NADPH Oxidase-Related Pathophysiology in Experimental Models of Stroke

1
Laboratory of Neurochemistry, National Hospital Organization Hizen Psychiatric Center, Saga 842-0192, Japan
2
Department of Medicine and Clinical Science, Graduate School of Medical Sciences, Kyushu University, Fukuoka 812-8582, Japan
3
Department of Functional Pathology, Shimane University School of Medicine, Izumo 693-8501, Japan
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2017, 18(10), 2123; https://doi.org/10.3390/ijms18102123
Submission received: 6 September 2017
/
Revised: 5 October 2017
/
Accepted: 6 October 2017
/
Published: 11 October 2017
(This article belongs to the Special Issue Oxidative Stress in Vascular Diseases)
Abstract
:Several experimental studies have indicated that nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (Nox) exert detrimental effects on ischemic brain tissue; Nox-knockout mice generally exhibit resistance to damage due to experimental stroke following middle cerebral artery occlusion (MCAO). Furthermore, our previous MCAO study indicated that infarct size and blood-brain barrier breakdown are enhanced in mice with pericyte-specific overexpression of Nox4, relative to levels observed in controls. However, it remains unclear whether Nox affects the stroke outcome directly by increasing oxidative stress at the site of ischemia, or indirectly by modifying physiological variables such as blood pressure or cerebral blood flow (CBF). Because of technical problems in the measurement of physiological variables and CBF, it is often difficult to address this issue in mouse models due to their small body size; in our previous study, we examined the effects of Nox activity on focal ischemic injury in a novel congenic rat strain: stroke-prone spontaneously hypertensive rats with loss-of-function in Nox. In this review, we summarize the current literature regarding the role of Nox in focal ischemic injury and discuss critical issues that should be considered when investigating Nox-related pathophysiology in animal models of stroke.
1. Introduction
The nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (Nox)—which function solely to generate superoxide—are the predominant source of reactive oxygen species [1,2]. Nox-derived reactive oxygen species are thought to exert detrimental effects on the brain following a stroke. Paradoxically, tissue injury in stroke is caused by both the depletion (ischemia) and replenishment (reperfusion) of oxygen [2]. To date, seven members of the Nox family have been identified: Nox1-5, dual oxidase 1, and dual oxidase 2. Nox1-3, and Nox1-5 primarily produce superoxide, which is subsequently dismutated into H2O2, whereas Nox4, dual oxidase 1 and dual oxidase 2 directly produce H2O2 [1,2]. With the exception of Nox5, all Nox family members are multi-subunit enzymes; stabilization of Nox1-4 requires the assistance of the membrane-bound subunit p22phox. Expressions of Nox3 and 5 in the brain have only been documented in inner ear and brain cancer, respectively, and Nox5 is absent in rats and mice [1,2]. Furthermore, dual oxidase 1 and dual oxidase 2 are most commonly associated with thyroid function [1,2]. Hence, the present review discusses the impact of Nox1, Nox2, and Nox4 on stroke pathophysiology.
The physiological functions of Nox family members are extremely diverse, as these enzymes play key roles in host defense and inflammation, cellular signaling, gene expression, cellular death, cellular senescence, cell growth, oxygen sensing, angiogenesis, and various other biological processes [1]. Among Nox family members, Nox2 was first identified, and originally named gp91phox because of its high expression and activity in phagocytes; phox indicates phagocyte oxidase. A recent review by Sorce et al. summarized the physiological and pathological roles of Nox in the central nervous system (CNS) [3]. Nox2 and Nox4 are the main Nox isoforms expressed in the CNS. Under physiological conditions, most Nox isoforms are expressed only to a negligible extent in the CNS. In the CNS, Nox2 is expressed mostly in microglia. High levels of Nox2 activity was associated with decreased survival in amyotrophic lateral sclerosis patients and autoimmune demyelination [3]. Nox2 activity in blood samples may be a useful biomarker for neuroinflammation that is associated with neurodegenerative diseases.
Research has demonstrated that Nox is also involved in the physiological functions of the vasculature as well as hypertension [1,4], the latter of which represents a major risk factor for stroke. Superoxide rapidly interacts with nitric oxide to form peroxynitrite, resulting in vasoconstriction and increased blood pressure [4]. Basal and stimulated generation of superoxide from Nox is one to two orders of magnitude higher in intracranial cerebral arteries than in systemic arteries, and activation of Nox in cerebral arteries can cause vasodilation under normal conditions [5]. In addition, Nox4 may function as the major catalytic component of an endothelial NADPH oxidase [6]. Previous studies have reported that rat basilar artery endothelial cells exhibit abundant expression of Nox1, while Nox1 expression in aortic endothelial cells is marginal, indicating that different vascular beds may contain different sets of Nox proteins [7]. Several studies have indicated that Nox4 activity is increased in cerebral arteries during chronic hypertension, and that this increase in expression is associated with greater production of superoxide and vasodilatation in spontaneously hypertensive rats [8].
Stroke has a secondary injury component following a primary ischemic event; secondary injury involves a cascade of injury pathways that exacerbate tissue damage including inflammation, excitotoxicity, oxidative stress, loss of ion homeostasis, and increased blood-brain barrier (BBB) permeability [9]. Neurally-elicited inflammatory process (i.e., neurogenic inflammation) plays a key role in the secondary injury cascades (e.g., enhanced permeability of the BBB after stroke) that evolve following a stroke. Excessive Nox-derived reactive oxygen species likely lead to neurodegeneration via breakdown of the BBB, and/or via neuronal apoptosis [10]. However, whether the effects of Nox on stroke outcomes are due to direct increases in oxidative stress at the site of ischemia or indirect increases due to the modification of physiological variables (e.g., blood pressure or cerebral blood flow (CBF)) remains unknown. Although the mechanisms underlying ischemia/reperfusion injury caused by Nox-related oxidative stress remain to be fully elucidated, Nox are promising therapeutic targets for the treatment of stroke.
In this review, we present a brief overview of experimental stroke models and pathophysiology associated with the “classic” concept of the ischemic penumbra. The ischemic penumbra is classically defined as an ischemic brain region with CBF between the upper threshold of electrical silence and the lower threshold of energy and ion pump failure (see legends for Figure 1). We highlight findings obtained in Nox knockout mouse models of experimental stroke, as well as the effects of Nox on cerebral blood vessels and the BBB, focusing on the role of pericytes. In addition, we discuss several confounding factors associated with experimental models of stroke based on our recent findings in stroke-prone spontaneously hypertensive rats (SHRSP), which exhibit Nox dysfunction.
2. Experimental Models of Stroke
Models of acute brain infarction associated with the occlusion of major cerebral arteries are developed by inducing focal ischemia or middle cerebral artery occlusion (MCAO) in animals [19]. Here, we summarize the characteristics of the most commonly utilized rat and mouse models of stroke (Table 1).
2.1. Distal Middle Cerebral Artery Occlusion
The pathophysiological consequences of distal MCAO distal to the lenticulostriate branches have been investigated in 35–40 day old Wistar Kyoto rats (WKY) and SHRSP. Such studies have revealed that, even in young SHRSP with mild hypertension, distal MCAO lesions are larger than those observed in normotensive WKY [20].
2.2. Proximal MCAO
Tamura et al. first described methods for MCAO proximal to the lenticulostriate arteries, which produces infarction of both the cortex and the lateral portion of the striatum [21]. Duverger and MacKenzie subsequently modified this method using SHRSP, SHR, and three normotensive strains (WKY, Sprague-Dawley rats, and Fischer-344 rats) [22]. Proximal MCAO results in a considerably larger infarction in hypertensive rats than in normotensive rats. Moreover, minimal variability was observed among the hypertensive strains [coefficient of variation (C.V.) = 7–8%] when compared with findings in the three normotensive strains (C.V. = 20–49%).
2.3. Intraluminal Suture Occlusion
The intraluminal suture model, developed by Koizumi et al. [23] and Longa et al. [24], is undoubtedly the most frequently used model of focal ischemia in rats and mice [25]. This method is advantageous in that it is easy to perform, minimally invasive, and—most importantly—does not require craniectomy. Although reperfusion of ischemic brain tissue is critical for restoring normal function, it can paradoxically result in secondary damage or reperfusion injury. Several lines of evidence have suggested that post-ischemic oxidative stress and inflammation contribute to brain injury [26]. As reperfusion injury is the condition of ischemia aggravated by the occurrence of reperfusion more so than in the case where reperfusion does not occur (i.e., permanent occlusion), in order to see reperfusion injury it is necessary to make a comparison with permanent occlusion. Furthermore, from a critical point of view, the intraluminal suture model is a top of the internal carotid artery occlusion model rather than a MCAO model [27]. Consequently, this model exhibits a wide zone of ischemia, and because the mortality rate is high in the case of permanent occlusion, the permanent occlusion group and the reperfusion group—that shared the same time lapse—cannot be compared.
2.4. Photothrombotic MCAO in Spontaneously Hypertensive Rats
Several studies have indicated that the effects of thrombotic stroke on compromised brain tissue may differ from those due to cerebral ischemia induced by mechanical occlusion of intracranial or extracranial brain arteries [28,29]. Prado et al. first used SHR as a model of a photothrombotic distal MCAO model without common carotid artery occlusion [30]. In our experience, the photothrombotic distal MCAO model in SHR yields a highly reproducible infarct volume (average C.V. = 21%) and does not entail extensive surgery or opening of the dura, thereby avoiding unacceptable local tissue trauma at the site of MCAO [31,32,33]. This model encompasses appropriate physiological monitoring, associated risk factors for stroke, and clinically relevant pathophysiology of thrombosis. Watson et al. achieved successful ultraviolet laser-induced reperfusion in animal models of photothrombotic stroke [34], which has also been applied in SHR [31,32].
