Emerging Therapeutics to Overcome Chemoresistance in Epithelial Ovarian Cancer: A Mini-Review

Department of Obstetrics and Gynecology, University of Virginia, Charlottesville, VA 22908, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2017, 18(10), 2171; https://doi.org/10.3390/ijms18102171
Submission received: 13 September 2017
/
Revised: 13 October 2017
/
Accepted: 16 October 2017
/
Published: 18 October 2017
(This article belongs to the Special Issue Gynecologic Oncology: From Molecular Mechanisms to Targeted Therapies)
Abstract
:Ovarian cancer is the fifth leading cause of cancer death among women and the most lethal gynecologic malignancy. One of the leading causes of death in high-grade serous ovarian cancer (HGSOC) is chemoresistant disease, which may present as intrinsic or acquired resistance to therapies. Here we discuss some of the known molecular mechanisms of chemoresistance that have been exhaustively investigated in chemoresistant ovarian cancer, including drug efflux pump multidrug resistance protein 1 (MDR1), the epithelial–mesenchymal transition, DNA damage and repair capacity. We also discuss novel therapeutics that may address some of the challenges in bringing approaches that target chemoresistant processes from bench to bedside. Some of these new therapies include novel drug delivery systems, targets that may halt adaptive changes in the tumor, exploitation of tumor mutations that leave cancer cells vulnerable to irreversible damage, and novel drugs that target ribosomal biogenesis, a process that may be uniquely different in cancer versus non-cancerous cells. Each of these approaches, or a combination of them, may provide a greater number of positive outcomes for a broader population of HGSOC patients.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Ovarian cancer remains a devastating diagnosis with an overall survival rate of ~40%, making it the fifth leading cause of cancer death in women and the most lethal gynecologic malignancy [1,2,3]. Over 220,000 women worldwide are diagnosed each year and an estimated 14,000 will die annually in the U.S. alone, a number that has only changed slightly after 30 years of research [4,5,6]. Histologically ovarian cancer is roughly composed of epithelial, germ cell and stromal tumors with epithelial ovarian cancer (EOC) being the most common and the most deadly. EOC can develop as high-grade serous, low-grade serous, endometrioid, clear cell and mucinous histotypes. It is now becoming clear that most serous cancers likely originate in the fallopian tube, although currently they are still referred to as ovarian cancers [7]. A more important distinction is high-grade versus low-grade cytologic subtypes, as low-grade serous ovarian cancers are more slow-growing, but more chemoresistant, than high-grade serous ovarian cancers (HGSOC). This review of chemoresistance will focus on HGSOC as it is the dominant subtype seen in the clinic. Importantly, until recently most preclinical studies have been performed on cell lines that are more of an endometrioid subtype than serous, and therefore may not be applicable to the tumor protein 53 (TP53)-mutant driven serous subtype [8].
First-line therapeutic interventions in ovarian cancer have evolved over the last few decades from a single nitrogen mustard alkylating agent to the current standard of care: cytoreductive surgery followed by combination taxane-platinum treatment [1,4,5]. About 60–80% of EOC patients receiving this combination after surgery will achieve complete remission, with ~80% of these having a chemoresistant recurrence [3,6,9]. Acquired platinum resistance remains a largely incurable condition and novel targeted therapeutics, new combination therapies, or innovative therapeutic strategies to specifically address the chemoresistant phenotype, are desperately needed [10,11,12]. Studies have elucidated many of the mechanisms that underlie the development of chemotherapy resistance in HGSOC (for reviews see [13,14,15,16,17]) and successfully targeting these systems in the clinic is critical in extending patient survival. This review will focus on HGSOC chemoresistance and emerging therapies that may show promise in mitigating and possibly defeating it.
2. Chemoresistance
Historically, the first major studies that encountered acquired resistance to therapy were the clinical trials in 1965 in pediatric hematopoietic malignancies by Frie et al. [18]. In using combinations of several cytotoxic agents, they saw the first real progress in extending the lives of children with leukemia. This success was cut short by recurrences where the leukemic cells had acquired the ability to resist treatment by hiding in a reservoir on the other side of the blood-brain barrier. The therapeutic agents used were unable to efficiently pass through the blood-brain barrier, leading to the patients eventually succumbing to multi-drug resistant disease. This gave the first clear evidence of dormant cancers taking advantage of our own defenses to resist treatment [19].
Broadly speaking, resistance to therapies is categorized into intrinsic or acquired resistance, although distinguishing between these two mechanisms can be difficult. Intrinsic resistance is the innate ability of the cancer cells to maintain and persist through their first exposure to treatment. Acquired resistance is the evolution of cancer cells, following treatment exposure, to an unaffected and persistent state whereby cells maintain and expand in the presence of subsequent therapies [13,20,21]. Acquired resistance can simply be thought of as microevolution: any survival advantage, whether geographic or molecular, will be clonally selected [22,23]. In terms of bacteria, this has been seen since the invention of antibiotics. In cancer cells, the overall story is similar but more complicated. One current hypothesis for chemoresistance is that a small percentage of a tumor consists of cancer stem cells or tumor initiating cells that are capable of self-renewal and recreating the full repertoire of cancer cells of the parental tumor as well as the expression of a distinctive set of surface biomarkers [13]. Terminology can vary here, as some would characterize this process as intrinsic resistance being present in a small population of cells. The presence of these cells creates unique challenges in addressing intrinsic and acquired resistance in chemoresistant tumors. For the purpose of this review, we will consider intrinsic resistance as being present when a patient progresses or incompletely responds to treatment, and acquired resistance when a patient achieves remission and later has a recurrence with resistant disease.
3. Intrinsic Versus Acquired Resistance
The inherent ability of cells to survive chemotherapy can be mediated through several distinct mechanisms. Drug efflux pumps (adenosine triphosphate (ATP) binding cassette (ABC) transporters) lower drug concentrations within cells (reviewed in [24] with follow-up [25]), and therapeutic agents can be biochemically degraded through detoxifying enzymes such as cytochrome p450 and glutathione transferases (reviewed in [26,27]). Poor vascularization can lead to decreased drug concentration at the tumor site (reviewed in [28,29]), and extracellular matrix (ECM) interactions and secreted factors can influence tumor environment mediated resistance (reviewed in [30]). These intrinsic mechanisms of resistance are critical in determining initial response to therapies and may influence subsequent outcomes that lead to acquired resistance (Figure 1).
Acquired resistance is developed by the step-wise, molecular evolution of tumor cells through natural selection of changes giving a survival advantage [31]. This can include modulation of the expression of genes that drive increased anti-apoptotic signaling (i.e., X-linked inhibitor of apoptosis protein/cellular inhibitor of apoptosis protein (xIAP/cIAP), B-cell lymphoma 2 (BCL-2), B-cell lymphoma-extra-large (BCL-XL), and myeloid cell leukemia 1 (MCL-1)), DNA repair capacity, and the tolerability of genetic damage. Other alterations can be decreased sensitivity to DNA damage checkpoint signaling, or changes to ECM-collagen VI surface proteins, and the genes responsible for interacting with the ECM and stromal cells surrounding the tumor [4,11,15,32,33,34,35]. Cells can also acquire changes to drug targets that are mediated through multiple mechanisms [36,37], or in the case of some targeted therapies, by simply bypassing the target through an alternate pathway [38,39]. The simplest example of this is seen in androgen therapy for prostate cancer. After androgen deprivation therapy, the cells amplify the level of androgen receptor to the point that the levels are too high to stoichiometrically defeat [40]. Thus, acquired resistance can dramatically alter tumor cells to a point that they can be resistant to one or several types of therapy, but may also become uniquely sensitive to other therapies that have no effect on treatment-naïve cancer cells.
Many cancers, particularly HGSOC, are characterized by marked intratumoral heterogeneity. Overall, cancer is a disease of heterogeneity. The inherent genetic instability that gave rise to the tumor cells gives each the ability to quickly respond to changes in its local molecular microenvironment. This gives tumors the ability to use multiple combinations of these adaptive intrinsic and acquired mechanisms of resistance to defeat most modern chemotherapies and targeted therapeutics. For HGSOC, studies have focused on drug efflux and enhanced DNA damage repair capacity as being the critical, and druggable, mediators of resistance.
4. Efflux Pumps and MDR1
The ability of cancer cells to lower the intracellular concentration of drugs is well documented and the ATP binding cassette (ABC) transporters have been extensively studied in cancer chemoresistance. They are a diverse group of ATP-dependent efflux pumps containing 48 different family members with a common structural theme: a variable transmembrane domain and a conserved nucleotide binding domain [32,41]. In action, the transmembrane domain binds its target which induces a conformation change, removing the bound substrate from the cell using the hydrolysis of ATP for energy [24,25,42]. In ovarian cancer chemoresistance, the most commonly targeted of these transporters are the ABCB subfamily, with multidrug resistance protein 1 (MDR1, also known as P-glycoprotein (P-gp) and ABCB1) being the most exhaustively studied (Figure 2). The other ABC transporters appear to be limited in their ability to use paclitaxel as a substrate, with MDR1 being the primary P-gp responsible for reducing its intracellular concentrations [5,37,43].
MDR1 is responsible for the efflux of a truly staggering number of toxic compounds with diverse properties [44]. Several large studies have been undertaken and identified single nucleotide polymorphisms in MDR1 that are predictive of response to therapy, [45,46] with several promising drugs emerging to reduce their effects [42,47]. Over the last 15 years, intense research has explored strategies to clinically inhibit MDR1 with small molecules. Resistance to taxane therapy in ovarian cancer appears to occur through multiple mechanisms depending on the genetic background of the cells, but MDR1 efflux is thought to be the primary mechanism [20,39]. Eliminating MDR1-mediated paclitaxel/cisplatin efflux specifically has been shown to reverse resistance in the laboratory, but translating this promising outcome to the clinic has been incredibly challenging [48,49,50]. Drugs that inhibit MDR1 can also inhibit some cytochrome p450 isoenzymes, which are necessary to detoxify non-cancerous cells of the cytotoxic chemotherapies typically used. Targeting proteins responsible for protecting cells from toxic molecules is problematic, and combining MDR1 inhibition with cytotoxic chemotherapy can cause unpredictable and dangerous side effects [51,52].
First generation small molecule inhibitors of MDR pumps have had limited success due to these unforeseen consequences and overall toxicity problems, with clinical trials being marred by poor tolerability [53,54,55]. Second generation inhibitors with minimal cytochrome p450 inhibition showed promise but generally targeted multiple ABC transporters, resulting in negative effects in a diverse set of tissues. Using this information, combined with advances in structural chemistry and drug design, third generation P-gp inhibitors appear to be more specific and better tolerated, with several currently being clinically investigated [56,57]. Apatinib has been found to reverse paclitaxel resistance in both in vitro and in vivo patient-derived xenograft (PDX) model systems [58]. These third generation agents, as well as Tariquidar, have shown significant improvement in potency, specificity and tolerability with clinical trials ongoing [59]. Ovarian cancer groups are also investigating these latest agents using novel encapsulation methods to deliver them preferentially to tumor cells, with some success [60]. Overall pharmacologic inhibition of the efflux pumps remains challenging due to limits in balancing toxicity risks against therapeutic gains.
Targeting the MDR1 mRNA for degradation with small interfering RNA/antisense oligonucleotide (siRNA/ASO) is also being investigated as a method to ensure target specificity, and combining this with nanoliposomes that target cancer cells has been attempted in an effort to limit toxicity [47,54]. Yang et al. published findings on self-assembling nanoparticles that target cluster of differentiation 44 (CD44) to deliver MDR1 siRNA specifically to cancer cells in a mouse PDX model of ovarian cancer. When used in combination with paclitaxel, the siRNA delivery was specific to the cancer cells as increased sensitivity to paclitaxel was demonstrated [61]. While these modifications in drug delivery and specificity may alleviate some toxicity hurdles observed in the treatment of chemoresistant disease, it is also important to consider therapies that do not directly overlap with primary mechanisms of chemoresistance.
The development of novel tubule stabilizers that are not substrates for MDR1 efflux, such as the epothilone family, has shown efficacy in cells and PDX models previously resistant to paclitaxel [62,63,64,65]. Ixabepilone has been shown to have inhibitory concentrations of 50% (IC50s) an order of magnitude lower than typical concentrations seen with paclitaxel, and also has less neuropathies and other side effects in patients [62]. Positive clinical outcomes have been seen using Ixabepilone in recurrent ovarian cancer in combination with bevacizumab, and studies are ongoing to determine optimum patient enrollment criteria [66]. These results may highlight the utility of combination therapies that are not directly targeting major mechanisms of chemoresistance in ovarian cancer [35].
