The Innate and Adaptive Immune System as Targets for Biologic Therapies in Inflammatory Bowel Disease

 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Components of the Innate Immune System
3. Components of the Adaptive Immune System
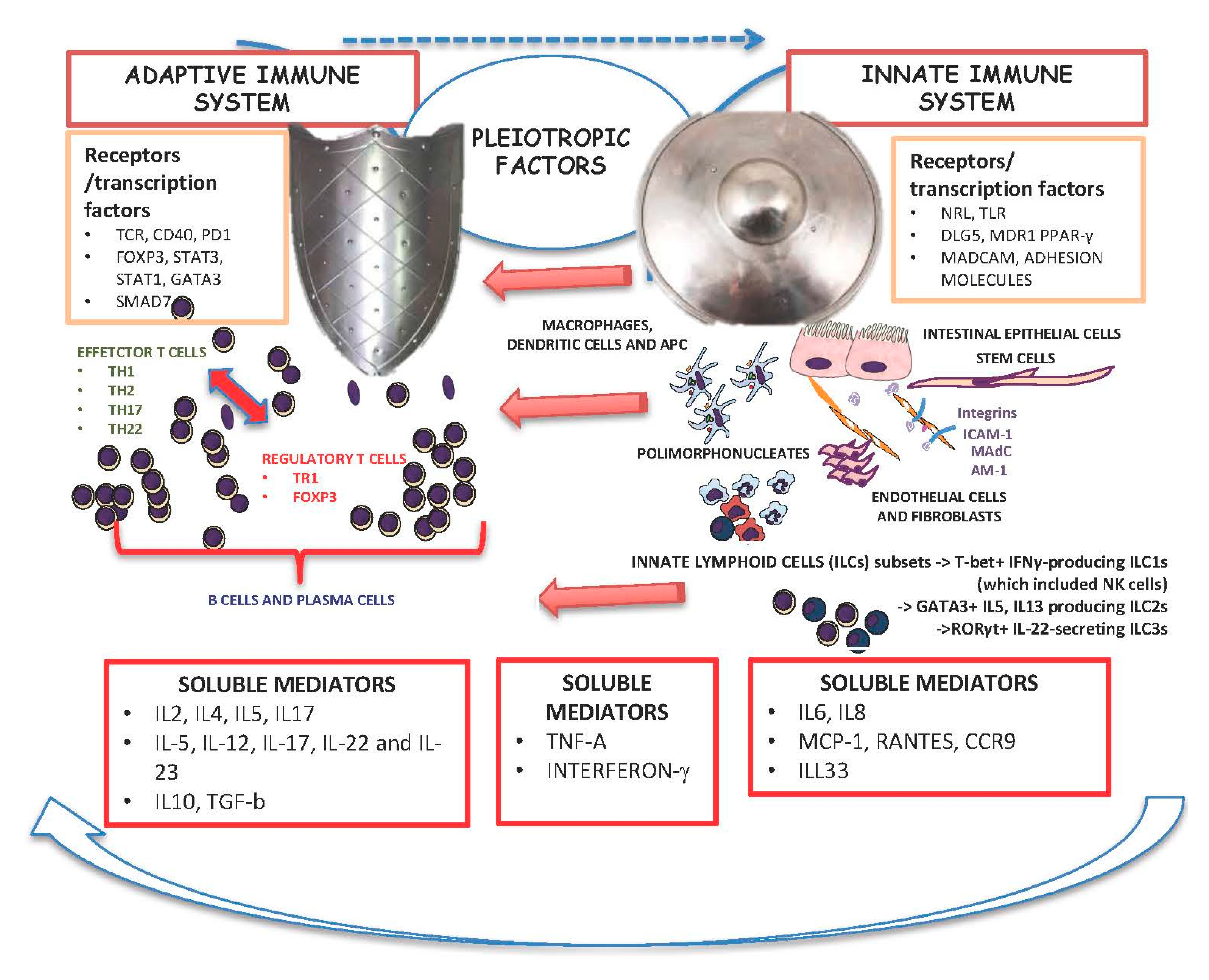
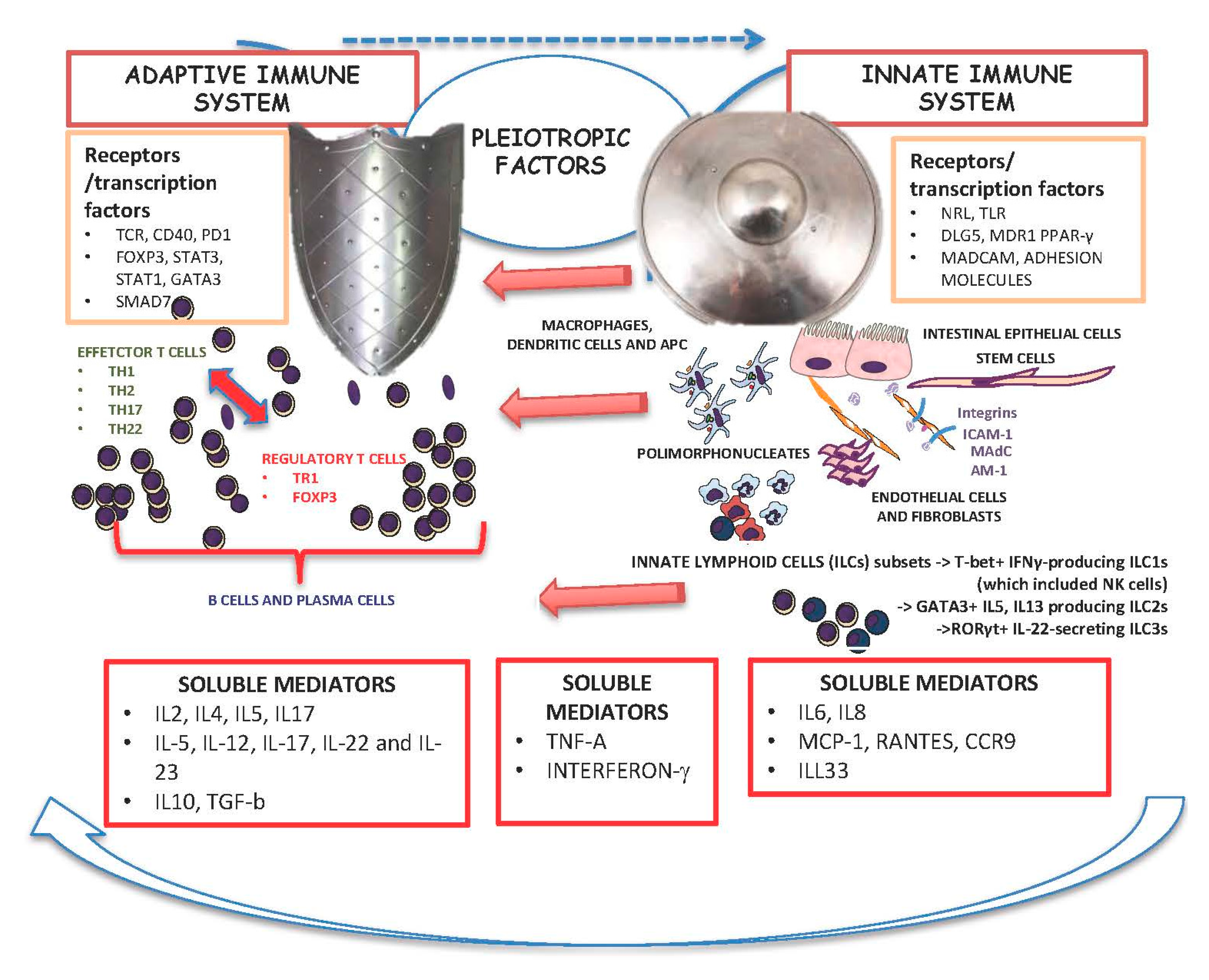
4. Interplay between the Innate and Adaptive Immune System in IBD and the Influence of Pleiotropic Factors as Targets for Current Treatment of IBD
5. Targeting the Adaptive Immune System
5.1. Inhibitor of Th1/Th17-Anti-Cytokine-IL 12/23
5.2. Anti-SMAD7—Mongersen
5.3. JAK1/JAK3 Inhibitors—Tofacitinib
5.4. IL-13—Tralokinumab
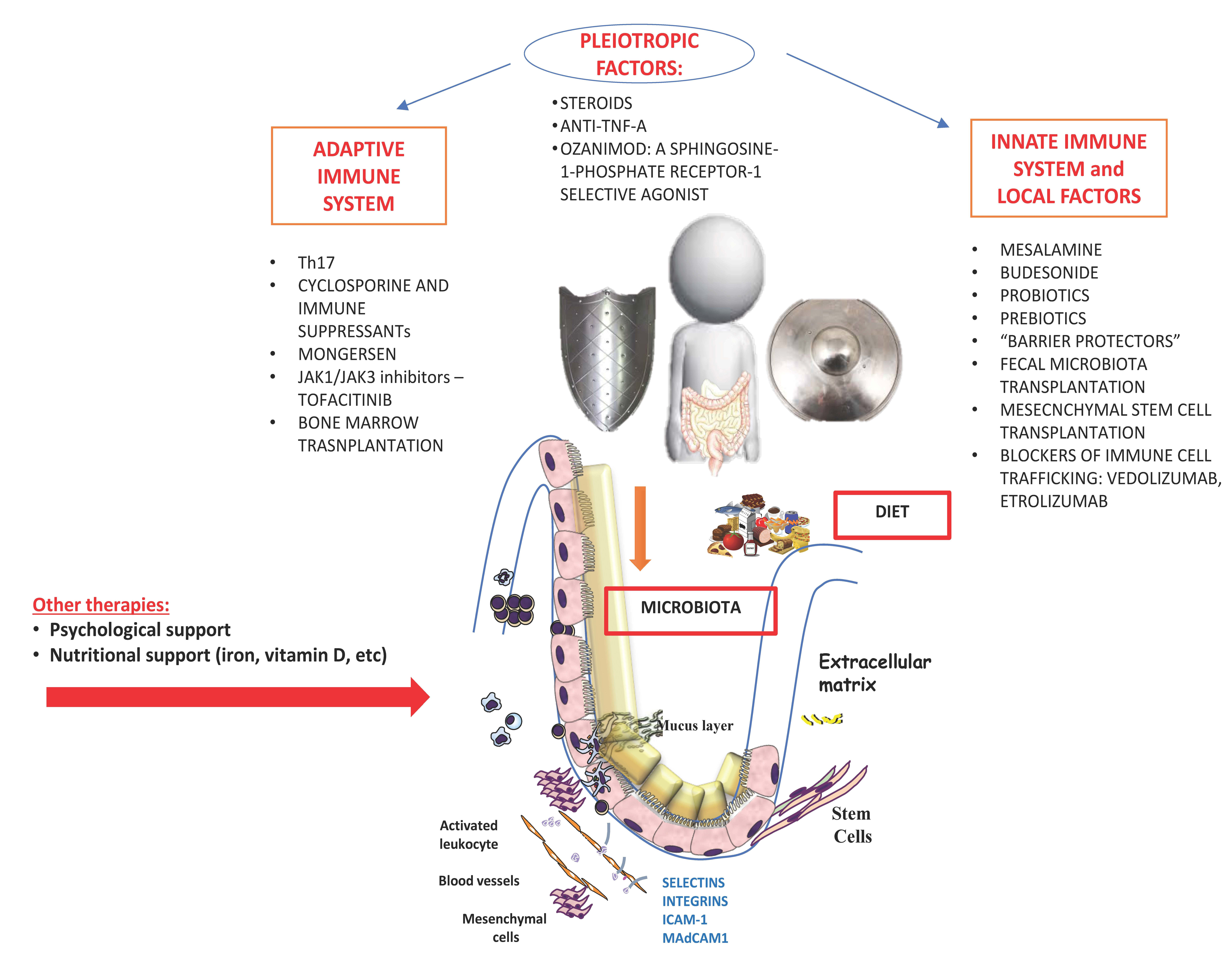
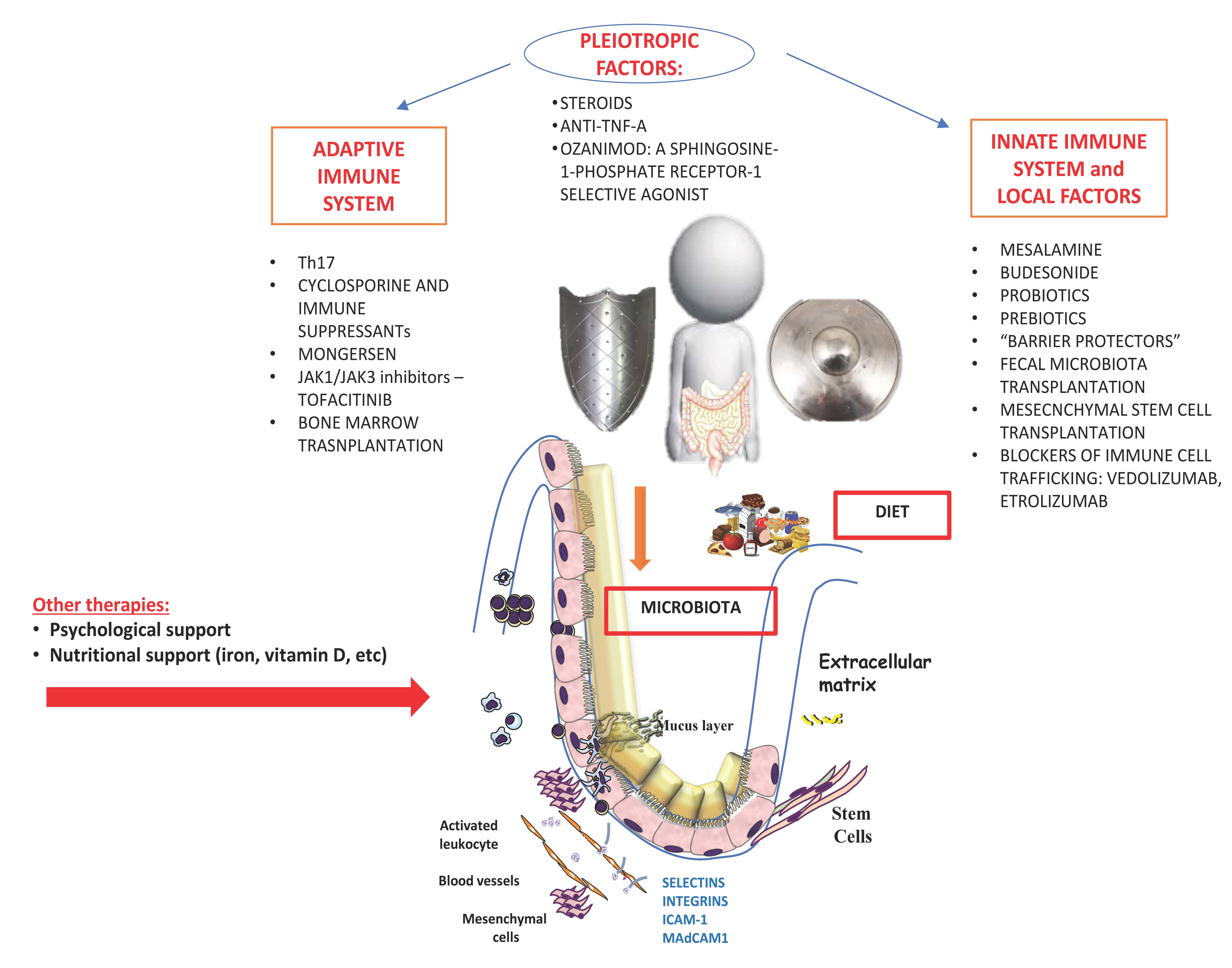
6. Targeting Pleiotropic Factors between the Adaptive and Innate Immune Systems
6.1. Anti-TNF: Infliximab, Adalimumab, Golimumab, and Certolizumab
6.2. Ozanimod: Yes a Sphingosine-1-Phosphate Receptor-1 Selective Agonist
6.3. Anti-IL-6: Tocilizumab Yes a Colon Is More Appropriate
6.4. Other Non-Biologic Therapies with a Pleiotropic Effect
7. Targeting the Innate Immune System and Local Factors
7.1. Leukocyte Trafficking: Anti-Integrins and Adhesion Molecules
7.2. Vedolizumab
7.3. Etrolizumab
7.4. Alicaforsen
7.5. Anti-MAdCAM-1 Antibody
7.6. Manipulation of the Intestinal Microbiome: Fecal Microbiota Transplantation and Bacteriotherapy
7.7. TLR Agonists and Antagonists
7.8. Kappaproct: TLR-9 Agonist
7.9. Potential Emerging Target: IL-33/ST2 Axis
7.10. Conventional Therapies Affecting the Innate Immune System and Local Factors
8. Immunogenicity
9. Biosimilars
10. Conclusions
Author Contributions
Conflicts of Interest
Abbreviations
| IBD | Inflammatory Bowel Disease |
| CD | Crohn’s Disease |
| UC | Ulcerative Colitis |
| TNF | Tumor Necrosis Factor |
| APC | Antigen Presenting Cells |
| TLR | Toll-like Receptors |
| NLR | Nod-like Receptors |
| DC | Dendritic Cells |
| Th | T helper |
| Treg | T regulatory |
| IL | Interleukin |
| IFN | Interferon |
| NK | Natural Killer |
| TGF | Transforming Growth Factor |
| ECCO | European Crohn’s and Colitis Organization |
| FDA | Food and Drug Association |
| TDM | Therapeutic Drug Monitoring |
| ADA | Anti-Drug Antibodies |
References
- Ye, Y.; Pang, Z.; Chen, W.; Ju, S.; Zhou, C. The epidemiology and risk factors of inflammatory bowel disease. Int. J. Clin. Exp. Med. 2015, 8, 22529–22542. [Google Scholar] [PubMed]
- Sartor, R.B. Current concepts of the etiology and pathogenesis of ulcerative colitis and Crohn’s disease. Gastroenterol. Clin. N. Am. 1995, 24, 475–507. [Google Scholar]
- Gomollón, F. 3rd European evidence-based consensus on the diagnosis and management of Crohn’s disease 2016: Part 1: Diagnosis and medical management. J. Crohns Colitis 2017, 11, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Harbord, M.; Rami, E.; Bettenworth, D.; Karmiris, K.; Katsanos, K.; Kopylov, U.; Kucharzik, T.; Molnar, T.; Raine, T.; Sebastian, S.; et al. Third European Evidence-based Consensus on Diagnosis and Management of Ulcerative Colitis. Part 2: Current Management. J. Crohns Colitis 2017, 11, 769–784. [Google Scholar] [PubMed]
- Panés, J.; García-Olmo, D.; Van Assche, G.; Colombel, J.F.; Reinisch, W.; Baumgart, D.C.; Dignass, A.; Nachury, M.; Ferrante, M.; Kazemi-Shirazi, L.; et al. Expanded allogeneic adipose-derived mesenchymal stem cells (Cx601) for complex perianal fistulas in Crohn’s disease: A phase 3 randomised, double-blind controlled trial. Lancet 2016, 388, 1281–1290. [Google Scholar] [CrossRef]
- Jauregui-Amezaga, A.; Rovira, M.; Marín, P.; Salas, A.; Pinó-Donnay, S.; Feu, F.; Elizalde, J.I.; Fernández-Avilés, F.; Martínez, C.; Gutiérrez, G.; et al. Improving safety of autologous haematopoietic stem cell transplantation in patients with Crohn’s disease. Gut 2016, 9, 1456–1462. [Google Scholar] [CrossRef] [PubMed]
- Ding, N.S.; Hart, A.; De Cruz, P. Systematic review: Predicting and optimising response to anti-TNF therapy in Crohn’s disease algorithm for practical management. Aliment. Pharmacol. Ther. 2016, 43, 30–51. [Google Scholar] [CrossRef] [PubMed]
- Geremia, A.; Biancheri, P.; Allan, P.; Corazza, G.R.; Di Sabatino, A. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun. Rev. 2014, 13, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Arseneau, K.O.; Tamagawa, H.; Pizarro, T.T.; Cominelli, F. Innate and adaptive immune responses related to IBD pathogenesis. Curr. Gastroenterol. Rep. 2007, 9, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141. [Google Scholar] [CrossRef] [PubMed]
- Gabbani, T.; Deiana, S.; Annese, A.L.; Lunardi, S.; Annese, V. The genetic burden of inflammatory bowel diseases: Implications for the clinic? Expert Rev. Gastroenterol. Hepatol. 2016, 10, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Katsanos, K.H.; Papadakis, K.A. Inflammatory Bowel Disease: Updates on Molecular Targets for Biologics. Gut liver 2017, 11, 455. [Google Scholar] [CrossRef] [PubMed]
- Van der Sluis, M.; De Koning, B.A.; De Bruijn, A.C.; Velcich, A.; Meijerink, J.P.; Van Goudoever, J.B.; Büller, H.A.; Dekker, J.; Van Seuningen, I.; Renes, I.B.; et al. MUC2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 2006, 131, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Larsson, J.M.; Karlsson, H.; Crespo, J.G.; Johansson, M.E.; Eklund, L.; Sjövall, H.; Hansson, G.C. Altered O-glycosylation profile of MUC2 mucin occurs in active ulcerative colitis and is associated with increased inflammation. Inflamm. Bowel Dis. 2011, 17, 2299–2307. [Google Scholar] [CrossRef] [PubMed]
