
Advances in Monitoring Cell-Based Therapies with Magnetic Resonance Imaging: Future Perspectives

Abstract
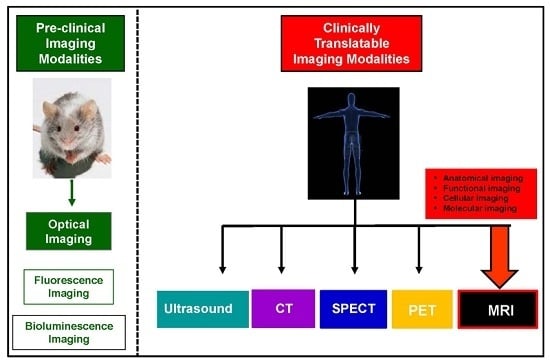
:
1. Introduction
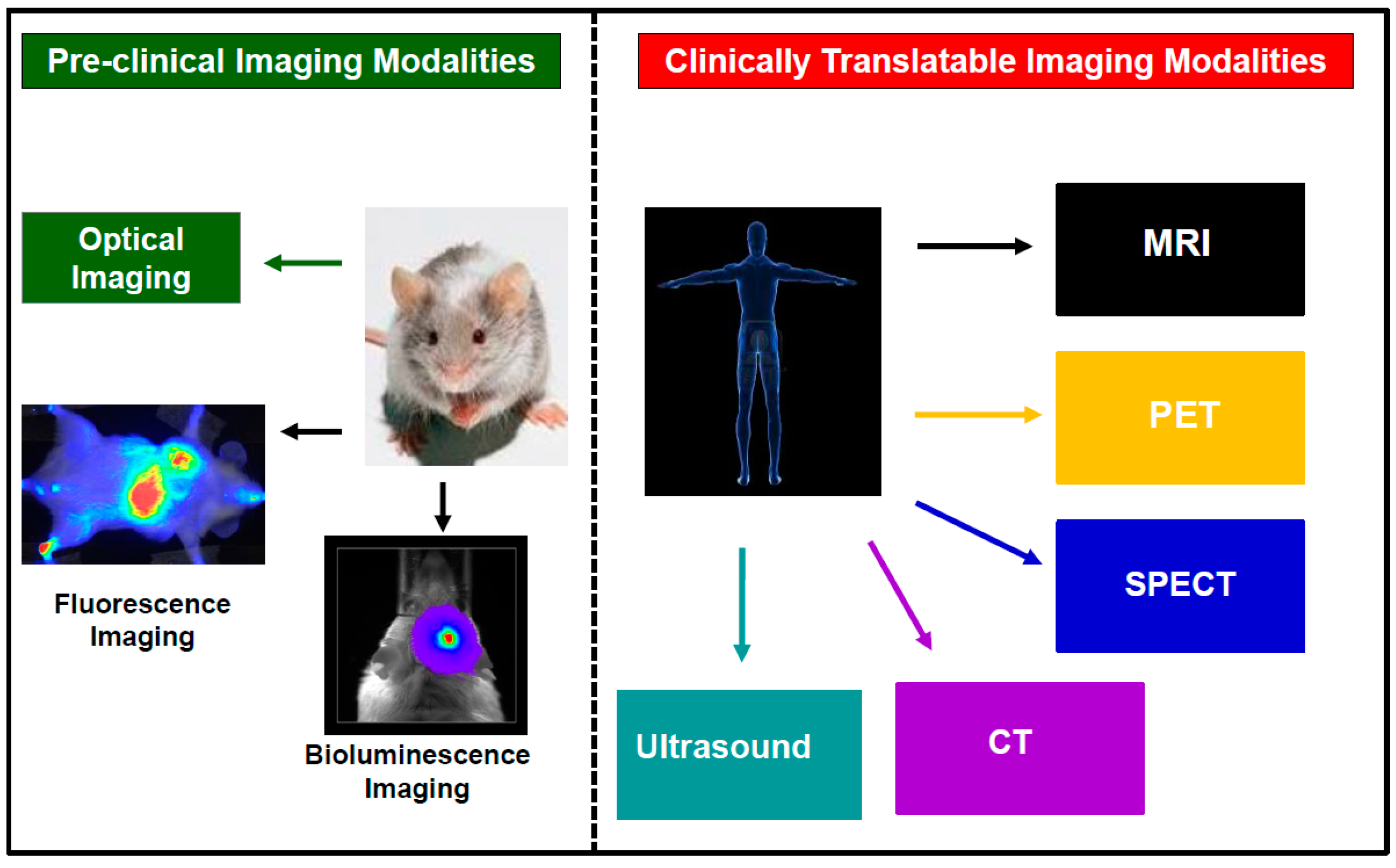
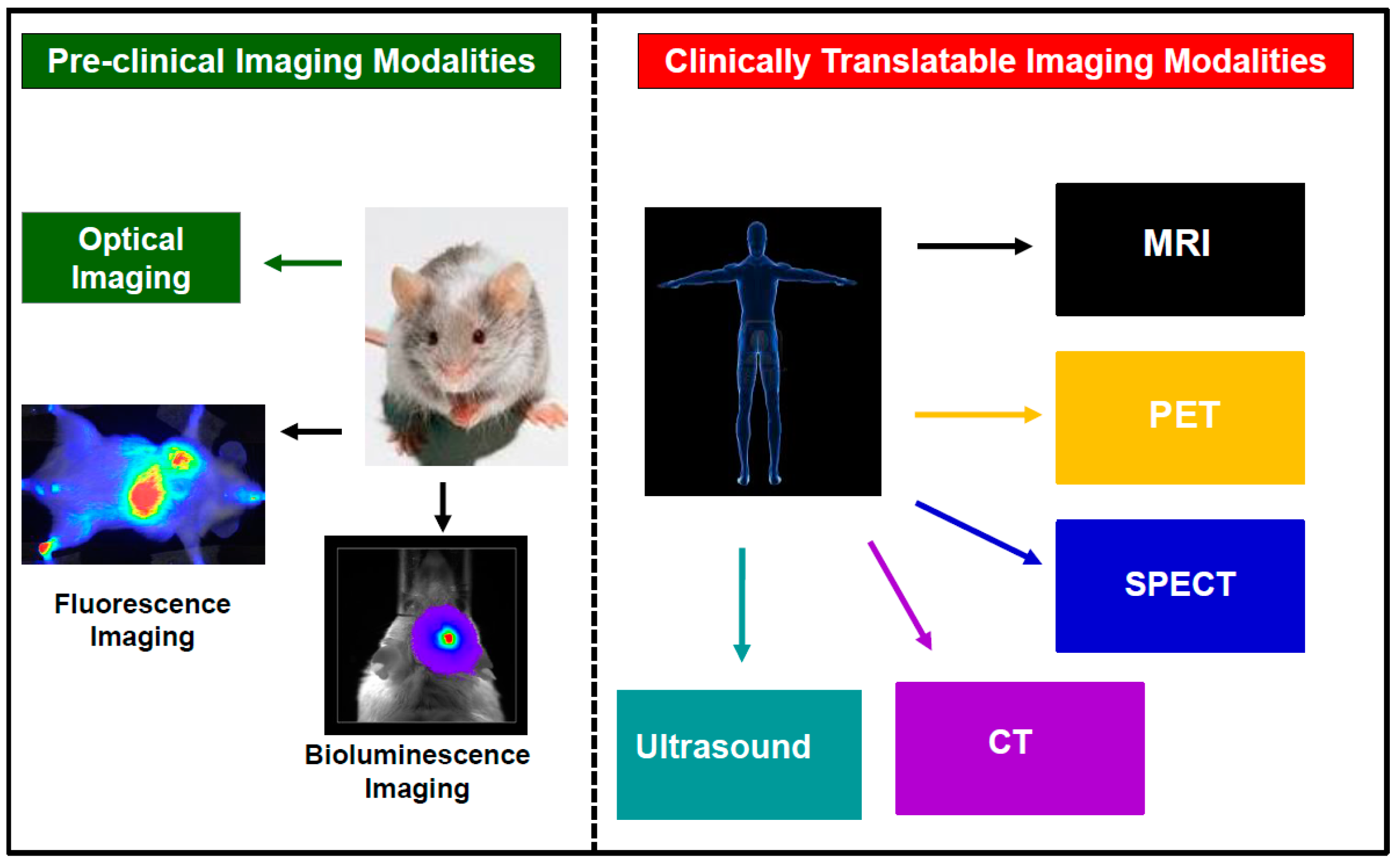
2. Current Trends in Cellular Imaging
3. The Principle of Cell Tracking with MRI
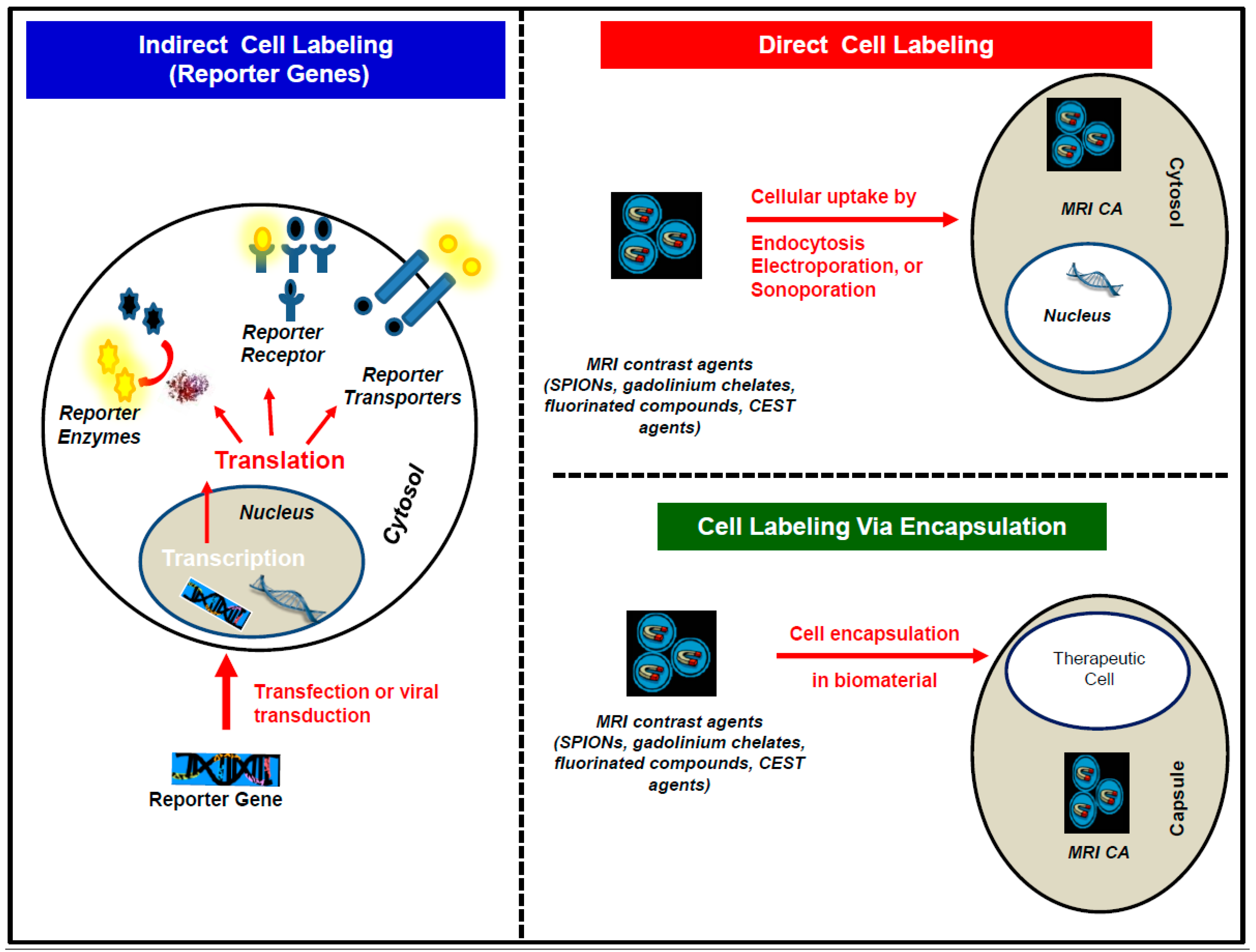
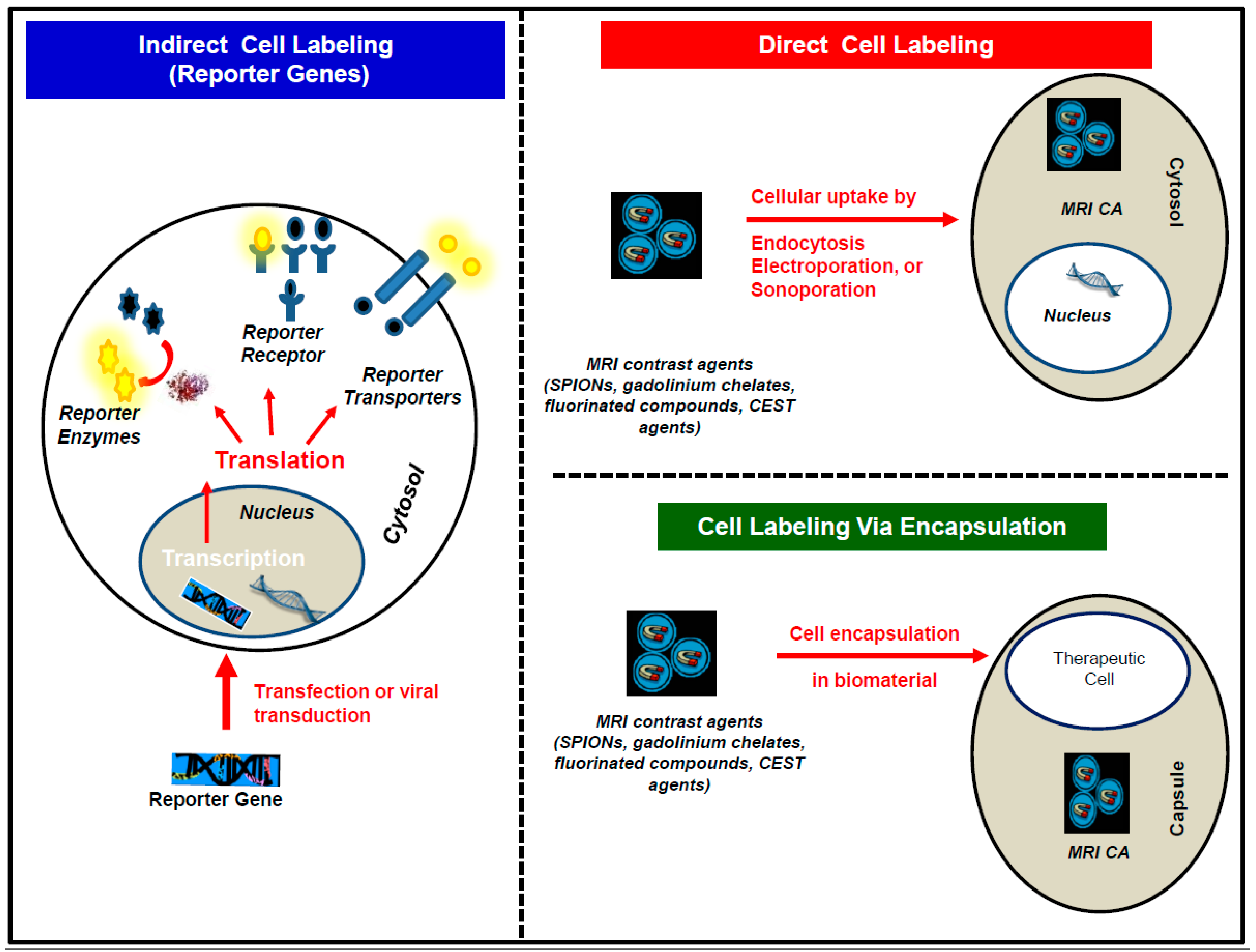
3.1. The Direct Cell Labeling Technique
3.2. The Indirect Cell Labeling Technique
3.3. The Encapsulation Cell Labeling Technique
4. MRI Contrast Generation Mechanisms
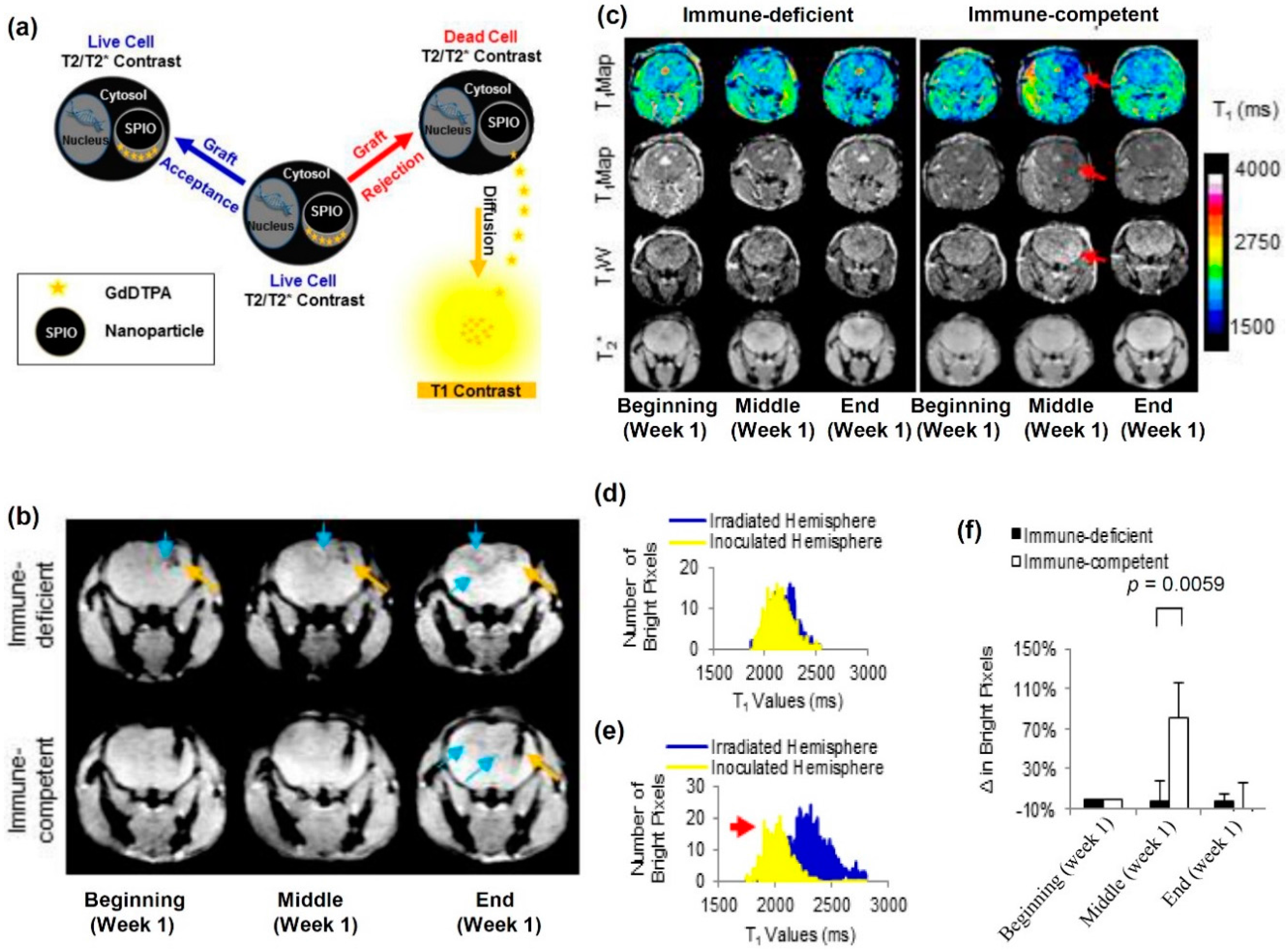
4.1. T2/T2* Contrast Agents
4.2. T1 Contrast Agents
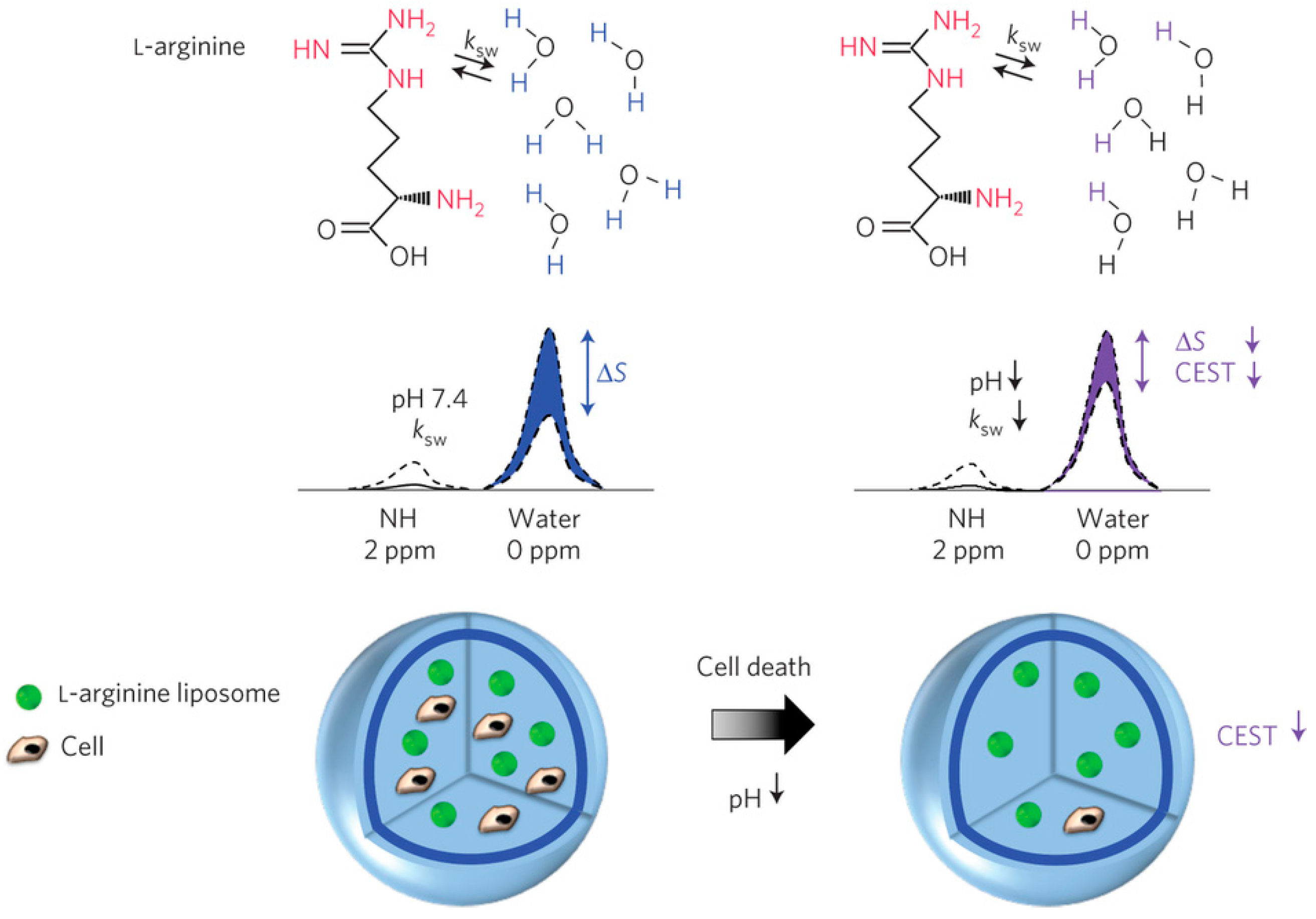
4.3. Chemical Exchange Saturation Transfer (CEST) Contrast Agents
4.4. Fluorine (19F) Contrast Agents
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ADSCs | Adipose-derived stem cells |
| ALS | Amyotrophic lateral sclerosis |
| CEST | Chemical exchange saturation transfer |
| CNS | Central nervous system |
| CPCs | Cardiac progenitor cells |
| CT | Computed tomography |
| DIACEST | Diamagnetic chemical exchange saturation transfer |
| EGCs | Embryonic germinal cells |
| EPCs | Epithelial progenitor cells |
| ESCs | Embryonic stem cells |
| FDA | Food and drug administration |
| 18F | Fluorine-18 isotope |
| 19F | Fluorine-19 isotope |
| HSCs | Hematopoietic stem cells |
| HyperCEST | Hyperpolarized chemical exchange saturation transfer |
| iN | Induced neuronal cells |
| iNPCs | Induced neuronal progeneitor cells |
| iPSCs | Induced pluripotent stem cells |
| LEPCs | Lens epithelial progenitor cells |
| LPSCs | Liver stem cells/progenitor cells |
| MRI | Magnetic Resonance Imaging |
| MS | Multiple sclerosis |
| MSCs | Mesenchymal stem cells |
| NIH | National Institute of Health |
| NPCs | Neural progenitor cells |
| NSCs | Neural stem cells |
| PARACEST | Paramagnetic chemical exchange saturation transfer |
| PET | Positron emission tomography |
| pH | Hydrogen potential |
| PLGA | Poly lactic-co-glycolic acid |
| PLL | Poly-l-lysine |
| R1 | Longitudinal relaxation rate |
| R2 | Transverse relaxation rate |
| SEPCs | Sinosoidal endothelial progenitor cells |
| SHPCs | Small hepatocytes-like progenitor cells |
| SPECT | Single photon emission computed tomography |
| SPIO | Superparamagnetic iron oxide |
| T1 | Longitudinal relaxation time |
| T2 | Transverse relaxation time |
References
- Schwarz, S.C.; Schwarz, J. Translation of stem cell therapy for neurological diseases. Transl. Res. 2010, 156, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Mimeault, M.; Batra, S.K. Concise review: Recent avances on the significance of stem cells in tissue regeneration and cancer therapies. Stem Cells 2006, 24, 2319–2345. [Google Scholar] [CrossRef]
- Mimeault, M.; Hauke, R.; Batra, S.K. Stem, Cells: A revolution in therapeutics—Recent advances in stem cell biology and their therapeutic applications in regenerative medicine and cancer therapies. Clin. Pharmacol. Ther. 2007, 82, 252–264. [Google Scholar] [CrossRef] [PubMed]
- NIH Cell Therapies: Completed. Available online: http://www.webcitation.org/6nRZoxwb4 (accessed on 16 January 2017).
- NIH Cell Therapies: Open. Available online: http://www.webcitation.org/6nRZkihz4 (accessed on 16 January 2017).