While some success has been observed in targeting MDR1 to overcome resistance, any real clinical gain has remained elusive. A recent whole genome sequencing study of a cohort of chemoresistant ovarian cancer has shown MDR1 to be rearranged in some chemoresistant populations, with promoter fusion driving upregulation. The promiscuous nature of the ABC transporters combined with the diverse side effects and toxicity problems has prompted some investigators to call for a change in tactics. Moving upstream of MDR1 to the epithelial–mesenchymal transition (EMT) pathway ties together multiple mechanisms associated with the acquisition of resistance, and EMT has a special position in ovarian cancer progression.
5. Epithelial–Mesenchymal Transition
EMT is the process of epithelial cells losing their apical-basal polarization, detaching through loss of adhesion, and becoming mesenchymal-like in both appearance and invasiveness [67,68]. There are myriad genetic changes involved in the process with cells typically losing expression of the adhesion protein E-cadherin, along with several of the tight junction proteins [67,69]. During EMT, cells begin expressing mesenchymal markers like vimentin and N-cadherin, as well as markers associated with stem cell populations such as aldehyde dehydrogenase-1 (ALDH1) and endothelin-A [13,70,71]. These changes are driven by the transcription factors Snail, Slug, Zeb1 and Twist, and many of the key pathways involved in ovarian cancer regulate their expression (Figure 3) [72,73,74,75]. EMT is seen after chemotherapy in surviving ovarian cancer cells and it’s been shown that Snail and Slug EMT transcription factors are upregulated [68,74,76,77]. Thus, factors altered in EMT are frequently overexpressed in ovarian cancer and are likely contributory to chemoresistance.
Chemoresistant phenotypic changes in ovarian cancer appear strongly correlated with EMT and the presence of subpopulations of cancer stem cells [10,68,71,74,78]. The pro-survival, anti-apoptotic signaling, efflux pump overexpression, and resistance to DNA damage seen in chemoresistant populations can all come from sustained EMT [69,75,79]. One relatively unique property of ovarian cancer is its fondness for metastasizing to the omentum, and interactions with this fatty compartment may play a critical role in the development of chemoresistance [3,80]. HGSOC does not require a hematologic route of metastasis as the entire peritoneal cavity is accessible from its site of origin, and HGSOC appears to readily shed and establish colonies throughout the chest cavity [80]. This shedding has been shown to involve EMT and the formation of tumor spheroids, which are protected by cancer-associated fibroblasts (CAFs) and hijacked immune system effector cells [3]. Ovarian cancer has been shown to be capable of responding to chemotherapy, with an acquired EMT phenotype giving rise to chemoresistance [67,81]. The endothelin receptor has been identified as a major source of EMT changes in response to chemotherapy, and blocking it has shown some reversal of chemoresistance [68]. Zibotentan is an antagonist of endothelin receptor A (ETA) [82]. After failing initial phase III trials, this drug is still being investigated, and its utility in chemoresistant disease in ovarian cancer is still promising. Targeting EMT in ovarian cancer may both stop the acquisition of chemoresistance and inhibit tumor progression and metastasis; however, cells that continue to evade death may have greater abilities to endure in the face of severe DNA damage.
6. DNA Damage Tolerance and Repair Capacity
DNA damage response pathways are very well characterized and generally signal through checkpoint kinase 1 and checkpoint kinase 2 (CHK1/CHK2) to determine the amount of damage present, amplify the repair signal to enhance repair efficiency, and delay the cell cycle to allow time for repair [83]. Damage is detected by the MRE11/RAD50/NBS1 (MRN complex), which signals through the phosphotidylinositol 3-kinase (PI3) family members ataxia telangiectasia mutated (ATM), Rad-3-related (ATR) and DNA-dependent protein kinase (DNA-pk) by phosphorylation of H2A histone family member X (H2AX). This in turn amplifies the repair signal, and enhances the buildup of DNA repair pathway members at the site of damage [84,85]. In order to allow sufficient time for repair, the cell cycle must be halted. The Gap2-Mitosis (G2-M) boundary is maintained through phosphorylation of cell-division cycle 2 (CDC2) on threonine 14 and tyrosine 15 by WEE1-like (WEE1) and myelin transcription factor 1 (MYT1) kinases, which sequester the CDC2/cyclin B complex in the cytoplasm [86,87,88,89]. For mitosis to proceed, CDC2 must be activated by cell-division cycle 25 (CDC25) dephosphorylation and translocation to the nucleus [90,91]. The delay is induced by ATM and ATR activation of the CHK2/CHK1 kinases, respectively. CHK2/CHK1 phosphorylate CDC25B, inhibiting its phosphatase activity on CDC2 and leaving the inactive complex in the cytoplasm [83]. Thus, the cell cycle is tightly regulated by several mechanisms, and sensitivity or resistance to treatments may involve alterations in these processes that promote imminent death or allow for recovery and repair.
Platinums are the drug of choice for treating many aggressive cancers, and most HGSOC are quite sensitive to its toxicity [92]. The mechanisms of platinum toxicity have yet to be fully elucidated, but it’s thought to act as a severe DNA damaging agent, creating inter- and intra-strand cross links from the formation of platinum:DNA adducts [93]. Ovarian cancers presenting with some form of homologous recombination (HR) deficiency generally respond better to DNA damaging therapies like cisplatin, and also may benefit from poly(ADP-ribose) polymerase-I (PARP I) inhibitor therapy [94,95]. PARP inhibitors exploit the broken DNA repair pathway to act as a synthetic lethal [96]. The deficient repair system also increases the efficacy of DNA damaging agents, depending on the specific gene mutation, and improves overall survival. Ovarian cancers where there are no HR deficiencies are less well-defined and may represent an early step in tumor initiation or chemoresistance [95].
TP53 mutant cells are thought to have a deficient G1 arrest ability with an enhanced G2 delay, allowing them to cope with genetic damage [97,98]. Cancer cells over time can become resistant to platinum-induced DNA damage and apoptosis through G2 delay and enhanced repair (Figure 4) [99,100]. The HGSOC combination of TP53 loss and HR repair deficiency has led many groups to ask the question: can we force cells with severe DNA damage through the cell cycle by attacking the delay mechanisms, and lead cells to death by apoptosis or mitotic catastrophe? CDC2 phosphorylation by Wee1 kinase is an attractive drug target for this question with Wee1 kinase specific inhibitors, such as MK-1775 (low nanomolar IC50), showing promising results [101,102]. PD0166285 is a newer Wee1 inhibitor and G2 checkpoint abrogator [103,104]. With HGSOC being almost entirely TP53 mutant, removing the G2 arrest has been shown to reverse platinum resistance, and targeting this may help alleviate chemoresistance in some subpopulations of ovarian cancer [105]. Other mechanisms of chemoresistance that may be required for the upregulation of any and all processes related to DNA damage repair, EMT, drug efflux, and the capacity of the cell to utilize intrinsic mechanisms of resistance, and develop acquired mechanisms of resistance, are currently under investigation as potential targeted therapies for HGSOC.
7. Angiogenesis Inhibitors
Angiogenesis has long been a hallmark of most malignancies and it is well characterized in HGSOC. Anti-angiogenic therapies for HGSOC have been FDA approved for use in the recurrent setting, and while showing an increase in progression free survival (PFS) there has been no change to overall survival (OS) [106,107]. Targeting vascular endothelial growth factor (VEGF) and VEGF receptor (VEGFR) in HGSOC showed only transient benefit with resistant disease occurring in many cases (reviewed in [108,109]). Alternative pathways were identified that cancer cells could use to bypass the VEGF blockade with angiopoietins being shown to play a critical role in mediating neovascularization and response to antiangiogenic therapies [110,111].
Recent phase III clinical trials in HGSOC have shown a significant increase in PFS with the same lack of OS increase in platinum resistant HGSOC with both bevacizumab and trebananib [112,113]. While the early excitement of angiogenesis inhibitors has diminished, the increase in PFS is not an insignificant endpoint in HGSOC. The continued investigation of combinations of angiogenesis inhibitors and identification of biomarkers for better screening criteria may still show clinical benefit.
8. PARP Inhibitors
Using poly(ADP-ribose) polymerase (PARP) inhibitors for treating breast cancer 1 (BRCA) or HR deficient HGSOC is a highly active area of research with several FDA-approved therapies in the recurrent and maintenance setting [114]. PARP is an enzyme responsible for the correct repair of DNA damage, and it does this through directly interacting with the repair machinery on the damaged site. PARP has a strong affinity for single strand DNA breaks and autoactivates, forming chains of PAR attached to the DNA adjacent to the break. These polymers act as a scaffolding system to recruit and assemble the various repair system effector molecules [115]. If PARP is functionally inhibited these single strand DNA breaks with attached PARP polymers will evolve into double strand breaks (DSBs) due to replication fork collision [116]. In cancer cells with deficient HR repair systems these DSB’s will build up over time whereas normal cells have the capacity to repair them [117]. By using small molecules to ‘trap’ PARP on the DNA, cancers deficient in DNA repair will be preferentially targeted in a synthetic lethal fashion as a result of being unable to clear the polymer chains [118,119,120].
Resistance to PARP inhibitors develops rapidly in some patients and occurs in surprising ways (reviewed in [121]). Reversion of mutated DNA repair genes back to functionally normal, full length forms being one method commonly seen [122,123,124]. Other mechanisms alter the complex balance between non-homologous end joining and HR repair through microRNAs (miRNAs) [125], epigenetics [126] or nascent, truncated BRCA mutants [127,128]. Upregulation of the MDR1 efflux pumps has also been shown to induce resistance to PARP inhibitors [129] with drugs identified that can specifically reverse this [130].
9. Exosomes and Cancer
Exosomes are extracellular vesicles <100 nm in diameter that were initially thought of as cellular waste disposal units [131]. Research has shown a diverse array of functions for exosomes (reviewed in [132]) and they generally act as a form of an intracellular communication system [133]. Cancer cells are generally seen as releasing more exosomes than normal cells and evidence is building that exosomes play a crucial role in cancer progression, metastasis, and chemoresistance [134,135,136,137].
Exosomes have been found to play a role in resistance to chemotherapy [138], and in HGSOC have been shown to transmit platinum resistance to sensitive cells through intracellular communication of EMT signals [139]. Cao et al. showed exosomes mediating platinum resistance through DNA methyltransferase I (DNMT1) [140]. DNMT1 is a regulator of a large array of developmental processes and plays a critical role in regulating DNA damage response and repair [141]. If exosomes are capable of controlling such critical aspects of cellular responses to chemotherapy, then exosomal inhibitors may be used as a targeted therapeutic [134,135].
The idea that exosomes are also capable of sending out tumor specific signals begs the question: can we use exosomes as a cancer specific targeting system for drug delivery? Evidence is building for this being a promising way to deliver toxic therapies directly to the tumor [142] with Hadla et al. illustrating their use in ovarian cancer [143].
10. Checkpoint Inhibitors
The most commonly-used therapeutic strategy in HGSOC today is combined platinum/taxane treatment after surgery. Several other options are becoming more clinically viable. The discovery of immune system checkpoint inhibition, like programmed cell death-1 (PD-1), has revolutionized the treatment of several types of cancers and is being actively explored in HGSOC [144,145].
The immune system has a dedicated surveillance network for identifying and destroying cells displaying neoplastic changes, neoantigens or other signals of dysfunction. Tumors, as they form, generally set off various checkpoint alarms and inflammatory processes that signal to the immune effector cells. For any growing lesion to form a viable tumor it has to evade immune system detection. PD-1 is one of a host of cell surface receptors used to identify cells to the immune system as “self” to stop normal tissues from being attacked. Many types of tumors have used this as a mechanism to hide from immune system detection, and blocking this escape tactic has shown efficacy in a diverse set of cancers. In HGSOC, trials have shown positive results using both anti-PD-1 antibody therapies as well as antibodies directed at programmed death-ligand 1 (PD-L1), its normal ligand [146,147]. HGSOC has been identified as having high expression of the PD-1 receptor and PD-1 inhibitors such as pembrolizumab and nivolumab are in FDA trials [146]. Cytotoxic T-lymphocyte associated protein 4 (CTLA-4) is another checkpoint inhibitor that is in trials and ipilumab is currently under investigation in HGSOC [148].
Resistance to checkpoint inhibitors is both intrinsic and acquired, with intense research underway determining sensitivities in many tumor types. The recognition system appears to have multiple compensatory factors interacting, and blockade of a single receptor has been shown to compensate with upregulation of parallel pathways [145]. Several examples have been identified of tumors making dramatic changes to their cell surface markers to evade the immune system entirely. Deletion of major histocompatibility complex (MHC) class II has been seen and presents a difficult resistance mechanism to overcome [149]. However, as broad use of checkpoint inhibitors is relatively recent, more information on resistance mechanisms is anticipated.