- Sartor, R.B. Mechanisms of disease: Pathogenesis of Crohn’s disease and ulcerative colitis. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 390–407. [Google Scholar] [CrossRef] [PubMed]
- Parkes, M. Evidence from genetics for a role of autophagy and innate immunity in IBD pathogenesis. Dig. Dis. 2012, 30, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Rakoff-Nahoum, S.; Paglino, J.; Eslami-Varzaneh, F.; Edberg, S.; Medzhitov, R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 2014, 118, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Wallace, K.L.; Zheng, L.B.; Kanazawa, Y.; Shih, D.Q. Immunopathology of inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Hart, A.L.; Al-Hassi, H.O.; Rigby, R.J.; Bell, S.J.; Emmanuel, A.V.; Knight, S.C.; Kamm, M.A.; Stagg, A.J. Characteristics of intestinal dendritic cells in inflammatory bowel diseases. Gastroenterology 2005, 129, 50–65. [Google Scholar] [CrossRef] [PubMed]
- Van Furth, R.; Cohn, Z.A.; Hirsch, J.G.; Humphrey, J.H.; Spector, W.G.; Langevoort, H.L. The mononuclear phagocyte system: A new classification of macrophages, monocytes, and their precursor cells. Bull. World Health Organ. 1972, 46, 845. [Google Scholar] [PubMed]
- Maloy, K.J.; Powrie, F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 2011, 474, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Rioux, J.D.; Xavier, R.J.; Taylor, K.D.; Silverberg, M.S.; Goyette, P.; Huett, A.; Green, T.; Kuballa, P.; Barmada, M.M.; Datta, L.W.; et al. Genome-wide association study identifies five novel susceptibility loci for Crohn’s disease and implicates a role for autophagy in disease pathogenesis. Nat. Genet. 2007, 39, 596. [Google Scholar] [CrossRef] [PubMed]
- Juelke, K.; Romagnani, C. Differentiation of human innate lymphoid cells (ILCs). Curr. Opin. Immunol. 2016, 45, 2171–2182. [Google Scholar] [CrossRef] [PubMed]
- Fort, M.M.; Cheung, J.; Yen, D.; Li, J.; Zurawski, S.M.; Lo, S.; Menon, S.; Clifford, T.; Hunte, B.; Lesley, R.; et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity 2001, 15, 985–995. [Google Scholar] [CrossRef]
- Takatori, H.; Kanno, Y.; Watford, W.T.; Tato, C.M.; Weiss, G.; Ivanov, I.I.; Littman, D.R.; O’Shea, J.J. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J. Exp. Med. 2009, 206, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Geremia, A.; Arancibia-Cárcamo, C.V.; Fleming, M.P.; Rust, N.; Singh, B.; Mortensen, N.J.; Travis, S.P.; Powrie, F. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J. Exp. Med. 2011, 208, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Medzhitov, R. Regulation of adaptive immunity by the innate immune system. Science 2010, 327, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427. [Google Scholar] [CrossRef] [PubMed]
- Mora, J.R.; Bono, M.R.; Manjunath, N.; Weninger, W. Selective imprinting of gut-homing T cells by Peyer’s patch dendritic cells. Nature 2003, 424, 88. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, M.; King, N.; Lobb, R.; Benjamin, C.; Podolsky, D.K. Expression of vascular adhesion molecules in inflammatory bowel disease. Gastroenterology 1992, 103, 840–847. [Google Scholar] [CrossRef]
- Meenan, J.; Spaans, J.; Grool, T.A.; Pals, S.T.; Tytgat, G.N.; Van Deventer, S.J. Altered expression of alpha 4 beta 7, a gut homing integrin, by circulating and mucosal T cells in colonic mucosal inflammation. Gut 1997, 40, 241–246. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, T.T.; Monteleone, G. Immunity, inflammation, and allergy in the gut. Science 2005, 307, 1920–1925. [Google Scholar] [CrossRef] [PubMed]
- Sakuraba, A.; Sato, T.; Kamada, N.; Kitazume, M.; Sugita, A.; Hibi, T. Th1/Th17 immune response is induced by mesenteric lymph node dendritic cells in Crohn’s disease. Gastroenterology 2009, 137, 1736–1745. [Google Scholar] [CrossRef] [PubMed]
- Caza, T.; Landas, S. Functional and phenotypic plasticity of CD4(+) T cell subsets. BioMed Res. Int. 2015, 2015, 521957. [Google Scholar] [CrossRef] [PubMed]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, S. Lymphokine production by human T cells in disease states. Annu. Rev. Immunol. 1994, 12, 227–257. [Google Scholar] [CrossRef] [PubMed]
- Kmieć, Z.; Cyman, M.; Ślebioda, T.J. Cells of the innate and adaptive immunity and their interactions in inflammatory bowel disease. Adv. Med. Sci. 2017, 62, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.T.; Xu, A.T.; Shen, J.; Ran, Z.H. Crosstalk between intestinal epithelial cell and adaptive immune cell in intestinal mucosal immunity. J. Gastroenterol. Hepatol. 2017, 32, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Peyrin-Biroulet, L.; Eisenhut, M.; Shin, J.I. IBD immunopathogenesis: A comprehensive review of inflammatory molecules. Autoimmun. Rev. 2017, 16, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Engel, T.; Kopylov, U. Ustekinumab in Crohn’s disease: Evidence to date and place in therapy. Ther. Adv. Chronic Dis. 2016, 7, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Strober, W.; Fuss, I. Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology 2011, 140, 1756–1767. [Google Scholar] [CrossRef] [PubMed]
- Tuskey, A.; Behm, B. Profile of ustekinumab and its potential in patients with moderate-to-severe Crohn’s disease. Clin. Exp. Gastroenterol. 2014, 7, 173–179. [Google Scholar] [PubMed]
