3. Oxidative Stress in Genetic Epilepsies
Epilepsy syndromes that are associated with genetic mutations are relatively rare, representing only a small fraction of all cases; however, this number is likely to increase as genetic testing becomes more commonplace. The genetic epilepsies are comprised of a heterogeneous group of disorders associated with diverse genetic mechanisms that can include de novo mutations to ion channel genes (channelopathies), chromosomal abnormalities, alterations to genes associated with brain malformations, and inherited or acquired mutations in mitochondrial DNA. In the following sections, we review genetic epilepsies that are associated with increased oxidative stress or metabolic dysfunction (summarized in Table 1).
3.1. Mitochondrial Encephalopathies
Mitochondrial DNA (mtDNA) mutations that result in epileptic phenotypes represent the strongest evidence for a link between epilepsy and oxidative stress or mitochondrial dysfunction. The most common of these include myoclonic epilepsy with ragged-red fibers (MERRF), Leigh syndrome, and Alpers–Huttenlocher syndrome. MERFF is caused by alterations to the tRNALys gene of the mtDNA, which affects complex I of the electron transport chain [32]. In cells cultured from patients with MERRF, there is evidence of increased ROS production, decreased ATP production, alterations to antioxidant gene expression, and oxidative damage [33]. Additionally, in cells harboring the genetic mutation associated with MERRF, mitochondrial calcium homeostasis was significantly altered, which could contribute to neuronal hyperexcitability [34]. Leigh syndrome is a neurodegenerative disease characterized by severe encephalopathy and refractory seizures. It is a genetically heterogeneous disorder that has been associated with over 35 mutations to mtDNA affecting any of the respiratory chain complexes. As such, those mutations affecting complex I result in increased ROS production and those mutations affecting complex V result in decreased ATP production [35,36,37]. In an animal model of Leigh syndrome, treatment with rapamycin attenuated neuropathological abnormalities and improved survival which the authors attributed to a metabolic shift away from glycolysis [38]. Another study suggested that genetic and pharmacological targeting of oxidative stress can attenuate neurodegeneration associated with Leigh syndrome [39]. Alpers–Huttenlocher syndrome (AHS) is caused by mutations to the mtDNA replicase, POLG, which disrupts mtDNA replication resulting in decreased mtDNA [40]. Neurologically, this disorder is characterized by refractory seizures, neuronal loss, and cognitive decline [41]. Decreased mtDNA can result in ATP depletion leading to energy failure and activation of apoptotic or necrotic cell pathways, which is evident in patients with AHS [42,43]. Additionally, ATP depletion is sufficient to induce neuronal hyperexcitability by altering sodium–potassium ATPase activity and decreasing neuronal membrane potential [44,45]. Thus, it would appear that oxidative stress contributes to seizures associated with mtDNA mutations or at the very least contributes to neuropathology.
3.2. Genetic Epilepsies Associated with Metabolic Dysfunction
Dravet syndrome (DS) is a rare and catastrophic epileptic encephalopathy that is characterized by intractable seizures and neurocognitive decline. In approximately 80% of patients, the disorder can be traced to de novo mutations in the SCN1A gene [46]. This gene encodes the sodium channel Nav1.1, and altered function of this channel seems to specifically affect GABAergic interneurons leading to decreased GABAergic tone and therefore increased hyperexcitability. Although direct evidence suggesting that oxidative stress contributes to seizure susceptibility or progression in DS is lacking, some evidence suggests that metabolism and mitochondrial defects may play a role. Perhaps the best evidence implicating metabolism as a therapeutic target in DS is the high degree of clinical efficacy of the ketogenic diet (KD). The KD provides alternative mitochondrial fuels and several studies have reported a greater than 50% reduction in seizure frequency with some patients attaining seizure freedom [47,48]. Additionally, stiripentol, in addition to its anticonvulsant action, inhibits a key enzyme for glycolysis and has shown promise as a novel medication to attenuate seizures in DS when given alone or in conjunction with other anticonvulsant drugs [49,50,51]. Although the mechanism(s) by which metabolically targeted therapies exert anticonvulsant effects are incompletely understood, one straightforward mechanism is by improving altered metabolic pathways. Indeed, in a small pilot study, fibroblasts obtained from patients with DS showed mitochondrial defects including severe reduction in mitochondrial complex III enzymatic activity and overall poor ATP output [52]. In a zebrafish model of DS, metabolic pathways were found to be perturbed leading to decreased glycolytic and oxygen consumption rates as well as downregulation of 5 genes associated with the glycolytic pathway [53]. Treatment with the KD returned altered metabolism to control levels in zebrafish and has been shown to attenuate seizure activity in this model [53,54]. Thus, a deeper understanding of how deficits in metabolism confer risk for seizures may prove useful in the development of novel therapeutics to treat genetic epilepsies.
Glucose transporter type 1 (GLUT1) deficiency syndrome is a rare metabolic disorder characterized by the appearance of recurrent seizures in early infancy. This disorder is caused by mutations to the SLC2A1 gene, which encodes the protein required for glucose transport across the blood–brain barrier and into neurons. In addition to a wide array of seizure types, patients with this disorder typically present with cognitive delays and motor abnormalities. Exactly how GLUT1 deficiency results in seizures is unknown; however, as glucose is the main source of ATP in the brain, one obvious possibility is alterations to ATP production [55]. As mentioned previously, ATP depletion can alter neuronal membrane potential, leading to neuronal hyperexcitability [56]. Indeed, supplying the brain with alternative fuels, by use of the ketogenic or modified Atkins diet, is the most effective treatment to reduce seizures [57]. Although the effects of these diets on indices of oxidative stress have not been commonly reported in the literature, at least one study found that treatment with the modified Atkins diet significantly reduced oxidative damage to DNA and lipid peroxidation [58].