- NIH Cell Therapies in Oncology: Completed. Available online: http://www.webcitation.org/6nRainDln (accessed on 16 January 2017).
- NIH Cell Therapies in Oncology: Open. Available online: http://www.webcitation.org/6nRb1iPab (accessed on 16 January 2017).
- Hedlund, E.; Perlmann, T. Neuronal cell replacement in Parkinson’s disease. J. Int. Med. 2009, 266, 358–371. [Google Scholar] [CrossRef] [PubMed]
- Baraniak, P.R.; McDevitt, T.C. Stem cell paracrine actions and tissue regeneration. Regen. Med. 2010, 5, 121–143. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, J.; Leyh, B. Paracrine effects of stem cells in wound healing and cancer progression. Int. J. Oncol. 2014, 44, 1789–1798. [Google Scholar] [CrossRef] [PubMed]
- Grealish, S.; Drouin-Ouellet, J.; Parmar, M. Brain repair and reprogramming: The route to clinical translation. J. Int. Med. 2016, 280, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.U.; Lee, H.J.; Kim, Y.B. Neural stem cell-based treatment for neurodegenerative diseases. Neuropathology 2013, 33, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Narayanan, K.; Chaudhary, R. K.; Mishra, S.; Kumar, S.; Vinoth, K.J.; Padmanabhan, P.; Gulyás, B. Current perspective of stem cell therapy in neurodegenerative and metabolic diseases. Mol. Neurobiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Lescaudron, L.; Naveilhan, P.; Neveu, I. The use of stem cells in regenerative medicine for Parkinson’s and Huntington’s diseases. Curr. Med. Chem. 2012, 19, 6018–6035. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, S.; Yang, D.; Le, W.D. Stem cell transplantation: A promising therapy for Parkinson’s disease. J. Neuroimmune. Pharmacol. 2007, 2, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Francisco, P.M.; Richard, H.W.F. Implications of parkinson’s disease pathophysiology for the development of cell replacement strategies and drug discovery in neurodegenerative diseases. CNS Neurol. Disord. Drug Targets 2012, 11, 907–920. [Google Scholar]
- Gibson, S.A.J.; Gao, G.D.; McDonagh, K.; Shen, S. Progress on stem cell research towards the treatment of Parkinson’s disease. Stem Cell Res. Ther. 2012, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Tincer, G.; Mashkaryan, V.; Bhattarai, P.; Kizil, C. Neural stem/progenitor cells in Alzheimer’s disease. Yale J. Biol. Med. 2016, 89, 23–35. [Google Scholar]
- Ager, R.R.; Davis, J.L.; Agazaryan, A.; Benavente, F.; Poon, W.W.; LaFerla, F.M.; Blurton-Jones, M. Human neural stem cells improve cognition and promote synaptic growth in two complementary transgenic models of Alzheimer’s disease and neuronal loss. Hippocampus 2015, 25, 813–826. [Google Scholar] [CrossRef] [PubMed]
- McGinley, L.M.; Sims, E.; Lunn, J.S.; Kashlan, O.N.; Chen, K.S.; Bruno, E.S.; Pacut, C.M.; Hazel, T.; Johe, K.; Sakowski, S.A.; et al. Human cortical neural stem cells expressing insulin-like growth factor-I: A novel cellular therapy for Alzheimer’s disease. Stem Cells Transl. Med. 2016, 5, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Di Ruscio, A.; Patti, F.; Welner, R.S.; Tenen, D.G.; Amabile, G. Multiple sclerosis: Getting personal with induced pluripotent stem cells. Cell Death Dis. 2015, 6, e1806. [Google Scholar]
