The Role of Gap Junction-Mediated Endothelial Cell–Cell Interaction in the Crosstalk between Inflammation and Blood Coagulation


Abstract
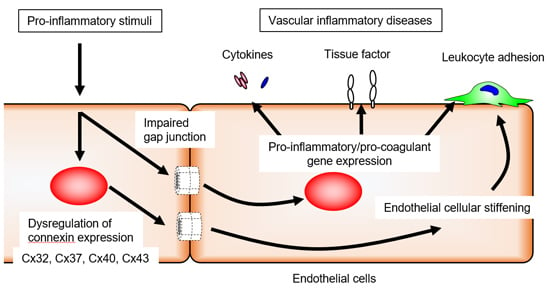
:
1. Introduction
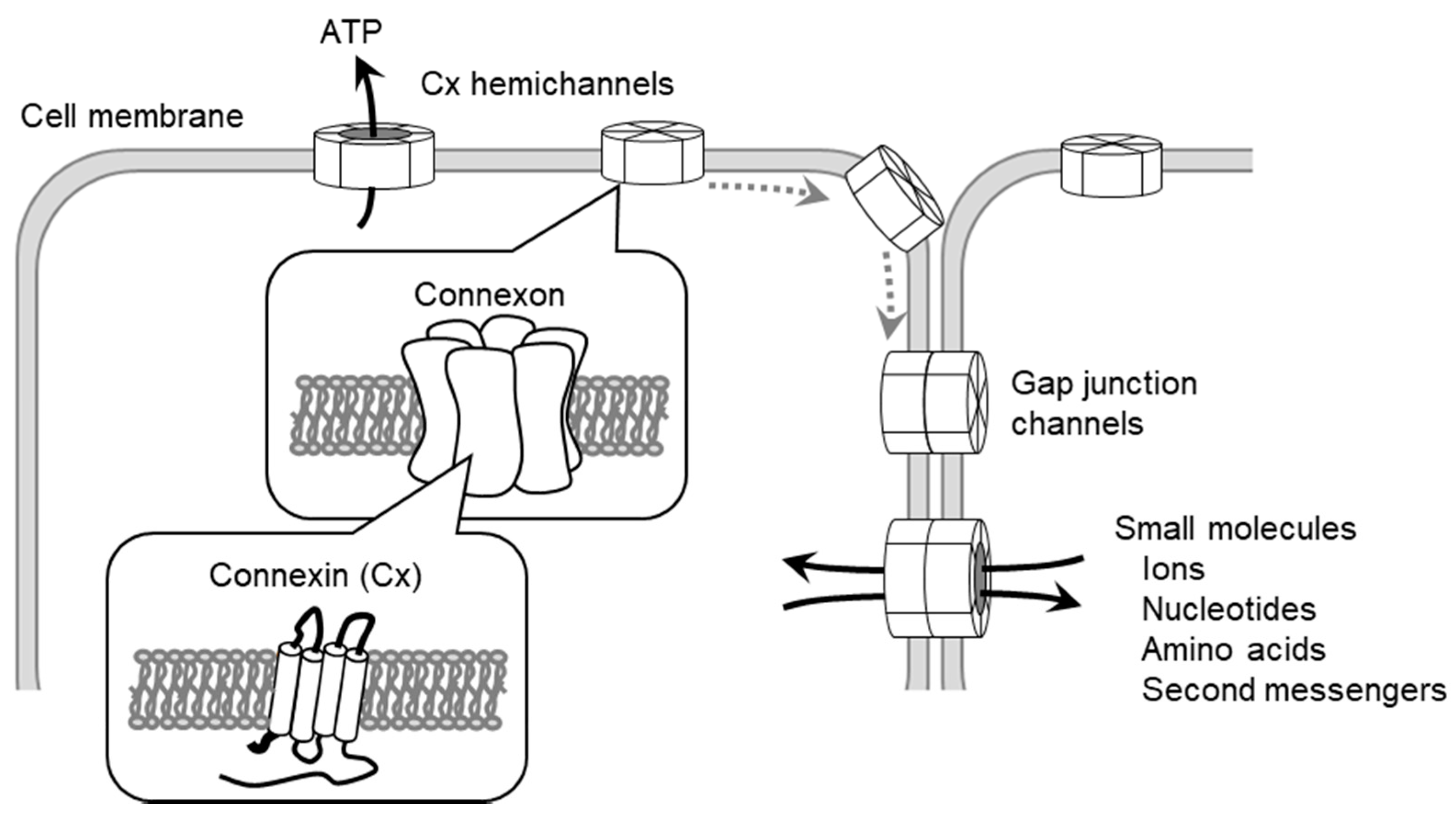
2. The Patho/Physiological Role of Connexin (Cx) in Progression of Cardiovascular Diseases
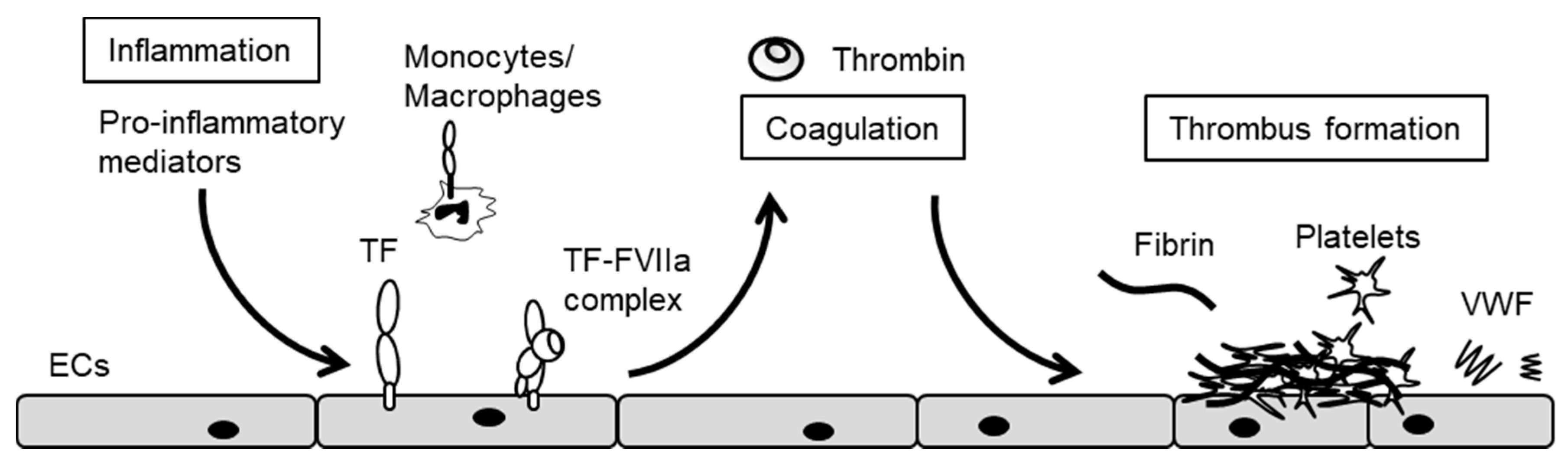
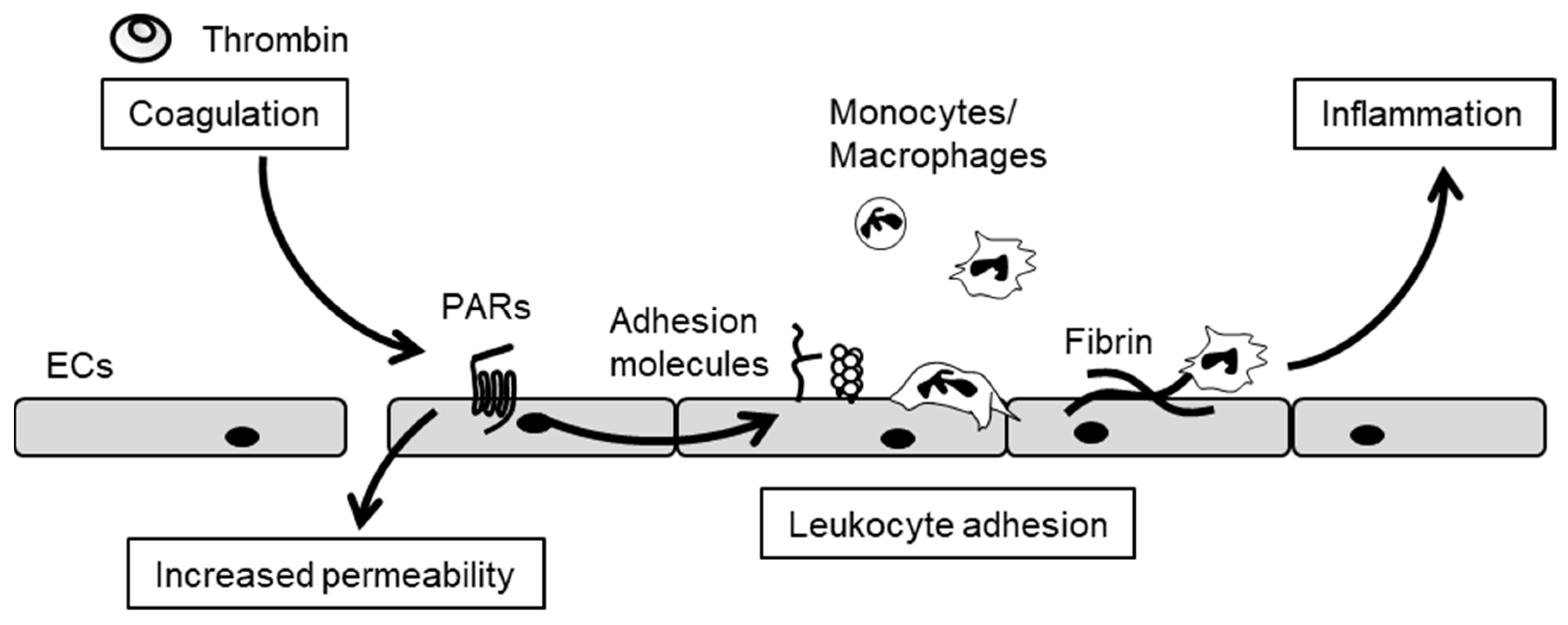
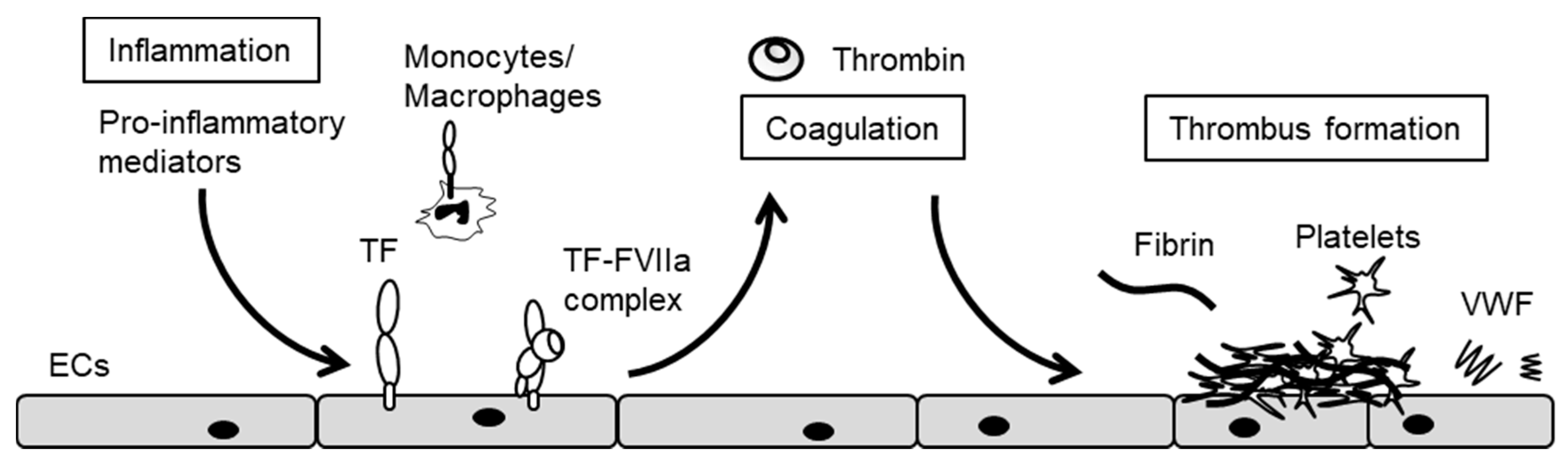
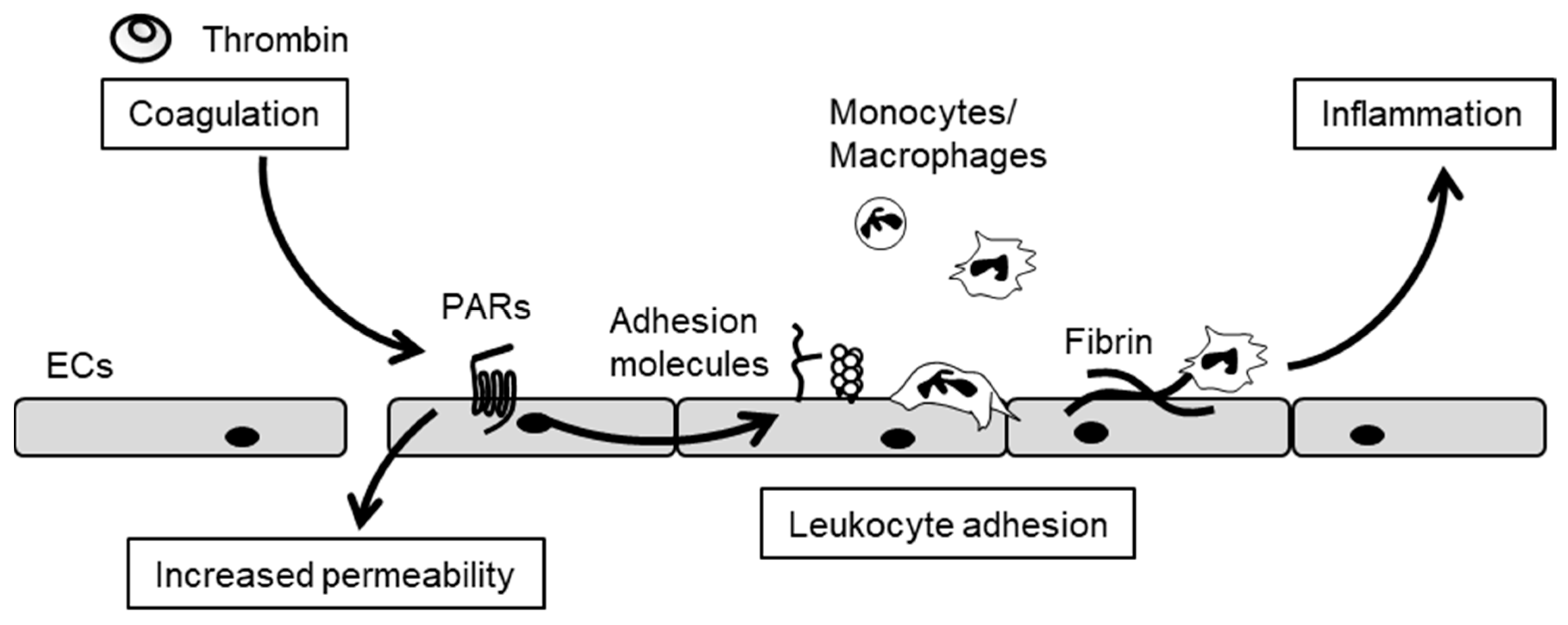
3. The Crosstalk between Inflammation and Blood Coagulation
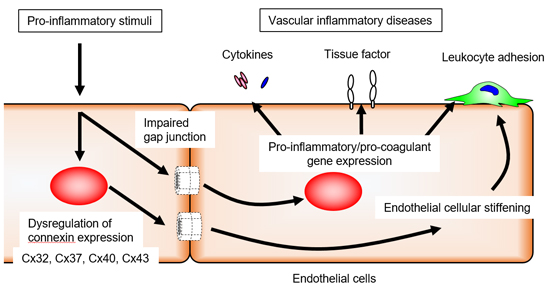
4. Endothelial Cxs and Inflammation
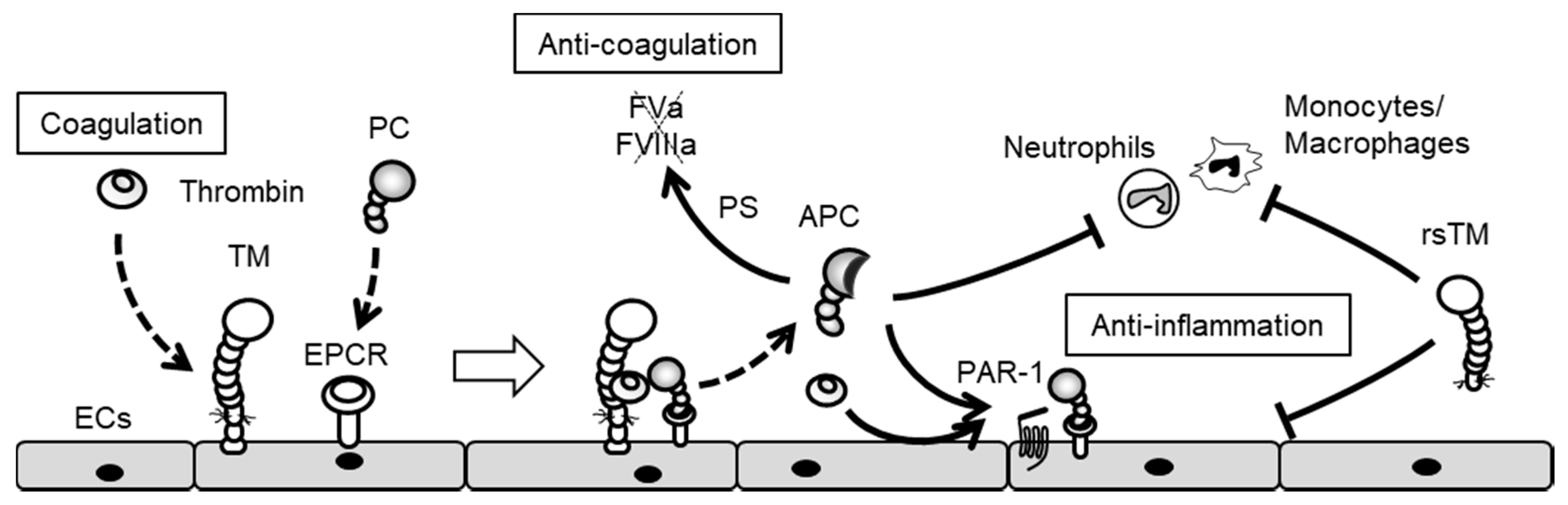
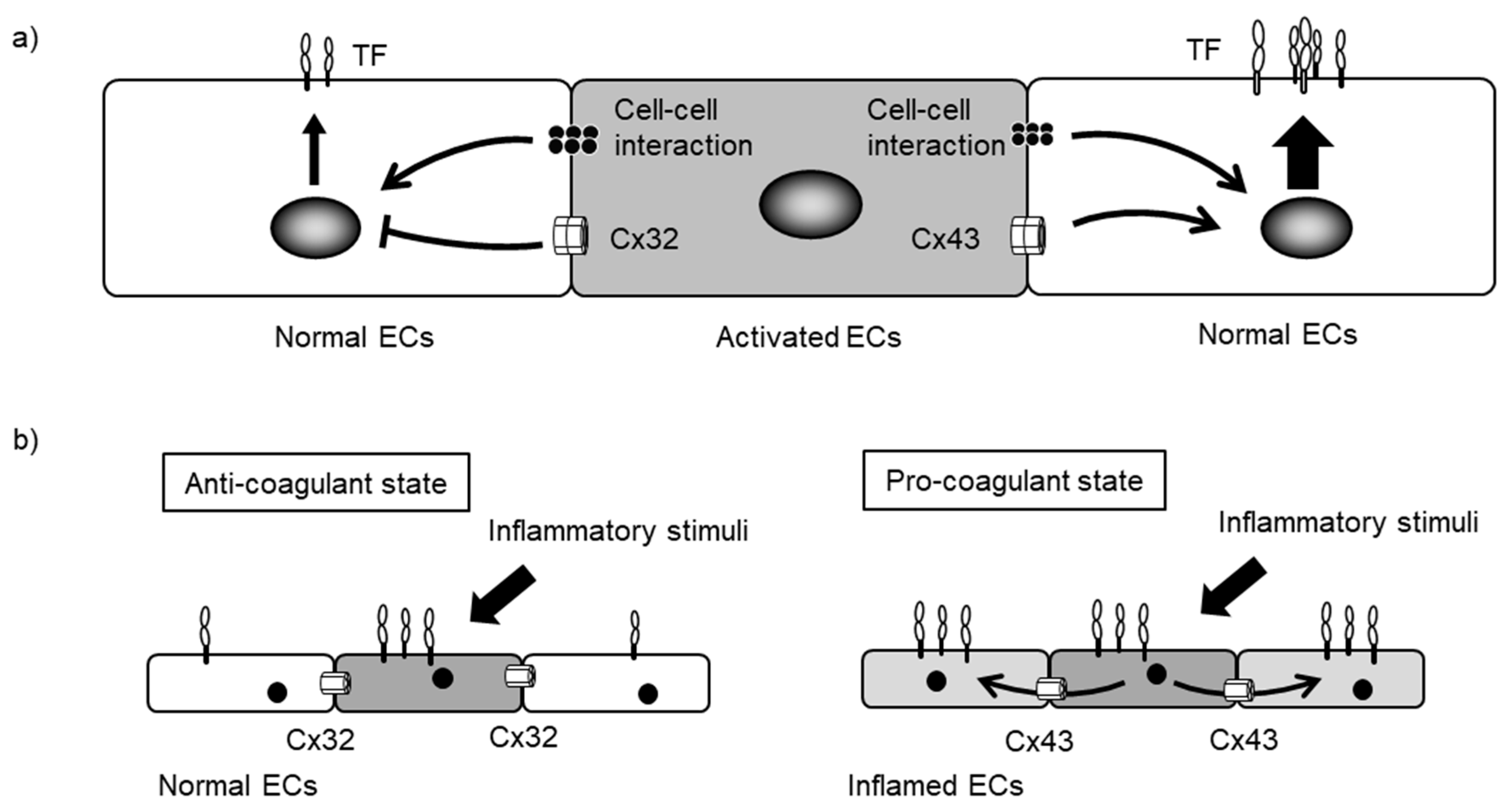
5. Endothelial Cxs and Blood Coagulation
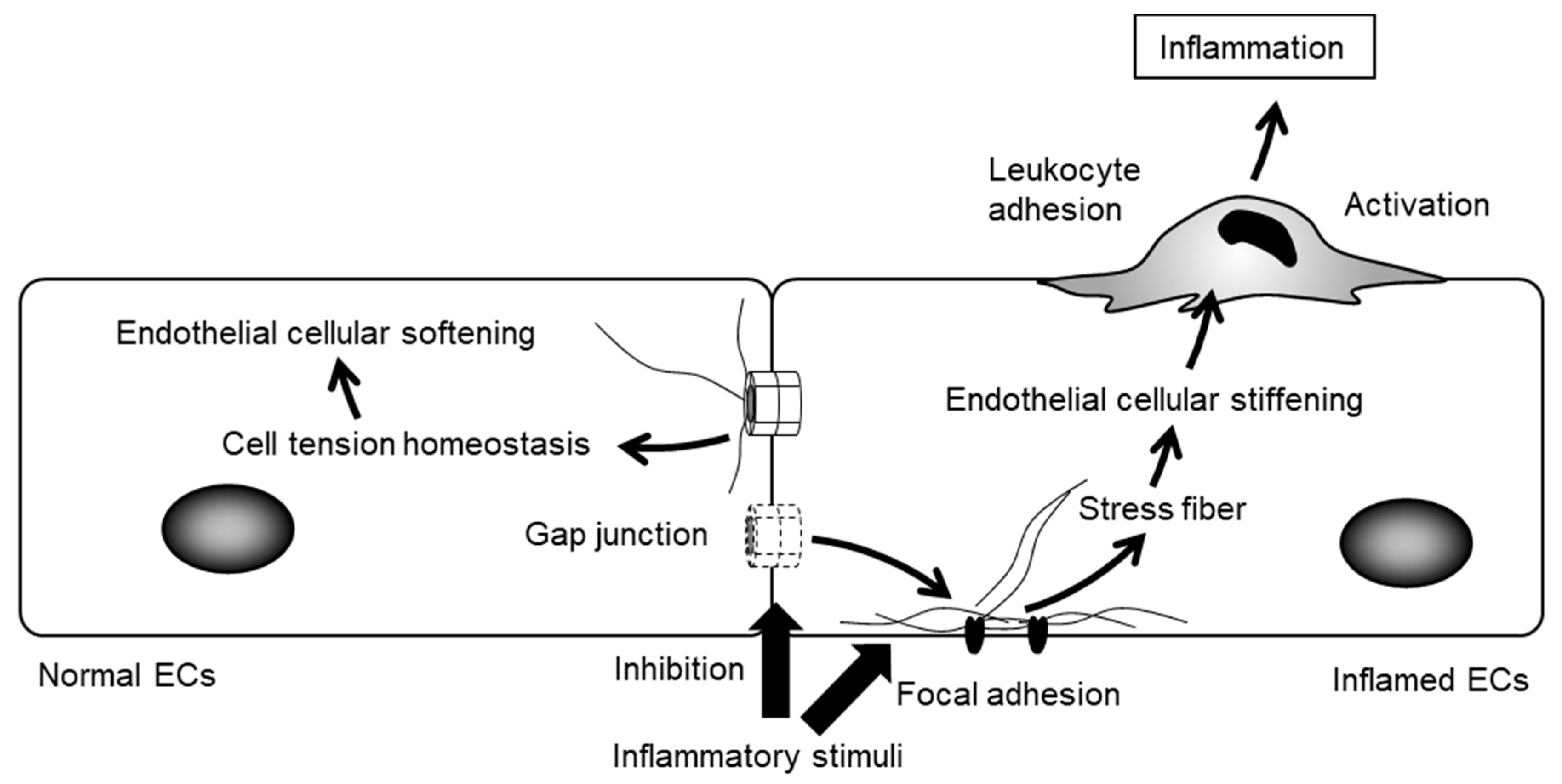
6. Cxs and Leukocyte Adhesion
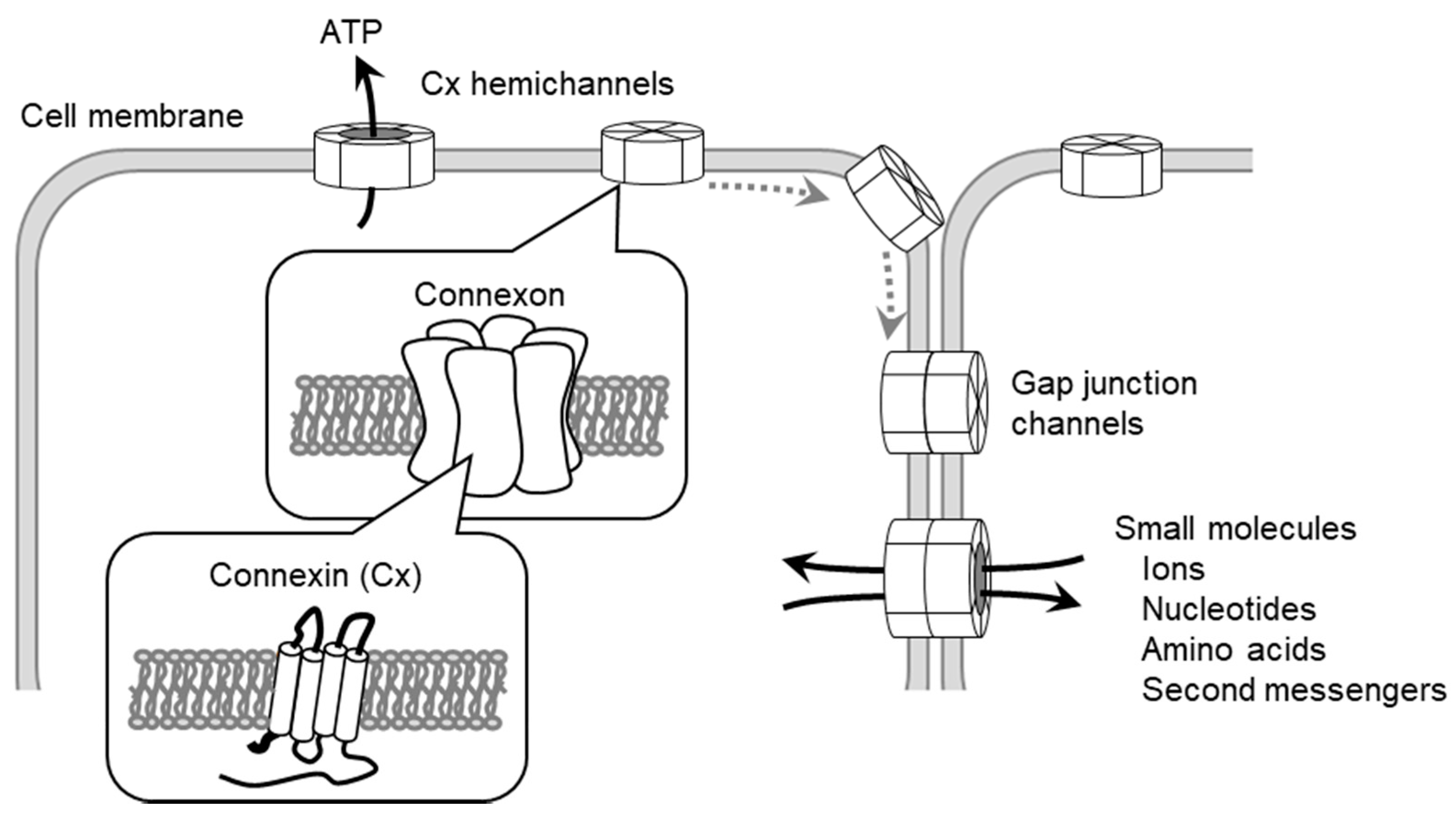
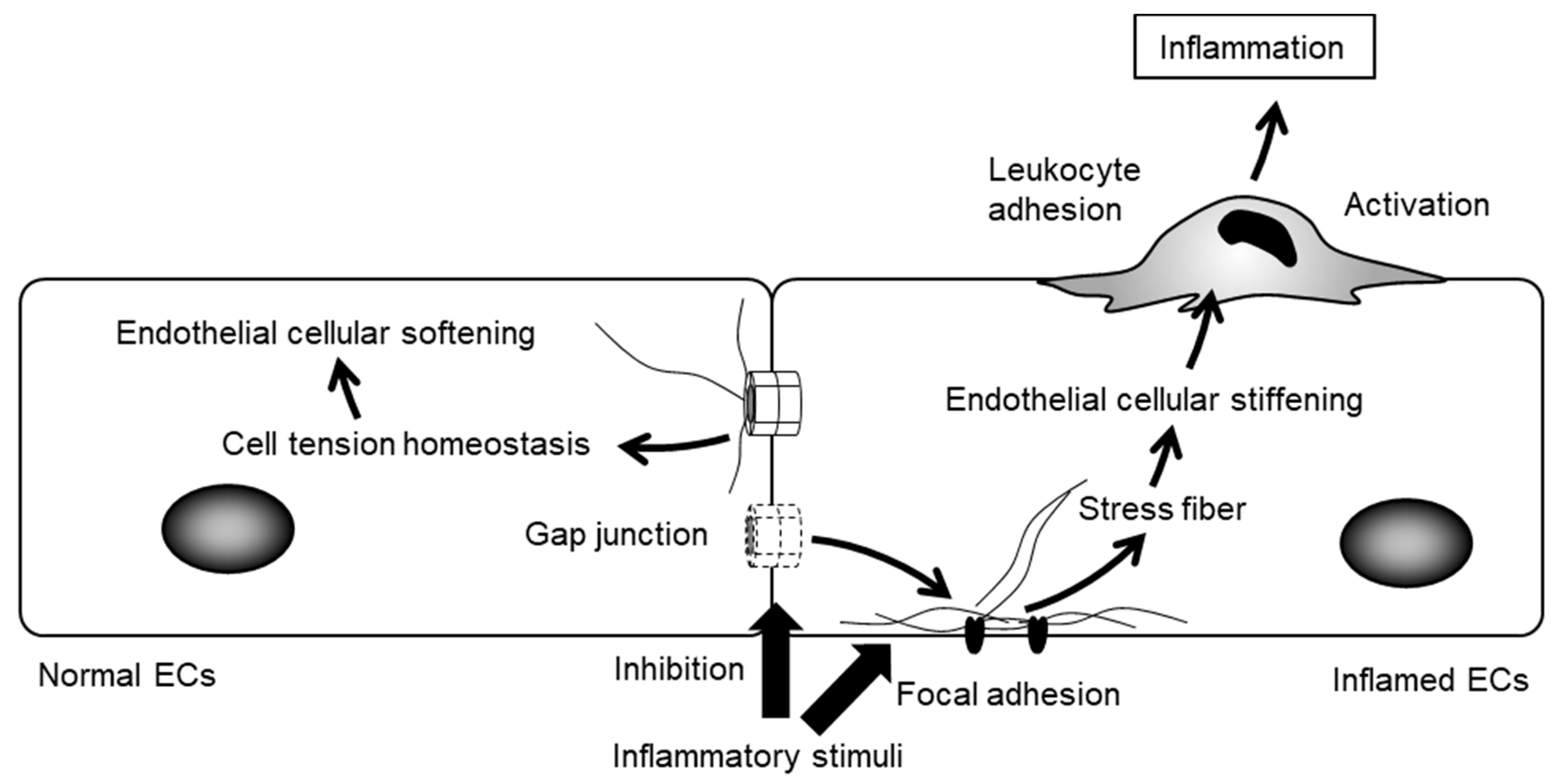
7. The Regulation of Cellular Function by Gap Junctions (GJs) and Cx Channels
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| EC | Endothelial cell |
| GJ | Gap junction |
| Cx | Connexin |
| GJIC | Gap junction intercellular communication |
| SMC | Smooth muscle cell |
| TF | Tissue factor |
| FVIIa | Factor VIIa |
| VWF | von Willebrand factor |
| TM | Thrombomodulin |
| PAR | Protease-activated receptor |
| IL-6 | Interleukin-6 |
| EPCR | Endothelial cell protein C receptor |
| APC | Activated protein C |
| NF-κB | Nuclear factor-κB |
| LPS | Lipopolysaccharide |
| TNF-α | Tumor necrosis factor-α |
| MCP-1 | Monocyte chemotactic protein-1 |
| HUVEC | Human umbilical vein endothelial cell |
| VCAM-1 | Vascular cell adhesion molecule-1 |
| IP3 | Inositol trisphosphate |
| eNOS | Endothelial nitric oxide synthase |
References
- Kumar, N.M.; Gilula, N.B. The gap junction communication channel. Cell 1996, 84, 381–388. [Google Scholar] [CrossRef]
- Saez, J.C.; Berthoud, V.M.; Branes, M.C.; Martinez, A.D.; Beyer, E.C. Plasma membrane channels formed by connexins: Their regulation and functions. Physiol. Rev. 2003, 83, 1359–1400. [Google Scholar] [CrossRef] [PubMed]
- Willecke, K.; Eiberger, J.; Degen, J.; Eckardt, D.; Romualdi, A.; Guldenagel, M.; Deutsch, U.; Sohl, G. Structural and functional diversity of connexin genes in the mouse and human genome. Biol. Chem. 2002, 383, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Sohl, G.; Willecke, K. Gap junctions and the connexin protein family. Cardiovasc. Res. 2004, 62, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Eastman, S.D.; Chen, T.H.; Falk, M.M.; Mendelson, T.C.; Iovine, M.K. Phylogenetic analysis of three complete gap junction gene families reveals lineage-specific duplications and highly supported gene classes. Genomics 2006, 87, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Oshima, A.; Tani, K.; Hiroaki, Y.; Fujiyoshi, Y.; Sosinsky, G.E. Three-dimensional structure of a human connexin26 gap junction channel reveals a plug in the vestibule. Proc. Natl. Acad. Sci. USA 2007, 104, 10034–10039. [Google Scholar] [CrossRef] [PubMed]
- Oshima, A. Structure and closure of connexin gap junction channels. FEBS Lett. 2014, 588, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Yeager, M.; Nicholson, B.J. Structure of gap junction intercellular channels. Curr. Opin. Struct. Biol. 1996, 6, 183–192. [Google Scholar] [CrossRef]
- Hebert, C.; Stains, J.P. An intact connexin43 is required to enhance signaling and gene expression in osteoblast-like cells. J. Cell. Biochem. 2013, 114, 2542–2550. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.J.; Xu, X.; Lo, C.W. Connexins and cell signaling in development and disease. Annu. Rev. Cell. Dev. Biol. 2004, 20, 811–838. [Google Scholar] [CrossRef] [PubMed]