- Benson, J.M.; Peritt, D.; Scallon, B.J. Discovery and mechanism of ustekinumab: A human monoclonal antibody targeting interleukin-12 and interleukin-23 for treatment of immune-mediated disorders. MAbs 2011, 7, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Gasink, C.; Gao, L.L. Ustekinumab induction and maintenance therapy in refractory Crohn’s disease. N. Engl. J. Med. 2012, 367, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Feagan, B.G.; Sandborn, W.J.; Gasink, C.; Jacobstein, D.; Lang, Y.; Friedman, J.R.; Blank, M.A.; Johanns, J.; Gao, L.L.; Miao, Y.; et al. Ustekinumab as Induction and Maintenance Therapy for Crohn’s Disease. N. Engl. J. Med. 2016, 375, 1946–1960. [Google Scholar] [CrossRef] [PubMed]
- Feagan, B.G.; Sandborn, W.; Panés, J. Efficacy and safety of induction therapy with the selective IL-23 inhibitor BI 655066, in patients with moderate-to-severe Crohn’s disease: Results of a randomized, double-blind, placebo-controlled phase II study. Gastroenterology 2016, 150, S1266. [Google Scholar] [CrossRef]
- Sands, B.E.; Chen, J.; Penney, M. Initial evaluation of MEDI2070 (specific anti-IL-23 antibody) in patients with active Crohn’s disease who have failed anti-TNF antibody therapy: A randomized, double-blind placebo-controlled phase 2A induction study. Gastroenterology 2015, 148, S163–S164. [Google Scholar] [CrossRef]
- Panaccione, R.; Sandborn, W.J.; Gordon, G.L.; Lee, S.D.; Safdi, A.; Sedqhi, S.; Feaqan, B.G.; Hanauer, S.; Reinisch, W.; Valentine, J.F.; et al. Briakinumab for treatment of Crohn’s disease: Results of a randomized trial. Inflamm. Bowel Dis. 2015, 21, 1329–1340. [Google Scholar] [PubMed]
- Monteleone, G.; Kumberova, A.; Croft, N.M.; McKenzie, C.; Steer, H.W.; MacDonald, T.T. Blocking Smad7 restores TGF-beta1 signaling in chronic inflammatory bowel disease. J. Clin. Investig. 2001, 108, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, G.; Neurath, M.F.; Ardizzone, S.; Di Sabatino, A.; Fantini, M.C.; Castiglione, F. Mongersen, an oral SMAD7 antisense oligonucleotide, and Crohn’s disease. N. Engl. J. Med. 2015, 372, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Dulai, P.S.; Sandborn, W.J. Next-Generation Therapeutics for Inflammatory Bowel Disease. Curr. Gastroenterol. Rep. 2016, 18, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Bravatà, I.; Fiorino, G.; Allocca, M.; Repici, A.; Danese, S. New targeted therapies such as anti-adhesion molecules, anti-IL-12/23 and anti-Janus kinases are looking toward a more effective treatment of inflammatory bowel disease. Scand. J. Gastroenterol. 2015, 50, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Fiocchi, C. Ulcerative colitis. N. Engl. J. Med. 2011, 365, 1713–1725. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, O.H.; Seidelin, J.B.; Ainsworth, M.; Coskun, M. Will novel oral formulations change the management of inflammatory bowel disease? Expert Opin. Investig. Drugs 2016, 25, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Ghoreschi, K.; Jesson, M.I.; Li, X. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J. Immunol. 2011, 186, 4234–4243. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Ghosh, S.; Panes, J. Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis. N. Engl. J. Med. 2012, 367, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Sands, B.E.; D’Haens, G. Efficacy and safety of oral tofacitinib as induction therapy in patients with moderate-to-severe ulcerative colitis: Results from 2 phase 3 randomised controlled trials. J. Crohns Colitis 2016, 10, S157. [Google Scholar] [CrossRef]
- Panés, J.; Sandborn, W.J.; Schreiber, S.; Sands, B.E.; Vermeire, S.; D’Haens, G.; Panaccione, R.; Higgins, P.D.; Colombel, J.F.; Feagan, B.G.; et al. Tofacitinib for induction and maintenance therapy of Crohn’s disease: Results of two phase IIb randomised placebo-controlled trials. Gut 2017, 66, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Vermeire, S.; Schreiber, S.; Petryka, R. Filgotinib (GLPG0634), an oral JAK1 selective inhibitor, induces clinical remission in patients with moderate-to-severe Crohn’s disease: Results from the phase 2 FITZROY study interim analysis. Gastroenterology 2016, 150, S1267. [Google Scholar] [CrossRef]
- Heller, F.; Florian, P.; Bojarski, C.; Richter, J.; Christ, M.; Hillenbrand, B.; Mankertz, J.; Gitter, A.H.; Bürgel, N.; Fromm, M.; et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 2005, 129, 550–564. [Google Scholar] [CrossRef] [PubMed]
- Fuss, I.J.; Heller, F.; Boirivant, M.; Leon, F.; Yoshida, M.; Fichtner-Feigl, S.; Yang, Z.; Exley, M.; Kitani, A.; Blumberg, R.S.; et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J. Clin. Investig. 2004, 113, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Jovani, M.; Fiorino, G.; Danese, S. Anti-IL-13 in inflammatory bowel disease: From the bench to the bedside. Curr. Drug Targets 2013, 14, 1444–1452. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Rudziński, J.; Brandt, W.; Dupas, J.L.; Peyrin-Biroulet, L.; Bouhnik, Y.; Kleczkowski, D.; Uebel, P.; Lukas, M.; Knutsson, M.; et al. Tralokinumab for moderate-to-severe UC: A randomised, double-blind, placebo-controlled, phase IIa study. Gut 2015, 64, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Rutgeerts, P.; Van Assche, G.; Vermeire, S. Optimizing anti-TNF treatment in inflammatory bowel disease. Gastroenterology 2004, 126, 1593–1610. [Google Scholar] [CrossRef] [PubMed]