- Xiao, J.; Yang, R.; Biswas, S.; Qin, X.; Zhang, M.; Deng, W. Mesenchymal, Stem Cells and Induced, Pluripotent Stem, Cells as Therapies for Multiple, Sclerosis. Int. J. Mol. Sci. 2015, 16, 9283–9302. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.; Chen, Y. Stem cell transplantation for treating spinal cord injury: A literature comparison between studies of stem cells obtained from various sources. Neural. Regen. Res. 2012, 7, 1256–1263. [Google Scholar] [PubMed]
- Boido, M.; Garbossa, D.; Fontanella, M.; Ducati, A.; Vercelli, A. Mesenchymal stem cell transplantation reduces glial cyst and improves functional outcome after spinal cord compression. World Neurosurg. 2014, 81, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Chau, M.J.; Deveau, T.C.; Song, M.; Gu, X.; Chen, D.; Wei, L. iPSC transplantation increases regeneration and functional recovery after ischemic stroke in neonatal rats. Stem Cells 2014, 32, 3075–3087. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.J.; Lee, N.; Park, I.H.; Choi, C.; Jeon, I.; Kwon, J.; Oh, S.H.; Shin, D.A.; Do, J.T.; Lee, D.R.; et al. Therapeutic potential of human induced pluripotent stem cells in experimental stroke. Cell Transplant. 2013, 22, 1427–1440. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.; Zou, Z.; Tian, H.; Zhang, Y.; Zhou, H.; Liu, L. Stem cell-based therapies for ischemic stroke. BioMed Res. Int. 2014. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Sun, J.; Sheng, C.; Wang, Z.; Wang, Y.; Zhang, C.; Fan, R. Systematic review and meta-analysis of efficacy of mesenchymal stem cells on locomotor recovery in animal models of traumatic brain injury. Stem Cell Res. Ther. 2015, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Wang, X.; Xiong, W.; Chen, J. In vivo reprogramming reactive glia into iPSCs to produce new neurons in the cortex following traumatic brain injury. Sci. Rep. 2016, 6, 22490. [Google Scholar] [CrossRef] [PubMed]
- Savitz, S.I.; Cox, C.S. Concise review: Cell therapies for stroke and traumatic brain injury: Targeting microglia. Stem Cells 2016, 34, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Ngen, E.J.; Wang, L.; Gandhi, N.; Kato, Y.; Armour, M.; Zhu, W.; Wong, J.; Gabrielson, K.L.; Artemov, D. A preclinical murine model for the early detection of radiation-induced brain injury using magnetic resonance imaging and behavioral tests for learning and memory: With applications for the evaluation of possible stem cell imaging agents and therapies. J. Neuro Oncol. 2016, 128, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Acharya, M.M.; Martirosian, V.; Christie, L.A.; Limoli, C.L. Long-term cognitive effects of human stem cell transplantation in the irradiated brain. Int. J. Radiat. Biol. 2014, 90, 816–820. [Google Scholar] [CrossRef] [PubMed]
- Acharya, M.M.; Rosi, S.; Jopson, T.; Limoli, C.L. Human neural stem cell transplantation provides long-term restoration of neuronal plasticity in the irradiated hippocampus. Cell Transplant. 2015, 24, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Greene-Schloesser, D.; Robbins, M.E. Radiation-induced cognitive impairment-from bench to bedside. Neuro Oncol. 2012, 14, IV37–IV44. [Google Scholar] [CrossRef] [PubMed]