- Oyamada, M.; Oyamada, Y.; Takamatsu, T. Regulation of connexin expression. Biochim. Biophys. Acta 2005, 1719, 6–23. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Connexin channel permeability to cytoplasmic molecules. Prog. Biophys. Mol. Biol. 2007, 94, 120–143. [Google Scholar] [CrossRef] [PubMed]
- Esseltine, J.L.; Laird, D.W. Next-Generation Connexin and Pannexin Cell Biology. Trends Cell. Biol. 2016, 26, 944–955. [Google Scholar] [CrossRef] [PubMed]
- Pfenniger, A.; Chanson, M.; Kwak, B.R. Connexins in atherosclerosis. Biochim. Biophys. Acta 2013, 1828, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Scheckenbach, K.E.; Crespin, S.; Kwak, B.R.; Chanson, M. Connexin channel-dependent signaling pathways in inflammation. J. Vasc. Res. 2011, 48, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Esmon, C.T. The interactions between inflammation and coagulation. Br. J. Haematol. 2005, 131, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Tanigami, H.; Suzuki, K.; Shimaoka, M. Thrombomodulin: A bifunctional modulator of inflammation and coagulation in sepsis. Crit. Care Res. Pract. 2012, 2012, 614545. [Google Scholar] [CrossRef] [PubMed]
- Reglero-Real, N.; Marcos-Ramiro, B.; Millan, J. Endothelial membrane reorganization during leukocyte extravasation. Cell. Mol. Life Sci. 2012, 69, 3079–3099. [Google Scholar] [CrossRef] [PubMed]
- Naldini, A.; Carraro, F. Role of inflammatory mediators in angiogenesis. Curr. Drug Targets Inflamm. Allergy 2005, 4, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Gabriels, J.E.; Paul, D.L. Connexin43 is highly localized to sites of disturbed flow in rat aortic endothelium but connexin37 and connexin40 are more uniformly distributed. Circ. Res. 1998, 83, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.I.; Dupont, E.; Coppen, S.; Rothery, S.; Severs, N.J. Gap junction localization and connexin expression in cytochemically identified endothelial cells of arterial tissue. J. Histochem. Cytochem. 1997, 45, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.S.; Coppen, S.R.; Dupont, E.; Rothery, S.; Severs, N.J. Regional differentiation of desmin, connexin43, and connexin45 expression patterns in rat aortic smooth muscle. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Little, T.L.; Beyer, E.C.; Duling, B.R. Connexin 43 and connexin 40 gap junctional proteins are present in arteriolar smooth muscle and endothelium in vivo. Am. J. Physiol. 1995, 268, H729–H739. [Google Scholar] [PubMed]
- Kilarski, W.M.; Dupont, E.; Coppen, S.; Yeh, H.I.; Vozzi, C.; Gourdie, R.G.; Rezapour, M.; Ulmsten, U.; Roomans, G.M.; Severs, N.J. Identification of two further gap-junctional proteins, connexin40 and connexin45, in human myometrial smooth muscle cells at term. Eur. J. Cell. Biol. 1998, 75, 1–8. [Google Scholar] [CrossRef]
- Li, X.; Simard, J.M. Multiple connexins form gap junction channels in rat basilar artery smooth muscle cells. Circ. Res. 1999, 84, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.W.; Christen, T.; Roth, I.; Chadjichristos, C.E.; Derouette, J.P.; Foglia, B.F.; Chanson, M.; Goodenough, D.A.; Kwak, B.R. Connexin37 protects against atherosclerosis by regulating monocyte adhesion. Nat. Med. 2006, 12, 950–954. [Google Scholar] [CrossRef] [PubMed]
- Eugenin, E.A.; Branes, M.C.; Berman, J.W.; Saez, J.C. TNF-alpha plus IFN-gamma induce connexin43 expression and formation of gap junctions between human monocytes/macrophages that enhance physiological responses. J. Immunol. 2003, 170, 1320–1328. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Regan, C.P.; Manabe, I.; Owens, G.K.; Day, K.H.; Damon, D.N.; Duling, B.R. Smooth muscle-targeted knockout of connexin43 enhances neointimal formation in response to vascular injury. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Majesky, M.W. Adventitia and perivascular cells. Arterioscler. Thromb. Vasc. Biol. 2015, 35, e31–e35. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Izawa, H.; Ichihara, S.; Takatsu, F.; Ishihara, H.; Hirayama, H.; Sone, T.; Tanaka, M.; Yokota, M. Prediction of the risk of myocardial infarction from polymorphisms in candidate genes. N. Engl. J. Med. 2002, 347, 1916–1923. [Google Scholar] [CrossRef] [PubMed]