2.5. Ischemic Penumbra
The term “penumbra” refers to the salvageable tissue surrounding the ischemic core (Figure 1). Focal ischemia essentially consists of an ischemic core and penumbra, although it remains unclear which model of focal ischemia is most relevant for investigating phenomena in the penumbra. Proximal MCAO produces two ischemic vascular territories associated with the lenticulostriate arteries and cortical branches of the MCA, the former of which are end-arteries, and therefore treatment-resistant (i.e., ischemic core without collateral perfusion). The intraluminal suture model involves internal carotid artery occlusion rather than pure MCAO [27]. In clinical settings, the arterial obstruction site strongly predicts infarct growth and clinical outcome; internal carotid artery occlusion has been associated with uniformly poor prognosis and poor response to tissue plasminogen activator therapy [35]. Although the infarcts produced by distal MCAO in normotensive rats are small with a presumably limited size of penumbra, distal MCAO alone—occlusion of one major cerebral artery with collateral perfusion—in SHR results in substantial infarct size and an acceptable amount of penumbra [31,32].
Taken together, these findings indicate that distal MCAO in SHR represents the best choice for an experimental model of human stroke. In our experience with distal MCAO in SHR, early restoration of perfusion confers advantages on the ischemic brain; our findings indicated that early reperfusion attenuated damage in the cortical region, which was regarded as the penumbral zone [36]. Although reperfusion after a relatively long period of ischemia did not decrease final infarct size or fodrin breakdown, as an index of evolving ischemic injury was attenuated in the penumbra [37]. These results support the notion that distal MCAO in SHR provides a rational approach for investigation phenomena associated with the ischemic penumbra.
2.6. Nox in Experimental Models of Stroke
Because neither Nox-specific nor isoform-specific inhibitors for NADPH oxidases are not available, Nox knock-out mice are widely utilized to assess the effects of Nox on stroke pathophysiology (Table 2).
2.7. Nox2
Among the Nox isoforms, Nox2 has been the most widely studied in the context of focal ischemia or stroke. Transient focal ischemia in Nox2 knock-out mice results in smaller infarct volume than that induced in wild-type mice, suggesting that superoxide generated from NADPH oxidase plays a major role in mediating ischemia-reperfusion injury [38,39,40,41]. Apocynin is currently the most selective inhibitor of the Nox family and is thought to inhibit activation of the enzyme by preventing translocation of p47phox. In several earlier studies, both Nox2 depletion and apocynin treatment significantly reduced infarct volume compared to that of untreated wild-type mice [39,40]. Furthermore, pre-treatment with apocynin also reduced neurological impairments and mortality in wild-type but not Nox2 knockout mice. The authors further reported that pre-treatment with apocynin significantly reduced superoxide production as detected using in situ dihydroethidium methods following ischemia-reperfusion [41]. However, Nox2 knockouts failed to attenuate infarct size in the absence of reperfusion (i.e., permanent occlusion) [42]. Additional studies indicated that the larger infarct region observed in male mice when compared with female mice after transient occlusion (but not permanent suture occlusion) was dependent on Nox2 [43].
Oxidative stress induced by ischemia-reperfusion causes endothelial dysfunction via increased expression and activity of Nox2 in the cerebral artery. This endothelial dysfunction appears to be due to impaired NO-mediated vasodilation most likely via the direct inactivation of NO and the formation of peroxynitrite. A study reported that Nox2 knockout decreased superoxide production in the MCA, attenuated Nω-Nitro-L-arginine methyl ester-induced vasoconstriction, and reduced infarct size compared with the wild-type mice [44]. In accordance with the effects of Nox2 on vascular function, genetic depletion of Nox2 attenuates suture occlusion-induced infarction and early BBB dysfunction [45]. Nox2 knockout showed transient nature of protection (i.e., reduced infarct volume at 24 h, but not at 6 or 72 h post-stroke), suggesting that other factors mediate infarct size in acute stroke. Furthermore, Nox2 deletion increased re-vascularization of the damaged brain by 72 h [46]. Taken together, these findings indicate that Nox2 knockout generally exerts protective effects against focal ischemia-reperfusion but not against permanent MCAO.
2.8. Nox1
Angiotensin II-stimulated increases in superoxide generation in intact cerebral arteries were threefold higher in wild-type than in Nox1 knockout mice; although cortical (i.e., penumbra) infarct volume was fourfold greater in Nox1 knockout mice than in wild-type mice, no such effects were observed on subcortical infarct volume, suggesting that Nox1 exerts protective effects against focal ischemic injury [47]. After 1 h of ischemia and 23 h of reperfusion, infarct volume was reduced by 44% in Nox1 knock-out mice relative to levels observed in wild-type mice; however, no difference in infarct size was observed between Nox1 knockout and wild-type mice with permanent occlusion (2 h of occlusion time) [48]. Both infarct size and neuronal death after 90 min of suture occlusion in male Wistar rats were significantly reduced by adeno-associated virus-mediated transduction of Nox1 short hairpin RNA (i.e., Nox1 knockdown) [49].
2.9. Nox4
Nox4 knockout—but not Nox1 or Nox2 knockout—exerted protective effects against both transient and permanent focal ischemia; mice deficient in Nox4 were also protected from oxidative stress, BBB disruption, and neuronal apoptosis [50]. Surprisingly, in this study, photothrombosis-induced, not photothrombotic MCAO-induced, cortical infarction was attenuated in Nox4 knock-out mice when compared to wild-type mice. Nox4 is highly expressed in the endothelium and contributes to hydrogen peroxide (H2O2) formation [1,2,4]. Although H2O2—the dismutation product of O2−—elicits adverse effects on the vascular system (e.g., smooth muscle cell hypertrophy, metalloproteinase activation, and NOS inhibition, etc.), Nox4-derived H2O2 also induces positive endothelial effects. For example, Nox4 depletion attenuates ischemia-induced angiogenesis (femoral artery ligation) and exacerbates angiotensin II-induced vascular dysfunction [51].
2.10. Pericyte Nox4 in Focal Brain Ischemia
Pericytes have been recognized as a key component of the BBB or neurovascular unit [52]. Although endothelial cells primarily contribute to the formation of the BBB by expressing tight junction-related proteins (e.g., claudins and occludins) on their plasma membranes, high coverage ratios of pericytes to endothelial cells and the interaction between the two cell types are indispensable for maintaining the BBB [53]. In addition, the pericytes localized in pre-capillary arterioles may be responsible for the neuronal activation-coupled vasodilatation that increases CBF (i.e., neurovascular coupling) [54]. Therefore, dysfunction or functional loss of pericytes can directly disrupt the BBB and impair CBF regulation, thereby leading to neuronal dysfunction or death.
Among the Nox family proteins, cultured brain pericytes specifically express Nox4 [55]. Hypoxia is a significant up-regulator of Nox4 in the pericytes, as well as in other types of cells [55,56]. Several studies have reported a close association between hypoxia-inducible factor-1 and Nox4, suggesting that Nox4 functions at multiple sites, including both upstream and downstream of hypoxia-inducible factor-1 [57,58]. Another important target of Nox4 may be nuclear factor kappa B (NFκB). Previous studies have indicated that Nox4 overexpression leads to the activation of IκB kinase (IKK), which in turn induces the degradation of IκB and the subsequent stabilization and phosphorylation of NFκB in cultured brain pericytes [59]. Because NFκB is a pro-inflammatory transcription factor, microvascular cells with upregulated Nox4 expression may produce pro-inflammatory cytokines/chemokines, thereby enhancing inflammation. However, NFκB has also been observed to enhance the survival of microvascular cells under hypoxic conditions [60]. Furthermore, Nox4 overexpression enhances the proliferation of cultured pericytes, while Nox4 knockdown attenuates such proliferation [55].
Nox4 is expressed ubiquitously in various cell types; however, immunohistochemistry experiments have demonstrated that the levels of Nox4 expression in the brain are very low under physiological conditions [50,59]. Indeed, Nox4 was originally identified as one of the Nox isoforms specific for the kidney, which senses anemia and systemic hypoxia to produce erythropoietin under physiological conditions [61]. However, the expression of Nox4 is markedly upregulated in microvascular cells (including pericytes), particularly in peri-infarct areas, with peak expression occurring approximately 3–5 days after the onset of both permanent and transient MCAO [59]. Interestingly, the extent of Nox4 upregulation was greater in permanent than in transient MCAO with 1–2 h of ischemia [59], which may support the notion that Nox4 is upregulated in response to hypoxia/ischemia [56].
As mentioned above, research has demonstrated that infarct volumes were smaller and accompanied by decreased BBB leakage and neuronal apoptosis in systemic Nox4 knockout mice than in wild-type mice after both permanent and transient MCAO [50]. In this study, Nox4 deletion had a greater impact on phenotypic changes (i.e., reduction of neuronal damages after MCAO) than deletion of Nox1 or Nox2 in mice [50]. Furthermore, we demonstrated that infarct volume was significantly larger with enhanced BBB breakdown in mice with pericyte-specific overexpression of Nox4 (Tg-Nox4) than in their littermate controls in a permanent MCAO model [59]. These findings indicate that upregulation of Nox4 in microvascular pericytes exerts a detrimental effect in the acute phase of ischemic stroke. This study reported that the activation of NFκB in the ischemic hemisphere following permanent MCAO was significantly greater in Tg-Nox4 mice than in wild-type controls, as predicted by in vitro experiments, and enzymatic activity of matrix metalloproteinase-9 (MMP-9)—known to be involved in the breakdown of the BBB—was significantly greater and prolonged in Tg-Nox4 [59]. Thus, Nox4-mediated increases in the activation of NFκB may account in part for the phenotypic changes observed in Tg-Nox4 mice. In addition, previous researchers have suggested that reactive oxygen species may cause contraction of pericytes, thereby impairing capillary reperfusion after ischemia and aggravating brain ischemia [62]. It is also possible that Nox4 in pericytes is associated with ROS-induced capillary constriction during ischemia-reperfusion.