11. Ribosomal Biogenesis
One of the first identified hallmarks of cancer, dating back to the late 1800’s, was enlarged and/or pronounced nucleoli [150]. The size of the nucleoli is generally thought to reflect the amount of ribosomal biogenesis happening in the nucleolus, and proliferating cells typically require a large amount of ribosomes to progress through the cell cycle [151]. Ribosomes are critical molecular machines common to all eukaryotic cells that allow the conversion of RNA messages to nascent proteins. Ribosomal synthesis and processing is one of the most energetically demanding activities cells undergo, and consequently their synthesis is also one of the most regulated systems in eukaryotic life [152]. Altering the rate of ribosomal biogenesis can lead to TP53 stabilization, representing a phenotype known as nucleolar stress [153,154,155]. Given that ribosomal biogenesis is central to life and appears to play a prominent role in cancer, it’s important to consider if and how this system can be targeted in cancer cells specifically.
Recently the idea of targeting ribosomal biogenesis has come forward as a viable strategy for treating various forms of cancer [156,157] and specifically for HGSOC [158]. Ribosomal synthesis begins with the formation of the RNA polymerase I (RNA Pol I) pre-initiation complex, followed by transcription of the 47 s pre-ribosomal RNA. The pre-initiation complex is set up first by binding of the high mobility group protein upstream binding transcription factor (UBTF) within the ribosomal DNA (rDNA) promoter region, ejecting the H1 histone. This interaction is then stabilized by the binding of the selectivity complex selective factor 1 (SL1), which commits the promoter for transcription by multiple rounds of RNA polymerase I. After transcription, a complex series of modifications spanning several subcellular compartments leads to the assembly of functional ribosomes [159,160]. Although there are several key players in ribosomal biogenesis, there may be mechanisms that are specific and can be targeted in cancer cells without negatively impacting the majority of normal cells.
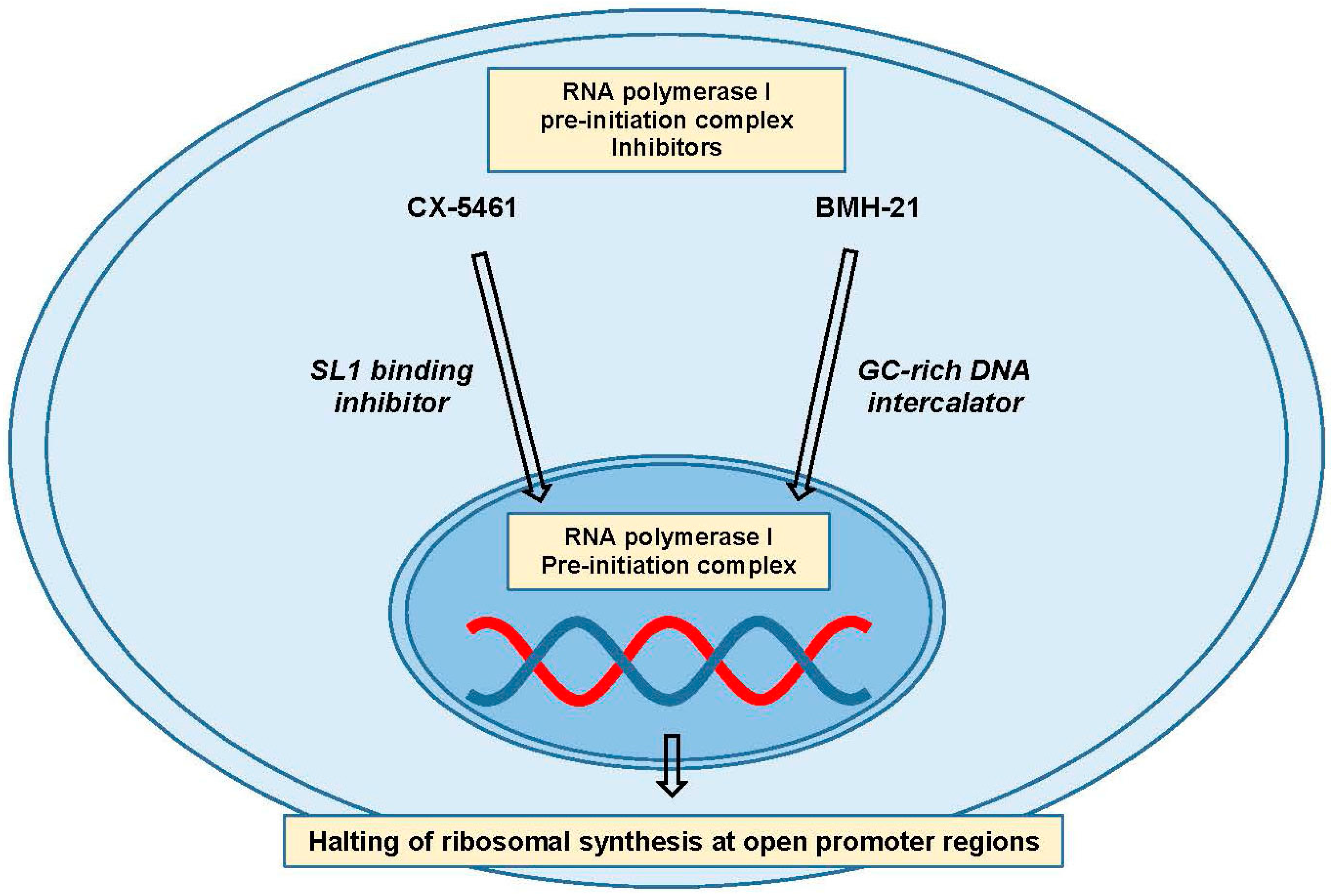
RNA polymerase I is only responsible for the transcription of the pre-ribosomal RNA, so targeting it with small molecule inhibitors appears to be the most appealing strategy for specifically shutting down ribosomal synthesis [156,161]. Two specific pol I inhibitors, CX-5461 and BMH-21, have been developed, and have slightly different substrates (Figure 5). CX-5461 is thought to inhibit the binding of SL1 to the rDNA promoter, leading to pre-initiation complex failure and possibly an abnormal chromatin structure picked up as a form of damage [162,163,164]. BMH-21, on the other hand, is a GC-rich DNA intercalating agent that inhibits formation of the pre-initiation complex leading to RNA polymerase I subunit A (RPA194) degradation, the major subunit of the RNA pol I holoenzyme [165,166]. These inhibitors may begin to shed light on the possibility and efficacy of targeting ribosomal biogenesis in cancer, and differences in their activity may shed light on the most crucial aspects of RNA Pol I inhibition.
12. Summary
In summary, while the understanding of mechanisms underlying intrinsic and acquired chemoresistance in ovarian cancer is expanding rapidly, overall therapeutic options remain limited. Promising results generated in vitro showing the therapeutic potential of targeting MDR1 have fallen short in practical application in the clinic due to toxicity. Alterations in EMT targets have had minimal success given there are several mechanisms remaining active that allow the tumor to adapt. As well, agents that try to exploit high levels of DNA damage may only be clinically applicable in contexts where tumors are also HR deficient.
The pitfalls and shortcomings of translating these novel targets and associated drugs to the clinic further highlight the essential need for approaches to treatment that go beyond targeting single mechanisms. Mechanisms targeted previously only represent a handful of players in a complex and heterogeneous system that comprises chemoresistance in ovarian cancer. With all the new information on mechanisms of resistance to cytotoxic agents, specific resistance mechanisms to emerging targeted therapies and whole genome characterization of HGSOC personalized medicine may be on the horizon, and required for durable cures. Molecular characterization of the tumor followed by prediction of optimal therapeutic combinations with inhibitors blocking the expected paths of resistance may be the only way to achieve an increase in overall cure rates. The novel approach of targeting ribosomal biogenesis may prove more useful than previous approaches in improving outcomes for a larger proportion of patients with chemoresistant ovarian cancer in the future; however, previous research provides hope that it is feasible to identify the most important mediators of chemoresistance, while exploring the therapeutic potentials of current novel targets, so a broader population of patients with chemoresistant ovarian cancer will experience positive outcomes. As the understanding of resistance with tumor heterogeneity is enhanced, it is hopeful that targeting the specific chemoresistant population with upfront or consolidation therapy will lead to absolute cures, rather than just modest responses to therapy.
Acknowledgments
Funding support provided in part by the Norma Livingston Ovarian Cancer Foundation, the Research Scientist Development Program (K12 HD00849), the Department of Defense Ovarian Cancer Research Academy (OC093443), and UVA Cancer Center Support Grant (P30 CA44579).
Author Contributions
All authors contributed to the composition and writing of this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bowtell, D.D.; Böhm, S.; Ahmed, A.A.; Aspuria, P.-J.; Bast, R.C.; Beral, V.; Berek, J.S.; Birrer, M.J.; Blagden, S.; Bookman, M.A.; et al. Rethinking ovarian cancer II: Reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 2015, 15, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Bast, R.C.; Hennessy, B.; Mills, G.B.; Mills, G.B. The biology of ovarian cancer: New opportunities for translation. Nat. Rev. Cancer 2009, 9, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E. Ovarian cancer development and metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Harries, M.; Kaye, S.B. Recent advances in the treatment of epithelial ovarian cancer. Expert Opin. Investig. Drugs 2001, 10, 1715–1724. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Kaye, S.B. Ovarian cancer: Strategies for overcoming resistance to chemotherapy. Nat. Rev. Cancer 2003, 3, 502–516. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-Y.; Zhang, P.-Y. Recent perspectives of epithelial ovarian carcinoma (Review). Oncol. Lett. 2016, 12, 3055–3058. [Google Scholar] [CrossRef] [PubMed]
- Erickson, B.K.; Conner, M.G.; Landen, C.N. The role of the fallopian tube in the origin of ovarian cancer. Am. J. Obstet. Gynecol. 2013, 209, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef] [PubMed]
- Cannistra, S.A. Cancer of the Ovary. N. Engl. J. Med. 2004, 35124351, 2519–2529. [Google Scholar] [CrossRef] [PubMed]
- Abubaker, K.; Latifi, A.; Luwor, R.; Nazaretian, S.; Zhu, H.; Quinn, M.A.; Thompson, E.W.; Findlay, J.K.; Ahmed, N. Short-term single treatment of chemotherapy results in the enrichment of ovarian cancer stem cell-like cells leading to an increased tumor burden. Mol. Cancer 2013, 12, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coward, J.I.; Middleton, K.; Murphy, F. New perspectives on targeted therapy in ovarian cancer. Int. J. Women’s Health 2015, 7, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Bagnoli, M.; Granata, A.; Nicoletti, R.; Krishnamachary, B.; Bhujwalla, Z.M.; Canese, R.; Podo, F.; Canevari, S.; Iorio, E.; Mezzanzanica, D. Choline Metabolism Alteration: A Focus on Ovarian Cancer. Front. Oncol. 2016, 6, 153. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, L.N.; Chow, E.K.-H. Mechanisms of chemoresistance in cancer stem cells. Clin. Transl. Med. 2013, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef]
- Liu, X.; Chan, D.; Ngan, H. Mechanisms of Chemoresistance in Human Ovarian Cancer at a Glance. Gynecol. Obstet. 2012, 2, 1000e104. [Google Scholar] [CrossRef]
- Krzystyniak, J.; Ceppi, L.; Dizon, D.S.; Birrer, M.J. Epithelial ovarian cancer: The molecular genetics of epithelial ovarian cancer. Ann. Oncol. 2016, 27, i4–i10. [Google Scholar] [CrossRef] [PubMed]
- Dobbin, Z.C.; Katre, A.A.; Steg, A.D.; Erickson, B.K.; Shah, M.M.; Alvarez, R.D.; Conner, M.G.; Schneider, D.; Chen, D.; Landen, C.N. Using heterogeneity of the patient-derived xenograft model to identify the chemoresistant population in ovarian cancer. Oncotarget 2014, 5, 8750–8764. [Google Scholar] [CrossRef] [PubMed]
- Frei, E.; Karon, M.; Levin, R.H.; Freireich, E.J.; Taylor, R.J.; Hananian, J.; Selawry, O.; Holland, J.F.; Hoogstraten, B.; Wolman, I.J.; et al. The effectiveness of combinations of antileukemic agents in inducing and maintaining remission in children with acute leukemia. Blood 1965, 26, 642–656. [Google Scholar] [PubMed]
- Zahreddine, H.; Borden, K.L.B. Mechanisms and insights into drug resistance in cancer. Front. Pharmacol. 2013, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- Ling, K.-S.; Chen, G.-D.; Tsai, H.-J.; Lee, M.-S.; Wang, P.-H.; Liu, F.-S. Mechanisms Involved in Chemoresistance in Ovarian Cancer. Taiwan J. Obstet. Gynecol. 2005, 44, 209–217. [Google Scholar] [CrossRef]
- Tapia, G.; Diaz-padilla, I. Molecular Mechanisms of Platinum Resistance in Ovarian Cancer. Ovarian Cancer A Clin. Transl. Update 2013, 371. [Google Scholar] [CrossRef]
- Cooke, S.L.; Brenton, J.D. Evolution of platinum resistance in high-grade serous ovarian cancer. Lancet Oncol. 2011, 12, 1169–1174. [Google Scholar] [CrossRef]
- Salomon-Perzyński, A.; Salomon-Perzyńska, M.; Michalski, B.; Skrzypulec-Plinta, V. High-grade serous ovarian cancer: The clone wars. Arch. Gynecol. Obstet. 2017, 295, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Pastan, I.H. The Role of Multidrug Resistance Efflux Pumps in Cancer: Revisiting a JNCI Publication Exploring Expression of the MDR1 (P-glycoprotein) Gene. J. Natl. Cancer Inst. 2015, 107, djv222. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Antona, C.; Ingelman-Sundberg, M. Cytochrome P450 pharmacogenetics and cancer. Oncogene 2006, 25, 1679–1691. [Google Scholar] [CrossRef] [PubMed]