- Firouzi, M.; Kok, B.; Spiering, W.; Busjahn, A.; Bezzina, C.R.; Ruijter, J.M.; Koeleman, B.P.; Schipper, M.; Groenewegen, W.A.; Jongsma, H.J.; de Leeuw, P.W. Polymorphisms in human connexin40 gene promoter are associated with increased risk of hypertension in men. J. Hypertens. 2006, 24, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.; de Wit, C.; Kurtz, L.; Grunberger, C.; Kurtz, A.; Schweda, F. Connexin40 is essential for the pressure control of renin synthesis and secretion. Circ. Res. 2007, 100, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Derouette, J.P.; Wong, C.; Burnier, L.; Morel, S.; Sutter, E.; Galan, K.; Brisset, A.C.; Roth, I.; Chadjichristos, C.E.; Kwak, B.R. Molecular role of Cx37 in advanced atherosclerosis: A micro-array study. Atherosclerosis 2009, 206, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Chadjichristos, C.E.; Scheckenbach, K.E.; van Veen, T.A.; Richani Sarieddine, M.Z.; de Wit, C.; Yang, Z.; Roth, I.; Bacchetta, M.; Viswambharan, H.; Foglia, B.; et al. Endothelial-specific deletion of connexin40 promotes atherosclerosis by increasing CD73-dependent leukocyte adhesion. Circulation 2010, 121, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Kwak, B.R.; Veillard, N.; Pelli, G.; Mulhaupt, F.; James, R.W.; Chanson, M.; Mach, F. Reduced connexin43 expression inhibits atherosclerotic lesion formation in low-density lipoprotein receptor-deficient mice. Circulation 2003, 107, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Billaud, M.; Lohman, A.W.; Johnstone, S.R.; Biwer, L.A.; Mutchler, S.; Isakson, B.E. Regulation of cellular communication by signaling microdomains in the blood vessel wall. Pharmacol. Rev. 2014, 66, 513–569. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; van der Poll, T. Inflammation and coagulation. Crit. Care Med. 2010, 38, S26–S34. [Google Scholar] [CrossRef] [PubMed]
- Antoniak, S.; Mackman, N. Editorial Commentary: Tissue factor expression by the endothelium: Coagulation or inflammation? Trends Cardiovasc. Med. 2016, 26, 304–305. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N. The many faces of tissue factor. J. Thromb. Haemost. 2009, 7, 136–139. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.D.; Bonfanti, R. von Willebrand factor and the endothelium. Mayo Clin. Proc. 1991, 66, 621–627. [Google Scholar] [CrossRef]
- Ait-Oufella, H.; Maury, E.; Lehoux, S.; Guidet, B.; Offenstadt, G. The endothelium: Physiological functions and role in microcirculatory failure during severe sepsis. Intensive Care Med. 2010, 36, 1286–1298. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, M.; Monroe, D.M.; Roberts, H.R. Cellular interactions in hemostasis. Haemostasis 1996, 26, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Popovic, M.; Smiljanic, K.; Dobutovic, B.; Syrovets, T.; Simmet, T.; Isenovic, E.R. Thrombin and vascular inflammation. Mol. Cell. Biochem. 2012, 359, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, S.R. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J. Thromb. Haemost. 2005, 3, 1800–1814. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.; Choi, Y.; DeGroot, E.; Samuels, I.; Creasey, A.; Aarden, L. Potential mechanisms for a proinflammatory vascular cytokine response to coagulation activation. J. Immunol. 1998, 160, 5130–5135. [Google Scholar] [PubMed]
- Bizios, R.; Lai, L.; Fenton, J.W., 2nd; Malik, A.B. Thrombin-induced chemotaxis and aggregation of neutrophils. J. Cell. Physiol. 1986, 128, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Lum, H.; Malik, A.B. Mechanisms of increased endothelial permeability. Can. J. Physiol. Pharmacol. 1996, 74, 787–800. [Google Scholar] [CrossRef] [PubMed]