MMP-9 activation occurs biphasically following acute brain ischemia, while immediate MMP-9 activation occurs within one day after ischemic stroke due to the infiltration of inflammatory cells, and delayed activation is derived from vascular cells in the brain, contributing to angiogenesis and subsequent tissue repair [63]. Based on the profile of Nox4 expression in the ischemic brain [59], pericyte-Nox4 may participate in the latter process. For example, in a hindlimb ischemia model, Nox4 overexpression in endothelial cells promoted the angiogenesis and beneficial restoration of blood flow in the chronic phase [64]. Poststroke fibrotic changes within infarct areas occurring sequentially after angiogenesis may play important roles in the tissue repair process leading to functional recovery [53,54,55,56,57,58,59,60,61,62,63,64,65,66,67]. The involvement of Nox4 in fibrotic responses has been demonstrated in some diseases out of central nervous system [68]. Thus, there is a possibility that microvascular Nox4 may promote angiogenic and/or fibrotic responses in a chronic phase even in brain ischemia. In this context, the effects of Nox4 may be similar to those of vascular endothelial growth factor (VEGF) during brain ischemia, although VEGF is established as an angiogenic and neuroprotective molecule in the brain, it appeared to function detrimentally in the very acute phases of brain ischemia due to enhancement of BBB breakdown [69]. These findings indicate that the effects of Nox4 may vary in different organs or tissues and according to the type of injury or timing of disease states.
2.11. Stroke-Prone Spontaneously Hypertensive Rats (SHRSP) with Loss-of-Function in Nox
As mentioned above, although knockout of Nox attenuated focal ischemic injury in most but not all studies, the mechanisms by which Nox attenuates ischemic injury are not clear. Nox may exert direct effects on stroke outcome by increasing oxidative stress at the site of ischemia, or indirect effects by modifying physiological variables such as blood pressure and/or cerebral blood flow (CBF) [19]. In particular, blood pressure levels and antihypertensive therapy critically affects CBF via collateral and infarct size in hypertensive rats [70,71]. Because measurement of physiological variables and CBF in mice is often difficult due to their small body size, we attempted to construct a new rat model lacking Nox activities. The Matsumoto Eosinophilia Shinshu (MES) rat was found to harbor a deletion of four nucleotides in Intron 4 of the cytochrome b-245, alpha polypeptide (Cyba) gene, which resulted in an abnormal splicing [72]. As Cyba encodes p22phox, this strain lacked expression of the p22phox protein and showed loss of Nox activities. By introgressing the mutated Cyba gene from the MES strain, we constructed a congenic strain (referred to as SP.MES hereafter). Because Nox1-4 need to form heterodimers with the membrane-bound p22phox subunit [1], SP.MES lacks Nox activities on the background of the SHRSP. Although we have reported the results of photothrombotic distal MCAO in this new congenic strain [73], the findings are summarized below.
Resting MABP in SP.MES decreased slightly but significantly compared with that in the control PM0/SHRSP. In our previous study, CBF decreased to 37 ± 13% in SP.MES and 35 ± 17% in PM0/SHRSP at 10 min after MCA occlusion. The changes in CBF from 10 to 60 min after MCA occlusion were 7 ± 8% and −4 ± 8% in SP.MES and PM0/SHRSP, respectively (p = 0.006). The distal MCA pattern was more complex in SP.MES when compared to PM0/SHRSP. Although infarct volume in SP.MES was not significantly different from that in the PM0/SHRSP (89 ± 39 mm3 vs. 83 ± 35 mm3, respectively), we had revealed that the infarct volume with complex MCA had been larger than that in simple MCA [33]. We then adjusted infarct volume for the branching pattern as a covariate in an analysis of covariance; the adjusted mean of infarct volume was significantly smaller in SP.MES than in PM0/SHRSP (67 (95% CI 46 to 87) mm3 vs. 100 (95% CI 82 to 118) mm3, p = 0.032).
Functional loss of Nox activity associated with the absence of the p22phox protein did not mitigate the size of infarction produced by distal MCA occlusion as mentioned above. We speculated that decreased superoxide production and lower resting blood pressure would decrease the infarct size in SP.MES, whereas more complex MCA—as observed in SP.MES—would result in larger infarction. These divergent phenotypic effects caused by Nox dysfunction may explain the similarity in infarct size between SP.MES and PM0/SHRSP. Such divergence represents a major limitation of knockout strategies in stroke research (Figure 2).
2.12. Nox and Branching Morphogenesis
The distal MCA is considered to be more complicated when the number of classification of the distal MCA pattern increases from Type I to Type VII (i.e., more branches accompanied by tortuosity) [33]. In our previous study, we observed that distal MCA patterns were more complex in SP.MES (median = 3, interquartile range (IQR) = 3–5) than in PM0/SHRSP (median = 2, IQR = 1–3), indicating that oxidative stress may alter complex MCA into simple MCA. In a retrospective analysis of the distal MCA, simple patterns were more prevalent in SHRSP, while more complicated patterns were observed in WKY; SHR exhibited intermediate features relative to those observed between SHRSP and WKY. Because SHR were initially obtained by selective inbreeding of WKY with the highest blood pressure, and SHRSP were established from the A substrain of the SHR, these three substrains are genetically very close [19]. Nonetheless, distal MCA patterns among these three substrains were considerably different.
The vascular system is probably the most prominent instance of hierarchical tubular network characterized by a single topological feature (i.e., branching) [74,75]. To our knowledge, a direct involvement of reactive oxygen species or of the Nox activity has not been suggested in the context of branching morphogenesis of blood vessels. Reactive oxygen species exert profound effects on vascular smooth muscle cell growth and migration. Earlier studies have indicated that angiotensin II infusion significantly increased aortic hypertrophy in transgenic mice overexpressing Nox1 when compared with wild-type littermates [76]. Nox4-derived reactive oxygen species have also proven critical to the maintenance of the differentiated phenotype of vascular smooth muscle cells [77]. A theory—age-related loss of complexity—was proposed based on nonlinear analyses [i.e., fractal-like (self-similar) branching architecture] [78]. Interestingly, decreased branching complexity of retinal vessels was associated with increasing age and lacunar infarction in stroke patients, which suggested that more pathological status brought less complex blood vessels or loss of complexity [79]. SHRSP dominantly showed “loss of complexity” of MCA compared with its normotensive counterpart (WKY). Since it is widely believed that SHRSP had a higher level of the oxidative stress [80], the hypothesis seems intuitively attractive that increased levels of oxidative stress in SHRSP induced the simple pattern of MCA.
2.13. Effects of Nox on Blood Pressure
Under physiological conditions, superoxide and hydrogen peroxide produced by Nox participate in cardiovascular regulation [81]. Physiological variables such as blood pressure are critically important in experimental stroke; for example, congenic removal of a blood pressure quantitative trait locus (decreased blood pressure by 12% or −29 mmHg) attenuated infarct size produced by MCAO [70]. Our previous study mentioned above showed slightly but significantly decreased blood pressure in SP.MES (i.e., Nox dysfunction due to the absence of p22phox protein) when compared with SHRSP/PM0, suggesting that decreased levels of resting or pre-stroke blood pressure in hypertensive rats would attenuate infarct size produced by MCAO [73]. Therefore, decreases in blood pressure due to the absence of p22phox may explain at least in part the protective effects against focal ischemic injury observed in SP.MES. Nox is an important sources of reactive oxygen species in blood pressure regulation. Although Nox1 was involved in the late phase of the response to angiotensin II (i.e., after seven days of continuous infusion of angiotensin II), no significant difference in baseline blood pressure was observed between Nox1 knockout and wild-type mice [82,83]. While one study reported that baseline blood pressure was lower in Nox2 knockout mice than in wild-type mice [84], another study reported that Nox2 deficiency did not protect against the development of hypertension in another study [85]. Nox2 is expected to impair NO-mediated relaxation and lead to vascular contraction, while endothelial Nox4 enhances vasodilatation and reduce blood pressure [86]. These results highlight the complexities regarding the interactions between Nox activity and blood pressure regulation. Furthermore, one of the authors’ group reported that blood pressure response to glutamate microinjection into the rostral ventrolateral medulla was significantly greater in SHRSP than in SP.MES, SHR and WKY, suggesting that higher oxidative stress or Nox activity in the regulatory center of the sympathetic nerve activity in SHRSP [87].
3. Perspectives—Nox Knockout in Rats
As four NOX subtypes rely on p22phox to maintain their activity, interpretation of the observations on SP.MES described above is not straightforward. In this context, it is of interest to study phenotypes of SHRSP depleted with each Nox subtype. Recently, a groundbreaking method, genome editing using CRISPR/CAS9, has been introduced to modify genomes in any types of animals [88].
This method has many advantages such as high efficiency, low cost, and is less time-consuming. In addition, the most important feature for us is that it can be applied to any strains of rats, particularly genetic model rats for human diseases such as SHRSP.
To address the questions raised in above sections, we planned to construct SHRSP-depleted Nox subtypes using the CRISPR/CAS9 method. We have successfully established knockout lines each for Nox2 and Nox4, respectively. Preliminary results suggested that Nox2 and Nox4 knockout decreased and increased baseline blood pressure in SHRSP, respectively. We will employ these new knockout rats in MCAO experiments to reveal the roles of different Nox subtypes in the development of cerebral infarction.
4. Concluding Comments
Although distal MCAO in SHR is considered to be the best choice for an experimental model of human stroke, the suture model using knockout mice are widely utilized to assess the effects of Nox on stroke pathophysiology. However, due to the technical problems in the measurement of physiological variables or CBF in mice with their small body size, it remains unclear whether Nox affects the stroke outcome via direct increases in oxidative stress at the site of ischemia, or via indirect effects by modifying physiological variables such as blood pressure or CBF. Nonetheless, Nox knockout generally conferred protection against focal experimental stroke, suggesting that Nox exerts detrimental effects of Nox on stroke outcome. Notably, the pericyte-specific overexpression of Nox4 played a detrimental role in the acute phase of ischemic stroke by increasing the size of the infarct area and enhancing breakdown of the BBB.
To date, evidence further suggests that the effects of Nox isoforms on focal ischemic injury vary according to cell type (neuron, glia, microglia, blood vessels, or pericytes), the presence of ischemia with or without reperfusion, and disease state (acute or chronic). The absence of p22phox resulted in divergent effects (presumably beneficial vs. detrimental phenomena), representing a substantial limitation in experimental stroke research. As such, knockout strategies cannot provide a simple answer regarding whether a specific gene or protein is beneficial or detrimental for stroke. SHRSP with a higher level of the oxidative stress showed simple MCA pattern or “loss of complexity” of MCA compared with its normotensive counterpart. Based on this finding, we propose that increased levels of oxidative stress induce decreases in the complexity of the MCA.