- Sueyoshi, T.; Negishi, M. Phenobarbital response elements of cytochrome P450 genes and nuclear receptors. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 123–143. [Google Scholar] [CrossRef] [PubMed]
- Saggar, J.K.; Yu, M.; Tan, Q.; Tannock, I.F. The tumor microenvironment and strategies to improve drug distribution. Front. Oncol. 2013, 3, 154. [Google Scholar] [CrossRef] [PubMed]
- Minchinton, A.I.; Tannock, I.F. Drug penetration in solid tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Lu, A.Y. Interindividual variability in inhibition and induction of cytochrome P450 enzymes. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 535. [Google Scholar] [CrossRef] [PubMed]
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Carden, C.P.; Kaye, S.B. Beyond chemotherapy: Targeted therapies in ovarian cancer. Nat. Rev. Cancer 2009, 9, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Hu, D.; Qiu, J.; Xie, X.; Ye, F.; Lu, W.-G. Overexpression of glycogen synthase kinase-3 in ovarian carcinoma cells with acquired paclitaxel resistance. Int. J. Gynecol. Cancer 2011, 21, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Patch, A.-M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Groenendijk, F.H.; Bernards, R. Drug resistance to targeted therapies: Déjà vu all over again. Mol. Oncol. 2014, 8, 1067–1083. [Google Scholar] [CrossRef] [PubMed]
- Orr, G.A.; Verdier-Pinard, P.; McDaid, H.; Horwitz, S.B. Mechanisms of Taxol resistance related to microtubules. Oncogene 2003, 22, 7280–7295. [Google Scholar] [CrossRef] [PubMed]
- Cree, I.A.; Charlton, P. Molecular chess? Hallmarks of anti-cancer drug resistance. BMC Cancer 2017, 17, 10. [Google Scholar] [CrossRef] [PubMed]
- Kavallaris, M.; Kuo, D.Y.; Burkhart, C.A.; Regl, D.L.; Norris, M.D.; Haber, M.; Horwitz, S.B. Taxol-resistant epithelial ovarian tumors are associated with altered expression of specific beta-tubulin isotypes. J. Clin. Investig. 1997, 100, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.D.; Welsbie, D.S.; Tran, C.; Baek, S.H.; Chen, R.; Vessella, R.; Rosenfeld, M.G.; Sawyers, C.L. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 2004, 10, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Lee, K. Targeting multidrug resistance with small molecules for cancer therapy. Biomol. Ther. 2010, 18, 375–385. [Google Scholar] [CrossRef]
- Vaidyanathan, A.; Sawers, L.; Gannon, A.-L.; Chakravarty, P.; Scott, A.L.; Bray, S.E.; Ferguson, M.J.; Smith, G. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br. J. Cancer 2016, 115, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Hedditch, E.L.; Gao, B.; Russell, A.J.; Lu, Y.; Emmanuel, C.; Beesley, J.; Johnatty, S.E.; Chen, X.; Harnett, P.; George, J.; et al. ABCA transporter gene expression and poor outcome in epithelial ovarian cancer. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.H. P-glycoprotein, a gatekeeper in the blood-brain barrier. Adv. Drug Deliv. Rev. 1999, 36, 179–194. [Google Scholar] [CrossRef]
- Johnatty, S.E.; Beesley, J.; Paul, J.; Fereday, S.; Spurdle, A.B.; Webb, P.M.; Byth, K.; Marsh, S.; McLeod, H.; Harnett, P.R.; et al. ABCB1 (MDR 1) Polymorphisms and Progression-Free Survival among Women with Ovarian Cancer following Paclitaxel/Carboplatin Chemotherapy. Clin. Cancer Res. 2008, 14, 5594–5601. [Google Scholar] [CrossRef] [PubMed]
- Johnatty, S.E.; Beesley, J.; Gao, B.; Chen, X.; Lu, Y.; Law, M.H.; Henderson, M.J.; Russell, A.J.; Hedditch, E.L.; Emmanuel, C.; et al. ABCB1 (MDR1) polymorphisms and ovarian cancer progression and survival: A comprehensive analysis from the Ovarian Cancer Association Consortium and The Cancer Genome Atlas. Gynecol. Oncol. 2013, 131, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Iyer, A.K.; Singh, A.; Choy, E.; Hornicek, F.J.; Amiji, M.M.; Duan, Z. MDR1 siRNA loaded hyaluronic acid-based CD44 targeted nanoparticle systems circumvent paclitaxel resistance in ovarian cancer. Sci. Rep. 2015, 5, 8509. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Li, X.; Meng, X.-N.; Yan, J.; Zong, Z. MicroRNA-873 mediates multidrug resistance in ovarian cancer cells by targeting ABCB1. Tumor Biol. 2016, 37, 10499–10506. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, S.; Meng, X.; Shang, H.; Guan, Y. Inhibition of mdr1 by G-quadruplex oligonucleotides and reversal of paclitaxel resistance in human ovarian cancer cells. Tumor Biol. 2015, 36, 6433–6443. [Google Scholar] [CrossRef] [PubMed]
- Planting, A.S.T.; Sonneveld, P.; van der Gaast, A.; Sparreboom, A.; van der Burg, M.E.L.; Luyten, G.P.M.; de Leeuw, K.; de Boer-Dennert, M.; Wissel, P.S.; Jewell, R.C.; et al. A phase I and pharmacologic study of the MDR converter GF120918 in combination with doxorubicin in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2005, 55, 91–99. [Google Scholar] [CrossRef] [PubMed]
- El-Awady, R.; Saleh, E.; Hashim, A.; Soliman, N.; Dallah, A.; Elrasheed, A.; Elakraa, G. The Role of Eukaryotic and Prokaryotic ABC Transporter Family in Failure of Chemotherapy. Front. Pharmacol. 2016, 7, 535. [Google Scholar] [CrossRef] [PubMed]
- Wandel, C.; Kim, R.B.; Kajiji, S.; Guengerich, P.; Wilkinson, G.R.; Wood, A.J. P-glycoprotein and cytochrome P-450 3A inhibition: Dissociation of inhibitory potencies. Cancer Res. 1999, 59, 3944–3948. [Google Scholar] [PubMed]
- Seiden, M.V.; Swenerton, K.D.; Matulonis, U.; Campos, S.; Rose, P.; Batist, G.; Ette, E.; Garg, V.; Fuller, A.; Harding, M.W.; et al. A phase II study of the MDR inhibitor biricodar (INCEL, VX-710) and paclitaxel in women with advanced ovarian cancer refractory to paclitaxel therapy. Gynecol. Oncol. 2002, 86, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Kapse-Mistry, S.; Govender, T.; Srivastava, R.; Yergeri, M. Nanodrug delivery in reversing multidrug resistance in cancer cells. Front. Pharmacol. 2014, 5, 159. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Ohnuma, S.; Ambudkar, S.V. Improving cancer chemotherapy with modulators of ABC drug transporters. Curr. Drug Targets 2011, 12, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Binkhathlan, Z.; Lavasanifar, A. P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: Current status and future perspectives. Curr. Cancer Drug Targets 2013, 13, 326–346. [Google Scholar] [CrossRef] [PubMed]
- Weidner, L.D.; Fung, K.L.; Kannan, P.; Moen, J.K.; Kumar, J.S.; Mulder, J.; Innis, R.B.; Gottesman, M.M.; Hall, M.D. Tariquidar Is an Inhibitor and Not a Substrate of Human and Mouse P-glycoprotein. Drug Metab. Dispos. 2016, 44, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Mi, Y.-J.; Liang, Y.-J.; Huang, H.-B.; Zhao, H.-Y.; Wu, C.-P.; Wang, F.; Tao, L.-Y.; Zhang, C.-Z.; Dai, C.-L.; Tiwari, A.K.; et al. Apatinib (YN968D1) reverses multidrug resistance by inhibiting the efflux function of multiple ATP-binding cassette transporters. Cancer Res. 2010, 70, 7981–7991. [Google Scholar] [CrossRef] [PubMed]
- Chung, F.S.; Santiago, J.S.; De Jesus, M.F.M.; Trinidad, C.V.; See, M.F.E. Disrupting P-glycoprotein function in clinical settings: What can we learn from the fundamental aspects of this transporter? Am. J. Cancer Res. 2016, 6, 1583–1598. [Google Scholar] [PubMed]
- Zhang, Y.; Sriraman, S.K.; Kenny, H.A.; Luther, E.; Torchilin, V.; Lengyel, E. Reversal of Chemoresistance in Ovarian Cancer by Co-Delivery of a P-glycoprotein Inhibitor and Paclitaxel in a Liposomal Platform. Mol. Cancer Ther. 2016, 15, 2282–2293. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Iyer, A.K.; Singh, A.; Milane, L.; Choy, E.; Hornicek, F.J.; Amiji, M.M.; Duan, Z. Cluster of Differentiation 44 Targeted Hyaluronic Acid Based Nanoparticles for MDR1 siRNA Delivery to Overcome Drug Resistance in Ovarian Cancer. Pharm. Res. 2015, 32, 2097–2109. [Google Scholar] [CrossRef] [PubMed]
- Rivera, E.; Gomez, H. Chemotherapy resistance in metastatic breast cancer: The evolving role of ixabepilone. Breast Cancer Res. 2010, 12 (Suppl. 2), S2. [Google Scholar] [CrossRef] [PubMed]
- Vishnu, P.; Colon-Otero, G.; Kennedy, G.T.; Marlow, L.A.; Kennedy, W.P.; Wu, K.J.; Santoso, J.T.; Copland, J.A. RhoB mediates antitumor synergy of combined ixabepilone and sunitinib in human ovarian serous cancer. Gynecol. Oncol. 2012, 124, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Padilla, I.; Oza, A.M. Epothilones in the treatment of ovarian cancer. Future Oncol. 2011, 7, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Zagouri, F.; Sergentanis, T.; Chrysikos, D.; Bamias, A.; Dimopoulos, A.-M. Epothilones in epithelial ovarian, fallopian tube, or primary peritoneal cancer: A systematic review. Oncol. Targets Ther. 2015, 8, 2187. [Google Scholar] [CrossRef] [PubMed]
- Roque, D.M.; Ratner, E.S.; Silasi, D.-A.; Azodi, M.; Rutherford, T.J.; Schwartz, P.E.; Nelson, W.K.; Santin, A.D. Weekly ixabepilone with or without biweekly bevacizumab in the treatment of recurrent or persistent uterine and ovarian/primary peritoneal/fallopian tube cancers: A retrospective review. Gynecol. Oncol. 2015, 137, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B.; Tropé, C.G.; Reich, R. Epithelial-Mesenchymal Transition in Ovarian Carcinoma. Front. Oncol. 2012, 2, 33. [Google Scholar] [CrossRef] [PubMed]
- Rosanò, L.; Cianfrocca, R.; Spinella, F.; Di Castro, V.; Nicotra, M.R.; Lucidi, A.; Ferrandina, G.; Natali, P.G.; Bagnato, A. Acquisition of chemoresistance and EMT phenotype is linked with activation of the endothelin A receptor pathway in ovarian carcinoma cells. Clin. Cancer Res. 2011, 17, 2350–2360. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Young, M.-J.; Wu, Y.-H.; Chiu, W.-T.; Weng, T.-Y.; Huang, Y.-F.; Chou, C.-Y. All-trans retinoic acid downregulates ALDH1-mediated stemness and inhibits tumour formation in ovarian cancer cells. Carcinogenesis 2015, 36, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Bagnato, A.; Rosanò, L. Understanding and overcoming chemoresistance in ovarian cancer: Emerging role of the endothelin axis. Curr. Oncol. 2012, 19, 36–38. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Kobayashi, S.; Yamada, D.; Nagano, H.; Tomokuni, A.; Tomimaru, Y.; Noda, T.; Gotoh, K.; Asaoka, T.; Wada, H.; et al. A Histone Deacetylase Inhibitor Suppresses Epithelial-Mesenchymal Transition and Attenuates Chemoresistance in Biliary Tract Cancer. PLoS ONE 2016, 11, e0145985. [Google Scholar] [CrossRef] [PubMed]