- Kopec, A.K.; Abrahams, S.R.; Thornton, S.; Palumbo, J.S.; Mullins, E.S.; Divanovic, S.; Weiler, H.; Owens, A.P.; Mackman, N.; Goss, A.; et al. Thrombin promotes diet-induced obesity through fibrin-driven inflammation. J. Clin. Investig. 2017, 127, 3152–3166. [Google Scholar] [CrossRef] [PubMed]
- Conway, E.M. Thrombomodulin and its role in inflammation. Semin. Immunopathol. 2012, 34, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Adams, T.E.; Huntington, J.A. Thrombin-cofactor interactions: Structural insights into regulatory mechanisms. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1738–1745. [Google Scholar] [CrossRef] [PubMed]
- Stearns-Kurosawa, D.J.; Kurosawa, S.; Mollica, J.S.; Ferrell, G.L.; Esmon, C.T. The endothelial cell protein C receptor augments protein C activation by the thrombin-thrombomodulin complex. Proc. Natl. Acad. Sci. USA 1996, 93, 10212–10216. [Google Scholar] [CrossRef] [PubMed]
- Bouwens, E.A.; Stavenuiter, F.; Mosnier, L.O. Mechanisms of anticoagulant and cytoprotective actions of the protein C pathway. J. Thromb. Haemost. 2013, 11, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Feistritzer, C.; Riewald, M. Endothelial barrier protection by activated protein C through PAR1-dependent sphingosine 1-phosphate receptor-1 crossactivation. Blood 2005, 105, 3178–3184. [Google Scholar] [CrossRef] [PubMed]
- Yuksel, M.; Okajima, K.; Uchiba, M.; Horiuchi, S.; Okabe, H. Activated protein C inhibits lipopolysaccharide-induced tumor necrosis factor-alpha production by inhibiting activation of both nuclear factor-kappa B and activator protein-1 in human monocytes. Thromb. Haemost. 2002, 88, 267–273. [Google Scholar] [PubMed]
- White, B.; Schmidt, M.; Murphy, C.; Livingstone, W.; O’Toole, D.; Lawler, M.; O’Neill, L.; Kelleher, D.; Schwarz, H.P.; Smith, O.P. Activated protein C inhibits lipopolysaccharide-induced nuclear translocation of nuclear factor kappaB (NF-kappaB) and tumour necrosis factor alpha (TNF-alpha) production in the THP-1 monocytic cell line. Br. J. Haematol. 2000, 110, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.S.; Rezaie, A.R. Thrombin inhibits nuclear factor kappaB and RhoA pathways in cytokine-stimulated vascular endothelial cells when EPCR is occupied by protein C. Thromb. Haemost. 2009, 101, 513–520. [Google Scholar] [PubMed]
- Murakami, K.; Okajima, K.; Uchiba, M.; Johno, M.; Nakagaki, T.; Okabe, H.; Takatsuki, K. Activated protein C attenuates endotoxin-induced pulmonary vascular injury by inhibiting activated leukocytes in rats. Blood 1996, 87, 642–647. [Google Scholar] [PubMed]
- Elphick, G.F.; Sarangi, P.P.; Hyun, Y.M.; Hollenbaugh, J.A.; Ayala, A.; Biffl, W.L.; Chung, H.L.; Rezaie, A.R.; McGrath, J.L.; Topham, D.J.; et al. Recombinant human activated protein C inhibits integrin-mediated neutrophil migration. Blood 2009, 113, 4078–4085. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, E.; Okamoto, T.; Takagi, Y.; Honda, G.; Suzuki, K.; Imai, H.; Shimaoka, M. LFA-1 and Mac-1 integrins bind to the serine/threonine-rich domain of thrombomodulin. Biochem. Biophys. Res. Commun. 2016, 473, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Fink, K.; Busch, H.J.; Bourgeois, N.; Schwarz, M.; Wolf, D.; Zirlik, A.; Peter, K.; Bode, C.; von Zur Muhlen, C. Mac-1 directly binds to the endothelial protein C-receptor: A link between the protein C anticoagulant pathway and inflammation? PLoS ONE 2013, 8, e53103. [Google Scholar] [CrossRef] [PubMed]