3.3. Genetic Epilepsies and Antioxidant Systems
Although research into the role of oxidative stress in human genetic epilepsies is rather limited, genetic animal models of epilepsy offer further evidence of oxidative stress in epileptic models. In genetically epilepsy-prone rats (GEPRs), endogenous antioxidant systems are perturbed which appears to contribute to an increased oxidative burden. Specifically, glutathione peroxidase (GPx) was found to be decreased and therefore less likely to protect against oxidative insults, resulting in decreased glutathione redox status, increased lipid peroxidation, and protein oxidization [59]. These oxidative markers were further exacerbated by kainate, and seizure activity was found to positively correlate with increased hippocampal oxidative stress markers [59].
Important information can also be gleaned by mouse models that are deficient in or overexpress endogenous antioxidants. For example, mice that are deficient in mitochondrial superoxide dismutase (SOD2) suffer from extensive mitochondrial dysfunction that culminates in increased mitochondrial oxidative stress, decreased ATP production, ataxia, and epilepsy before early postnatal death [60,61]. This suggests that oxidative stress and mitochondrial dysfunction can produce epilepsy and not be merely a consequence of seizures. Confirming the role of mitochondrial oxidative stress in the etiology of seizures, treatment with a catalytic antioxidant attenuated oxidative stress and seizure activity [61]. Even partial deficiency of SOD2 (SOD2−/+) was sufficient to increase the incidence of spontaneous and handling-induced seizure as a function of age, which correlated with increased mitochondrial oxidative stress [62]. Oxidation of targets that increase excitability is the most direct link between oxidative stress and epilepsy, and two such targets are the glial glutamate transporters, GLT-1 and GLAST. The uptake of extracellular glutamate by these transporters is driven by various electrochemical gradients, however specific “redox-sensing” elements have been identified, making these integral transporters sensitive to oxidation [63]. GLT-1 and GLAST were shown to be downregulated as a factor of age in SOD2−/+ mice, which could account for their increased seizure vulnerability [62,63]. Overexpression of SOD2 was associated with decreased mitochondrial oxidative stress and protection from KA-induced hippocampal neurodegeneration [10]. These studies suggest an important role of excessive mitochondrial superoxide production as a contributing factor to the development of seizures and seizure-related neuropathology.
While no human epilepsy syndrome resulting from SOD2 alterations has been identified, genetic mutations in another important antioxidant pathway, the thioredoxin system, have been associated with generalized epilepsy. Along with the glutathione system, the thioredoxin system serves as the cells major detoxifier of H2O2 (Figure 1) [64]. In fact, brain mitochondria have been shown to predominantly possess and utilize the thioredoxin–peroxiredoxin system for H2O2 detoxification [65]. Kudin and colleagues report a mutation in thioredoxin reductase 1 (TXNRD1) and its association with genetic generalized epilepsy [66]. In addition to reduced TXNRD1 activity, patient fibroblasts were less resistant to an H2O2 challenge [66]. Another study identified a patient suffering from an infantile-onset neurodegenerative disorder of which seizures were a prominent symptom [67]. Exome sequencing in this patient, uncovered a homozygous stop mutation in the mitochondrial form of thioredoxin (TXN2) [67]. Fibroblasts exhibited increased RS levels, inhibited oxidative phosphorylation and depletion of ATP, while restoration of TXN2 or treatment with antioxidants protected cells against oxidative stress and damage [67]. These human studies suggest that epilepsy can arise due to reduced activity of the thioredoxin system. Interestingly, overexpression of thioredoxin in mice attenuates kainate-induced seizure severity and neuronal damage [68]. Thus, the proper functioning of this system may have important implications for epilepsy. Supporting this idea are numerous studies that demonstrate an association between selenium, an essential component of GPx and TXNRD, and seizures. Specifically, low selenium levels are associated with seizures in humans, whereas selenium supplementation in animal models attenuates seizure activity [69,70,71,72,73,74]. These antioxidant systems may therefore represent therapeutic targets.
4. Oxidative Stress in Acquired Epilepsies
The acquired epilepsy syndromes are those whose cause can be reasonably inferred from known or unknown neurological insults such as trauma, infection, ischemic stroke, or status epilepticus (SE). The precipitating brain injury leads to a process known as epileptogenesis in which cellular and biochemical changes occur and spontaneous recurrent seizures (SRSs) arise after a latent or seizure-free period of weeks to years. These initiating brain injuries particularly stroke, trauma and SE are known to increase oxidative stress [75,76,77]. Oxidative stress can contribute to neuronal hyperexcitability and neuronal cell death through numerous mechanisms including oxidative damage to membrane proteins such as neurotransmitter receptors and ion channels [28]. The acquired epilepsies and animal models thereof have consistently shown that oxidative stress can result from seizure activity and exacerbate consequences of seizures such as neuronal loss and cognitive impairment. The role of oxidative stress in acquired epilepsies is reviewed briefly here; however, for a more detailed review please see [78]. In patients with temporal lobe epilepsy (TLE), antioxidant systems such as glutathione and SOD are altered, indicative of ongoing oxidative stress [79,80,81]. Indeed, oxidative damage to biomolecules has been detected in surgically resected epileptic brain tissue and suggested to contribute to neuronal hyperexcitability and degeneration [13,80,82,83]. Animal models of acquired epilepsy have shown seizure-induced oxidative damage to vulnerable mitochondrial and hippocampal proteins (complex I and aconitase, etc.), mitochondrial DNA (8-OhdG), and various lipids in the hippocampus, the site of characteristic and selective seizure-induced neurodegeneration at times preceding overt neuronal death [10,11,14,84,85,86,87]. In epilepsy, key mediators of neuronal death are necrosis initiated by glutamate excitotoxicity and apoptosis [88,89,90,91,92,93]. Oxidative stress and mitochondria are contributors to glutamate excitotoxicity as well as apoptotic cell death [92,93,94,95]. The intrinsic (mitochondrial) pathway of apoptosis is initiated by intracellular abnormalities, such as DNA damage, calcium overload, and oxidative stress [96,97]. If sufficient to induce loss of the mitochondrial membrane potential, this signals the subsequent release of pro-apoptotic proteins (cytochrome c, apoptosis-inducing factor, etc.) and downstream activation of caspase-dependent and caspase-independent cell death [98]. The role of oxidative stress in seizure-induced neuronal cell death is well established [99,100,101,102,103,104]. In fact, treatment with various compounds that act to decrease oxidative stress (antioxidants, NADPH oxidase inhibitors, etc.) have been demonstrated to protect against seizure-induced neuronal death [24,99,100,101,105,106]. Neuronal death, particularly in the hippocampus, is a common feature of acquired epilepsy and is thought to contribute to cognitive dysfunction [107]. Importantly, attenuation of oxidative damage in models of acquired epilepsy appears to protect against cognitive dysfunction as well as confer neuroprotection [85,105,106]. Thus, oxidative damage results from seizure activity in acquired epilepsy and appears to contribute to seizure-induced neuronal loss and cognitive dysfunction.