Because hypertension is one of the major risk factors for stroke, the Nox-related mechanisms involved in blood pressure control are of great interest. Such Nox-related mechanisms in vascular morphogenesis and blood pressure control should be further examined in Nox isoform-specific “knockout rat” in SHRSP.
In conclusion, this review indicates that Nox is a potential therapeutic target in acute ischemic stroke. Furthermore, inhibition of Nox activity may play a role of stroke prevention through blood pressure lowering or attenuated vascular pathophysiology. The mechanisms by which Nox exert protection against stroke should be further elucidated.
Acknowledgments
This work was supported by Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Number 25461098 to Hiroshi Yao, 26461145 to Tetsuro Ago, 16H05439 to Takanari Kitazono and Tetsuro Ago. We would like to thank Editage (www.editage.jp) for English language editing. We thank Ms. Sachiko Kawasaki-Tsuchida for technical support.
Author Contributions
Hiroshi Yao conceived and designed this review. Hiroshi Yao, Tetsuro Ago, Takanari Kitazono, and Toru Nabika wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.W.; Wang, J.; Zhang, Q.; Wang, R.; Dhandapani, K.M.; Vadlamudi, R.K.; Brann, D.W. NADPH oxidase in brain injury and neurodegenerative disorders. Mol. Neurodegener. 2017, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Sorce, S.; Stocker, R.; Seredenina, T.; Holmdahl, R.; Aguzzi, A.; Chio, A.; Depaulis, A.; Heitz, F.; Olofsson, P.; Olsson, T.; et al. NADPH oxidases as drug targets and biomarkers in neurodegenerative diseases: What is the evidence? Free Radic. Biol. Med. 2017, 112, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Amanso, A.M.; Griendling, K.K. Differential roles of NADPH oxidases in vascular physiology and pathophysiology. Front. Biosci. 2012, 4, 1044–1064. [Google Scholar]
- Miller, A.A.; Drummond, G.R.; Schmidt, H.H.; Sobey, C.G. NADPH oxidase activity and function are profoundly greater in cerebral versus systemic arteries. Circ. Res. 2005, 97, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Ago, T.; Kitazono, T.; Ooboshi, H.; Iyama, T.; Han, Y.H.; Takada, J.; Wakisaka, M.; Ibayashi, S.; Utsumi, H.; Iida, M. Nox4 as the major catalytic component of an endothelial NADPH oxidase. Circulation 2004, 109, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Ago, T.; Kitazono, T.; Kuroda, J.; Kumai, Y.; Kamouchi, M.; Ooboshi, H.; Wakisaka, M.; Kawahara, T.; Rokutan, K.; Ibayashi, S.; et al. NADPH oxidases in rat basilar arterial endothelial cells. Stroke 2005, 36, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- Paravicini, T.M.; Chrissobolis, S.; Drummond, G.R.; Sobey, C.G. Increased NADPH-oxidase activity and Nox4 expression during chronic hypertension is associated with enhanced cerebral vasodilatation to NADPH in vivo. Stroke 2004, 35, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Sorby-Adams, A.J.; Marcoionni, A.M.; Dempsey, E.R.; Woenig, J.A.; Turner, R.J. The role of neurogenic inflammation in blood-brain barrier disruption and development of cerebral oedema following acute central nervous system (CNS) Injury. Int. J. Mol. Sci. 2017, 18, 1788. [Google Scholar] [CrossRef] [PubMed]
- Radermacher, K.A.; Wingler, K.; Kleikers, P.; Altenhöfer, S., Jr.; Hermans, J.; Kleinschnitz, C.; Schmidt, H.H.H.W. The 1027th target candidate in stroke: Will NADPH oxidase hold up? Exp. Transl. Stroke Med. 2012, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Astrup, J.; Siesjö, B.K.; Symon, L. Thresholds in cerebral ischemia—The ischemic penumbra. Stroke 1981, 12, 723–725. [Google Scholar] [CrossRef] [PubMed]
- Astrup, J.; Symon, L.; Branston, N.M.; Lassen, N.A. Cortical evoked potential and extracellular K+ and H+ at critical levels of brain ischemia. Stroke 1977, 8, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Belayev, L.; Ginsberg, M.D. Transient middle cerebral artery occlusion by intraluminal suture: II. Neurological deficits, and pixel-based correlation of histopathology with local blood flow and glucose utilization. J. Cereb. Blood Flow Metab. 1997, 17, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Hossmann, K.A. Viability thresholds and the penumbra of focal ischemia. Ann. Neurol. 1994, 36, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.Z.; Zhou, J.; Sun, W.; Huang, J.; van Zijl, P.C. Detection of the ischemic penumbra using pH-weighted MRI. J. Cereb. Blood Flow Metab. 2007, 27, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Warach, S.J.; Luby, M.; Albers, G.W.; Bammer, R.; Bivard, A.; Campbell, B.C.; Derdeyn, C.; Heit, J.J.; Khatri, P.; Lansberg, M.G.; et al. Acute Stroke Imaging Research Roadmap III Imaging Selection and Outcomes in Acute Stroke Reperfusion Clinical Trials. Stroke 2016, 47, 1389–1398. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Kudo, K.; Christensen, S.; Yamashita, F.; Goodwin, J.; Higuchi, S.; Ogawa, A. Penumbral imaging by using perfusion computed tomography and perfusion-weighted magnetic resonance imaging: Current concepts. J. Stroke Cerebrovasc. Dis. 2013, 22, 1212–1215. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.C.; Purushotham, A.; Christensen, S.; Desmond, P.M.; Nagakane, Y.; Parsons, M.W.; Lansberg, M.G.; Mlynash, M.; Straka, M.; de Silva, D.A.; et al. The infarct core is well represented by the acute diffusion lesion: Sustained reversal is infrequent. J. Cereb. Blood Flow Metab. 2012, 32, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Nabika, T. Standards and pitfalls of focal ischemia models in spontaneously hypertensive rats: With a systematic review of recent articles. J. Transl. Med. 2012, 10, 139. [Google Scholar] [CrossRef] [PubMed]
- Coyle, P.; Jokelainen, P.T. Differential outcome to middle cerebral artery occlusion in spontaneously hypertensive stroke-prone rats (SHRSP) and Wistar Kyoto (WKY) rats. Stroke 1983, 14, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Tamura, A.; Graham, D.I.; McCulloch, J.; Teasdale, G.M. Focal cerebral ischaemia in the rat: 1. Description of technique and early neuropathological consequences following middle cerebral artery occlusion. J. Cereb. Blood Flow Metab. 1981, 1, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Duverger, D.; MacKenzie, E.T. The quantification of cerebral infarction following focal ischemia in the rat: Influence of strain, arterial pressure, blood glucose concentration, and age. J. Cereb. Blood Flow Metab. 1988, 8, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, J.; Yoshida, Y.; Nakazawa, T.; Ooneda, G. Experimental studies of ischemic brain edema. I: A new experimental model of cerebral embolism in rats in which recirculation can be introduced in the ischemic area. Jpn. J. Stroke 1986, 8, 1–8. [Google Scholar] [CrossRef]
- Longa, E.Z.; Weinstein, P.R.; Carlson, S.; Cummins, R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989, 20, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Howells, D.W.; Porritt, M.J.; Rewell, S.S.; O’Collins, V.; Sena, E.S.; van der Worp, H.B.; Traystman, R.J.; Macleod, M.R. Different strokes for different folks: The rich diversity of animal models of focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2010, 30, 1412–1431. [Google Scholar] [CrossRef] [PubMed]
- Lakhan, S.E.; Kirchgessner, A.; Hofer, M. Inflammatory mechanisms in ischemic stroke: Therapeutic approaches. J. Transl. Med. 2009, 7, 97. [Google Scholar] [CrossRef] [PubMed]
- Kanemitsu, H.; Nakagomi, T.; Tamura, A.; Tsuchiya, T.; Kono, G.; Sano, K. Differences in the extent of primary ischemic damage between middle cerebral artery coagulation and intraluminal occlusion models. J. Cereb. Blood Flow Metab. 2002, 22, 1196–1204. [Google Scholar] [CrossRef] [PubMed]
- Watson, B.D.; Dietrich, W.D.; Busto, R.; Wachtel, M.S.; Ginsberg, M.D. Induction of reproducible brain infarction by photochemically initiated thrombosis. Ann. Neurol. 1985, 17, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Watson, B.D.; Dietrich, W.D.; Prado, R.; Nakayama, H.; Kanemitsu, H.; Futrell, N.; Yao, H.; Markgraf, C.G.; Wester, P. Concepts and techniques of experimental stroke induced by cerebrovascular photothrombosis. In Central Nervous System Trauma: Laboratory Techniques and Recent Advancement; Ohnishi, S.T., Ohnishi, T., Eds.; CRC Press: Boca Raton, FL, USA, 1995; pp. 169–194. [Google Scholar]