- Meens, M.J.; Kutkut, I.; Rochemont, V.; Dubrot, J.; Kaladji, F.R.; Sabine, A.; Lyons, O.; Hendrikx, S.; Bernier-Latmani, J.; Kiefer, F.; et al. Cx47 fine-tunes the handling of serum lipids but is dispensable for lymphatic vascular function. PLoS ONE 2017, 12, e0181476. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Akiyama, M.; Takeda, M.; Gabazza, E.C.; Hayashi, T.; Suzuki, K. Connexin32 is expressed in vascular endothelial cells and participates in gap-junction intercellular communication. Biochem. Biophys. Res. Commun. 2009, 382, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Thuringer, D.; Berthenet, K.; Cronier, L.; Solary, E.; Garrido, C. Primary tumor- and metastasis-derived colon cancer cells differently modulate connexin expression and function in human capillary endothelial cells. Oncotarget 2015, 6, 28800–28815. [Google Scholar] [CrossRef] [PubMed]
- Chadjichristos, C.E.; Derouette, J.P.; Kwak, B.R. Connexins in atherosclerosis. Adv. Cardiol. 2006, 42, 255–267. [Google Scholar] [PubMed]
- Severs, N.J.; Coppen, S.R.; Dupont, E.; Yeh, H.I.; Ko, Y.S.; Matsushita, T. Gap junction alterations in human cardiac disease. Cardiovasc. Res. 2004, 62, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Van Rijen, H.V.; van Kempen, M.J.; Postma, S.; Jongsma, H.J. Tumour necrosis factor alpha alters the expression of connexin43, connexin40, and connexin37 in human umbilical vein endothelial cells. Cytokine 1998, 10, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Akiyama, M.; Takeda, M.; Akita, N.; Yoshida, K.; Hayashi, T.; Suzuki, K. Connexin32 protects against vascular inflammation by modulating inflammatory cytokine expression by endothelial cells. Exp. Cell. Res. 2011, 317, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Lidington, D.; Tyml, K.; Ouellette, Y. Lipopolysaccharide-induced reductions in cellular coupling correlate with tyrosine phosphorylation of connexin 43. J. Cell. Physiol. 2002, 193, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Bolon, M.L.; Kidder, G.M.; Simon, A.M.; Tyml, K. Lipopolysaccharide reduces electrical coupling in microvascular endothelial cells by targeting connexin40 in a tyrosine-, ERK1/2-, PKA-, and PKC-dependent manner. J. Cell. Physiol. 2007, 211, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Tyml, K. Role of connexins in microvascular dysfunction during inflammation. Can. J. Physiol. Pharmacol. 2011, 89, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rennick, R.E.; Connat, J.L.; Burnstock, G.; Rothery, S.; Severs, N.J.; Green, C.R. Expression of connexin43 gap junctions between cultured vascular smooth muscle cells is dependent upon phenotype. Cell. Tissue Res. 1993, 271, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Arishiro, K.; Hoshiga, M.; Ishihara, T.; Kondo, K.; Hanafusa, T. Connexin 43 expression is associated with vascular activation in human radial artery. Int. J. Cardiol. 2010, 145, 270–272. [Google Scholar] [CrossRef] [PubMed]
- Kwak, B.R.; Mulhaupt, F.; Veillard, N.; Gros, D.B.; Mach, F. Altered pattern of vascular connexin expression in atherosclerotic plaques. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Polacek, D.; Lal, R.; Volin, M.V.; Davies, P.F. Gap junctional communication between vascular cells. Induction of connexin43 messenger RNA in macrophage foam cells of atherosclerotic lesions. Am. J. Pathol. 1993, 142, 593–606. [Google Scholar] [PubMed]
- Nieman, M.T. Protease-activated receptors in hemostasis. Blood 2016, 128, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Van Zeijl, L.; Ponsioen, B.; Giepmans, B.N.; Ariaens, A.; Postma, F.R.; Varnai, P.; Balla, T.; Divecha, N.; Jalink, K.; Moolenaar, W.H. Regulation of connexin43 gap junctional communication by phosphatidylinositol 4,5-bisphosphate. J. Cell. Biol. 2007, 177, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Postma, F.R.; Hengeveld, T.; Alblas, J.; Giepmans, B.N.; Zondag, G.C.; Jalink, K.; Moolenaar, W.H. Acute loss of cell-cell communication caused by G protein-coupled receptors: A critical role for c-Src. J. Cell. Biol. 1998, 140, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.M.; Kim, N.; Gumpert, A.M.; Segretain, D.; Falk, M.M. Acute internalization of gap junctions in vascular endothelial cells in response to inflammatory mediator-induced G-protein coupled receptor activation. FEBS Lett. 2008, 582, 4039–4046. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.J., 3rd; Birukova, A.A.; Beyer, E.C.; Birukov, K.G. Gap junction protein connexin43 exacerbates lung vascular permeability. PLoS ONE 2014, 9, e100931. [Google Scholar] [CrossRef] [PubMed]