- Reimund, J.M.; Wittersheim, C.; Dumont, S.; Muller, C.D.; Kenney, J.S.; Baumann, R.; Poindron, P.; Duclos, B. Increased production of tumour necrosis factor-alpha interleukin-1 beta, and interleukin-6 by morphologically normal intestinal biopsies from patients with Crohn’s disease. Gut 1996, 39, 684–689. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, T.; Mitoma, H.; Harashima, S.; Tsukamoto, H.; Shimoda, T. Transmembrane TNF-α: Structure, function and interaction with anti-TNF agents. Rheumatology 2010, 49, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Lichtenstein, G.R. Comprehensive review: Antitumor necrosis factor agents in inflammatory bowel disease and factors implicated in treatment response. Ther. Adv. Gastroenterol. 2013, 6, 269–293. [Google Scholar] [CrossRef] [PubMed]
- Olesen, C.M.; Coskun, M.; Peyrin-Biroulet, L.; Nielsen, O.H. Mechanisms behind efficacy of tumor necrosis factor inhibitors in inflammatory bowel diseases. Pharmacol. Ther. 2016, 159, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Rutella, S.; Fiorino, G.; Vetrano, S.; Correale, C.; Spinelli, A.; Pagano, N.; Arena, V.; Maggiano, N.; Repici, A.; Malesci, A.; et al. Infliximab therapy inhibits inflammation-induced angiogenesis in the mucosa of patients with Crohn’s disease. Am. J. Gastroenterol. 2011, 106, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Nusrat, A. The life and death of epithelia during inflammation: Lessons learned from the gut. Annu. Rev. Pathol. 2012, 7, 35–60. [Google Scholar] [CrossRef] [PubMed]
- Guidi, L.; Felice, C.; Procoli, A.; Bonanno, G.; Martinelli, E.; Marzo, M. FOXP3(+) T regulatory cell modifications in inflammatory bowel disease patients treated with anti-TNFα agents. Biomed Res. Int. 2013, 2013, 286368. [Google Scholar] [CrossRef] [PubMed]
- Scaldaferri, F.; Pecere, S.; D’Ambrosio, D.; Bibbò, S.; Petito, P. Emerging Mechanisms of Action and Loss of Response to Infliximab in Ibd: A Broader Picture. Biochem. Pharmacol. 2016, 5, 206. [Google Scholar] [CrossRef]
- Hanauer, S.B.; Feagan, B.G.; Lichtenstein, G.R.; Mayer, L.F.; Schreiber, S.; Colombel, J.F.; Rachmilewitz, D.; Wolf, D.C.; Olson, A.; Bao, W.; et al. Maintenance infliximab for Crohn’s disease: The ACCENT I randomised trial. Lancet 2002, 359, 1541–1549. [Google Scholar] [CrossRef]
- Rutgeerts, P.; Sandborn, W.J.; Feagan, B.G.; Reinisch, W.; Olson, A.; Johanns, J.; Travers, S.; Rachmilewitz, D.; Hanauer, S.B.; Lichtenstein, G.R.; et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 2005, 353, 2462–2476. [Google Scholar] [CrossRef] [PubMed]
- Dulai, P.S.; Siegel, C.A.; Colombel, J.F.; Sandborn, W.J.; Peyrin-Biroulet, L. Systematic review: Monotherapy with antitumour necrosis factor alpha agents versus combination therapy with an immunosuppressive for IBD. Gut 2014, 63, 1843–1853. [Google Scholar] [CrossRef] [PubMed]
- Khanna, R.; Bressler, B.; Levesque, B.G.; Zou, G.; Stitt, L.W.; Greenberg, G.R.; Panaccione, R.; Bitton, A.; Paré, P.; Vermeire, S.; et al. Early combined immunosuppression for the management of Crohn’s disease (REACT): A cluster randomised controlled trial. Lancet 2015, 386, 1825–1834. [Google Scholar] [CrossRef]
- Hanauer, S.B.; Sandborn, W.J.; Rutgeerts, P.; Fedorak, R.N.; Lukas, M.; MacIntosh, D.; Panaccione, R.; Wolf, D.; Pollack, P. Human anti-tumor necrosis factor monoclonal antibody (adalimumab) in Crohn’s disease: The CLASSIC-I trial. Gastroenterology 2006, 130, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Hanauer, S.B.; Rutgeerts, P.; Fedorak, R.N.; Lukas, M.; MacIntosh, D.G.; Panaccione, R.; Wolf, D.; Kent, J.D.; Bittle, B.; et al. Adalimumab for maintenance treatment of Crohn’s disease: Results of the CLASSIC II trial. Gut 2007, 56, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Colombel, J.F.; Sandborn, W.J.; Ghosh, S.; Wolf, D.C.; Panaccione, R.; Feagan, B.; Reinisch, W.; Robinson, A.M.; Lazar, A.; Kron, M.; et al. Four-year maintenance treatment with adalimumab in patients with moderately to severely active ulcerative colitis: Data from ULTRA 1, 2, and 3. Am. J. Gastroenterol. 2014, 109, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Lowenberg, M.; de Boer, N.; Hoentjen, F. Golimumab for the treatment of ulcerative colitis. Clin. Exp. Gastroenterol. 2014, 7, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Feagan, B.G.; Marano, C. Subcutaneous golimumab induces clinical response and remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology 2014, 146, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Rutgeerts, P.; Feagan, B.G.; Marano, C.W. Randomised clinical trial: A placebo-controlled study of intravenous golimumab induction therapy for ulcerative colitis. Aliment. Pharmacol. Ther. 2015, 42, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Nesbitt, A.; Fossati, G.; Bergin, M.; Stephens, P.; Stephens, S.; Foulkes, R.; Brown, D.; Robinson, M.; Bourne, T. Mechanism of action of certolizumab pegol (CDP870): In vitro comparison with other anti-tumor necrosis factor alpha agents. Inflamm. Bowel Dis. 2007, 11, 1323–1332. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.; Ceska, T.; Henry, A.; Heads, J.; Turner, A.; Heywood, S.; O’Hara, J.; Nesbitt, A. Detailing the novel structure of the biopharmaceutical certolizumab pegol. Am. J. Gastroenterol. 2008, 103, s430. [Google Scholar] [CrossRef]