- Kajiyama, H.; Shibata, K.; Terauchi, M.; Yamashita, M.; Ino, K.; Nawa, A.; Kikkawa, F. Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int. J. Oncol. 2007, 31, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Haslehurst, A.M.; Koti, M.; Dharsee, M.; Nuin, P.; Evans, K.; Geraci, J.; Childs, T.; Chen, J.; Li, J.; Weberpals, J.; et al. EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer 2012, 12, 91. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Xu, Z.; El-Sehemy, A.; Steed, H.; Fu, Y. Notch3 induces epithelial-mesenchymal transition and attenuates carboplatin-induced apoptosis in ovarian cancer cells. Gynecol. Oncol. 2013, 130, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Latifi, A.; Abubaker, K.; Castrechini, N.; Ward, A.C.; Liongue, C.; Dobill, F.; Kumar, J.; Thompson, E.W.; Quinn, M.A.; Findlay, J.K.; et al. Cisplatin treatment of primary and metastatic epithelial ovarian carcinomas generates residual cells with mesenchymal stem cell-like profile. J. Cell. Biochem. 2011, 112, 2850–2864. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.P.; Dupre, T.V.; Siskind, L.J.; Beverly, L.J. Common cytotoxic chemotherapeutics induce epithelial-mesenchymal transition (EMT) downstream of ER stress. Oncotarget 2017, 8, 22625–22639. [Google Scholar] [CrossRef] [PubMed]
- Rosano, L.; Cianfrocca, R.; Tocci, P.; Spinella, F.; Di Castro, V.; Caprara, V.; Semprucci, E.; Ferrandina, G.; Natali, P.G.; Bagnato, A. Endothelin A Receptor/-Arrestin Signaling to the Wnt Pathway Renders Ovarian Cancer Cells Resistant to Chemotherapy. Cancer Res. 2014, 74, 7453–7464. [Google Scholar] [CrossRef] [PubMed]
- Miow, Q.H.; Tan, T.Z.; Ye, J.; Lau, J.A.; Yokomizo, T.; Thiery, J.; Mori, S. Epithelial-mesenchymal status renders differential responses to cisplatin in ovarian cancer. Oncogene 2015, 34, 1899–1907. [Google Scholar] [CrossRef] [PubMed]
- Pradeep, S.; Kim, S.; Wu, S.; Nishimura, M.; Chaluvally-Raghavan, P.; Miyake, T.; Pecot, C.; Kim, S.J.; Choi, H.; Bischoff, F.; et al. Hematogenous Metastasis of Ovarian Cancer: Rethinking Mode of Spread. Cancer Cell 2014, 26, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Marchini, S.; Fruscio, R.; Clivio, L.; Beltrame, L.; Porcu, L.; Nerini, I.F.; Cavalieri, D.; Chiorino, G.; Cattoretti, G.; Mangioni, C.; et al. Resistance to platinum-based chemotherapy is associated with epithelial to mesenchymal transition in epithelial ovarian cancer. Eur. J. Cancer 2013, 49, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Tomkinson, H.; Kemp, J.; Oliver, S.; Swaisland, H.; Taboada, M.; Morris, T. Pharmacokinetics and tolerability of zibotentan (ZD4054) in subjects with hepatic or renal impairment: Two open-label comparative studies. BMC Clin. Pharmacol. 2011, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Paull, T.T. ATM Activation by DNA Double-Strand Breaks through the Mre11-Rad50-Nbs1 Complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Fattaey, A.; Booher, R.N. Myt1: A Wee1-type kinase that phosphorylates Cdc2 on residue Thr14. Prog. Cell Cycle Res. 1997, 3, 233–240. [Google Scholar] [PubMed]
- Pines, J.; Hunter, T. The differential localization of human cyclins A and B is due to a cytoplasmic retention signal in cyclin B. EMBO J. 1994, 13, 3772–3781. [Google Scholar] [PubMed]
- McGowan, C.H.; Russell, P. Human Wee1 kinase inhibits cell division by phosphorylating p34cdc2 exclusively on Tyr15. EMBO J. 1993, 12, 75–85. [Google Scholar] [PubMed]
- Wells, N.J.; Watanabe, N.; Tokusumi, T.; Jiang, W.; Verdecia, M.A.; Hunter, T. The C-terminal domain of the Cdc2 inhibitory kinase Myt1 interacts with Cdc2 complexes and is required for inhibition of G(2)/M progression. J. Cell Sci. 1999, 112, 3361–3371. [Google Scholar] [PubMed]
- Atherton-Fessler, S.; Liu, F.; Gabrielli, B.; Lee, M.S.; Peng, C.Y.; Piwnica-Worms, H. Cell cycle regulation of the p34cdc2 inhibitory kinases. Mol. Biol. Cell 1994, 5, 989–1001. [Google Scholar] [CrossRef] [PubMed]
- Hunter, T. Protein kinases and phosphatases: The yin and yang of protein phosphorylation and signaling. Cell 1995, 80, 225–236. [Google Scholar] [CrossRef]
- Ho, G.Y.; Woodward, N.; Coward, J.I.G. Cisplatin versus carboplatin: Comparative review of therapeutic management in solid malignancies. Crit. Rev. Oncol. Hematol. 2016, 102, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Nojima, K.; Hochegger, H.; Saberi, A.; Fukushima, T.; Kikuchi, K.; Yoshimura, M.; Orelli, B.J.; Bishop, D.K.; Hirano, S.; Ohzeki, M.; et al. Multiple repair pathways mediate tolerance to chemotherapeutic cross-linking agents in vertebrate cells. Cancer Res. 2005, 65, 11704–11711. [Google Scholar] [CrossRef] [PubMed]
- Reinbolt, R.E.; Hays, J.L. The Role of PARP Inhibitors in the Treatment of Gynecologic Malignancies. Front. Oncol. 2013, 3, 237. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, A. A Synthetic Lethal Therapeutic Approach: Poly(ADP) Ribose Polymerase Inhibitors for the Treatment of Cancers Deficient in DNA Double-Strand Break Repair. J. Clin. Oncol. 2008, 26, 3785–3790. [Google Scholar] [CrossRef] [PubMed]
- Bache, M.; Pigorsch, S.; Dunst, J.; Würl, P.; Meye, A.; Bartel, F.; Schmidt, H.; Rath, F.W.; Taubert, H. Loss of G2/M arrest correlates with radiosensitization in two human sarcoma cell lines with mutant p53. Int. J. Cancer 2001, 96, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Matheson, C.J.; Backos, D.S.; Reigan, P. Targeting WEE1 Kinase in Cancer. Trends Pharmacol. Sci. 2016, 37, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Venezia, T.A.; Merchant, A.A.; Ramos, C.A.; Whitehouse, N.L.; Young, A.S.; Shaw, C.A.; Goodell, M.A. Molecular signatures of proliferation and quiescence in hematopoietic stem cells. PLoS Biol. 2004, 2, e301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvakumaran, M.; Pisarcik, D.A.; Bao, R.; Yeung, A.T.; Hamilton, T.C. Enhanced cisplatin cytotoxicity by disturbing the nucleotide excision repair pathway in ovarian cancer cell lines. Cancer Res. 2003, 63, 1311–1316. [Google Scholar] [PubMed]
- Leijen, S.; Beijnen, J.H.; Schellens, J.H.M. Abrogation of the G2 checkpoint by inhibition of Wee-1 kinase results in sensitization of p53-deficient tumor cells to DNA-damaging agents. Curr. Clin. Pharmacol. 2010, 5, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Osman, A.A.; Monroe, M.M.; Ortega Alves, M.V.; Patel, A.A.; Katsonis, P.; Fitzgerald, A.L.; Neskey, D.M.; Frederick, M.J.; Woo, S.H.; Caulin, C.; et al. Wee-1 Kinase Inhibition Overcomes Cisplatin Resistance Associated with High-Risk TP53 Mutations in Head and Neck Cancer through Mitotic Arrest Followed by Senescence. Mol. Cancer Ther. 2015, 14, 608–619. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, O.; Shinkawa, M.; Torimura, T.; Nakamura, T.; Selvendiran, K.; Sakamoto, M.; Koga, H.; Ueno, T.; Sata, M. Cell cycle regulation by the Wee1 Inhibitor PD0166285, Pyrido [2,3-d] pyimidine, in the B16 mouse melanoma cell line. BMC Cancer 2006, 6, 292. [Google Scholar] [CrossRef] [PubMed]
- PosthumaDeBoer, J.; Würdinger, T.; Graat, H.C.; van Beusechem, V.W.; Helder, M.N.; van Royen, B.J.; Kaspers, G.J. WEE1 inhibition sensitizes osteosarcoma to radiotherapy. BMC Cancer 2011, 11, 156. [Google Scholar] [CrossRef] [PubMed]
- Heijink, A.M.; Blomen, V.A.; Bisteau, X.; Degener, F.; Matsushita, F.Y.; Kaldis, P.; Foijer, F.; van Vugt, M.A.T.M. A haploid genetic screen identifies the G1/S regulatory machinery as a determinant of Wee1 inhibitor sensitivity. Proc. Natl. Acad. Sci. USA 2015, 112, 15160–15165. [Google Scholar] [CrossRef] [PubMed]
- Trimbos, J.B.; Parmar, M.; Vergote, I.; Guthrie, D.; Bolis, G.; Colombo, N.; Vermorken, J.B.; Torri, V.; Mangioni, C.; Pecorelli, S.; et al. International Collaborative Ovarian Neoplasm trial 1 and Adjuvant ChemoTherapy In Ovarian Neoplasm trial: Two parallel randomized phase III trials of adjuvant chemotherapy in patients with early-stage ovarian carcinoma. J. Natl. Cancer Inst. 2003, 95, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Reinthaller, A. Antiangiogenic therapies in ovarian cancer. Memo 2016, 9, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Han, E.S.; Monk, B.J. Bevacizumab in the treatment of ovarian cancer. Expert Rev. Anticancer Ther. 2007, 7, 1339–1345. [Google Scholar] [CrossRef] [PubMed]
- Aghajanian, C. The role of bevacizumab in ovarian cancer—An evolving story. Gynecol. Oncol. 2006, 102, 131–133. [Google Scholar] [CrossRef] [PubMed]
- Rigamonti, N.; Kadioglu, E.; Keklikoglou, I.; Wyser Rmili, C.; Leow, C.C.; De Palma, M. Role of Angiopoietin-2 in Adaptive Tumor Resistance to VEGF Signaling Blockade. Cell Rep. 2014, 8, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Brunckhorst, M.K.; Xu, Y.; Lu, R.; Yu, Q. Angiopoietins Promote Ovarian Cancer Progression by Establishing a Procancer Microenvironment. Am. J. Pathol. 2014, 184, 2285–2296. [Google Scholar] [CrossRef] [PubMed]
- Pujade-Lauraine, E.; Hilpert, F.; Weber, B.; Reuss, A.; Poveda, A.; Kristensen, G.; Sorio, R.; Vergote, I.; Witteveen, P.; Bamias, A.; et al. Bevacizumab Combined With Chemotherapy for Platinum-Resistant Recurrent Ovarian Cancer: The AURELIA Open-Label Randomized Phase III Trial. J. Clin. Oncol. 2014, 32, 1302–1308. [Google Scholar] [CrossRef] [PubMed]
- Monk, B.J.; Poveda, A.; Vergote, I.; Raspagliesi, F.; Fujiwara, K.; Bae, D.-S.; Oaknin, A.; Ray-Coquard, I.; Provencher, D.M.; Karlan, B.Y.; et al. Anti-angiopoietin therapy with trebananib for recurrent ovarian cancer (TRINOVA-1): A randomised, multicentre, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2014, 15, 799–808. [Google Scholar] [CrossRef]
- McLachlan, J.; George, A.; Banerjee, S. The current status of PARP inhibitors in ovarian cancer. Tumori J. 2016, 102, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Curtin, N.J. The role of PARP in DNA repair and its therapeutic exploitation. Br. J. Cancer 2011, 105, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Dockery, L.E.; Gunderson, C.C.; Moore, K.N. Rucaparib: The past, present, and future of a newly approved PARP inhibitor for ovarian cancer. Onco Targets Ther. 2017, 10, 3029–3037. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.G.; Sarkaria, J.N.; Kaufmann, S.H. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3406–3411. [Google Scholar] [CrossRef] [PubMed]