- Godecke, S.; Roderigo, C.; Rose, C.R.; Rauch, B.H.; Godecke, A.; Schrader, J. Thrombin-induced ATP release from human umbilical vein endothelial cells. Am. J. Physiol. Cell. Physiol. 2012, 302, C915–C923. [Google Scholar] [CrossRef] [PubMed]
- Seminario-Vidal, L.; Kreda, S.; Jones, L.; O’Neal, W.; Trejo, J.; Boucher, R.C.; Lazarowski, E.R. Thrombin promotes release of ATP from lung epithelial cells through coordinated activation of rho- and Ca2+-dependent signaling pathways. J. Biol. Chem. 2009, 284, 20638–20648. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Akita, N.; Hayashi, T.; Shimaoka, M.; Suzuki, K. Endothelial connexin 32 regulates tissue factor expression induced by inflammatory stimulation and direct cell-cell interaction with activated cells. Atherosclerosis 2014, 236, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Vaiyapuri, S.; Flora, G.D.; Gibbins, J.M. Gap junctions and connexin hemichannels in the regulation of haemostasis and thrombosis. Biochem. Soc. Trans. 2015, 43, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Vaiyapuri, S.; Jones, C.I.; Sasikumar, P.; Moraes, L.A.; Munger, S.J.; Wright, J.R.; Ali, M.S.; Sage, T.; Kaiser, W.J.; Tucker, K.L.; et al. Gap junctions and connexin hemichannels underpin hemostasis and thrombosis. Circulation 2012, 125, 2479–2491. [Google Scholar] [CrossRef] [PubMed]
- Vaiyapuri, S.; Moraes, L.A.; Sage, T.; Ali, M.S.; Lewis, K.R.; Mahaut-Smith, M.P.; Oviedo-Orta, E.; Simon, A.M.; Gibbins, J.M. Connexin40 regulates platelet function. Nat. Commun. 2013, 4, 2564. [Google Scholar] [CrossRef] [PubMed]
- Angelillo-Scherrer, A.; Fontana, P.; Burnier, L.; Roth, I.; Sugamele, R.; Brisset, A.; Morel, S.; Nolli, S.; Sutter, E.; Chassot, A.; et al. Connexin 37 limits thrombus propensity by downregulating platelet reactivity. Circulation 2011, 124, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Park, E.J.; Yuki, Y.; Kiyono, H.; Shimaoka, M. Structural basis of blocking integrin activation and deactivation for anti-inflammation. J. Biomed. Sci. 2015, 22, 51. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Garlanda, C.; Locati, M. Macrophage diversity and polarization in atherosclerosis: A question of balance. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1419–1423. [Google Scholar] [CrossRef] [PubMed]
- Zukowska, P.; Kutryb-Zajac, B.; Toczek, M.; Smolenski, R.T.; Slominska, E.M. The role of ecto-5′-nucleotidase in endothelial dysfunction and vascular pathologies. Pharmacol. Rep. 2015, 67, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, E.; Palomo, I. Extracellular ATP metabolism on vascular endothelial cells: A pathway with pro-thrombotic and anti-thrombotic molecules. Vascul. Pharmacol. 2015, 75, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zernecke, A.; Bidzhekov, K.; Ozuyaman, B.; Fraemohs, L.; Liehn, E.A.; Luscher-Firzlaff, J.M.; Luscher, B.; Schrader, J.; Weber, C. CD73/ecto-5′-nucleotidase protects against vascular inflammation and neointima formation. Circulation 2006, 113, 2120–2127. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Wu, M.H.; Yuan, S.Y. Nonmuscle myosin light-chain kinase deficiency attenuates atherosclerosis in apolipoprotein E-deficient mice via reduced endothelial barrier dysfunction and monocyte migration. Circulation 2011, 124, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Stroka, K.M.; Aranda-Espinoza, H. Endothelial cell substrate stiffness influences neutrophil transmigration via myosin light chain kinase-dependent cell contraction. Blood 2011, 118, 1632–1640. [Google Scholar] [CrossRef] [PubMed]