- Stein, A.C.; Rubin, D.T.; Hanauer, S.B.; Cohen, R.D. Incidence and predictors of clinical response, re-induction dose, and maintenance dose escalation with certolizumab pegol in Crohn’s disease. Inflamm. Bowel Dis. 2014, 20, 1722–1728. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Feagan, B.G.; Stoinov, S.; Honiball, P.J.; Rutgeerts, P.; Mason, D.; Bloomfield, R.; Schreiber, S. Certolizumab pegol for the treatment of Crohn’s disease. N. Engl. J. Med. 2007, 357, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Lee, S.D.; Randall, C.; Gutierrez, A.; Schwartz, D.A.; Ambarkhane, S.; Kayhan, C.; Pierre-Louis, B.; Schreiber, S.; Lichtenstein, G.R. Long-term safety and efficacy of certolizumab pegol in the treatment of Crohn’s disease: 7-year results from the PRECiSE 3 study. Aliment. Pharmacol. Ther. 2014, 8, 903–916. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.; Khaliq-Kareemi, M.; Lawrance, I.C.; Thomsen, O.Ø.; Hanauer, S.B.; McColm, J.; Bloomfield, R.; Sandborn, W.J.; PRECISE 2 Study Investigators. Maintenance therapy with certolizumab pegol for Crohn’s disease. N. Engl. J. Med. 2007, 3, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Hanauer, S.B.; Katz, S.; Safdi, M.; Wolf, D.G.; Baerg, R.D.; Tremaine, W.J.; Johnson, T.; Diehl, N.N.; Zinsmeister, A.R. Etanercept for active Crohn’s disease: A randomized, double-blind, placebo-controlled trial. Gastroenterology 2001, 5, 1088–1094. [Google Scholar] [CrossRef]
- Perrier, C.; de Hertogh, G.; Cremer, J.; Vermeire, S.; Rutgeerts, P.; Van Assche, G.; Szymkowski, D.E.; Ceuppens, J.L. Neutralization of membrane TNF, but not soluble TNF, is crucial for the treatment of experimental colitis. Inflamm. Bowel Dis. 2013, 19, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Vincent, F.B.; Morand, E.F.; Murphy, K.; Mackay, F.; Mariette, X.; Marcelli, C. Antidrug antibodies (ADAb) to tumour necrosis factor (TNF)-specific neutralising agents in chronic inflammatory diseases: A real issue, a clinical perspective. Ann. Rheum. Dis. 2013, 72, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Biancheri, P.; Brezski, R.J.; Di Sabatino, A.; Greenplate, A.R.; Soring, K.L.; Corazza, G.R.; Kok, K.B.; Rovedatti, L.; Vossenkämper, A.; Ahmad, N.; et al. Proteolytic cleavage and loss of function of biologic agents that neutralize tumor necrosis factor in the mucosa of patients with inflammatory bowel disease. Gastroenterology 2015, 149, 1564–1574. [Google Scholar] [CrossRef] [PubMed]
- Papamichael, K.; Gils, A.; Rutgeerts, P.; Levesque, B.G.; Vermeire, S.; Sandborn, W.J.; Vande Casteele, N. Role for therapeutic drug monitoring during induction therapy with TNF an-tagonists in IBD: Evolution in the definition and management of primary nonre- sponse. Inflamm. Bowel Dis. 2015, 21, 182–197. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Feagan, B.G.; Wolf, D.C.; D’Haens, G.; Vermeire, S.; Hanauer, S.B. Ozanimod induction and maintenance treatment for ulcerative colitis. N. Engl. J. Med. 2016, 374, 1754–1762. [Google Scholar] [CrossRef] [PubMed]
- Karuppuchamy, T.; Behrens, E.H.; González-Cabrera, P.; Sarkisyan, G.; Gima, L.; Boyer, J.D.; Bamias, G.; Jedlicka, P.; Veny, M.; Clark, D.; et al. Sphingosine-1-phosphate receptor-1 (S1P1) is expressed by lymphocytes, dendritic cells, and endothelium and modulated during inflammatory bowel disease. Mucosal Immunol. 2017, 10, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Allocca, M.; Jovani, M.; Fiorino, G.; Schreiber, S.; Danese, S. Anti-IL-6 treatment for inflammatory bowel diseases: Next cytokine, next target. Curr. Drug Targets 2013, 14, 1508–1521. [Google Scholar] [CrossRef] [PubMed]
- Atreya, R.; Mudter, J.; Finotto, S.; Müllberg, J.; Jostock, T.; Wirtz, S.; Schütz, M.; Bartsch, B.; Holtmann, M.; Becker, C.; et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: Evidence in crohn disease and experimental colitis in vivo. Nat. Med. 2000, 6, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Hyams, J.S.; Fitzgerald, J.E.; Treem, W.R.; Wyzga, N.; Kreutzer, D.L. Relationship of functional and antigenic interleukin 6 to disease activity in inflammatory bowel disease. Gastroenterology 1993, 104, 1285–1292. [Google Scholar] [CrossRef]
- Umehara, Y.; Kudo, M.; Nakaoka, R.; Kawasaki, T.; Shiomi, M. Serum proinflammatory cytokines and adhesion molecules in ulcerative colitis. Hepatogastroenterology 2006, 53, 879–882. [Google Scholar] [PubMed]
- Helbling, R.; Nyddeger, A.; Angelini, F.; Gete, A.V.S.; Hofer, M. P03-024—Early onset IBD treated by tocilizumab. Pediatr. Rheumatol. 2013, 11, A222. [Google Scholar] [CrossRef]
- Szeto, M.C.; Yalçın, M.D.; Khan, A.; Piotrowicz, A. Successful Use of Tocilizumab in a Patient with Coexisting Rheumatoid Arthritis and Ulcerative Colitis. Case Rep. Immunol. 2016, 2016, 7562123. [Google Scholar] [CrossRef] [PubMed]
- Shipman, L. Rheumatoid arthritis: Tocilizumab and the risk of intestinal perforation. Nat. Rev. Rheumatol. 2016, 12, 499. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Bouman-Thio, E.; Comisar, C.; Frederick, B.; Van Hartingsveldt, B.; Marini, J.C.; Davis, H.M.; Zhou, H. Pharmacokinetics, pharmacodynamics and safety of a human anti-IL-6 monoclonal antibody (sirukumab) in healthy subjects in a first-in-human study. Br. J. Clin. Pharmacol. 2011, 72, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Shaw, S.; Bourne, T.; Meier, C.; Carrington, B.; Gelinas, R.; Henry, A.; Popplewell, A.; Adams, R.; Baker, T.; Rapecki, S.; et al. Discovery and characterization of olokizumab: A humanized antibody targeting interleukin-6 and neutralizing gp130-signaling. MAbs 2014, 6, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Ungar, B.; Kopylov, U. Advances in the development of new biologics in inflammatory bowel disease. Ann. Gastroenterol. 2016, 29, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Fedyk, E.R.; Wyant, T.; Yang, L.L. Exclusive antagonism of the α4β7 integrin by vedolizumab confirms the gut-selectivity of this pathway in primates. Inflamm. Bowel Dis. 2012, 18, 2107–2119. [Google Scholar] [CrossRef] [PubMed]