5. Oxidative Stress as a Therapeutic Target
Targeting of oxidative stress to attenuate seizures and related sequalae has largely been limited to studies of animal models. Vitamin E has been shown to have antioxidant properties and has shown promise in attenuating oxidative stress and seizure parameters in various animal models but not others [108,109]. This animal data is supported by clinical data suggesting that Vitamin E can significantly reduce oxidative stress markers and seizure frequency [110,111]. Treatment with Vitamin E is somewhat controversial, however, as other studies have found no significant benefits on seizure frequency, and the dosage needed to achieve therapeutic concentrations is relatively high. Indeed, clinical data may be sparse, as it is relatively difficult for large antioxidant molecules to pass the blood–brain barrier, and antioxidants that act stoichiometrically to quench free radicals need to be given frequently and in large doses. One way around this is to treat with compounds that act to bolster the endogenous antioxidant systems (Figure 1). For example, a recent study demonstrated that co-treatment with N-acetylcysteine and sulforaphane, which act to increase glutathione levels, protected against brain oxidative stress, reduced seizure frequency, and attenuated neuronal loss and cognitive deficits [105]. Importantly, these compounds are already approved for human use in other conditions. Another potential therapeutic option is to use catalytic antioxidants that act enzymatically to scavenge free radicals to decrease oxidative stress (Figure 1). Although these types of compounds have not been shown to attenuate seizure activity, likely due to short treatment periods in animal models, they have shown excellent neuroprotective profiles and attenuation of seizure-induced cognitive dysfunction and may have potential as adjunctive therapies to current anti-seizure medications [61,106]. Similarly, inhibition of NADPH oxidase has been demonstrated to attenuate seizure-induced cell death [23,24]. Given that ROS may play a physiological role in cell signaling, any therapies targeting these pathways need to be thoroughly evaluated for deleterious side effects [112].
6. Conclusions
In this review, we have presented an overview of the evidence suggesting that oxidative stress plays a key role in genetic and acquired epilepsies. Targeting oxidative stress may provide a novel therapeutic to attenuate seizure activity and associated sequalae such as neuronal loss and cognitive impairment. Future research in the area will further elucidate the contribution of specific reactive species and mechanisms that transform the normal brain to one prone to hyperexcitability, thus providing novel therapeutics in conjunction with or in replacement of current anti-seizure medications.
Acknowledgments
This work is supported by grants from the National Institute of Health (NIHRO1NS039587, NIHRO1NS086423, UO1NS083422).
Author Contributions
Jennifer N. Pearson-Smith and Manisha Patel conceptualized this review, as well as drafted and edited the manuscript. Both authors approve of its submission.
Conflicts of Interest
Manisha Patel is a consultant for Aeolus Pharmaceuticals, which develops catalytic antioxidants for human diseases, and Epigenyx, which develops therapies for Dravet Syndrome.
References
- Institute of Medicine. Epilepsy across the Spectrum: Promoting Health and Understanding; National Academies Press: Washington, DC, USA, 2012. [Google Scholar]
- Porter, R.J.; Dhir, A.; Macdonald, R.L.; Rogawski, M.A. Chapter 39—Mechanisms of action of antiseizure drugs. In Handbook of Clinical Neurology; Hermann, S., William, H.T., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; Volume 108, pp. 663–681. [Google Scholar]
- Helmstaedter, C.; Kurthen, M.; Lux, S.; Reuber, M.; Elger, C.E. Chronic epilepsy and cognition: A longitudinal study in temporal lobe epilepsy. Ann. Neurol. 2003, 54, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex III Releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [PubMed]
- Boveris, A. Determination of the production of superoxide radicals and hydrogen peroxide in mitochondria. Methods Enzymol. 1984, 105, 429–435. [Google Scholar] [PubMed]
- Gardner, P.R.; Fridovich, I. Inactivation-reactivation of aconitase in Escherichia coli. A sensitive measure of superoxide radical. J. Biol. Chem. 1992, 267, 8757–8763. [Google Scholar] [PubMed]
- Flint, D.H.; Tuminello, J.F.; Emptage, M.H. The inactivation of Fe-S cluster containing hydro-lyases by superoxide. J. Biol. Chem. 1993, 268, 22369–22376. [Google Scholar] [PubMed]
- Liang, L.P.; Ho, Y.S.; Patel, M. Mitochondrial superoxide production in kainate-induced hippocampal damage. Neuroscience 2000, 101, 563–570. [Google Scholar] [CrossRef]
- Jarrett, S.G.; Liang, L.-P.; Hellier, J.L.; Staley, K.J.; Patel, M. Mitochondrial DNA damage and impaired base excision repair during epileptogenesis. Neurobiol. Dis. 2008, 30, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Cock, H.R.; Tong, X.; Hargreaves, I.P.; Heales, S.J.R.; Clark, J.B.; Patsalos, P.N.; Thom, M.; Groves, M.; Schapira, A.H.V.; Shorvon, S.D.; et al. Mitochondrial dysfunction associated with neuronal death following status epilepticus in rat. Epilepsy Res. 2002, 48, 157–168. [Google Scholar] [CrossRef]