- Haeger, A.; Wolf, K.; Zegers, M.M.; Friedl, P. Collective cell migration: Guidance principles and hierarchies. Trends Cell. Biol. 2015, 25, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Kawamoto, E.; Takagi, Y.; Akita, N.; Hayashi, T.; Park, E.J.; Suzuki, K.; Shimaoka, M. Gap junction-mediated regulation of endothelial cellular stiffness. Sci. Rep. 2017, 7, 6134. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, S.V.; Pasapera, A.M.; Sabass, B.; Waterman, C.M. Force fluctuations within focal adhesions mediate ECM-rigidity sensing to guide directed cell migration. Cell 2012, 151, 1513–1527. [Google Scholar] [CrossRef] [PubMed]
- Huveneers, S.; Daemen, M.J.; Hordijk, P.L. Between Rho(k) and a hard place: The relation between vessel wall stiffness, endothelial contractility, and cardiovascular disease. Circ. Res. 2015, 116, 895–908. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Ciciliano, J.; Myers, D.R.; Tran, R.; Lam, W.A. Platelets and physics: How platelets “feel” and respond to their mechanical microenvironment. Blood Rev. 2015, 29, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Brown, A.C.; Myers, D.R.; Sakurai, Y.; Mannino, R.G.; Tran, R.; Ahn, B.; Hardy, E.T.; Kee, M.F.; Kumar, S.; et al. Platelet mechanosensing of substrate stiffness during clot formation mediates adhesion, spreading, and activation. Proc. Natl. Acad. Sci. USA 2014, 111, 14430–14435. [Google Scholar] [CrossRef] [PubMed]
- Oviedo-Orta, E.; Errington, R.J.; Evans, W.H. Gap junction intercellular communication during lymphocyte transendothelial migration. Cell. Biol. Int. 2002, 26, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Zahler, S.; Hoffmann, A.; Gloe, T.; Pohl, U. Gap-junctional coupling between neutrophils and endothelial cells: A novel modulator of transendothelial migration. J. Leukoc. Biol. 2003, 73, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, V.H.; Bortolozzi, M.; Pertegato, V.; Beltramello, M.; Giarin, M.; Zaccolo, M.; Pantano, S.; Mammano, F. Unitary permeability of gap junction channels to second messengers measured by FRET microscopy. Nat. Methods 2007, 4, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, G.S.; Lampe, P.D.; Nicholson, B.J. Selective transfer of endogenous metabolites through gap junctions composed of different connexins. Nat. Cell. Biol. 1999, 1, 457–459. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Sin, W.C.; Harris, A.L.; Naus, C.C. Gap junctions modulate glioma invasion by direct transfer of microRNA. Oncotarget 2015, 6, 15566–15577. [Google Scholar] [CrossRef] [PubMed]
- Kizana, E.; Cingolani, E.; Marban, E. Non-cell-autonomous effects of vector-expressed regulatory RNAs in mammalian heart cells. Gene Ther. 2009, 16, 1163–1168. [Google Scholar] [CrossRef] [PubMed]
- Lemcke, H.; Steinhoff, G.; David, R. Gap junctional shuttling of miRNA—A novel pathway of intercellular gene regulation and its prospects in clinical application. Cell. Signal. 2015, 27, 2506–2514. [Google Scholar] [CrossRef] [PubMed]
- Brink, P.R.; Valiunas, V.; Gordon, C.; Rosen, M.R.; Cohen, I.S. Can gap junctions deliver? Biochim. Biophys. Acta 2012, 1818, 2076–2081. [Google Scholar] [CrossRef] [PubMed]
- Giepmans, B.N. Gap junctions and connexin-interacting proteins. Cardiovasc. Res. 2004, 62, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Duffy, H.S.; Iacobas, I.; Hotchkiss, K.; Hirst-Jensen, B.J.; Bosco, A.; Dandachi, N.; Dermietzel, R.; Sorgen, P.L.; Spray, D.C. The gap junction protein connexin32 interacts with the Src homology 3/hook domain of discs large homolog 1. J. Biol. Chem. 2007, 282, 9789–9796. [Google Scholar] [CrossRef] [PubMed]