- Feagan, B.G.; Rutgeerts, P.; Sands, B.E.; Hanauer, S.; Colombel, J.F.; Sandborn, W.J.; Van Assche, G.; Axler, J.; Kim, H.J.; Danese, S.; et al. Vedoli-zumab as induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 2013, 8, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Feagan, B.G.; Rutgeerts, P.; Hanauer, S.; Colombel, J.F.; Sands, B.E. Vedolizumab as induction and maintenance therapy for Crohn’s disease. N. Engl. J. Med. 2013, 369, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Sands, B.E.; Feagan, B.G.; Rutgeerts, P.; Colombel, J.F.; Sandborn, W.J.; Sy, R.; D’Haens, G.; Ben-Horin, S.; Xu, J.; Rosario, M.; et al. Effects of vedolizumab induction therapy for patients with Crohn’s disease in whom tumor necrosis factor antagonist treatment failed. Gastroenterology 2014, 147, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Loftus, E.V., Jr.; Colombel, J.F.; Feagan, B.G.; Vermeire, S.; Sandborn, W.J.; Sands, B.E.; Danese, S.; D’Haens, G.R.; Kaser, A.; Panaccione, R.; et al. Long-term Efficacy of Vedolizumab for Ulcerative Colitis. J. Crohns Colitis 2017, 11, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Vermeire, S.; Loftus, E.V., Jr.; Colombel, J.F.; Feagan, B.G.; Sandborn, W.J.; Sands, B.E.; Danese, S.; D’Haens, G.R.; Kaser, A.; Panaccione, R.; et al. Long-term Efficacy of Vedolizumab for Crohn’s Disease. J. Crohns Colitis 2017, 11, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Colombel, J.; Sands, B.E.; Rutgeerts, P. The safety of vedolizumab for ulcerative colitis and Crohn’s disease. Gut 2017, 66, 839–851. [Google Scholar] [CrossRef] [PubMed]
- Van Assche, G.; Van Ranst, M.; Sciot, R. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn’s disease. N. Engl. J. Med. 2005, 353, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Bravatà, I.; Allocca, M.; Fiorino, G.; Danese, S. Integrins and adhesion molecules as targets to treat inflammatory bowel disease. Curr. Opin. Pharmacol. 2015, 25, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Stefanich, E.; Danilenko, D.; Wang, H.; O’Byrne, S.; Erickson, R.; Gelzleichter, T. A humanized monoclonal antibody targeting the beta7 integrin selectively blocks intestinal homing of T lymphocytes. Br. J. Pharmacol. 2011, 162, 1855–1870. [Google Scholar] [CrossRef] [PubMed]
- Vermeire, S.; O’Byrne, S.; Keir, M.; Williams, M.; Lu, T.; Mansfield, J. Etrolizumab as induction therapy for ulcerative colitis: A randomised, controlled, phase 2 trial. Lancet 2014, 384, 309–318. [Google Scholar] [CrossRef]
- Gewirtz, A.T.; Sitaraman, S. Alicaforsen. Curr. Opin. Investig. Drugs 2001, 2, 1401–1406. [Google Scholar] [PubMed]
- Patel, R.T.; Pall, A.A.; Adu, D.; Keighley, M.R. Circulating soluble adhesion molecules in inflammatory bowel disease. Eur. J. Gastroenterol. Hepatol. 1995, 7, 1037–1041. [Google Scholar] [CrossRef] [PubMed]
- Greuter, T.; Biedermann, L.; Rogler, G.; Sauter, B.; Seibold, F. Alicaforsen, an antisense inhibitor of ICAM-1, as treatment for chronic refractory pouchitis after proctocolectomy: A case series. United Eur. Gastroenterol. J. 2016, 4, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Reinisch, W. A Randomized, Multicenter Double-Blind, Placebo-Controlled Study of the Safety and Efficacy of Anti-MAdCAM Antibody PF-00547659 (PF) in Patients with Moderate to Severe Ulcerative Colitis: Results of the TURANDOT Study. Gastroenterology 2015, 148, S-1193. [Google Scholar] [CrossRef]
- Sandborn, W.; Lee, S.D.; Tarabar, D.; Louis, E. Anti-MAdCAM-1 Antibody (PF-00547659) for Active Refractory Crohn’s Disease: Results of the OPERA Study. Gastroenterology 2015, 148, S162. [Google Scholar] [CrossRef]
- Elson, C.O.; Cong, Y.; McCracken, V.J.; Dimmitt, R.A.; Lorenz, R.G.; Weaver, C.T. Experimental models of inflammatory bowel disease reveal innate, adaptive, and regulatory mechanisms of host dialogue with the microbiota. Immunol. Rev. 2005, 206, 260–276. [Google Scholar] [CrossRef] [PubMed]
- Boltin, D.; Perets, T.T.; Vilkin, A.; Niv, Y. Mucin function in inflammatory bowel disease: An update. J. Clin. Gastroenterol. 2013, 47, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Birchenough, G.M.; Johansson, M.E.; Stabler, R.A.; Dalgakiran, F.; Hansson, G.C.; Wren, B.W.; Luzio, J.P.; Taylor, P.W. Altered innate defenses in the neonatal gastrointestinal tract in response to colonization by neuropathogenic Escherichia coli. Infect. Immun. 2013, 81, 3264–3267. [Google Scholar] [CrossRef] [PubMed]
- Prosberg, M.; Bendtsen, F.; Vind, I.; Petersen, A.M.; Gluud, L.L. The association between the gut microbiota and the inflammatory bowel disease activity: A systematic review and meta-analysis. Scand. J. Gastroenterol. 2016, 51, 1407–1415. [Google Scholar] [CrossRef] [PubMed]
- Sachs, R.E.; Edelstein, C.A. Ensuring the safe and effective FDA regulation of fecal microbiota transplantation. J. Law Biosci. 2015, 2, 396–415. [Google Scholar] [CrossRef] [PubMed]
- Surawicz, C.M.; Brandt, L.J.; Binion, D.G. Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. Am. J. Gastroenterol. 2013, 108, 478–498. [Google Scholar] [CrossRef] [PubMed]