- Prado, R.; Watson, B.D.; Zhao, W.; Yao, H.; Busto, R.; Dietrich, W.D.; Ginsberg, M.D. l-arginine does not improve cortical perfusion or histopathological outcome in spontaneously hypertensive rats subjected to distal middle cerebral artery photothrombotic occlusion. J. Cereb. Blood Flow Metab. 1996, 16, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Sugimori, H.; Fukuda, K.; Takada, J.; Ooboshi, H.; Kitazono, T.; Ibayashi, S.; Iida, M. Photothrombotic middle cerebral artery occlusion and reperfusion laser system in spontaneously hypertensive rats. Stroke 2003, 34, 2716–2721. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Ibayashi, S.; Sugimori, H.; Fujii, K.; Fujishima, M. Simplified model of krypton laser-induced thrombotic distal middle cerebral artery occlusion in spontaneously hypertensive rats. Stroke 1996, 27, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Yao, H.; Ibayashi, S.; Uchimura, H.; Fujishima, M. Photothrombotic middle cerebral artery occlusion in spontaneously hypertensive rats: Influence of substrain, gender, and distal middle cerebral artery patterns on infarct size. Stroke 1998, 29, 1982–1986. [Google Scholar] [CrossRef] [PubMed]
- Watson, B.D.; Prado, R.; Veloso, A.; Brunschwig, J.P.; Dietrich, W.D. Cerebral blood flow restoration and reperfusion injury after ultraviolet laser-facilitated middle cerebral artery recanalization in rat thrombotic stroke. Stroke 2002, 33, 428–434. [Google Scholar] [CrossRef] [PubMed]
- De Silva, D.A.; Brekenfeld, C.; Ebinger, M.; Christensen, S.; Barber, P.A.; Butcher, K.S.; Levi, C.R.; Parsons, M.W.; Bladin, C.F.; Donnan, G.A.; et al. Echoplanar Imaging Thrombolytic Evaluation Trial (EPITHET) Investigators: The benefits of intravenous thrombolysis relate to the site of baseline arterial occlusion in the Echoplanar Imaging Thrombolytic Evaluation Trial (EPITHET). Stroke 2010, 41, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Okada, Y.; Ibayashi, S. Therapeutic time window for YAG laser-induced reperfusion of thrombotic stroke in hypertensive rats. Neuroreport 2002, 13, 1005–1008. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Yoshii, N.; Akira, T.; Nakahara, T. Reperfusion-induced temporary appearance of therapeutic window in penumbra after 2 h of photothrombotic middle cerebral artery occlusion in rats. J. Cereb. Blood Flow Metab. 2009, 29, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Walder, C.E.; Green, S.P.; Darbonne, W.C.; Mathias, J.; Rae, J.; Dinauer, M.C.; Curnutte, J.T.; Thomas, G.R. Ischemic stroke injury is reduced in mice lacking a functional NADPH oxidase. Stroke 1997, 28, 2252–2258. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Song, Y.S.; Chan, P.H. Inhibition of NADPH oxidase is neuroprotective after ischemia-reperfusion. J. Cereb. Blood Flow Metab. 2009, 29, 1262–1272. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Kim, G.S.; Okami, N.; Narasimhan, P.; Chan, P.H. NADPH oxidase is involved in post-ischemic brain inflammation. Neurobiol. Dis. 2011, 42, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Jackman, K.A.; Miller, A.A.; de Silva, T.M.; Crack, P.J.; Drummond, G.R.; Sobey, C.G. Reduction of cerebral infarct volume by apocynin requires pretreatment and is absent in Nox2-deficient mice. Br. J. Pharmacol. 2009, 156, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.A.; Brait, V.H.; Lee, S.; de Silva, T.M.; Diep, H.; Eisenhardt, A.; Drummond, G.R.; Sobey, C.G. Brain infarct volume after permanent focal ischemia is not dependent on Nox2 expression. Brain Res. 2012, 1483, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Brait, V.H.; Jackman, K.A.; Walduck, A.K.; Selemidis, S.; Diep, H.; Mast, A.E.; Guida, E.; Broughton, B.R.; Drummond, G.R.; Sobey, C.G. Mechanisms contributing to cerebral infarct size after stroke: Gender, reperfusion, T lymphocytes, and Nox2-derived superoxide. J. Cereb. Blood Flow Metab. 2010, 30, 1306–1317. [Google Scholar] [CrossRef] [PubMed]
- De Silva, T.M.; Brait, V.H.; Drummond, G.R.; Sobey, C.G.; Miller, A.A. Nox2 oxidase activity accounts for the oxidative stress and vasomotor dysfunction in mouse cerebral arteries following ischemic stroke. PLoS ONE 2011, 6, e28393. [Google Scholar] [CrossRef] [PubMed]
- Kahles, T.; Luedike, P.; Endres, M.; Galla, H.J.; Steinmetz, H.; Busse, R.; Neumann-Haefelin, T.; Brandes, R.P. NADPH oxidase plays a central role in blood-brain barrier damage in experimental stroke. Stroke 2007, 38, 3000–3006. [Google Scholar] [CrossRef] [PubMed]
- McCann, S.K.; Dusting, G.J.; Roulston, C.L. Nox2 knockout delays infarct progression and increases vascular recovery through angiogenesis in mice following ischaemic stroke with reperfusion. PLoS ONE 2014, 9, e110602. [Google Scholar] [CrossRef] [PubMed]
- Jackman, K.A.; Miller, A.A.; Drummond, G.R.; Sobey, C.G. Importance of NOX1 for angiotensin II-induced cerebrovascular superoxide production and cortical infarct volume following ischemic stroke. Brain Res. 2009, 1286, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Kahles, T.; Kohnen, A.; Heumueller, S.; Rappert, A.; Bechmann, I.; Liebner, S.; Wittko, I.M.; Neumann-Haefelin, T.; Steinmetz, H.; Schroeder, K.; et al. NADPH oxidase Nox1 contributes to ischemic injury in experimental stroke in mice. Neurobiol. Dis. 2010, 40, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.H.; Kim, J.H.; Lee, K.H.; Kim, H.Y.; Kim, Y.S.; Choi, W.S.; Lee, J. Role of neuronal NADPH oxidase 1 in the peri-infarct regions after stroke. PLoS ONE 2015, 10, e0116814. [Google Scholar] [CrossRef] [PubMed]
- Kleinschnitz, C.; Grund, H.; Wingler, K.; Armitage, M.E.; Jones, E.; Mittal, M.; Barit, D.; Schwarz, T.; Geis, C.; Kraft, P.; et al. Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol. 2010, 8, e1000479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schröder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Schröder, K.; Weissmann, N.; Brandes, R.P. Organizers and activators: Cytosolic Nox proteins impacting on vascular function. Free Radic. Biol. Med. 2017, 109, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Genove, G.; Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Attwell, D.; Mishra, A.; Hall, C.N.; O’Farrell, F.M.; Dalkara, T. What is a pericyte? J. Cereb. Blood Flow Metab. 2016, 36, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, J.; Ago, T.; Nishimura, A.; Nakamura, K.; Matsuo, R.; Wakisaka, Y.; Kamouchi, M.; Kitazono, T. Nox4 is a major source of superoxide production in human brain pericytes. J. Vasc. Res. 2014, 51, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Ago, T.; Kuroda, J.; Kamouchi, M.; Sadoshima, J.; Kitazono, T. Pathophysiological roles of NADPH oxidase/nox family proteins in the vascular system.-Review and perspective. Circ. J. 2011, 75, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Bonello, S.; Zahringer, C.; BelAiba, R.S.; Djordjevic, T.; Hess, J.; Michiels, C.; Kietzmann, T.; Gorlach, A. Reactive oxygen species activate the HIF-1α promoter via a functional NFκB site. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Diebold, I.; Petry, A.; Hess, J.; Gorlach, A. The NADPH oxidase subunit NOX4 is a new target gene of the hypoxia-inducible factor-1. Mol. Biol. Cell 2010, 21, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, A.; Ago, T.; Kuroda, J.; Arimura, K.; Tachibana, M.; Nakamura, K.; Wakisaka, Y.; Sadoshima, J.; Iihara, K.; Kitazono, T. Detrimental role of pericyte Nox4 in the acute phase of brain ischemia. J. Cereb. Blood Flow Metab. 2016, 36, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Irani, K. Oxidant Signaling in Vascular Cell Growth, Death, and Survival: A Review of the Roles of Reactive Oxygen Species in Smooth Muscle and Endothelial Cell Mitogenic and Apoptotic Signaling. Circ. Res. 2000, 87, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Shiose, A.; Kuroda, J.; Tsuruya, K.; Hirai, M.; Hirakata, H.; Naito, S.; Hattori, M.; Sakaki, Y.; Sumimoto, H. A novel superoxide-producing NADPH oxidase in kidney. J. Biol. Chem. 2001, 276, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Yemisci, M.; Gursoy-Ozdemir, Y.; Vural, A.; Can, A.; Topalkara, K.; Dalkara, T. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat. Med. 2009, 15, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Jickling, G.C.; Liu, D.; Stamova, B.; Ander, B.P.; Zhan, X.; Lu, A.; Sharp, F.R. Hemorrhagic transformation after ischemic stroke in animals and humans. J. Cereb. Blood Flow Metab. 2014, 34, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Craige, S.M.; Chen, K.; Pei, Y.; Li, C.; Huang, X.; Chen, C.; Shibata, R.; Sato, K.; Walsh, K.; Keaney, J.F., Jr. NADPH oxidase 4 promotes endothelial angiogenesis through endothelial nitric oxide synthase activation. Circulation 2011, 124, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Goritz, C.; Dias, D.O.; Tomilin, N.; Barbacid, M.; Shupliakov, O.; Frisen, J. A pericyte origin of spinal cord scar tissue. Science 2011, 333, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Makihara, N.; Arimura, K.; Ago, T.; Tachibana, M.; Nishimura, A.; Nakamura, K.; Matsuo, R.; Wakisaka, Y.; Kuroda, J.; Sugimori, H.; et al. Involvement of platelet-derived growth factor receptor beta in fibrosis through extracellular matrix protein production after ischemic stroke. Exp. Neurol. 2015, 264, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Ago, T.; Wakisaka, Y.; Kuroda, J.; Shijo, M.; Yoshikawa, Y.; Komori, M.; Nishimura, A.; Makihara, N.; Nakamura, K.; et al. Early reperfusion after brain ischemia has beneficial effects beyond rescuing neurons. Stroke 2017, 48, 2222–2230. [Google Scholar] [CrossRef] [PubMed]
- Hecker, L.; Vittal, R.; Jones, T.; Jagirdar, R.; Luckhardt, T.R.; Horowitz, J.C.; Pennathur, S.; Martinez, F.J.; Thannickal, V.J. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009, 15, 1077–1081. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, L.; Jiang, Q.; Zhang, R.; Davies, K.; Powers, C.; Bruggen, N.; Chopp, M. VEGF enhances angiogenesis and promotes blood–brain barrier leakage in the ischemic brain. J. Clin. Investig. 2000, 106, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Cui, Z.H.; Masuda, J.; Nabika, T. Congenic removal of a QTL for blood pressure attenuates infarct size produced by middle cerebral artery occlusion in hypertensive rats. Physiol. Genom. 2007, 30, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Fujii, K.; Weno, B.L.; Baumbach, G.L.; Heistad, D.D. Effect of antihypertensive treatment on focal cerebral infarction. Hypertension 1992, 19, 713–716. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Li, G.; Hashimoto, M.; Nishio, A.; Tomozawa, H.; Suzuki, N.; Usami, S.; Higuchi, K.; Matsumoto, K. Pivotal Advance: Eosinophilia in the MES rat strain is caused by a loss-of-function mutation in the gene for cytochrome b(-245), alpha polypeptide (Cyba). J. Leukoc. Biol. 2009, 86, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Ferdaus, M.Z.; Zahid, H.M.; Ohara, H.; Nakahara, T.; Nabika, T. Focal ischemic injury with complex middle cerebral artery in stroke-prone spontaneously hypertensive rats with with loss-of-function in NADPH Oxidases. PLoS ONE 2015, 10, e0138551. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, A.; Simons, M. Branching morphogenesis. Circ. Res. 2009, 104, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Mettouchi, A. The role of extracellular matrix in vascular branching morphogenesis. Cell Adhes. Migr. 2012, 6, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.; Clempus, R.; Lassègue, B.; Cheng, G.; McCoy, J.; Dikalov, S.; San Martin, A.; Lyle, A.; Weber, D.S.; Weiss, D.; et al. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation 2005, 112, 2668–2676. [Google Scholar] [CrossRef] [PubMed]
- Clempus, R.E.; Sorescu, D.; Dikalova, A.E.; Pounkova, L.; Jo, P.; Sorescu, G.P.; Schmidt, H.H.; Lassègue, B.; Griendling, K.K. Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Lipsitz, L.A.; Goldberger, A.L. Loss of “complexity” and aging. Potential applications of fractals and chaos theory to senescence. JAMA 1992, 267, 1806–1809. [Google Scholar] [CrossRef] [PubMed]
- Doubal, F.N.; MacGillivray, T.J.; Patton, N.; Dhillon, B.; Dennis, M.S.; Wardlaw, J.M. Fractal analysis of retinal vessels suggests that a distinct vasculopathy causes lacunar stroke. Neurology 2010, 74, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- Kerr, S.; Brosnan, M.J.; McIntyre, M.; Reid, J.L.; Dominiczak, A.F.; Hamilton, C.A. Superoxide anion production is increased in a model of genetic hypertension: Role of the endothelium. Hypertension 1999, 33, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Lassègue, B.; San Martín, A.; Griendling, K.K. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ. Res. 2012, 110, 1364–1390. [Google Scholar]
- Matsuno, K.; Yamada, H.; Iwata, K.; Jin, D.; Katsuyama, M.; Matsuki, M.; Takai, S.; Yamanishi, K.; Miyazaki, M.; Matsubara, H.; et al. Nox1 is involved in angiotensin II-mediated hypertension: A study in Nox1-deficient mice. Circulation 2005, 112, 2677–2685. [Google Scholar] [CrossRef] [PubMed]
- Landmesser, U.; Cai, H.; Dikalov, S.; McCann, L.; Hwang, J.; Jo, H.; Holland, S.M.; Harrison, D.G. Role of p47phox in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension 2002, 40, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.D.; Xu, S.; Johns, D.G.; Du, Y.; Quinn, M.T.; Cayatte, A.J.; Cohen, R.A. Role of NADPH oxidase in the vascular hypertrophic and oxidative stress response to angiotensin II in mice. Circ. Res. 2001, 88, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Mercure, C.; He, Y.; Javeshghani, D.; Yao, G.; Callera, G.E.; Yogi, A.; Lochard, N.; Reudelhuber, T.L. Angiotensin II-dependent chronic hypertension and cardiac hypertrophy are unaffected by gp91phox-containing NADPH oxidase. Hypertension 2005, 45, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.; Murdoch, C.E.; Wang, M.; Santos, C.X.; Zhang, M.; Alom-Ruiz, S.; Anilkumar, N.; Ouattara, A.; Cave, A.C.; Walker, S.J.; et al. Endothelial Nox4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1368–1376. [Google Scholar] [CrossRef] [PubMed]
- Zahid, H.M.; Ferdaus, M.Z.; Ohara, H.; Isomura, M.; Nabika, T. Effect of p22phox depletion on sympathetic regulation of blood pressure in SHRSP: Evaluation in a new congenic strain. Sci. Rep. 2016, 6, 36739. [Google Scholar] [CrossRef] [PubMed]
- Geurts, A.M.; Moreno, C. Zinc-finger nucleases: New strategies to target the rat genome. Clin. Sci. 2010, 119, 303–311. [Google Scholar] [CrossRef] [PubMed]
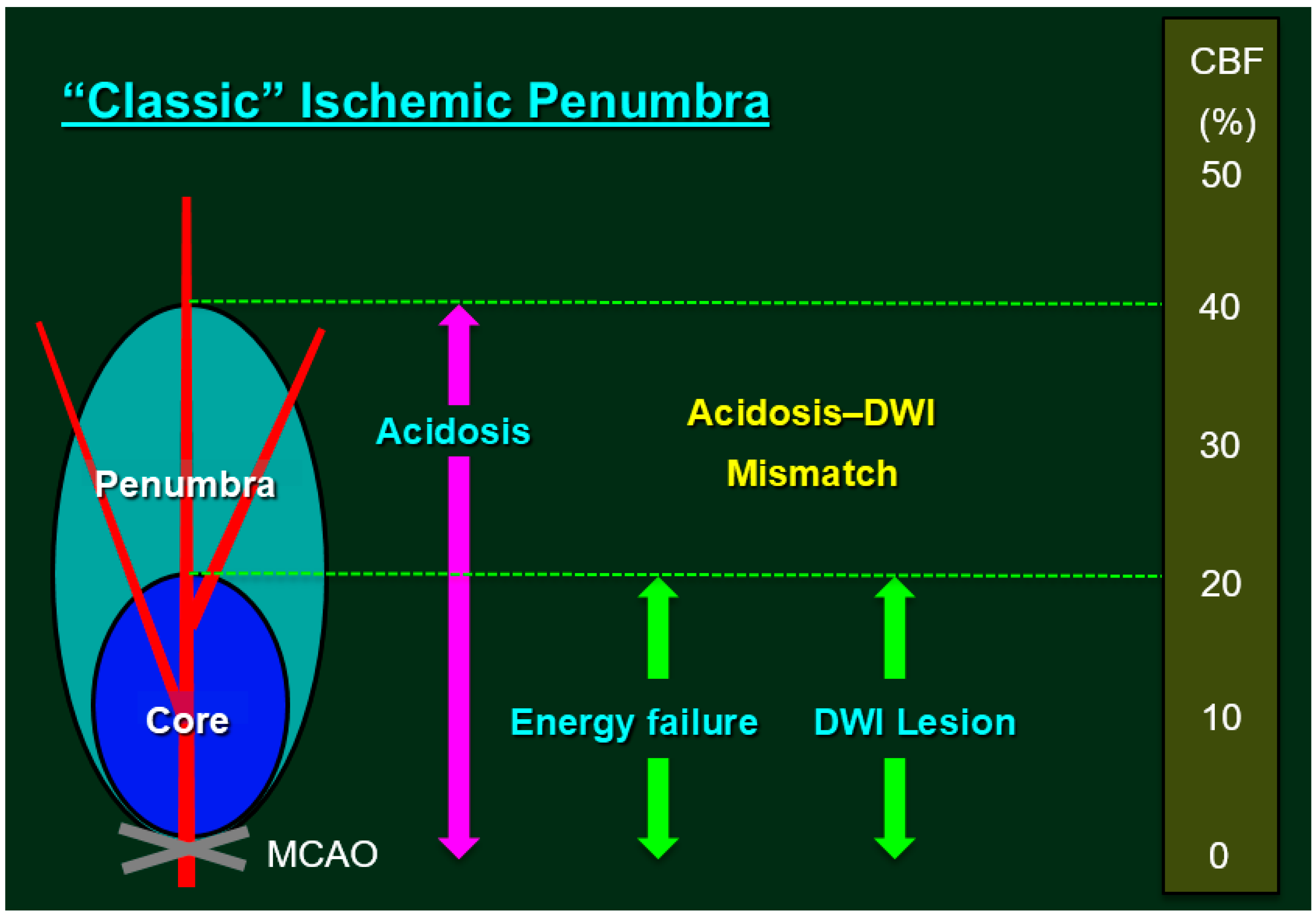
Figure 1.
Classic Ischemic Penumbra. The ischemic penumbra is classically defined as an ischemic brain region with cerebral blood flow (CBF) between the upper threshold of electrical silence and the lower threshold of energy and ion pump failure [11,12]; the green dashed lines indicate the upper and the lower thresholds. Regions exhibiting CBF in the ischemic core range (CBF <20% of control values) had a 96% probability of undergoing infarction, while zones with higher CBF (>40% of control) were largely spared from infarction [13]. Under experimental conditions, the most reliable method for localizing the ischemic core involves inducing a loss of adenosine triphosphate (ATP), although tissue acidosis can be used as a biochemical marker of the ischemic core plus the penumbra [14]. Research has demonstrated that pH-weighted magnetic resonance imaging and diffusion-weighted imaging (DWI) mismatch (i.e., cytotoxic edema due to ATP depletion) can provide a more comprehensive zone of penumbra [15]. In routine clinical settings, however, lesions detected via early DWI can be used to define the ischemic core, while the adjacent moderately ischemic brain tissue can be identified as the penumbra via perfusion-weighted imaging (i.e., perfusion-weighted imaging/DWI mismatch as a surrogate marker for penumbra) [16,17]. Previous studies have demonstrated that, even after early reperfusion with tissue plasminogen activator, sustained reversal of diffusion abnormalities was minimal, indicating that the infarct core is well represented by the acute DWI lesion [18].