- Kunz, W.S.; Kudin, A.P.; Vielhaber, S.; Blümcke, I.; Zuschratter, W.; Schramm, J.; Beck, H.; Elger, C.E. Mitochondrial complex I deficiency in the epileptic focus of patients with temporal lobe epilepsy. Ann. Neurol. 2000, 48, 766–773. [Google Scholar] [CrossRef]
- Ryan, K.; Backos, D.S.; Reigan, P.; Patel, M. Post-translational oxidative modification and inactivation of mitochondrial complex I in epileptogenesis. J. Neurosci. 2012, 32, 11250–11258. [Google Scholar] [CrossRef] [PubMed]
- Folbergrová, J.; Ješina, P.; Drahota, Z.; Lisý, V.; Haugvicová, R.; Vojtíšková, A.; Houštěk, J. Mitochondrial complex I inhibition in cerebral cortex of immature rats following homocysteic acid-induced seizures. Exp. Neurol. 2007, 204, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Folbergrová, J.; Ješina, P.; Haugvicová, R.; Lisý, V.; Houštěk, J. Sustained deficiency of mitochondrial complex I activity during long periods of survival after seizures induced in immature rats by homocysteic acid. Neurochem. Int. 2010, 56, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Tretter, L.; Adam-Vizi, V. Generation of reactive oxygen species in the reaction catalyzed by α-Ketoglutarate dehydrogenase. J. Neurosci. 2004, 24, 7771–7778. [Google Scholar] [CrossRef] [PubMed]
- Kudin, A.P.; Bimpong-Buta, N.Y.-B.; Vielhaber, S.; Elger, C.E.; Kunz, W.S. Characterization of superoxide-producing sites in isolated brain mitochondria. J. Biol. Chem. 2004, 279, 4127–4135. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cantu, D.; Schaack, J.; Patel, M. Oxidative inactivation of mitochondrial aconitase results in iron and H2O2-mediated neurotoxicity in rat primary mesencephalic cultures. PLoS ONE 2009, 4, e7095. [Google Scholar] [CrossRef] [PubMed]
- Fridovich, I. Quantitative aspects of the production of superoxide anion radical by milk xanthine oxidase. J. Biol. Chem. 1970, 245, 4053–4057. [Google Scholar] [PubMed]
- David, W.; Infanger, R.V.S.; Davisson, R.L. NADPH Oxidases of the brain: Distribution, regulation, and function. Antioxid. Redox Signal. 2006, 8, 1583–1596. [Google Scholar]
- Kovac, S.; Domijan, A.M.; Walker, M.C.; Abramov, A.Y. Seizure activity results in calcium- and mitochondria-independent ROS production via NADPH and xanthine oxidase activation. Cell Death Dis. 2014, 5, e1442. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Jang, B.G.; Choi, B.Y.; Kim, H.S.; Sohn, M.; Chung, T.N.; Choi, H.C.; Song, H.K.; Suh, S.W. Post-treatment of an NADPH oxidase inhibitor prevents seizure-induced neuronal death. Brain Res. 2013, 1499, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Li, Q.-Y.; Chang, L.-Y.; Crapo, J.; Liang, L.-P. Activation of NADPH oxidase and extracellular superoxide production in seizure-induced hippocampal damage. J. Neurochem. 2005, 92, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Pestana, R.R.F.; Kinjo, E.R.; Hernandes, M.S.; Britto, L.R.G. Reactive oxygen species generated by NADPH oxidase are involved in neurodegeneration in the pilocarpine model of temporal lobe epilepsy. Neurosci. Lett. 2010, 484, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Reactive oxygen species and the central nervous system. J. Neurochem. 1992, 59, 1609–1623. [Google Scholar] [CrossRef] [PubMed]
- Stadtman, E.R. Protein oxidation in aging and age-related diseases. Ann. N. Y. Acad. Sci. 2001, 928, 22–38. [Google Scholar] [CrossRef] [PubMed]
- Wong-ekkabut, J.; Xu, Z.; Triampo, W.; Tang, I.M.; Tieleman, D.P.; Monticelli, L. Effect of lipid peroxidation on the properties of lipid bilayers: A molecular dynamics study. Biophys. J. 2007, 93, 4225–4236. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, G.; Moraes, C.T.; Shanske, S.; Oh, S.J.; DiMauro, S. A new mtDNA mutation in the tRNA(Lys) gene associated with myoclonic epilepsy and ragged-red fibers (MERRF). Am. J. Hum. Genet. 1992, 51, 1213–1217. [Google Scholar] [PubMed]
- Wu, S.-B.; Ma, Y.-S.; Wu, Y.-T.; Chen, Y.-C.; Wei, Y.-H. Mitochondrial DNA mutation-elicited oxidative stress, oxidative damage, and altered gene expression in cultured cells of patients with MERRF syndrome. Mol. Neurobiol. 2010, 41, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Brini, M.; Pinton, P.; King, M.P.; Davidson, M.; Schon, E.A.; Rizzuto, R. A calcium signaling defect in the pathogenesis of a mitochondrial DNA inherited oxidative phosphorylation deficiency. Nat. Med. 1999, 5, 951–954. [Google Scholar] [PubMed]
- Ruhoy, I.S.; Saneto, R.P. The genetics of Leigh syndrome and its implications for clinical practice and risk management. Appl. Clin. Genet. 2014, 7, 221–234. [Google Scholar] [PubMed]
- Quintana, A.; Kruse, S.E.; Kapur, R.P.; Sanz, E.; Palmiter, R.D. Complex I deficiency due to loss of Ndufs4 in the brain results in progressive encephalopathy resembling Leigh syndrome. Proc. Natl. Acad. Sci. USA 2010, 107, 10996–11001. [Google Scholar] [CrossRef] [PubMed]