- Moayyedi, P.; Surette, M.G.; Kim, P.T.; Libertucci, J.; Wolfe, M.; Onischi, C.; Armstrong, D.; Marshall, J.K.; Kassam, Z.; Reinisch, W.; et al. Fecal Microbiota Transplantation Induces Remission in Patients With Active Ulcerative Colitis in a Randomized Controlled Trial. Gastroenterology 2015, 149, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.; Feng, Q.; Wang, H.; Wang, M.; Peng, Z.; Li, P. Fecal microbiota transplantation through mid-gut for refractory crohn’s disease: Safety, feasibility, and efficacy trial results. J. Gastroenterol. Hepatol. 2015, 30, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Ianiro, G.; Bibbò, G.; Scaldaferri, F. Fecal microbiota transplantation in inflammatory bowel disease: Beyond the excitement. Medicine 2014, 93, e97. [Google Scholar] [CrossRef] [PubMed]
- Paramsothy, S.; Kamm, M.; Walsh, A. Multi-donor intense faecal microbiota transplantation is an effective treatment for resistant ulcerative colitis: A randomised placebo-controlled trial. J. Crohns Colitis 2016, 10, S14. [Google Scholar] [CrossRef]
- Cammarota, G.; Ianiro, G.; Tilg, H. European consensus conference on faecal microbiota transplantation in clinical practice. Gut 2017, 66, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Boirivant, M.; Strober, W. The mechanism of action of probiotics. Curr. Opin. Gastroenterol. 2007, 23, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, A.D.; Thomas, A.G.; Akobeng, A.K. Probiotics for induction of remission in Crohn’s disease. Cochrane Database Syst. Rev. 2008, 16, CD006634. [Google Scholar]
- Cammarota, G.; Ianiro, G.; Cianci, R.; Bibbò, S.; Gasbarrini, A.; Currò, D. The involvement of gut microbiota in inflammatory bowel disease pathogenesis: Potential for therapy. Pharmacol. Ther. 2015, 149, 191–212. [Google Scholar] [CrossRef] [PubMed]
- Zocco, M.A.; dal Verme, L.Z.; Cremonini, F.; Piscaglia, A.C.; Nista, E.C.; Candelli, M.; Novi, M.; Rigante, D.; Cazzato, I.A.; Ojetti, V. Efficacy of Lactobacillus GG in maintaining remission of ulcerative colitis. Aliment. Pharmacol. Ther. 2006, 23, 1567–1574. [Google Scholar] [CrossRef] [PubMed]
- Kruis, W.; Fric, P.; Pokrotnieks, J.; Lukás, M.; Fixa, B.; Kascák, M.; Kamm, M.A.; Weismueller, J.; Beglinger, C.; Stolte, M.; et al. Maintaining remission of ulcerative colitis with the probiotic Escherichia coli Nissle 1917 is as effective as with standard mesalazine. Gut 2004, 53, 1617–1623. [Google Scholar] [CrossRef] [PubMed]
- Sang, L.X.; Chang, B.; Zhang, W.L.; Wu, X.M.; Li, X.H.; Jiang, M. Remission induction and maintenance effect of probiotics on ulcerative colitis: A meta-analysis. World J. Gastroenterol. 2010, 16, 1908–1915. [Google Scholar] [CrossRef] [PubMed]
- Mallon, P.; McKay, D.; Kirk, S.; Gardiner, K. Probiotics for induction of remission in ulcerative colitis. Cochrane Database Syst. Rev. 2007, 4, CD005573. [Google Scholar]
- Floch, M.H.; Walker, W.A.; Madsen, K. Recommendations for probiotic use—2011 update. J. Clin. Gastroenterol. 2011, 45, S168–S171. [Google Scholar] [CrossRef] [PubMed]
- Frosali, S.; Pagliari, D.; Gambassi, G.; Landolfi, R.; Pandolfi, F.; Cianci, R. How the intricate interaction between toll-like receptors, microbiota, and intestinal immunity can influence gastrointestinal pathology. J. Immunol. Res. 2015, 2015, 489821. [Google Scholar] [CrossRef] [PubMed]
- Ungaro, R.; Fukata, M.; Hsu, D. A novel Toll-like receptor 4 antagonist antibody ameliorates inflammation but impairs mucosal healing in murine colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G1167–G1179. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.M. Activation of intracellular TLR9 drives the production of numerous proinflammatory cytokines, including TNF, IL-6, and IL-12, leading to a strong induction of the Th1-immune response. Vaccine 2000, 19, 618–622. [Google Scholar] [CrossRef]
- Hall, J.A.; Bouladoux, N.; Sun, C.M. Commensal DNA Limits Regulatory T Cell Conversion and Is a Natural Adjuvant of Intestinal Immune Responses. Immunity 2008, 29, 637–649. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Muñoz, F.; Fonseca-Camarillo, G.C.; Villeda-Ramirez, M.A. TLR9 mRNA expression is upregulated in patients with active ulcerative colitis. Inflamm. Bowel. Dis. 2010, 16, 1267–1268. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, J.; Andresen, G.; Matthiessen, L.; Rask-Madsen, M.W.; Brynskov, J. Expression of Toll-like receptor 9 and response to bacterial CpG oligodeoxynucleotides in human intestinal epithelium. Clin. Exp. Immunol. 2005, 141, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Atreya, R.; Bloom, S.; Scaldaferri, F.; Gerardi, V.; Admyre, C.; Karlsson, Å.; Knittel, T.; Kowalski, J.; Lukas, M.; Löfberg, R.; et al. Clinical Effects of a Topically Applied Toll-like Receptor 9 Agonist in Active Moderate-to-Severe Ulcerative Colitis. J. Crohns Colitis 2016, 10, 1294–1302. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Pastorelli, L.; De Salvo, C.; Cominelli, M.A.; Vecchi, M.; Pizarro, T.T. Novel cytokine signaling pathways in inflammatory bowel disease: Insight into the dichotomous functions of IL-33 during chronic intestinal inflammation. Ther. Adv. Gastroenterol. 2011, 4, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Pastorelli, L.; Garg, R.R.; Hoang, S.B.; Spina, L.; Mattioli, B.; Scarpa, M.; Fiocchi, C.; Vecchi, M.; Pizarro, T.T. Epithelial-derived IL-33 and its receptor ST2 are dysregulated in ulcerative colitis and in experimental Th1/Th2 driven enteritis. Proc. Natl. Acad. Sci. USA 2010, 107, 8017–8022. [Google Scholar] [CrossRef] [PubMed]