- Evans, T.; Matulonis, U. PARP inhibitors in ovarian cancer: Evidence, experience and clinical potential. Ther. Adv. Med. Oncol. 2017, 9, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, S.; Beaver, J.A.; Horton, S.; Fernandes, L.L.; Tang, S.; Horne, H.N.; Liu, J.; Liu, C.; Schrieber, S.J.; Yu, J.; et al. FDA Approval Summary: Rucaparib for the treatment of patients with deleterious BRCA mutation-associated advanced ovarian cancer. Clin. Cancer Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, A.; Sharip, A.; Sharip, A.; Jiang, J.; Yang, Q.; Xie, Y. Reverse the Resistance to PARP Inhibitors. Int. J. Biol. Sci. 2017, 13, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, A. Drug resistance caused by reversion mutation. Cancer Res. 2008, 68, 10021–10023. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, K.K.; Taniguchi, T. Resistance to PARP Inhibitors Mediated by Secondary BRCA1/2 Mutations; Humana Press: Cham, Switzerland, 2015; pp. 431–452. [Google Scholar]
- Barber, L.J.; Sandhu, S.; Chen, L.; Campbell, J.; Kozarewa, I.; Fenwick, K.; Assiotis, I.; Rodrigues, D.N.; Reis-Filho, J.S.; Moreno, V.; et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J. Pathol. 2013, 229, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Neijenhuis, S.; Bajrami, I.; Miller, R.; Lord, C.J.; Ashworth, A. Identification of miRNA modulators to PARP inhibitor response. DNA Repair 2013, 12, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Ter Brugge, P.; Kristel, P.; van der Burg, E.; Boon, U.; de Maaker, M.; Lips, E.; Mulder, L.; de Ruiter, J.; Moutinho, C.; Gevensleben, H.; et al. Mechanisms of Therapy Resistance in Patient-Derived Xenograft Models of BRCA1-Deficient Breast Cancer. J. Natl. Cancer Inst. 2016, 108, djw148. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008, 451, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Krais, J.J.; Bernhardy, A.J.; Nicolas, E.; Cai, K.Q.; Harrell, M.I.; Kim, H.H.; George, E.; Swisher, E.M.; Simpkins, F.; et al. RING domain-deficient BRCA1 promotes PARP inhibitor and platinum resistance. J. Clin. Investig. 2016, 126, 3145–3157. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; van der Burg, E.; Nygren, A.O.H.; Zander, S.A.L.; Derksen, P.W.B.; de Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17084. [Google Scholar] [CrossRef] [PubMed]
- Issaeva, N.; Thomas, H.D.; Djurenovic, T.; Jaspers, J.E.; Stoimenov, I.; Kyle, S.; Pedley, N.; Gottipati, P.; Zur, R.; Sleeth, K.; et al. 6-Thioguanine Selectively Kills BRCA2-Defective Tumors and Overcomes PARP Inhibitor Resistance. Cancer Res. 2010, 70, 6268–6276. [Google Scholar] [CrossRef] [PubMed]
- Trams, E.G.; Lauter, C.J.; Salem, N.; Heine, U. Exfoliation of membrane ecto-enzymes in the form of micro-vesicles. Biochim. Biophys. Acta 1981, 645, 63–70. [Google Scholar] [CrossRef]
- Rashed, M.H.; Bayraktar, E.; Helal, G.K.; Abd-Ellah, M.; Amero, P.; Chavez-Reyes, A.; Rodriguez-Aguayo, C. Exosomes: From Garbage Bins to Promising Therapeutic Targets. Int. J. Mol. Sci. 2017, 18, 538. [Google Scholar] [CrossRef] [PubMed]
- Corrado, C.; Raimondo, S.; Chiesi, A.; Ciccia, F.; De Leo, G.; Alessandro, R. Exosomes as intercellular signaling organelles involved in health and disease: Basic science and clinical applications. Int. J. Mol. Sci. 2013, 14, 5338–5366. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, X. The emerging roles and therapeutic potential of exosomes in epithelial ovarian cancer. Mol. Cancer 2017, 16, 92. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zheng, Y.; Zhao, M. Exosome-Based Cancer Therapy: Implication for Targeting Cancer Stem Cells. Front. Pharmacol. 2016, 7, 533. [Google Scholar] [CrossRef] [PubMed]
- Bard, M.P.; Hegmans, J.P.; Hemmes, A.; Luider, T.M.; Willemsen, R.; Severijnen, L.-A.A.; van Meerbeeck, J.P.; Burgers, S.A.; Hoogsteden, H.C.; Lambrecht, B.N. Proteomic Analysis of Exosomes Isolated from Human Malignant Pleural Effusions. Am. J. Respir. Cell Mol. Biol. 2004, 31, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Schorey, J.S.; Bhatnagar, S. Exosome Function: From Tumor Immunology to Pathogen Biology. Traffic 2008, 9, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Bach, D.-H.; Hong, J.-Y.; Park, H.J.; Lee, S.K. The role of exosomes and miRNAs in drug-resistance of cancer cells. Int. J. Cancer 2017, 141, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Crow, J.; Atay, S.; Banskota, S.; Artale, B.; Schmitt, S.; Godwin, A.K. Exosomes as mediators of platinum resistance in ovarian cancer. Oncotarget 2017, 8, 11917–11936. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.-L.; Zhuang, T.; Xing, B.-H.; Li, N.; Li, Q. Exosomal DNMT1 mediates cisplatin resistance in ovarian cancer. Cell Biochem. Funct. 2017, 35, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.; Robertson, K.D. DNA methyltransferases, DNA damage repair, and cancer. Adv. Exp. Med. Biol. 2013, 754, 3–29. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.C.; Kim, O.Y.; Yoon, C.M.; Choi, D.-S.; Roh, T.-Y.; Park, J.; Nilsson, J.; Lötvall, J.; Kim, Y.-K.; Gho, Y.S. Bioinspired Exosome-Mimetic Nanovesicles for Targeted Delivery of Chemotherapeutics to Malignant Tumors. ACS Nano 2013, 7, 7698–7710. [Google Scholar] [CrossRef] [PubMed]
- Hadla, M.; Palazzolo, S.; Corona, G.; Caligiuri, I.; Canzonieri, V.; Toffoli, G.; Rizzolio, F. Exosomes increase the therapeutic index of doxorubicin in breast and ovarian cancer mouse models. Nanomedicine 2016, 11, 2431–2441. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Lang, J. Programmed death-1 pathway blockade produces a synergistic antitumor effect: Combined application in ovarian cancer. J. Gynecol. Oncol. 2017, 28, e64. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.-Y.; Francois, A.; McGray, A.R.; Miliotto, A.; Odunsi, K. Compensatory upregulation of PD-1, LAG-3, and CTLA-4 limits the efficacy of single-agent checkpoint blockade in metastatic ovarian cancer. Oncoimmunology 2017, 6, e1249561. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.; Huemer, F.; Mlineritsch, B.; Greil, R. Immune checkpoint blockade in ovarian cancer. Memo 2016, 9, 82–84. [Google Scholar] [CrossRef] [PubMed]
- Hamanishi, J.; Mandai, M.; Ikeda, T.; Minami, M.; Kawaguchi, A.; Murayama, T.; Kanai, M.; Mori, Y.; Matsumoto, S.; Chikuma, S.; et al. Safety and Antitumor Activity of Anti-PD-1 Antibody, Nivolumab, in Patients With Platinum-Resistant Ovarian Cancer. J. Clin. Oncol. 2015, 33, 4015–4022. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Aust, S.; Felix, S.; Auer, K.; Bachmayr-Heyda, A.; Kenner, L.; Dekan, S.; Meier, S.M.; Gerner, C.; Grimm, C.; Pils, D. Absence of PD-L1 on tumor cells is associated with reduced MHC I expression and PD-L1 expression increases in recurrent serous ovarian cancer. Sci. Rep. 2017, 7, 42929. [Google Scholar] [CrossRef] [PubMed]
- Derenzini, M.; Montanaro, L.; Treré, D. What the nucleolus says to a tumour pathologist. Histopathology 2009, 54, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Sirri, V.; Urcuqui-Inchima, S.; Roussel, P.; Hernandez-Verdun, D. Nucleolus: The fascinating nuclear body. Histochem. Cell Biol. 2008, 129, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Deisenroth, C.; Zhang, Y. The Ribosomal Protein-Mdm2-p53 Pathway and Energy Metabolism: Bridging the Gap between Feast and Famine. Genes Cancer 2011, 2, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Boulon, S.; Westman, B.J.; Hutten, S.; Boisvert, F.M.; Lamond, A.I. The Nucleolus under Stress. Mol. Cell 2010, 40, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Woods, S.J.; Hannan, K.M.; Pearson, R.B.; Hannan, R.D. The nucleolus as a fundamental regulator of the p53 response and a new target for cancer therapy. Biochim. Biophys. Acta 2015, 1849, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Quin, J.E.; Devlin, J.R.; Cameron, D.; Hannan, K.M.; Pearson, R.B.; Hannan, R.D. Targeting the nucleolus for cancer intervention. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 802–816. [Google Scholar] [CrossRef] [PubMed]
- Peltonen, K.; Colis, L.; Liu, H.; Trivedi, R.; Moubarek, M.S.; Moore, H.M.; Bai, B.; Rudek, M.A.; Bieberich, C.J.; Laiho, M. A targeting modality for destruction of RNA polymerase I that possesses anticancer activity. Cancer Cell 2014, 25, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Pickard, A.J.; Bierbach, U. The cell’s nucleolus: An emerging target for chemotherapeutic intervention. ChemMedChem 2013, 8, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Frank, D.; Son, J.; Hannan, K.; Hannan, R.; Chan, K.; Pearson, R.; Sanij, E. The Potential of Targeting Ribosome Biogenesis in High-Grade Serous Ovarian Cancer. Int. J. Mol. Sci. 2017, 18, 210. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.; Zomerdijk, J.C.B.M. The RNA polymerase I transcription machinery. Biochem. Soc. Symp. 2006, 203–216. [Google Scholar] [CrossRef]
- Ray, S.; Panova, T.; Miller, G.; Volkov, A.; Porter, A.C.G.; Russell, J.; Panov, K.I.; Zomerdijk, J.C.B.M. Topoisomerase IIα promotes activation of RNA polymerase I transcription by facilitating pre-initiation complex formation. Nat. Commun. 2013, 4, 1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poortinga, G.; Quinn, L.; Hannan, R. Targeting RNA polymerase I to treat MYC-driven cancer. Oncogene 2014, 34, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Haddach, M.; Schwaebe, M.K.; Michaux, J.; Nagasawa, J.; O’Brien, S.E.; Whitten, J.P.; Pierre, F.; Kerdoncuff, P.; Darjania, L.; Stansfield, R.; et al. Discovery of CX-5461, the first direct and selective inhibitor of RNA polymerase I, for cancer therapeutics. ACS Med. Chem. Lett. 2012, 3, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Di Antonio, M.; Mckinney, S.; Mathew, V.; Neil, N.J.O.; Dos Santos, N.; Wei, V.; Garcia, J.; Yap, D.; Le, D.; et al. CX-5461 and CX-3543 are G-quadruplex stabilizers with selective synthetic lethality in BRCA1/2 deficient tumours. Nat. Commun. 2017, 8, 14432. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Pang, S.; Zhang, W.; Guo, X.; Wang, J.; Zhang, Y.; Liu, Y.; Wu, X.; Jiang, F. Therapeutic Targeting of RNA Polymerase I With the Small-Molecule CX-5461 for Prevention of Arterial Injury-Induced Neointimal Hyperplasia. Arterioscler. Thromb. Vasc. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Peltonen, K.; Colis, L.; Liu, H.; Jaamaa, S.; Zhang, Z.; Af Hallstrom, T.; Moore, H.M.; Sirajuddin, P.; Laiho, M. Small Molecule BMH-Compounds That Inhibit RNA Polymerase I and Cause Nucleolar Stress. Mol. Cancer Ther. 2014, 13, 2537–2546. [Google Scholar] [CrossRef] [PubMed]
- Colis, L.; Peltonen, K.; Sirajuddin, P.; Liu, H.; Sanders, S.; Ernst, G.; Barrow, J.C.; Laiho, M. DNA intercalator BMH-21 inhibits RNA polymerase I independent of DNA damage response. Oncotarget 2014, 5, 4361–4369. [Google Scholar] [CrossRef] [PubMed]
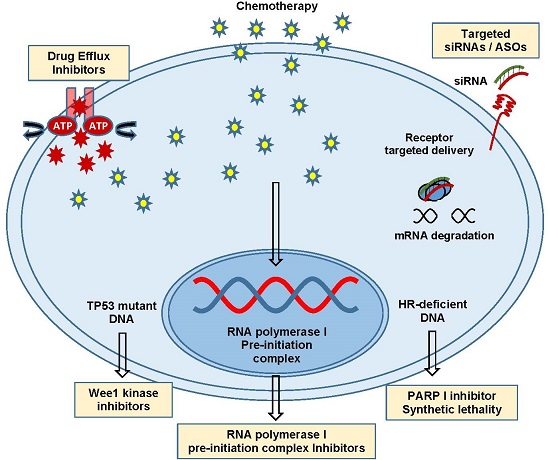
Figure 1.