- Wojtala, A.; Karkucinska-Wieckowska, A.; Sardao, V.A.; Szczepanowska, J.; Kowalski, P.; Pronicki, M.; Duszynski, J.; Wieckowski, M.R. Modulation of mitochondrial dysfunction-related oxidative stress in fibroblasts of patients with Leigh syndrome by inhibition of prooxidative p66Shc pathway. Mitochondrion 2017. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.C.; Yanos, M.E.; Kayser, E.-B.; Quintana, A.; Sangesland, M.; Castanza, A.; Uhde, L.; Hui, J.; Wall, V.Z.; Gagnidze, A.; et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of leigh syndrome. Science 2013, 342, 1524–1528. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, K.; Sandoval, H.; Yamamoto, S.; Jaiswal, M.; Sanz, E.; Li, Z.; Hui, J.; Graham, B.H.; Quintana, A.; et al. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 2015, 160, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Naviaux, R.K.; Nyhan, W.L.; Barshop, B.A.; Poulton, J.; Markusic, D.; Karpinski, N.C.; Haas, R.H. Mitochondrial DNA polymerase γ deficiency and mtDNA depletion in a child with Alpers’ syndrome. Ann. Neurol. 1999, 45, 54–58. [Google Scholar] [CrossRef]
- Saneto, R.P. Alpers-Huttenlocher syndrome: The role of a multidisciplinary health care team. J. Multidiscip. Healthc. 2016, 9, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Simonati, A.; Filosto, M.; Savio, C.; Tomelleri, G.; Tonin, P.; Dalla Bernardina, B.; Rizzuto, N. Features of cell death in brain and liver, the target tissues of progressive neuronal degeneration of childhood with liver disease (Alpers-Huttenlocher disease). Acta Neuropathol. 2003, 106, 57–65. [Google Scholar] [PubMed]
- Morris, A.A.M.; Singh-Kler, R.; Perry, R.H.; Griffiths, P.D.; Burt, A.D.; Wong, C.P.; Gardner-Medwin, D.; Tumbull, D.M. Respiratory chain dysfunction in progressive neuronal degeneration of childhood with liver disease. J. Child Neurol. 1996, 11, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Jamme, I.; Petit, E.; Divoux, D.; Alain, G.; Maixent, J.M.; Nouvelot, A. Modulation of mouse cerebral Na+, K+-ATPase activity by oxygen free radicals. Neuroreport 1995, 7, 333–337. [Google Scholar] [PubMed]
- Fighera, M.R.; Royes, L.F.F.; Furian, A.F.; Oliveira, M.S.; Fiorenza, N.G.; Frussa-Filho, R.; Petry, J.C.; Coelho, R.C.; Mello, C.F. GM1 ganglioside prevents seizures, Na+,K+-ATPase activity inhibition and oxidative stress induced by glutaric acid and pentylenetetrazole. Neurobiol. Dis. 2006, 22, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Depienne, C.; Trouillard, O.; Saint-Martin, C.; Gourfinkel-An, I.; Bouteiller, D.; Carpentier, W.; Keren, B.; Abert, B.; Gautier, A.; Baulac, S.; et al. Spectrum of SCN1A gene mutations associated with Dravet syndrome: Analysis of 333 patients. J. Med. Genet. 2009, 46, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Kinsman, S.L.; Vining, E.P.G.; Quaskey, S.A.; Mellits, D.; Freeman, J.M. Efficacy of the ketogenic diet for intractable seizure disorders: Review of 58 cases. Epilepsia 1992, 33, 1132–1136. [Google Scholar] [CrossRef] [PubMed]
- Veggiotti, P.; Burlina, A.; Coppola, G.; Cusmai, R.; De Giorgis, V.; Guerrini, R.; Tagliabue, A.; Bernardina, B.D. The ketogenic diet for Dravet syndrome and other epileptic encephalopathies: An Italian consensus. Epilepsia 2011, 52, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Wirrell, E.C.; Laux, L.; Franz, D.N.; Sullivan, J.; Saneto, R.P.; Morse, R.P.; Devinsky, O.; Chugani, H.; Hernandez, A.; Hamiwka, L.; et al. Stiripentol in dravet syndrome: Results of a retrospective U.S. study. Epilepsia 2013, 54, 1595–1604. [Google Scholar] [CrossRef] [PubMed]
- Chiron, C.; Marchand, M.C.; Tran, A.; Rey, E.; d’Athis, P.; Vincent, J.; Dulac, O.; Pons, G. Stiripentol in severe myoclonic epilepsy in infancy: A randomised placebo-controlled syndrome-dedicated trial. Lancet 2000, 356, 1638–1642. [Google Scholar] [CrossRef]
- Inoue, Y.; Ohtsuka, Y. Long-term safety and efficacy of stiripentol for the treatment of Dravet syndrome: A multicenter, open-label study in Japan. Epilepsy Res. 2015, 113, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Doccini, S.; Meschini, M.C.; Mei, D.; Guerrini, R.; Sicca, F.; Santorelli, F.M. Mitochondrial respiratory chain defects in skin fibroblasts from patients with Dravet syndrome. Neurol. Sci. 2015, 36, 2151–2155. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.G.; Rowley, S.; Fulton, R.; Dinday, M.T.; Baraban, S.C.; Patel, M. Altered Glycolysis and Mitochondrial Respiration in a zebrafish model of Dravet syndrome. eNeuro 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Baraban, S.C.; Dinday, M.T.; Hortopan, G.A. Drug screening in Scn1a zebrafish mutant identifies clemizole as a potential Dravet syndrome treatment. Nat. Commun. 2013, 4, 2410. [Google Scholar] [CrossRef] [PubMed]
- Fehm, H.L.; Kern, W.; Peters, A. The selfish brain: Competition for energy resources. In Progress in Brain Research; Kalsbeek, A., Fliers, E., Eds.; Elsevier: Amsterdam, The Netherlands, 2006; Volume 153, pp. 129–140. [Google Scholar]
- De Lores Arnaiz, G.R.; Ordieres, M.G.L. Brain Na(+), K(+)-ATPase activity in aging and disease. Int. J. Biomed. Sci. 2014, 10, 85–102. [Google Scholar] [PubMed]
- Klepper, J.; Diefenbach, S.; Kohlschütter, A.; Voit, T. Effects of the ketogenic diet in the glucose transporter 1 deficiency syndrome. Prostaglandins Leukot. Essent. Fatty Acids 2004, 70, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Okumura, A.; Hayashi, M.; Mori, H.; Takahashi, S.; Yanagihara, K.; Miyata, R.; Tanuma, N.; Mimaki, T.; Abe, S.; et al. Oxidative stress markers and phosphorus magnetic resonance spectroscopy in a patient with GLUT1 deficiency treated with modified Atkins diet. Brain Dev. 2012, 34, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.-J.; Ko, K.H.; Kim, W.-K.; Chae, J.S.; Yen, T.P.H.; Kim, H.J.; Wie, M.-B.; Kim, H.-C. Role of glutathione peroxidase in the ontogeny of hippocampal oxidative stress and kainate seizure sensitivity in the genetically epilepsy-prone rats. Neurochem. Int. 2008, 52, 1134–1147. [Google Scholar] [CrossRef] [PubMed]