- Piao, J.; Major, T.; Auyeung, G.; Policarpio, E.; Menon, J.; Droms, L.; Gutin, P.; Uryu, K.; Tchieu, J.; Soulet, D.; et al. Human embryonic stem cell-derived oligodendrocyte progenitors remyelinate the brain and rescue behavioral deficits following radiation. Cell Stem Cell 2015, 16, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Hocking, A.M.; Gibran, N.S. Mesenchymal stem cells: Paracrine signaling and differentiation during cutaneous wound repair. Exp. Cell Res. 2010, 316, 2213–2219. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhao, R.C.H.; Tredget, E.E. Concise review: Bone marrow-derived stem/progenitor cells in cutaneous repair and regeneration. Stem Cells 2010, 28, 905–915. [Google Scholar] [PubMed]
- Le, T.Y.L.; Chong, J.J.H. Cardiac progenitor cells for heart repair. Cell Death Discov. 2016, 2, 16052. [Google Scholar] [PubMed]
- Lerman, D.A.; Alotti, N.; Ume, K.L.; Péault, B. Cardiac repair and regeneration: The value of cell therapies. Eur. Cardiol. 2016, 11, 43–48. [Google Scholar] [CrossRef]
- Nguyen, P.K.; Rhee, J.; Wu, J.C. Adult stem cell therapy and heart failure, 2000 to 2016: A systematic review. JAMA Cardiol. 2016, 1, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Erbani, J.; Aberdam, D.; Larghero, J.; Vanneaux, V. Pluripotent stem cells and other innovative strategies for the treatment of ocular surface diseases. Stem Cell Rev. 2016, 12, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Gromova, A.; Voronov, D.A.; Yoshida, M.; Thotakura, S.; Meech, R.; Dartt, D.A.; Makarenkova, H.P. Lacrimal gland repair using progenitor cells. Stem Cells Transl. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Ouyang, H.; Zhu, J.; Huang, S.; Liu, Z.; Chen, S.; Cao, G.; Li, G.; Signer, R.A.J.; Xu, Y.; et al. Lens regeneration using endogenous stem cells with gain of visual function. Nature 2016, 531, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Omoto, M.; Katikireddy, K.R.; Rezazadeh, A.; Dohlman, T.H.; Chauhan, S.K. Mesenchymal stem cells home to inflamed ocular surface and suppress allosensitization in corneal transplantation M.S.C.s suppress corneal alloimmunity. Inves. Ophth. Vis. Sci. 2014, 55, 6631–6638. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.H.; Ren, L.N.; Wang, T.; Navarro-Alvarez, N.; Tang, L.J. The involving roles of intrahepatic and extrahepatic stem/progenitor cells (SPCs) to liver regeneration. Int. J. Biol. Sci. 2016, 12, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Winkler, S.; Hempel, M.; Brückner, S.; Tautenhahn, H.M.; Kaufmann, R.; Christ, B. Identification of pathways in liver repair potentially targeted by secretory proteins from human mesenchymal stem cells. Int. J. Mol. Sci. 2016, 17, 1099. [Google Scholar] [CrossRef] [PubMed]
- Brady, K.; Dickinson, S.C.; Guillot, P.V.; Polak, J.; Blom, A.W.; Kafienah, W.; Hollander, A.P. Human fetal and adult bone marrow-derived mesenchymal stem cells use different signaling pathways for the initiation of chondrogenesis. Stem Cells Dev. 2014, 23, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Gobbi, A.; Karnatzikos, G.; Sankineani, S.R. One-step surgery with multipotent stem cells for the treatment of large full-thickness chondral defects of the knee. Am. J. Sport Med. 2014, 42, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Ha, C.W.; Park, Y.B.; Chung, J.Y.; Park, Y.G. Cartilage repair using composites of human umbilical cord blood-derived mesenchymal stem cells and hyaluronic acid hydrogel in a minipig model. Stem Cells Trans. Med. 2015, 4, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, I.; Muneta, T.; Horie, M.; Koga, H. Arthroscopic transplantation of synovial stem cells improves clinical outcomes in knees with cartilage defects. Clin. Orthop. Relat. Res. 2015, 473, 2316–2326. [Google Scholar] [CrossRef] [PubMed]