Figure 1.
Classic Ischemic Penumbra. The ischemic penumbra is classically defined as an ischemic brain region with cerebral blood flow (CBF) between the upper threshold of electrical silence and the lower threshold of energy and ion pump failure [11,12]; the green dashed lines indicate the upper and the lower thresholds. Regions exhibiting CBF in the ischemic core range (CBF <20% of control values) had a 96% probability of undergoing infarction, while zones with higher CBF (>40% of control) were largely spared from infarction [13]. Under experimental conditions, the most reliable method for localizing the ischemic core involves inducing a loss of adenosine triphosphate (ATP), although tissue acidosis can be used as a biochemical marker of the ischemic core plus the penumbra [14]. Research has demonstrated that pH-weighted magnetic resonance imaging and diffusion-weighted imaging (DWI) mismatch (i.e., cytotoxic edema due to ATP depletion) can provide a more comprehensive zone of penumbra [15]. In routine clinical settings, however, lesions detected via early DWI can be used to define the ischemic core, while the adjacent moderately ischemic brain tissue can be identified as the penumbra via perfusion-weighted imaging (i.e., perfusion-weighted imaging/DWI mismatch as a surrogate marker for penumbra) [16,17]. Previous studies have demonstrated that, even after early reperfusion with tissue plasminogen activator, sustained reversal of diffusion abnormalities was minimal, indicating that the infarct core is well represented by the acute DWI lesion [18].
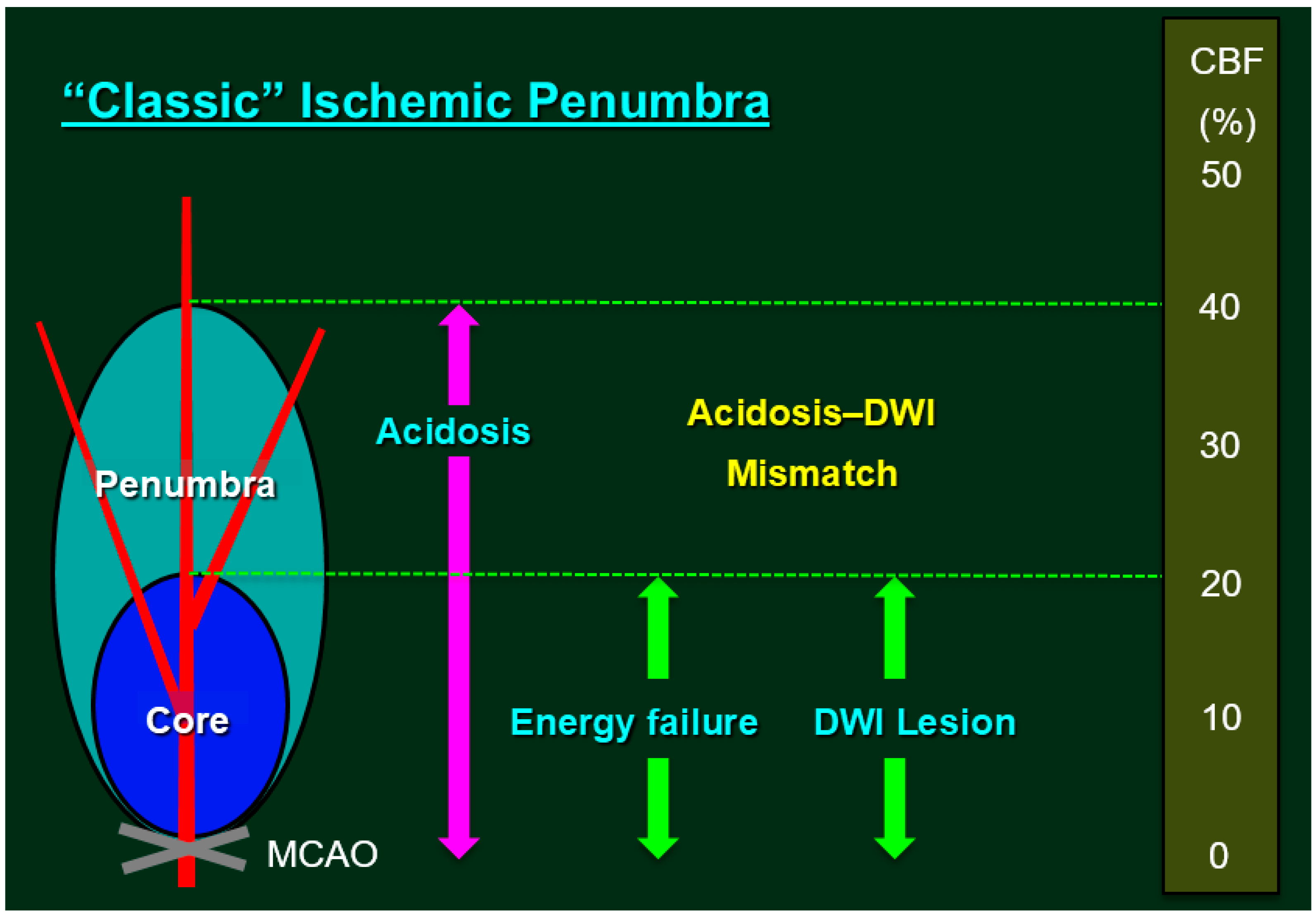
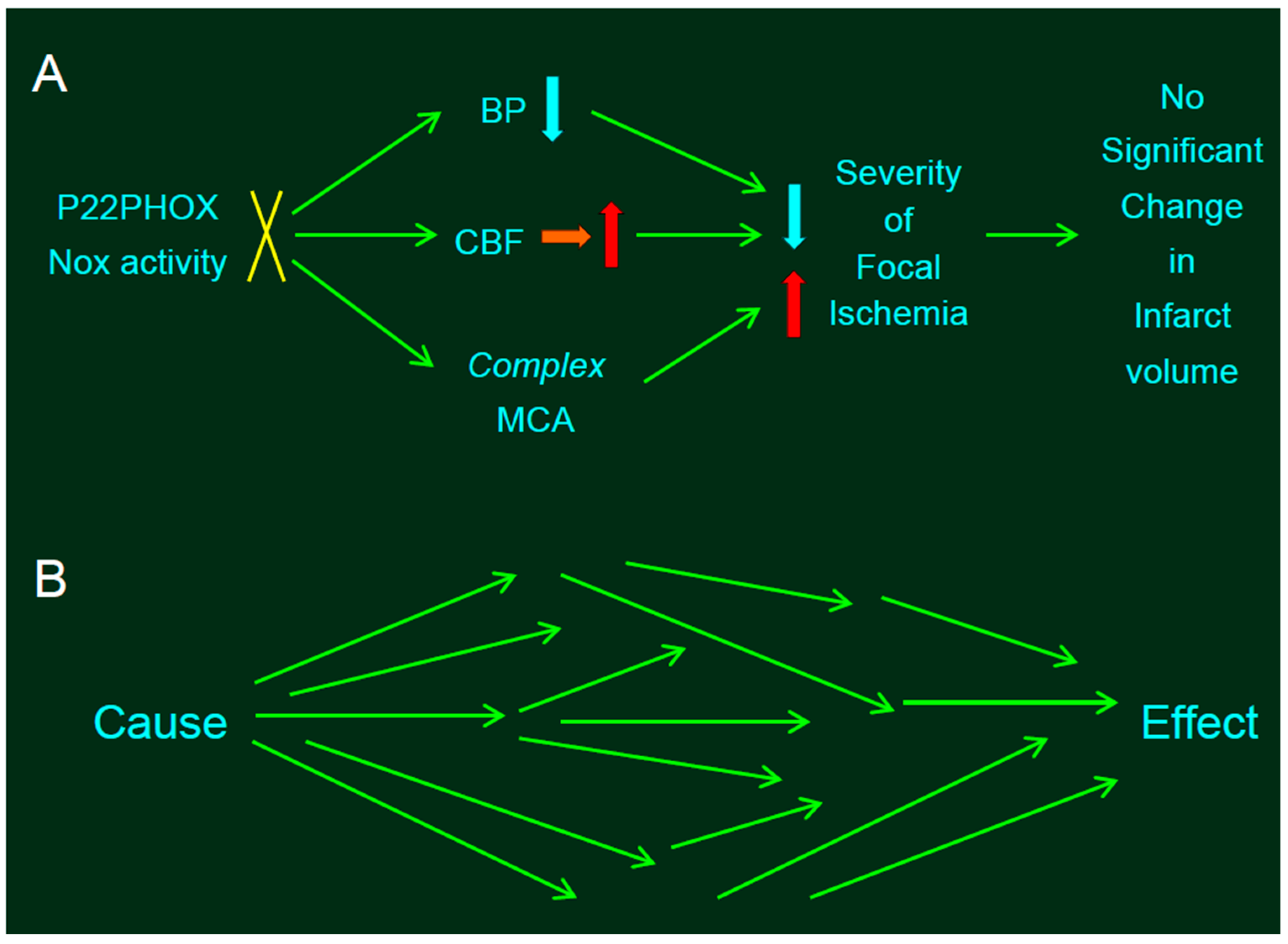
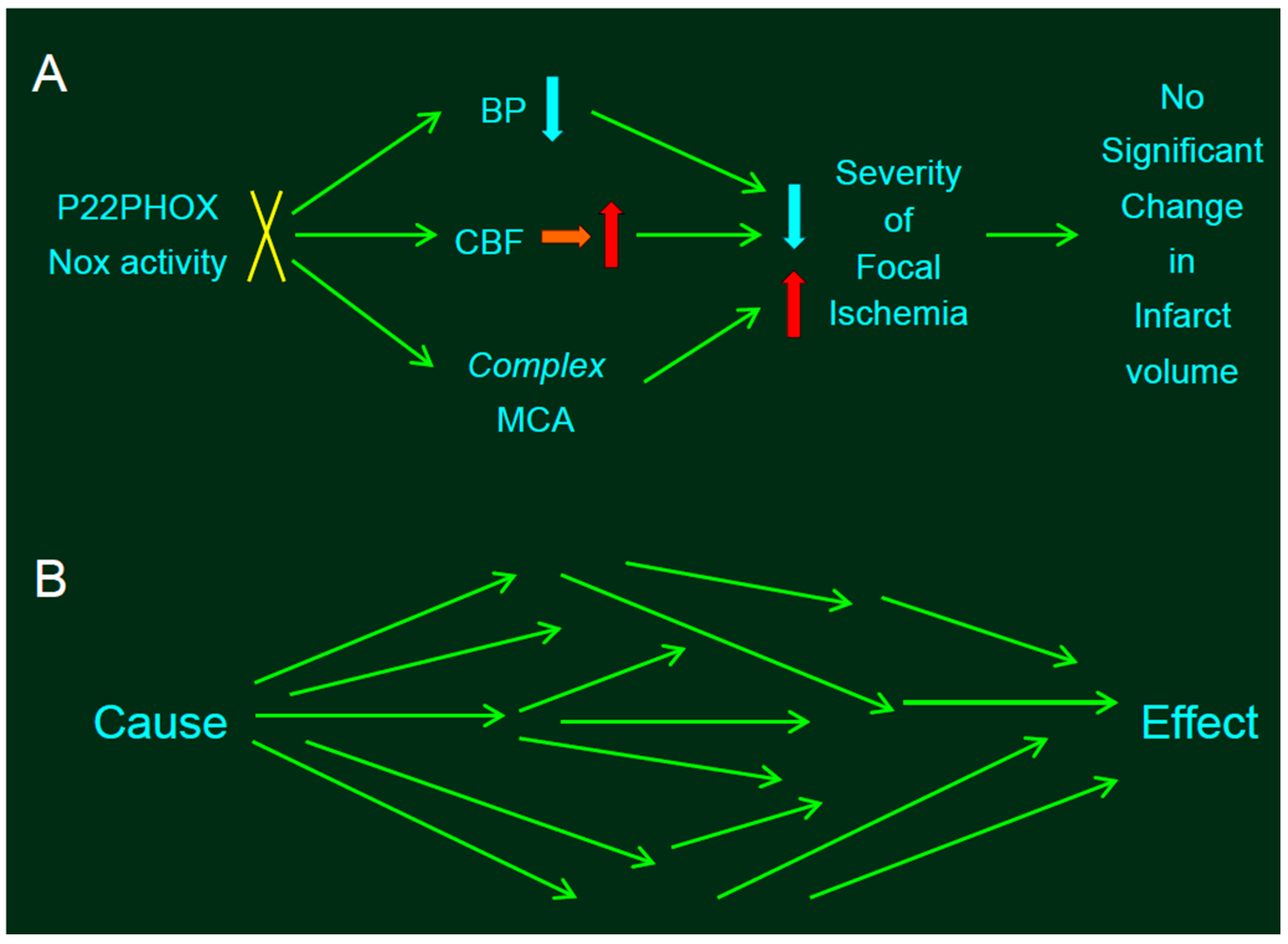
Figure 2.