- Melov, S.; Coskun, P.; Patel, M.; Tuinstra, R.; Cottrell, B.; Jun, A.S.; Zastawny, T.H.; Dizdaroglu, M.; Goodman, S.I.; Huang, T.-T.; et al. Mitochondrial disease in superoxide dismutase 2 mutant mice. Proc. Natl. Acad. Sci. USA 1999, 96, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.-P.; Waldbaum, S.; Rowley, S.; Huang, T.-T.; Day, B.J.; Patel, M. Mitochondrial oxidative stress and epilepsy in SOD2 deficient mice: Attenuation by a lipophilic metalloporphyrin. Neurobiol. Dis. 2012, 45, 1068–1076. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.-P.; Patel, M. Mitochondrial oxidative stress and increased seizure susceptibility in Sod2−/+ mice. Free Radic. Biol. Med. 2004, 36, 542–554. [Google Scholar] [CrossRef] [PubMed]
- Trotti, D.; Danbolt, N.C.; Volterra, A. Glutamate transporters are oxidant-vulnerable: A molecular link between oxidative and excitotoxic neurodegeneration? Trends Pharmacol. Sci. 1998, 19, 328–334. [Google Scholar] [CrossRef]
- Carmel-Harel, O.; Storz, G. Roles of the glutathione- and thioredoxin-dependent reduction systems in the Escherichia coli and saccharomyces cerevisiae responses to oxidative stress. Annu. Rev. Microbiol. 2000, 54, 439–461. [Google Scholar] [CrossRef] [PubMed]
- Drechsel, D.A.; Patel, M. Respiration-dependent H2O2 Removal in brain mitochondria via the thioredoxin/peroxiredoxin system. J. Biol. Chem. 2010, 285, 27850–27858. [Google Scholar] [CrossRef] [PubMed]
- Kudin, A.P.; Baron, G.; Zsurka, G.; Hampel, K.G.; Elger, C.E.; Grote, A.; Weber, Y.; Lerche, H.; Thiele, H.; Nürnberg, P.; et al. Homozygous mutation in TXNRD1 is associated with genetic generalized epilepsy. Free Radic. Biol. Med. 2017, 106, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Holzerova, E.; Danhauser, K.; Haack, T.B.; Kremer, L.S.; Melcher, M.; Ingold, I.; Kobayashi, S.; Terrile, C.; Wolf, P.; Schaper, J.; et al. Human thioredoxin 2 deficiency impairs mitochondrial redox homeostasis and causes early-onset neurodegeneration. Brain 2016, 139, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Takagi, Y.; Hattori, I.; Nozaki, K.; Mitsui, A.; Ishikawa, M.; Hashimoto, N.; Yodoi, J. Excitotoxic hippocampal injury is attenuated in thioredoxin transgenic mice. J. Cereb. Blood Flow Metab. 2000, 20, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Mahyar, A.; Ayazi, P.; Fallahi, M.; Javadi, A. Correlation between serum selenium level and febrile seizures. Pediatr. Neurol. 2010, 43, 331–334. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, M.R.; Shams, S.; Nouri, M.; Mohseni, M.; Shabanian, R.; Yekaninejad, M.S.; Chegini, N.; Khodadad, A.; Safaralizadeh, R. A Probable causative factor for an old problem: Selenium and glutathione peroxidase appear to play important roles in epilepsy pathogenesis. Epilepsia 2007, 48, 1750–1755. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, M.R.; Shabanian, R.; Abbaskhanian, A.; Nasirian, A.; Ghofrani, M.; Mohammadi, M.; Zamani, G.R.; Kayhanidoost, Z.; Ebrahimi, S.; Pourpak, Z. Selenium and intractable epilepsy: Is there any correlation? Pediatr. Neurol. 2007, 36, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Seven, M.; Basaran, S.Y.; Cengiz, M.; Unal, S.; Yuksel, A. Deficiency of selenium and zinc as a causative factor for idiopathic intractable epilepsy. Epilepsy Res. 2013, 104, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Nazıroğlu, M.; Kutluhan, S.; Yılmaz, M. Selenium and topiramate modulates brain microsomal oxidative stress values, Ca2+-ATPase activity, and EEG records in pentylentetrazol-induced seizures in rats. J. Membr. Biol. 2008, 225, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Rehni, A.K.; Singh, T.G. Selenium induced anticonvulsant effect: A potential role of prostaglandin E1 receptor activation linked mechanism. J. Trace Elem. Med. Biol. 2013, 27, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.L.; Bayraktutan, U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int. J. Stroke 2009, 4, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Ana, R.-R.; Juan Jose, E.-G.; Francisco, M.-C.; Antonio, C.-V. Oxidative stress in traumatic brain injury. Curr. Med. Chem. 2014, 21, 1201–1211. [Google Scholar]
- Puttachary, S.; Sharma, S.; Stark, S.; Thippeswamy, T. Seizure-induced oxidative stress in temporal lobe epilepsy. BioMed Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Kovac, S.; Dinkova Kostova, A.T.; Herrmann, A.M.; Melzer, N.; Meuth, S.G.; Gorji, A. Metabolic and homeostatic changes in seizures and acquired epilepsy—mitochondria, calcium dynamics and reactive oxygen species. Int. J. Mol. Sci. 2017, 18, 1935. [Google Scholar] [CrossRef] [PubMed]
- Ristić, A.J.; Savić, D.; Sokić, D.; Bogdanović Pristov, J.; Nestorov, J.; Baščarević, V.; Raičević, S.; Savić, S.; Spasojević, I. Hippocampal antioxidative system in mesial temporal lobe epilepsy. Epilepsia 2015, 56, 789–799. [Google Scholar] [CrossRef] [PubMed]