- Vonk, L.A.; de Windt, T.S.; Kragten, A.H.M.; Beekhuizen, M.; Mastbergen, S.C.; Dhert, W.J.A.; Lafeber, F.P.J.G.; Creemers, L.B.; Saris, D.B.F. Enhanced cell-induced articular cartilage regeneration by chondrons; the influence of joint damage and harvest site. Osteoarthr. Cartilage 2014, 22, 1910–1917. [Google Scholar] [CrossRef] [PubMed]
- Finocchiaro, G.; Pellegatta, S. Immunotherapy with dendritic cells loaded with glioblastoma stem cells: From preclinical to clinical studies. Cancer Immunol. Immun. 2016, 65, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Nava, S.; Lisini, D.; Pogliani, S.; Dossena, M.; Bersano, A.; Pellegatta, S.; Parati, E.; Finocchiaro, G.; Frigerio, S. Safe and reproducible preparation of functional dendritic cells for immunotherapy in glioblastoma patients. Stem Cells Transl. Med. 2015, 4, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Batich, K.A.; Swartz, A.M.; Sampson, J.H. Enhancing dendritic cell-based vaccination for highly aggressive glioblastoma. Expert Opin. Biol. Ther. 2015, 15, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Schaller, T.H.; Sampson, J.H. Advances and challenges: Dendritic cell vaccination strategies for glioblastoma. Expert Rev. Vaccines 2017, 16, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Desai, R.; Suryadevara, C.M.; Batich, K.A.; Farber, S.H.; Sanchez-Perez, L.; Sampson, J.H. Emerging immunotherapies for glioblastoma. Expert Opin. Emerg. Drugs 2016, 21, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Liu, G.; Yuan, X.; Xu, M.; Wang, H.; Ji, J.; Konda, B.; Black, K.L.; Yu, J.S. Antigen-specific T-cell response from dendritic cell vaccination using cancer stem-like cell-associated antigens. Stem Cells 2009, 27, 1734–1740. [Google Scholar] [CrossRef] [PubMed]
- Ikrame, A.; Elodie, P.; Catherine, S.B.; Audrey, D.; Rodney, M.; Géraldine, L.; Johanne, S.; Eric, T.; François, L.; Philippe, B.; et al. Engineered mesenchymal stem cells as vectors in a suicide gene therapy against preclinical murine models for solid tumors. J. Control. Release 2016, 239, 82–91. [Google Scholar]
- Amara, I.; Touati, W.; Beaune, P.; de Waziers, I. Mesenchymal stem cells as cellular vehicles for prodrug gene therapy against tumors. Biochimie 2014, 105, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Uchibori, R.; Tsukahara, T.; Ohmine, K.; Ozawa, K. Cancer gene therapy using mesenchymal stem cells. Int. J. Hematol. 2014, 99, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Montserrat, E.; Dreger, P. Treatment of chronic lymphocytic leukemia with del (17p)/ TP53 mutation: Allogeneic hematopoietic stem cell transplantation or BCR-signaling inhibitors? Clin. Lymphoma Myeloma Leuk. 2016, 16, S74–S81. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Saulle, E.; Castelli, G.; Pelosi, E. Endothelial progenitor cells in hematologic malignancies. Stem Cell Investig. 2016, 3, 26. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.F.; Gambhir, S.S.; Grimm, J. Noninvasive cell-tracking methods. Nat. Rev. Clin. Oncol. 2011, 8, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Naumova, A.V.; Modo, M.; Moore, A.; Murry, C.E.; Frank, J.A. Clinical imaging in regenerative medicine. Nat. Biotech. 2014, 32, 804–818. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Frank, J.A. Detection and quantification of magnetically labeled cells by cellular MRI. Eur. J. Radiol. 2009, 70, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, E.M.; Sharer, K.; Skrtic, S.; Koretsky, A.P. In vivo detection of single cells by MRI. Magn. Reson. Med. 2006, 55, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Makela, A.V.; Murrell, D.H.; Parkins, K.M.; Kara, J.; Gaudet, J.M.; Foster, P.J. Cellular, Imaging With, MRI. Top. Magn. Reson. Imaging 2016, 25, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Frank, J.A.; Miller, B.R.; Arbab, A.S.; Zywicke, H.A.; Jordan, E.K.; Lewis, B.K.; Bryant, L.H.; Bulte, J.W.M. Clinically applicable labeling of mammalian and stem cells by combining superparamagnetic iron oxides and transfection agents. Radiology 2003, 228, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Kalish, H.; Arbab, A.S.; Miller, B.R.; Lewis, B.K.; Zywicke, H.A.; Bulte, J.W.M.; Bryant, L.H.; Frank, J.A. Combination of transfection agents and magnetic resonance contrast agents for cellular imaging: Relationship between relaxivities, electrostatic forces, and chemical composition. Magn. Reson. Med. 2003, 50, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, E.T.; Flores, R.; Xu, H.; Morel, P.A. In vivo imaging platform for tracking immunotherapeutic cells. Nat. Biotech. 2005, 23, 983–987. [Google Scholar] [CrossRef]
- Qiu, B.; Xie, D.; Walczak, P.; Li, X.; Ruiz-Cabello, J.; Minoshima, S.; Bulte, J.W.M.; Yang, X. Magnetosonoporation: Instant magnetic labeling of stem cells. Magn. Reson. Med. 2010, 63, 1437–1441. [Google Scholar] [CrossRef] [PubMed]
- Walczak, P.; Kedziorek, D.A.; Gilad, A.A.; Lin, S.; Bulte, J.W.M. Instant, MR labeling of stem cells using magnetoelectroporation. Magn. Reson. Med. 2005, 54, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Walczak, P.; Ruiz-Cabello, J.; Kedziorek, D.A.; Gilad, A.A.; Lin, S.; Barnett, B.; Qin, L.; Levitsky, H.; Bulte, J.W.M. Magnetoelectroporation: Improved labeling of neural stem cells and leukocytes for cellular magnetic resonance imaging using a single FDA-approved agent. Nanomedicine 2006, 2, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Granot, D.; Scheinost, D.; Markakis, E.A.; Papademetris, X.; Shapiro, E.M. Serial monitoring of endogenous neuroblast migration by cellular MRI. Neuroimage 2011, 57, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Jefferson, A.; Wijesurendra, R.S.; McAteer, M.A.; Choudhury, R.P. Development and application of endothelium-targeted microparticles for molecular magnetic resonance imaging. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2012, 4, 247–256. [Google Scholar] [CrossRef] [PubMed]
- McAteer, M.A.; Schneider, J.E.; Ali, Z.A.; Warrick, N.; Bursill, C.A.; von zur Muhlen, C.; Greaves, D.R.; Neubauer, S.; Channon, K.M.; Choudhury, R.P. Magnetic resonance imaging of endothelial adhesion molecules in mouse atherosclerosis using dual-targeted microparticles of iron oxide. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 77–83. [Google Scholar] [CrossRef] [PubMed]