Statistical problems in experiments involving genetic manipulation. (A) The absence of p22phox protein did not affect infarct size produced by distal middle cerebral artery occlusion (MCAO). Resting mean blood pressure (MABP) in SP.MES rats was decreased slightly but significantly relative to that observed in control PM0/SHRSP rats, along with mitigated CBF. Thus, slightly but significantly decreased resting blood pressure (BP)—along with mitigated CBF—would decrease the infarct size in SP.MES, whereas more complex MCA might have resulted in larger infarction. The blue, red, and orange arrows indicate decrease, increase, and no significant change, respectively. (B) Such divergent effects of genetic manipulation provoke a critical problem. If the experiments were adequately randomized, we can conclude that the effect—significantly different between the groups—was due to a single cause, because the background factors between the groups were same; in other words, no factor except for a single cause can be the reason for the difference between the groups (i.e., the single effect) after randomization. However, when a single cause (e.g., knockout of a gene) induced multiple influences converging into a single effect (e.g., infarct volume), this assumption is invalid.
Figure 2.
Statistical problems in experiments involving genetic manipulation. (A) The absence of p22phox protein did not affect infarct size produced by distal middle cerebral artery occlusion (MCAO). Resting mean blood pressure (MABP) in SP.MES rats was decreased slightly but significantly relative to that observed in control PM0/SHRSP rats, along with mitigated CBF. Thus, slightly but significantly decreased resting blood pressure (BP)—along with mitigated CBF—would decrease the infarct size in SP.MES, whereas more complex MCA might have resulted in larger infarction. The blue, red, and orange arrows indicate decrease, increase, and no significant change, respectively. (B) Such divergent effects of genetic manipulation provoke a critical problem. If the experiments were adequately randomized, we can conclude that the effect—significantly different between the groups—was due to a single cause, because the background factors between the groups were same; in other words, no factor except for a single cause can be the reason for the difference between the groups (i.e., the single effect) after randomization. However, when a single cause (e.g., knockout of a gene) induced multiple influences converging into a single effect (e.g., infarct volume), this assumption is invalid.

{kind=link}
{kind=link}
Table 1.
Experimental models of stroke.
| Models | Anatomic Sites of MCAO | Duration | Characteristics | Disadvantage |
|---|---|---|---|---|
| Distal MCAO | MCAO distal to rhinal fissure | P | Distal MCAO was studied for the first time in SHRSP, revealing increased stroke sensitivity by hypertension. | The infarct size and penumbra are too small to evaluate the effects of pharmacotherapeutic agents in normotensive rats. |
| Proximal MCAO | MCAO proximal to lenticulostriate arteries | P (T) | The subtemporal approach method have emerged as a standard method of proximal MCAO. | The procedure is surgically demanding and may induce local traumatic effects. |
| Intraluminal suture occlusion | MCA origin and the proximal segment of ACA | T (P) | The procedure is easy to perform, minimally invasive, and does not require craniectomy. | This model has a wide ischemic zone, and the mortality rate is high in the case of PO. |
| Photothrombotic MCAO | Photochemical MCAO distal to rhinal fissure | P T | Photothrombotic MCAO in SHR yields a highly reproducible infarct volume, and does not requireopening of the dura. | Same as mentioned for distal MCAO. |
MCAO, middle cerebral artery occlusion; MCA, middle cerebral artery; ACA, anterior cerebral artery; P, permanent occlusion; T, transient occlusion; SHRSP, stroke-prone spontaneously hypertensive rats.
Table 2.
Effects of Nox knock-outs on experimental stroke.
| Author Ref. | Year | Mice (WT: C57 Bl/6J) | T/P | BP | CBF | Nox Isoform | Outcome | Protection by KO | |
|---|---|---|---|---|---|---|---|---|---|
| Age | Sex | ||||||||
| Walder [36] | 1997 | 8–10 wk | m | T | NA | NA | Nox2 | Infarct volume was reduced by 46% in KO mice compared with WT mice. | Yes |
| Kahles [43] | 2007 | 7–9 wk | m | T | NA | NA | Nox2 | BBB disruption and lesion volume were largely attenuated in KO mice. | Yes |
| Chen [37] | 2009 | NA | m | T | NA | NS | Nox2 | Mean infarct volume was 106.2 mm3 in WT mice, and 52.0 mm3 in KO mice. | Yes |
| Jackman [39] | 2009 | 5–9 wk WT 8–12 wk KO | m m | T | NA | NS | Nox2 | Protection by apocynin was found in WT mice but not in KO mice. | Yes |
| Brait [41] | 2010 | 6–8 wk | m+f | T/P | NA | NS | Nox2 | The larger infarction in male mice was dependent on both reperfusion and NOX2. | Yes (female) |
| De Silva [42] | 2011 | NA | m | T | NA | NS | Nox2 | Smaller infarct volume was observed in KO mice than in WT mice. | Yes |
| Chen [38] | 2011 | 12–16 wk | m | T | NA | NA | Nox2 | Brain infarction was 35–44% less in KO mice compared with WT mice. | Yes |
| Kim [40] | 2012 | 8–12 wk | m | P | NA | NS | Nox2 | No protection by KO was found in the absence of reperfusion. | NA |
| MaCann [44] | 2014 | 2–3 mo | NA | T | NA | NS | Nox2 | KO showed transient nature of protection and increased revascularization. | Yes |
| Jackman [45] | 2009 | 11–17 wk | m | T | NA | NS | Nox1 | Cortical but not total infarct was increased in KO mice. | No |
| Kahles [46] | 2010 | NA | m + f | T/P | NA | NA | Nox1 | Infarct volume was reduced by 44% after 1 h but not 2 h and pMCAO in KO mice. | Yes |
| Kleinschnitz [48] | 2010 | 6–8 wk | m | T | NA | NA | Nox1 | Deletion of NOX4 but not NOX1 or NOX2 prevented focal ischemic injury. | No |
| 6–8 wk | m | T | NA | NA | Nox2 | No | |||
| 6–8 wk | m + f | T/P | NA | NA | Nox4 | Yes | |||
| 18–20 wk | m | T | NA | NA | Nox4 | Yes | |||
WT, wild type; BP, blood pressure; CBF, cerebral blood flow; KO, knock out; T/P MCAO, transient/permanent middle cerebral artery occlusion; m, male; f, female; NA, not available; BBB, blood brain barrier; NS, not significant.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yao, H.; Ago, T.; Kitazono, T.; Nabika, T. NADPH Oxidase-Related Pathophysiology in Experimental Models of Stroke. Int. J. Mol. Sci. 2017, 18, 2123. https://doi.org/10.3390/ijms18102123
AMA Style
Yao H, Ago T, Kitazono T, Nabika T. NADPH Oxidase-Related Pathophysiology in Experimental Models of Stroke. International Journal of Molecular Sciences. 2017; 18(10):2123. https://doi.org/10.3390/ijms18102123
Chicago/Turabian StyleYao, Hiroshi, Tetsuro Ago, Takanari Kitazono, and Toru Nabika. 2017. "NADPH Oxidase-Related Pathophysiology in Experimental Models of Stroke" International Journal of Molecular Sciences 18, no. 10: 2123. https://doi.org/10.3390/ijms18102123
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.