- López, J.; González, M.E.; Lorigados, L.; Morales, L.; Riverón, G.; Bauzá, J.Y. Oxidative stress markers in surgically treated patients with refractory epilepsy. Clin. Biochem. 2007, 40, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Yüzbaşioğlu, A.; Karataş, H.; Gürsoy-Özdemir, Y.; Saygi, S.; Akalan, N.; Söylemezoğlu, F.; Dalkara, T.; Kocaefe, Y.Ç.; Özgüç, M. Changes in the expression of selenoproteins in mesial temporal lobe epilepsy patients. Cell. Mol. Neurobiol. 2009, 29, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Pecorelli, A.; Natrella, F.; Belmonte, G.; Miracco, C.; Cervellati, F.; Ciccoli, L.; Mariottini, A.; Rocchi, R.; Vatti, G.; Bua, A.; et al. NADPH oxidase activation and 4-hydroxy-2-nonenal/aquaporin-4 adducts as possible new players in oxidative neuronal damage presents in drug-resistant epilepsy. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Rumià, J.; Marmol, F.; Sanchez, J.; Giménez-Crouseilles, J.; Carreño, M.; Bargalló, N.; Boget, T.; Pintor, L.; Setoain, X.; Donaire, A.; et al. Oxidative stress markers in the neocortex of drug-resistant epilepsy patients submitted to epilepsy surgery. Epilepsy Res. 2013, 107, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Liang, L.-P.; Roberts Ii, L.J. Enhanced hippocampal F2-isoprostane formation following kainate-induced seizures. J. Neurochem. 2001, 79, 1065–1069. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.N.; Warren, E.; Liang, L.-P.; Roberts, L.J.; Patel, M. Scavenging of highly reactive gamma-ketoaldehydes attenuates cognitive dysfunction associated with epileptogenesis. Neurobiol. Dis. 2017, 98, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, A.; Kawamoto, Y.; Chiba, Y.; Takei, S.; Hasegawa-Ishii, S.; Kawamura, N.; Yoshikawa, K.; Hosokawa, M.; Oikawa, S.; Kato, M.; et al. Proteomic identification of hippocampal proteins vulnerable to oxidative stress in excitotoxin-induced acute neuronal injury. Neurobiol. Dis. 2011, 43, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Waldbaum, S.; Patel, M. Mitochondrial oxidative stress in temporal lobe epilepsy. Epilepsy Res. 2010, 88, 23–45. [Google Scholar] [CrossRef] [PubMed]
- Ankarcrona, M.; Dypbukt, J.M.; Bonfoco, E.; Zhivotovsky, B.; Orrenius, S.; Lipton, S.A.; Nicotera, P. Glutamate-induced neuronal death: A succession of necrosis or apoptosis depending on mitochondrial function. Neuron 1995, 15, 961–973. [Google Scholar] [CrossRef]
- Henshall, D.C. Apoptosis signalling pathways in seizure-induced neuronal death and epilepsy. Biochem. Soc. Trans. 2007, 35, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Niquet, J.; Liu, H.; Wasterlain, C.G. Programmed neuronal necrosis and status epilepticus. Epilepsia 2005, 46, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Henshall, D.C.; Engel, T. Contribution of apoptosis-associated signaling pathways to epileptogenesis: Lessons from Bcl-2 family knockouts. Front. Cell. Neurosci. 2013, 7, 110. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, I.; Hastings, T. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J. Neurosci. 1995, 15, 3318–3327. [Google Scholar] [PubMed]
- Henshall, D.C.; Murphy, B.M. Modulators of neuronal cell death in epilepsy. Curr. Opin. Pharm. 2008, 8, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Day, B.J.; Crapo, J.D.; Fridovich, I.; McNamara, J.O. Requirement for superoxide in excitotoxic cell death. Neuron 1996, 16, 345–355. [Google Scholar] [CrossRef]
- Lafon-Cazal, M.; Pietri, S.; Culcasi, M.; Bockaert, J. NMDA-dependent superoxide production and neurotoxicity. Nature 1993, 364, 535–537. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Blomgren, K.; Kroemer, G. Mitochondrial membrane permeabilization in neuronal injury. Nat. Rev. Neurosci. 2009, 10, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.G.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Strasser, A.; McDunn, J.E.; Swanson, P.E. Cell death in disease: Mechanisms and emerging therapeutic concepts. N. Engl. J. Med. 2009, 361, 1570–1583. [Google Scholar] [CrossRef] [PubMed]
- Frantseva, M.V.; Perez Velazquez, J.L.; Tsoraklidis, G.; Mendonca, A.J.; Adamchik, Y.; Mills, L.R.; Carlen, P.L.; Burnham, M.W. Oxidative stress is involved in seizure-induced neurodegeneration in the kindling model of epilepsy. Neuroscience 2000, 97, 431–435. [Google Scholar] [CrossRef]
- Rong, Y.; Doctrow, S.R.; Tocco, G.; Baudry, M. EUK-134, a synthetic superoxide dismutase and catalase mimetic, prevents oxidative stress and attenuates kainate-induced neuropathology. Proc. Natl. Acad. Sci. USA 1999, 96, 9897–9902. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.-Y.; Han, S.-H. Melatonin attenuates kainic acid-induced hippocampal neurodegeneration and oxidative stress through microglial inhibition. J. Pineal Res. 2003, 34, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.T.; Puttfarcken, P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993, 262, 689. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.-X.; Manchester, L.C.; Reiter, R.J.; Qi, W.; Kim, S.J.; El-Sokkary, G.H. Melatonin protects hippocampal neurons in vivo against kainic acid-induced damage in mice. J. Neurosci. Res. 1998, 54, 382–389. [Google Scholar] [CrossRef]