Intrinsic versus acquired resistance. Mechanisms of intrinsic and/or acquired chemoresistance include extracellular matrix proteins, P-glycoprotein (P-gp) efflux pumps and ATP binding cassette (ABC) transporters, cytochrome p450 and glutathione transferases, poor vascularization, DNA damage insensitivity and repair, drug target alteration and upregulation of pro-survival anti-apoptotic factors. Abbreviations: adenosine diphosphate (ADP), adenosine triphosphate (ATP), B-cell lymphoma 2 (BCL-2), B-cell lymphoma-extra large (BCL-XL), myeloid cell leukemia 1 (MCL-1), X-linked inhibitor of apoptosis protein/cellular inhibitor of apoptosis (xIAP/cIAP).
Figure 1.
Intrinsic versus acquired resistance. Mechanisms of intrinsic and/or acquired chemoresistance include extracellular matrix proteins, P-glycoprotein (P-gp) efflux pumps and ATP binding cassette (ABC) transporters, cytochrome p450 and glutathione transferases, poor vascularization, DNA damage insensitivity and repair, drug target alteration and upregulation of pro-survival anti-apoptotic factors. Abbreviations: adenosine diphosphate (ADP), adenosine triphosphate (ATP), B-cell lymphoma 2 (BCL-2), B-cell lymphoma-extra large (BCL-XL), myeloid cell leukemia 1 (MCL-1), X-linked inhibitor of apoptosis protein/cellular inhibitor of apoptosis (xIAP/cIAP).
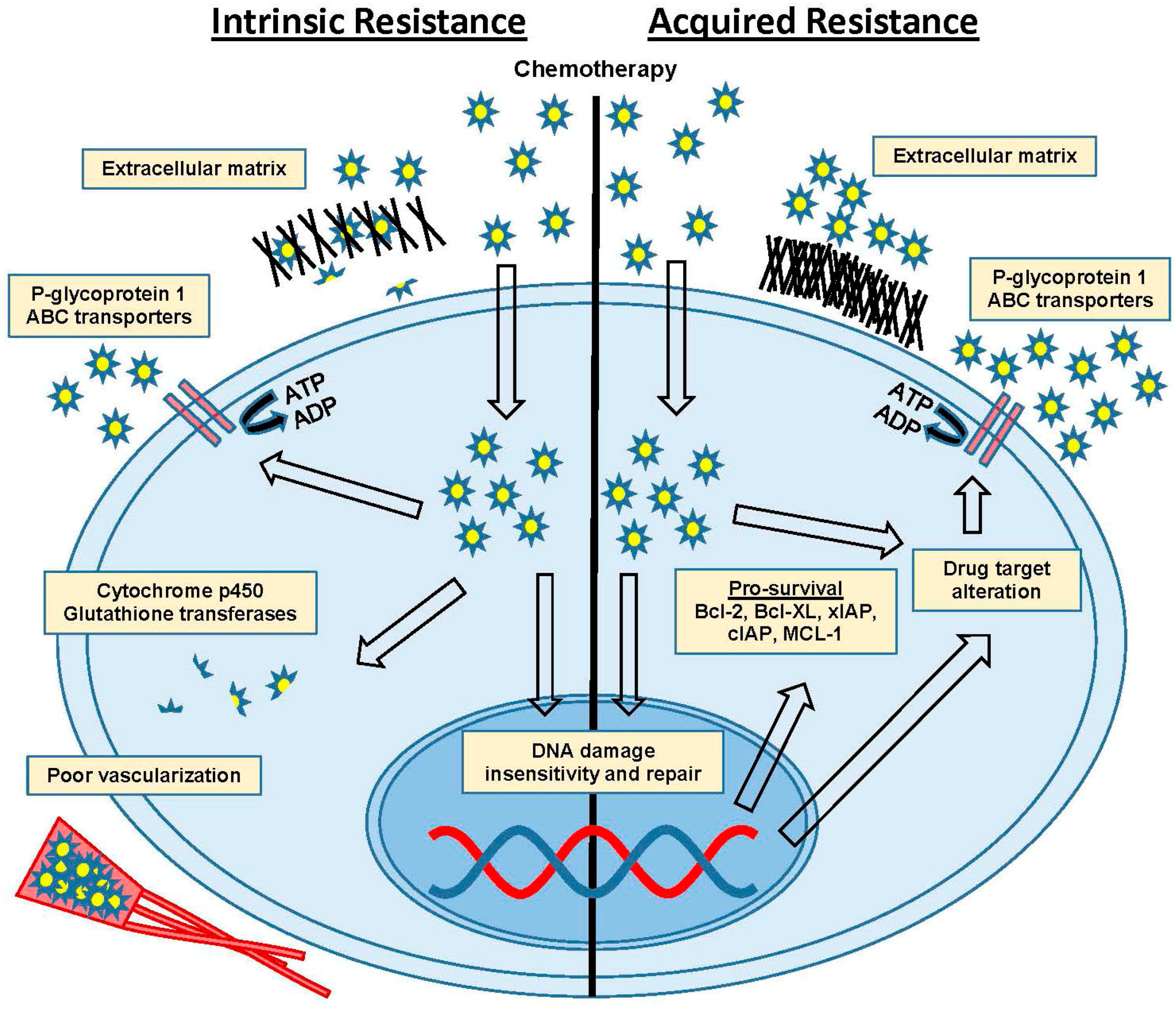
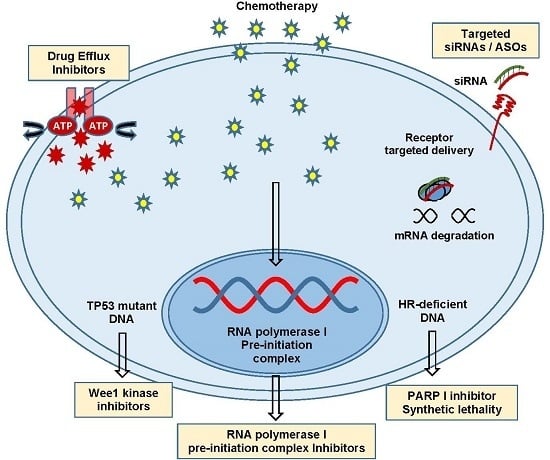
Figure 2.
Action and targeting of P-gp and MDR1 drug efflux pumps. Activation of MDR1/ABCB1 transcription leads to drug efflux and inhibition of cytochrome p450 isoenzymes. Chemosensitivity and cell death can be enhanced by inhibitors and siRNA targeted therapies. Abbreviations: adenosine triphosphate binding cassette-B1 (ABCB1) transporters; antisense oligonucleotide (ASO), cluster of differentiation 44 (CD44), multidrug resistance protein 1 (MDR1), small interfering RNA (siRNA).
Figure 2.
Action and targeting of P-gp and MDR1 drug efflux pumps. Activation of MDR1/ABCB1 transcription leads to drug efflux and inhibition of cytochrome p450 isoenzymes. Chemosensitivity and cell death can be enhanced by inhibitors and siRNA targeted therapies. Abbreviations: adenosine triphosphate binding cassette-B1 (ABCB1) transporters; antisense oligonucleotide (ASO), cluster of differentiation 44 (CD44), multidrug resistance protein 1 (MDR1), small interfering RNA (siRNA).
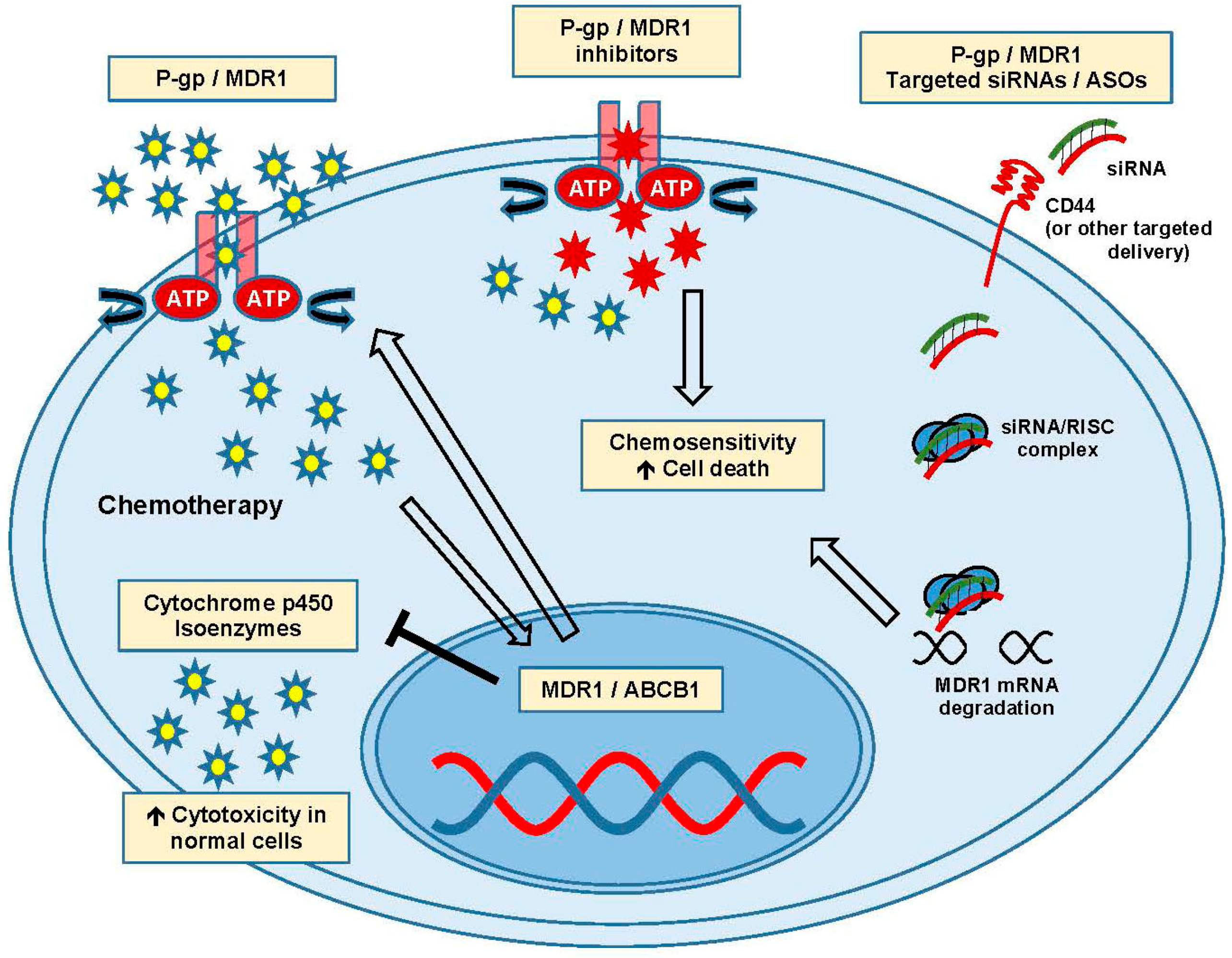
Figure 3.
Epithelial–mesenchymal transition. Upregulation of Snail, Slug, Zeb1 and Twist lead to changes in mesenchymal and stem cell markers including downregulation of E-cadherin, and upregulation of vimentin, N-cadherin, ALDH1 and endothelin-A. Abbreviations: aldehyde dehydrogenase-1 (ALDH1).
Figure 3.
Epithelial–mesenchymal transition. Upregulation of Snail, Slug, Zeb1 and Twist lead to changes in mesenchymal and stem cell markers including downregulation of E-cadherin, and upregulation of vimentin, N-cadherin, ALDH1 and endothelin-A. Abbreviations: aldehyde dehydrogenase-1 (ALDH1).