- Talhouk, R.S.; Mroue, R.; Mokalled, M.; Abi-Mosleh, L.; Nehme, R.; Ismail, A.; Khalil, A.; Zaatari, M.; El-Sabban, M.E. Heterocellular interaction enhances recruitment of alpha and beta-catenins and ZO-2 into functional gap-junction complexes and induces gap junction-dependant differentiation of mammary epithelial cells. Exp. Cell. Res. 2008, 314, 3275–3291. [Google Scholar] [CrossRef] [PubMed]
- Toyofuku, T.; Akamatsu, Y.; Zhang, H.; Kuzuya, T.; Tada, M.; Hori, M. c-Src regulates the interaction between connexin-43 and ZO-1 in cardiac myocytes. J. Biol. Chem. 2001, 276, 1780–1788. [Google Scholar] [CrossRef] [PubMed]
- Stroka, K.M.; Aranda-Espinoza, H. Effects of Morphology vs. Cell-Cell Interactions on Endothelial Cell Stiffness. Cell. Mol. Bioeng. 2011, 4, 9–27. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, A.; Te Riet, J.; Ritz, K.; Hoogenboezem, M.; Anthony, E.C.; Mul, F.P.; de Vries, C.J.; Daemen, M.J.; Figdor, C.G.; van Buul, J.D.; Hordijk, P.L. Actin-binding proteins differentially regulate endothelial cell stiffness, ICAM-1 function and neutrophil transmigration. J. Cell. Sci. 2014, 127, 4470–4482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, A.; Hordijk, P.L. Cell-stiffness-induced mechanosignaling—A key driver of leukocyte transendothelial migration. J. Cell. Sci. 2015, 128, 2221–2230. [Google Scholar] [CrossRef] [PubMed]
- Haidari, M.; Zhang, W.; Willerson, J.T.; Dixon, R.A. Disruption of endothelial adherens junctions by high glucose is mediated by protein kinase C-beta-dependent vascular endothelial cadherin tyrosine phosphorylation. Cardiovasc. Diabetol. 2014, 13, 105. [Google Scholar] [CrossRef] [PubMed]
- Meens, M.J.; Alonso, F.; Le Gal, L.; Kwak, B.R.; Haefliger, J.A. Endothelial Connexin37 and Connexin40 participate in basal but not agonist-induced NO release. Cell. Commun. Signal. 2015, 13, 34. [Google Scholar] [CrossRef] [PubMed]
- Pfenniger, A.; Derouette, J.P.; Verma, V.; Lin, X.; Foglia, B.; Coombs, W.; Roth, I.; Satta, N.; Dunoyer-Geindre, S.; Sorgen, P.; et al. Gap junction protein Cx37 interacts with endothelial nitric oxide synthase in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Alonso, F.; Boittin, F.X.; Beny, J.L.; Haefliger, J.A. Loss of connexin40 is associated with decreased endothelium-dependent relaxations and eNOS levels in the mouse aorta. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1365–H1373. [Google Scholar] [CrossRef] [PubMed]
- Siragusa, M.; Fleming, I. The eNOS signalosome and its link to endothelial dysfunction. Pflugers Arch. Eur. J. Physiol. 2016, 468, 1125–1137. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Cx Expression | Stimuli/Signaling/Process | ∆ Cx Expression/GJ Function |
|---|---|---|---|
| Endothelial cell | Cx32, Cx37, Cx40, Cx43 | TNF-α | Cx37↑, Cx40↓, Cx32↓, GJ↓ |
| LPS | Cx40↓, GJ↓ | ||
| Thrombin | Cx43↓, GJ↓ | ||
| Thrombin | Cx hemichannel↑ | ||
| Smooth muscle cell | Cx40, Cx43 | NF-κB | Cx43↑ |
| Monocyte/Macrophage | Cx37, Cx43 | TNF-α and IFN-γ | Cx43↑ |
| Foam cell | Cx37↑, Cx43↑ |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okamoto, T.; Suzuki, K. The Role of Gap Junction-Mediated Endothelial Cell–Cell Interaction in the Crosstalk between Inflammation and Blood Coagulation. Int. J. Mol. Sci. 2017, 18, 2254. https://doi.org/10.3390/ijms18112254
Okamoto T, Suzuki K. The Role of Gap Junction-Mediated Endothelial Cell–Cell Interaction in the Crosstalk between Inflammation and Blood Coagulation. International Journal of Molecular Sciences. 2017; 18(11):2254. https://doi.org/10.3390/ijms18112254
Chicago/Turabian StyleOkamoto, Takayuki, and Koji Suzuki. 2017. "The Role of Gap Junction-Mediated Endothelial Cell–Cell Interaction in the Crosstalk between Inflammation and Blood Coagulation" International Journal of Molecular Sciences 18, no. 11: 2254. https://doi.org/10.3390/ijms18112254