- MacGregor, D.G.; Higgins, M.J.; Jones, P.A.; Maxwell, W.L.; Watson, M.W.; Graham, D.I.; Stone, T.W. Ascorbate attenuates the systemic kainate-induced neurotoxicity in the rat hippocampus. Brain Res. 1996, 727, 133–144. [Google Scholar] [CrossRef]
- Pauletti, A.; Terrone, G.; Shekh-Ahmad, T.; Salamone, A.; Ravizza, T.; Rizzi, M.; Pastore, A.; Pascente, R.; Liang, L.-P.; Villa, B.R.; et al. Targeting oxidative stress improves disease outcomes in a rat model of acquired epilepsy. Brain 2017, 140, 1885–1899. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.N.; Rowley, S.; Liang, L.-P.; White, A.M.; Day, B.J.; Patel, M. Reactive oxygen species mediate cognitive deficits in experimental temporal lobe epilepsy. Neurobiol. Dis. 2015, 82, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Pitkänen, A.; Sutula, T.P. Is epilepsy a progressive disorder? Prospects for new therapeutic approaches in temporal-lobe epilepsy. Lancet Neurol. 2002, 1, 173–181. [Google Scholar] [CrossRef]
- Barros, D.O.; Xavier, S.M.L.; Barbosa, C.O.; Silva, R.F.; Freitas, R.L.M.; Maia, F.D.; Oliveira, A.A.; Freitas, R.M.; Takahashi, R.N. Effects of the vitamin E in catalase activities in hippocampus after status epilepticus induced by pilocarpine in Wistar rats. Neurosci. Lett. 2007, 416, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.L.; Burnham, W.M.; Hwang, P.A. An evaluation of the anticonvulsant effects of vitamin E. Epilepsy Res. 1990, 6, 12–17. [Google Scholar] [CrossRef]
- Ogunmekan, A.O.; Hwang, P.A. A Randomized, Double-Blind, Placebo-Controlled, Clinical Trial of D-α-Tocopheryl Acetate (Vitamin E), as Add-On Therapy, for Epilepsy in Children. Epilepsia 1989, 30, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Mehvari, J.; Motlagh, F.G.; Najafi, M.; Ghazvini, M.R.A.; Naeini, A.A.; Zare, M. Effects of Vitamin E on seizure frequency, electroencephalogram findings, and oxidative stress status of refractory epileptic patients. Adv. Biomed. Res. 2016, 5, 36. [Google Scholar] [PubMed]
- Rhee, S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Cellular sources of reactive species (RS). RSs are denoted by yellow starbursts.Antioxidant systems and their detoxification of RS are denoted in purple and purple arrows, respectively. Therapeutic strategies for detoxification of seizure-induced RSS are denoted by green arrows, indicating elevating GSH or t-bars indicating inhibition by antioxidants or NOX inhibitors. NOS: nitric oxide synthase; GSH: reduced glutathione; GR: Glutathione reductase; GSSG: oxidized glutathione; GPx: Glutathione peroxidase; SOD: superoxide dismutase; TXN: thioredoxin; TXNRD: thioredoxin reductase; Prxox: peroxiredoxin oxidized; Prxred: peroxiredoxin reduced; Aco2: aconitase; Α-KGDH: alpha-ketoglutarate dehydrogenase.
Figure 1.
Cellular sources of reactive species (RS). RSs are denoted by yellow starbursts.Antioxidant systems and their detoxification of RS are denoted in purple and purple arrows, respectively. Therapeutic strategies for detoxification of seizure-induced RSS are denoted by green arrows, indicating elevating GSH or t-bars indicating inhibition by antioxidants or NOX inhibitors. NOS: nitric oxide synthase; GSH: reduced glutathione; GR: Glutathione reductase; GSSG: oxidized glutathione; GPx: Glutathione peroxidase; SOD: superoxide dismutase; TXN: thioredoxin; TXNRD: thioredoxin reductase; Prxox: peroxiredoxin oxidized; Prxred: peroxiredoxin reduced; Aco2: aconitase; Α-KGDH: alpha-ketoglutarate dehydrogenase.

{kind=link}
{kind=link}
Table 1.
Select studies implicating oxidative stress and metabolic dysfunction in select epilepsies associated with a genetic cause.
Table 1.
Select studies implicating oxidative stress and metabolic dysfunction in select epilepsies associated with a genetic cause.
| Disorder | Gene Mutation | Resulting Dysfuntion | Finding | Citation |
|---|---|---|---|---|
| MERFF | tRNALys | Complex I | Decreased ATP, increased ROS, altered antioxidant gene expression, alterations to calcium homeostatsis | [32,33,34] |
| Leigh syndrome | Various mtDNA mutations | Complex I, V | Increased ROS, decreased ATP | [35,36,37] |
| AHS | POLG | Decreased mtDNA, Complex IV | Increased apoptosis and necrosis potentially modulated by mito pathways | [40,42] |
| Dravet Syndrome | SCN1A | Nav1.1 | In zebrafish—decreased glycolytic and oxygen consumption rates, downregulation of glycolytic pathway | [53] |
| Glut1 deficiency | SLC2A1 | Glucose transport into brain | Increased oxidative DNA damage, increased lipid peroxidation—attenuated by modified Atkins diet | [58] |
MERFF: Myoclonic epilepsy with ragged-red fibers; AHS: Alpers-Huttenlocher syndrome; ROS: reactive oxygen species.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pearson-Smith, J.N.; Patel, M. Metabolic Dysfunction and Oxidative Stress in Epilepsy. Int. J. Mol. Sci. 2017, 18, 2365. https://doi.org/10.3390/ijms18112365
AMA Style
Pearson-Smith JN, Patel M. Metabolic Dysfunction and Oxidative Stress in Epilepsy. International Journal of Molecular Sciences. 2017; 18(11):2365. https://doi.org/10.3390/ijms18112365
Chicago/Turabian StylePearson-Smith, Jennifer N., and Manisha Patel. 2017. "Metabolic Dysfunction and Oxidative Stress in Epilepsy" International Journal of Molecular Sciences 18, no. 11: 2365. https://doi.org/10.3390/ijms18112365
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.