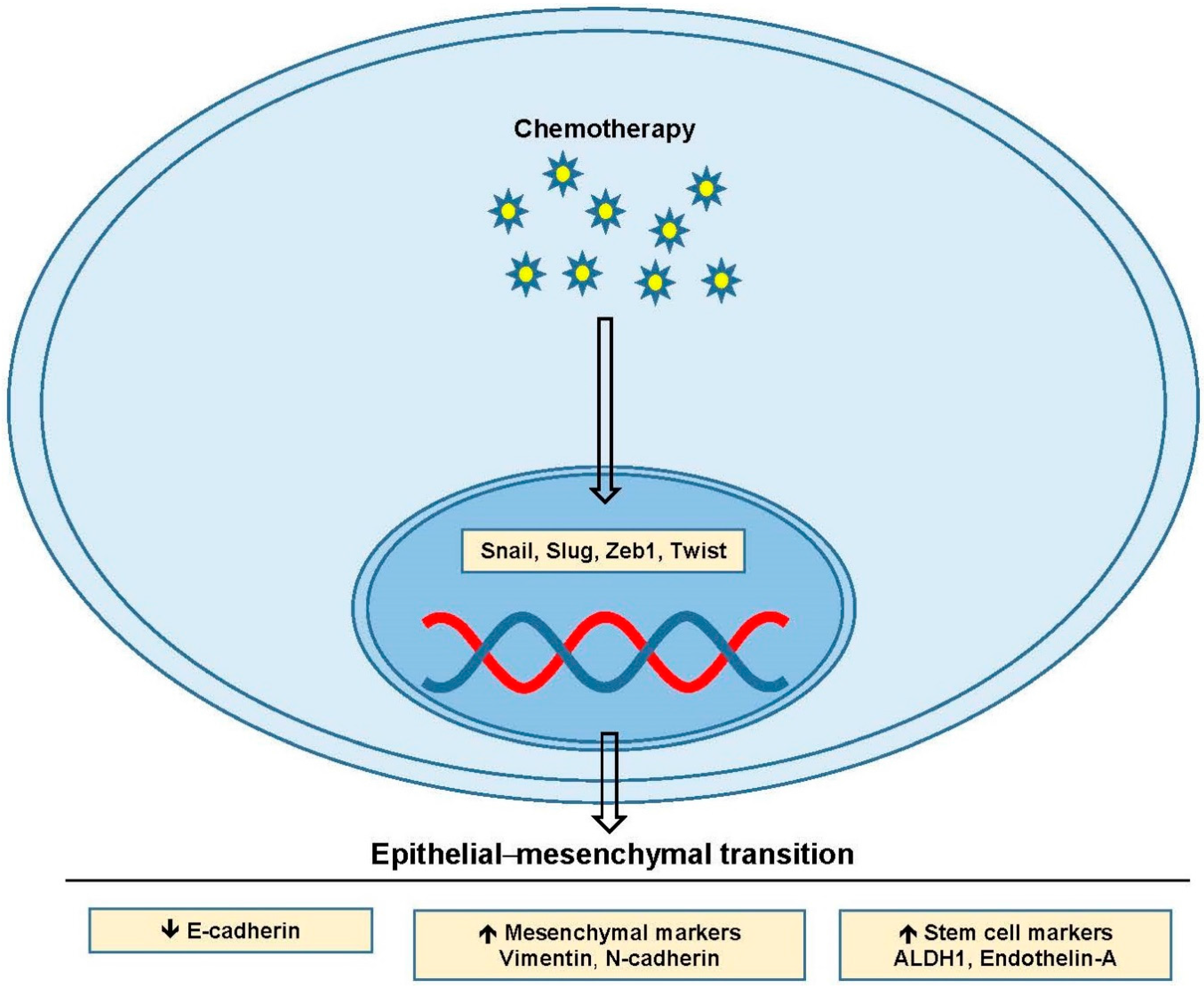
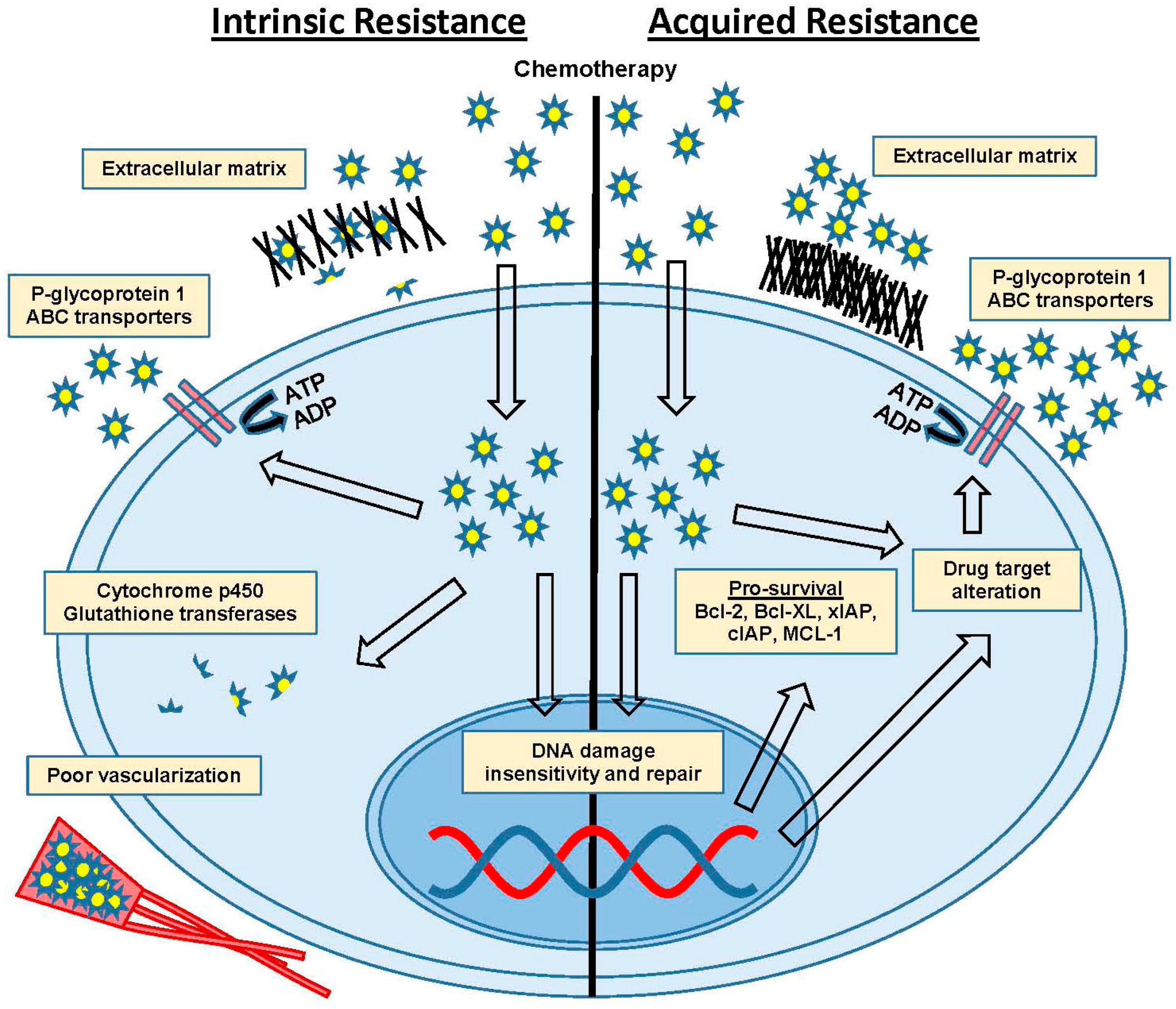
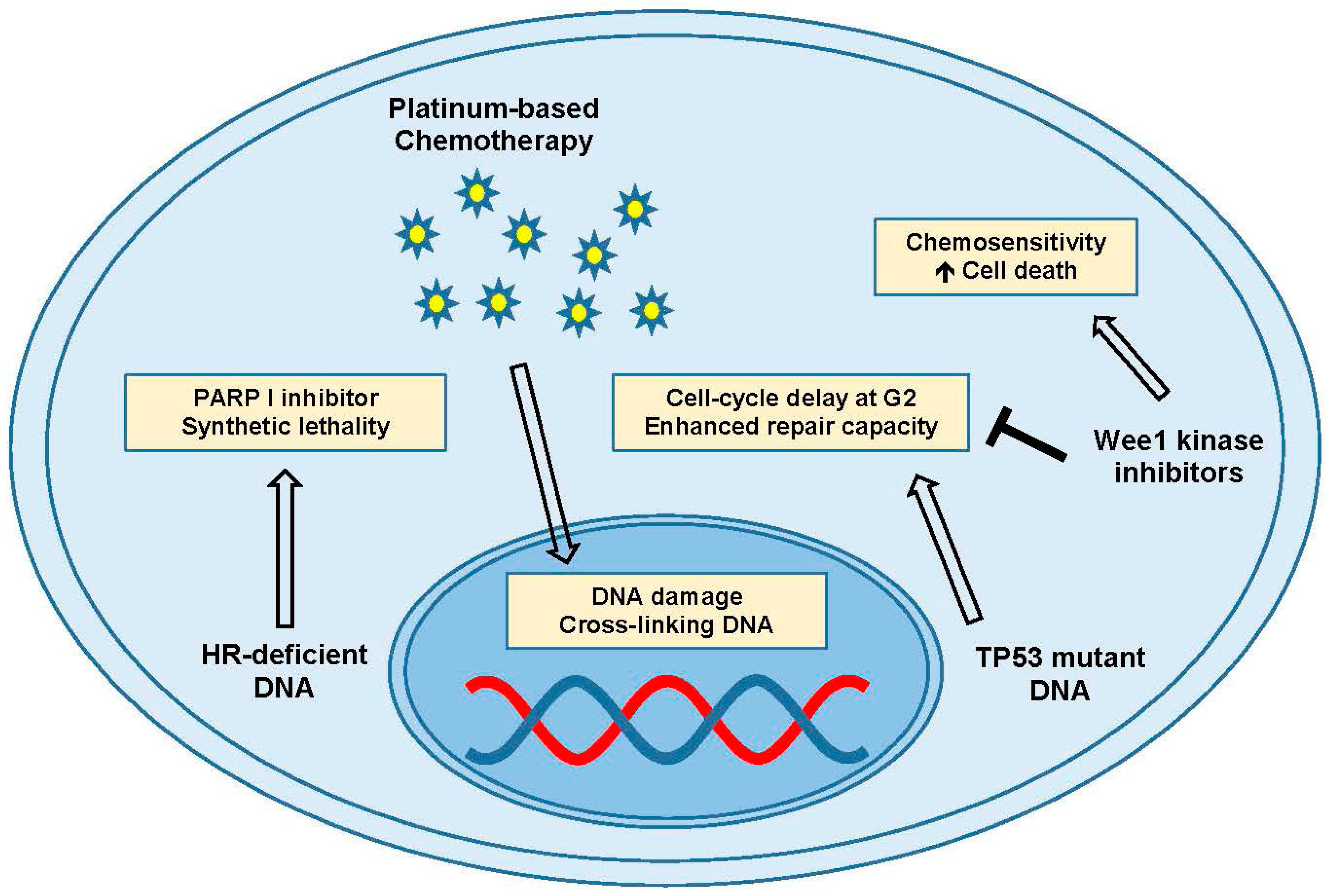
Figure 4.
DNA damage and repair. PARP inhibitors in the presence of platinum-based chemotherapy and homologous recombination (HR) deficient DNA can induce synthetic lethality. TP53 mutated DNA can show cell cycle delay at the G2 phase, which can be inhibited by Wee1 kinase inhibitors to promote cell death and enhance chemosensitivity. Abbreviations: Gap 2 (G2); poly(ADP-ribose) polymerase-I (PARP I); tumor protein 53 (TP53).
Figure 4.
DNA damage and repair. PARP inhibitors in the presence of platinum-based chemotherapy and homologous recombination (HR) deficient DNA can induce synthetic lethality. TP53 mutated DNA can show cell cycle delay at the G2 phase, which can be inhibited by Wee1 kinase inhibitors to promote cell death and enhance chemosensitivity. Abbreviations: Gap 2 (G2); poly(ADP-ribose) polymerase-I (PARP I); tumor protein 53 (TP53).

Figure 5.
Ribosomal biogenesis targeting. Inhibitors of the RNA polymerase I pre-initiation complex, such as CX-5461 and BMH-21, can inhibit ribosome production and availability. Some data suggests that cancer cells may be more susceptible to targeting this pathway than nontransformed cells. Abbreviations: selective factor 1 (SL1).
Figure 5.
Ribosomal biogenesis targeting. Inhibitors of the RNA polymerase I pre-initiation complex, such as CX-5461 and BMH-21, can inhibit ribosome production and availability. Some data suggests that cancer cells may be more susceptible to targeting this pathway than nontransformed cells. Abbreviations: selective factor 1 (SL1).

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cornelison, R.; Llaneza, D.C.; Landen, C.N. Emerging Therapeutics to Overcome Chemoresistance in Epithelial Ovarian Cancer: A Mini-Review. Int. J. Mol. Sci. 2017, 18, 2171. https://doi.org/10.3390/ijms18102171
AMA Style
Cornelison R, Llaneza DC, Landen CN. Emerging Therapeutics to Overcome Chemoresistance in Epithelial Ovarian Cancer: A Mini-Review. International Journal of Molecular Sciences. 2017; 18(10):2171. https://doi.org/10.3390/ijms18102171
Chicago/Turabian StyleCornelison, Robert, Danielle C. Llaneza, and Charles N. Landen. 2017. "Emerging Therapeutics to Overcome Chemoresistance in Epithelial Ovarian Cancer: A Mini-Review" International Journal of Molecular Sciences 18, no. 10: 2171. https://doi.org/10.3390/ijms18102171
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.