- McAteer, M.A.; Mankia, K.; Ruparelia, N.; Jefferson, A.; Nugent, H.B.; Stork, L.A.; Channon, K.M.; Schneider, J.E.; Choudhury, R.P. A leukocyte-mimetic magnetic resonance imaging contrast agent homes rapidly to activated endothelium and tracks with atherosclerotic lesion macrophage content. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.; Ng, T.S.C.; Sohi, H.; Palko, H.; House, A.; Jacobs, R.E.; Louie, A.Y. Receptor-targeted iron oxide nanoparticles for molecular MR imaging of inflamed atherosclerotic plaques. Biomaterials 2011, 32, 7209–7216. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Siow, T.Y.; Chou, C.H.; Lin, C.H.; Lin, M.H.; Chen, Y.C.; Hsieh, W.Y.; Wang, S.J.; Chang, C. Targeted superparamagnetic iron oxide nanoparticles for in vivo magnetic resonance imaging of T-cells in rheumatoid arthritis. Mol. Imaging Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ye, Q.; Wu, Y.; Hsieh, W.Y.; Chen, C.L.; Shen, H.H.; Wang, S.J.; Zhang, H.; Hitchens, T.K.; Ho, C. Tracking, T-cells in vivo with a new nano-sized MRI contrast agent. Nanomedicine 2012, 8, 1345–1354. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Duan, X.; Lu, L.; Zhang, X.; Zhong, X.; Mao, J.; Chen, M.; Shen, J. In vivo targeted MR imaging of endogenous neural stem cells in ischemic stroke. Molecules 2016, 21, 1143. [Google Scholar] [CrossRef] [PubMed]
- Khurana, A.; Chapelin, F.; Beck, G.; Lenkov, O.D.; Donig, J.; Nejadnik, H.; Messing, S.; Derugin, N.; Chan, R.C.F.; Gaur, A.; et al. Iron administration before stem cell harvest enables MR imaging tracking after transplantation. Radiology 2013, 269, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Bulte, J.W.M. Science to practice: Can stem cells be labeled inside the body instead of outside? Radiology 2013, 269, 1–3. [Google Scholar] [CrossRef] [PubMed]
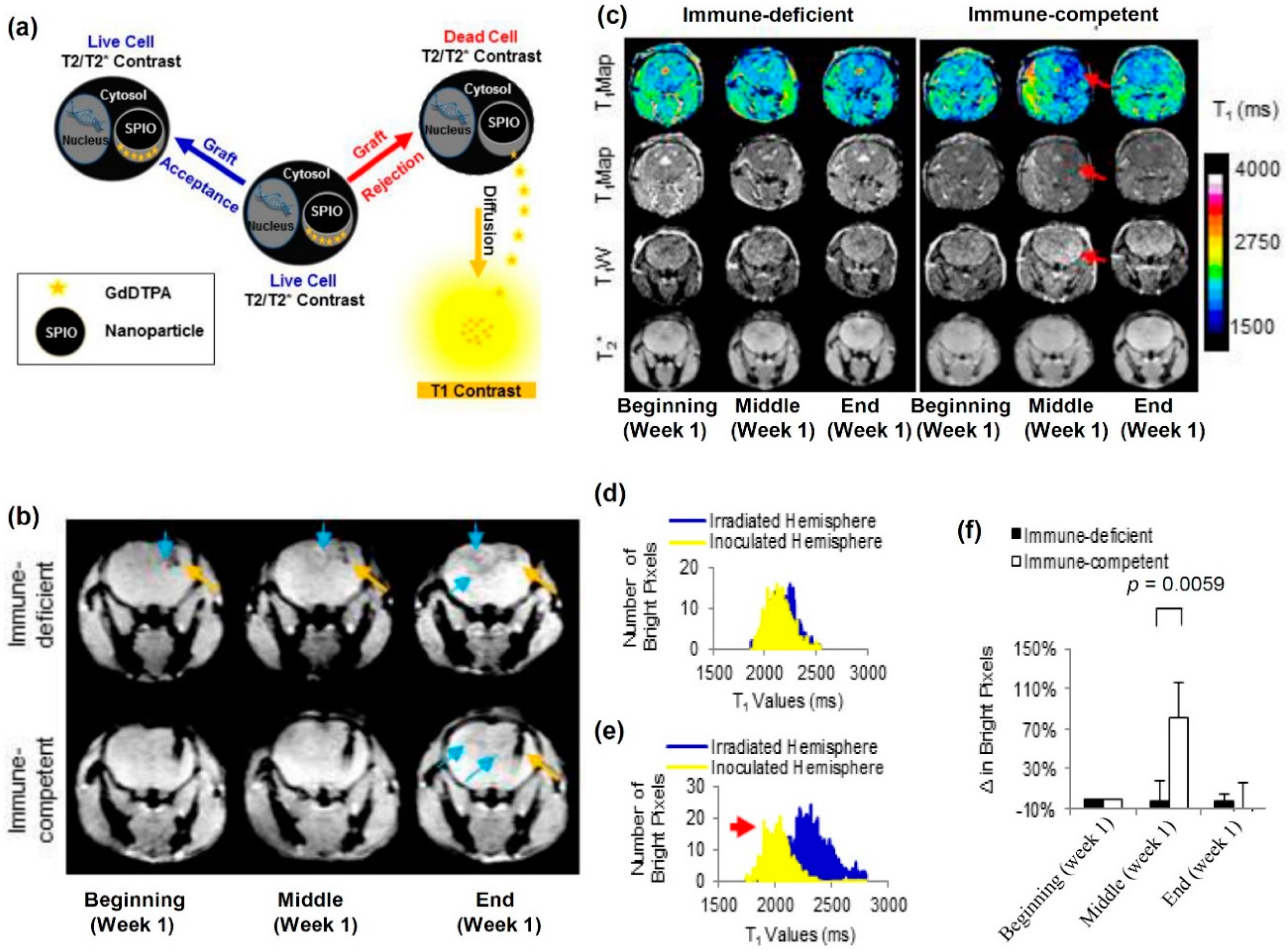
- Winter, E.M.; Hogers, B.; van der Graaf, L.M.; Gittenberger-de Groot, A.C.; Poelmann, R.E.; van der Weerd, L. Cell tracking using iron oxide fails to distinguish dead from living transplanted cells in the infarcted heart. Magn. Reson. Med. 2010, 63, 817–821. [Google Scholar] [CrossRef] [PubMed]
- Cianciaruso, C.; Pagani, A.; Martelli, C.; Bacigaluppi, M.; Leonardo Squadrito, M.; lo Dico, A.; de Palma, M.; Furlan, R.; Lucignani, G.; Falini, A.; et al. Cellular magnetic resonance with iron oxide nanoparticles: Long-term persistence of SPIO signal in the CNS after transplanted cell death. Nanomedicine 2014, 9, 1457–1474. [Google Scholar] [CrossRef] [PubMed]
- Naumova, A.V.; Balu, N.; Yarnykh, V.L.; Reinecke, H.; Murry, C.E.; Yuan, C. Magnetic resonance imaging tracking of graft survival in the infarcted heart: Iron oxide particles versus ferritin overexpression approach. J. Cardiovasc. Pharmacol. Ther. 2014, 19, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Terrovitis, J.; Stuber, M.; Youssef, A.; Preece, S.; Leppo, M.; Kizana, E.; Schär, M.; Gerstenblith, G.; Weiss, R.G.; Marbán, E.; et al. Magnetic resonance imaging overestimates ferumoxide-labeled stem cell survival after transplantation in the heart. Circulation 2008, 117, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- Ngen, E.J.; Wang, L.; Kato, Y.; Krishnamachary, B.; Zhu, W.; Gandhi, N.; Smith, B.; Armour, M.; Wong, J.; Gabrielson, K.; et al. Imaging transplanted stem cells in real time using an MRI dual-contrast method. Sci. Rep. 2015, 5, 13628. [Google Scholar] [CrossRef] [PubMed]