- Nunes, T.; Bernardazzi, C.; de Souza, H.S. Interleukin-33 and inflammatory bowel diseases: Lessons from human studies. Mediat. Inflamm. 2014, 2014, 423957. [Google Scholar] [CrossRef] [PubMed]
- Lopetuso, L.R.; Chowdhry, S.; Pizarro, T.T. Opposing Functions of Classic and Novel IL-1 Family Members in Gut Health and Disease. Front. Immunol. 2013, 4, 181. [Google Scholar] [CrossRef] [PubMed]
- Kobori, A.; Yagi, Y.; Imaeda, H.; Ban, H.; Bamba, S.; Tsujikawa, T. Interleukin-33 expression is specifically enhanced in inflamed mucosa of ulcerative colitis. J. Gastroenterol. 2010, 45, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Beltrán, C.J.; Núñez, L.E.; Díaz-Jiménez, D.; Farfan, N.; Candia, E.; Heine, C.; López, F.; González, M.J.; Quera, R.; Hermoso, M.A. Characterization of the novel ST2/IL-33 system in patients with inflammatory bowel disease. Inflamm. Bowel Dis. 2010, 16, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Sedhom, M.A.K.; Pichery, M.; Murdoch, J.R. Neutralisation of the interleukin-33/ST2 pathway ameliorates experimental colitis through enhancement of mucosal healing in mice. Gut 2013, 62, 1714–1723. [Google Scholar] [CrossRef] [PubMed]
- Oboki, K.; Ohno, T.; Kajiwara, N.; Saito, H.; Nakae, S. IL-33 and IL-33 receptors in host defense and diseases. Allergol. Int. 2010, 59, 143–160. [Google Scholar] [CrossRef] [PubMed]
- Rankin, A.L.; Mumm, J.B.; Murphy, E. IL-33 induces IL-13-dependent cutaneous fibrosis. J. Immunol. 2010, 184, 1526–1535. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Davis, H.M.; Zhou, H. Clinical impact of concomitant immunomodulators on biologic therapy: Pharmacokinetics, immunogenicity, efficacy and safety. J. Clin. Pharmacol. 2015, 55 (Suppl. 3), S60–S74. [Google Scholar] [CrossRef] [PubMed]
- Ben-Horin, S. Reversal of Immunogenicity in Inflammatory Bowel Disease. Gastroenterol. Hepatol. 2017, 13, 125–127. [Google Scholar]
- Sheasgreen, C.; Nguyen, G.C. The Evolving Evidence for Therapeutic Drug Monitoring of Monoclonal Antibodies in Inflammatory Bowel Disease. Curr. Gastroenterol. Rep. 2017, 19, 19. [Google Scholar] [CrossRef] [PubMed]
- Papamichael, K.; Cheifetz, A.S. Therapeutic Drug Monitoring in IBD: The New Standard-of-Care for Anti-TNF Therapy. Am. J. Gastroenterol. 2017, 112, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Rosario, M.; Wyant, T.; Leach, T.; Sankoh, S.; Scholz, C.; Parikh, A. Vedolizumab pharmacokinetics, pharmacodynamics, safety, and tolerability following administration of a single, ascending, intravenous dose to healthy volunteers. Clin. Drug Investig. 2016, 36, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Govani, S.M.; Waljee, A.K. Therapeutic Drug Monitoring in IBD: Prospective Promise Unfulfilled. Am. J. Gastroenterol. 2017, 112, 670–672. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Biologics Price Competition and Innovation Act. 2009. Available online: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/UCM216146.pdf (accessed on 8 September 2017).
- Blackstone, E.A.; Joseph, P.F. The economics of biosimilars. Am. Health Drug Benefits 2013, 6, 469–478. [Google Scholar] [PubMed]
- Jung, S.K.; Lee, K.H.; Jeon, J.W.; Lee, J.W.; Kwon, B.O.; Kim, Y.J.; Chang, S.J. Physicochemical characterization of Remsima®. MAbs 2014, 6, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, K.K.; Olsen, I.C.; Goll, G.L.; Lorentzen, M.; Bolstad, N.; Haavardsholm, E.A.; Lundin, K.E.A.; Mørk, C.; Jahnsen, J.; Kvien, T.K.; et al. Switching from originator infliximab to biosimilar CT-P13 compared with maintained treatment with originator infliximab (NOR-SWITCH): A 52-week, randomised, double-blind, non-inferiority trial. Lancet 2017, 389, 2304–2316. [Google Scholar] [CrossRef]
- Razanskaite, V.; Bettey, M.; Downey, L.; Wright, J.; Callaghan, J.; Rush, M.; Whiteoak, S.; Ker, S.; Perry, K.; Underhill, C.; et al. Biosimilar Infliximab in Inflammatory Bowel Disease: Outcomes of a Managed Switching Programme. J. Crohns Colitis 2017, 11, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Fiorino, G.; Raine, T.; Ferrante, M.; Kemp, K.; Kierkus, J.; Lakatos, P.L.; Mantzaris, G.; van der Woude, J.; Panes, J.; et al. ECCO Position Statement on the Use of Biosimilars for Inflammatory Bowel Disease—An Update. J. Crohns Colitis 2017, 11, 26–34. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holleran, G.; Lopetuso, L.; Petito, V.; Graziani, C.; Ianiro, G.; McNamara, D.; Gasbarrini, A.; Scaldaferri, F. The Innate and Adaptive Immune System as Targets for Biologic Therapies in Inflammatory Bowel Disease. Int. J. Mol. Sci. 2017, 18, 2020. https://doi.org/10.3390/ijms18102020
Holleran G, Lopetuso L, Petito V, Graziani C, Ianiro G, McNamara D, Gasbarrini A, Scaldaferri F. The Innate and Adaptive Immune System as Targets for Biologic Therapies in Inflammatory Bowel Disease. International Journal of Molecular Sciences. 2017; 18(10):2020. https://doi.org/10.3390/ijms18102020
Chicago/Turabian StyleHolleran, Grainne, Loris Lopetuso, Valentina Petito, Cristina Graziani, Gianluca Ianiro, Deirdre McNamara, Antonio Gasbarrini, and Franco Scaldaferri. 2017. "The Innate and Adaptive Immune System as Targets for Biologic Therapies in Inflammatory Bowel Disease" International Journal of Molecular Sciences 18, no. 10: 2020. https://doi.org/10.3390/ijms18102020