- Henning, T.D.; Wendland, M.F.; Golovko, D.; Sutton, E.J.; Sennino, B.; Malek, F.; Bauer, J.S.; McDonald, D.M.; Daldrup-Link, H. Relaxation effects of ferucarbotran-labeled mesenchymal stem cells at 1.5 T and 3 T: Discrimination of viable from lysed cells. Magn. Reson. Med. 2009, 62, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Guenoun, J.; Ruggiero, A.; Doeswijk, G.; Janssens, R.C.; Koning, G.A.; Kotek, G.; Krestin, G.P.; Bernsen, M.R. In vivo quantitative assessment of cell viability of gadolinium or iron-labeled cells using MRI and bioluminescence imaging. Contrast Media Mol. 2013, 8, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, Y.; Enmi, J.; Agudelo, C.A.; Iida, H.; Yamaoka, T. Long-Term/Bioinert, Labeling of Rat, Mesenchymal Stem, Cells with PVA-Gd, Conjugates and MRI Monitoring of the Labeled, Cell Survival after Intramuscular, Transplantation. Bioconjug. Chem. 2014, 25, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Ribot, E.J.; Foster, P.J. In vivo MRI discrimination between live and lysed iron-labelled cells using balanced steady state free precession. Eur. Radiol. 2012, 22, 2027–2034. [Google Scholar] [CrossRef] [PubMed]
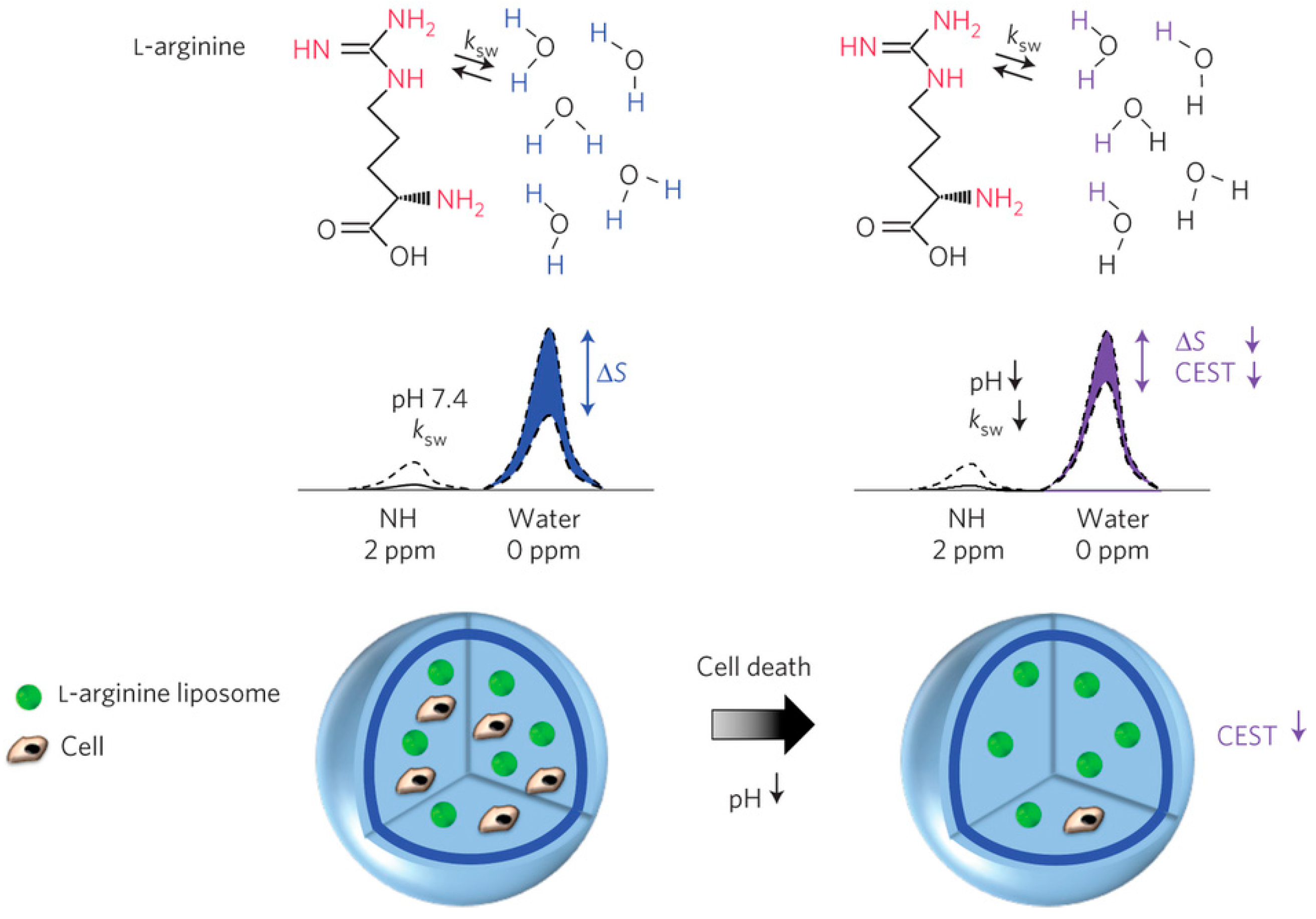
- Chan, K.W.Y.; Liu, G.; Song, X.; Kim, H.; Yu, T.; Arifin, D.R.; Gilad, A.A.; Hanes, J.; Walczak, P.; van Zijl, P.C.M.; et al. MRI-detectable pH nanosensors incorporated into hydrogels for in vivo sensing of transplanted-cell viability. Nat. Mater. 2013, 12, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Ngen, E.J.; Bar-Shir, A.; Jablonska, A.; Liu, G.; Song, X.; Ansari, R.; Bulte, J.W.M.; Janowski, M.; Pearl, M.; Walczak, P.; et al. Imaging the DNA alkylator melphalan by CEST MRI An advanced approach to theranostics. Mol. Pharm. 2016, 13, 3043–3053. [Google Scholar] [CrossRef] [PubMed]
- Berman, S.C.; Galpoththawela, C.; Gilad, A.A.; Bulte, J.W.M.; Walczak, P. Long-term MR cell tracking of neural stem cells grafted in immunocompetent versus immunodeficient mice reveals distinct differences in contrast between live and dead cells. Magn. Reson. Med. 2011, 65, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Gilad, A.A.; Winnard, P.T.; van Zijl, P.C.M.; Bulte, J.W.M. Developing, MR reporter genes: Promises and pitfalls. N.M.R. Biomed. 2007, 20, 275–290. [Google Scholar] [CrossRef] [PubMed]
- Gilad, A.A.; Ziv, K.; McMahon, M.T.; van Zijl, P.C.M.; Neeman, M.; Bulte, J.W.M. MRI reporter genes. J. Nucl. Med. 2008, 49, 1905–1908. [Google Scholar] [CrossRef] [PubMed]
- Cohen, B.; Ziv, K.; Plaks, V.; Israely, T.; Kalchenko, V.; Harmelin, A.; Benjamin, L.E.; Neeman, M. MRI detection of transcriptional regulation of gene expression in transgenic mice. Nat. Med. 2007, 13, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Genove, G.; DeMarco, U.; Xu, H.; Goins, W.F.; Ahrens, E.T. A new transgene reporter for in vivo magnetic resonance imaging. Nat. Med. 2005, 11, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cheng, E.C.H.; Long, R.C.; Yang, S.H.; Wang, L.; Cheng, P.H.; Yang, J.; Wu, D.; Mao, H.; Chan, A.W.S. Noninvasive monitoring of embryonic stem cells in vivo with MRI transgene reporter. Tissue Eng. Part C Methods 2009, 15, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Louie, A.Y.; Huber, M.M.; Ahrens, E.T.; Rothbacher, U.; Moats, R.; Jacobs, R.E.; Fraser, S.E.; Meade, T.J. In vivo visualization of gene expression using magnetic resonance imaging. Nat. Biotechnol. 2000, 18, 321–325. [Google Scholar] [PubMed]
- Bar-Shir, A.; Liang, Y.; Chan, K.W.Y.; Gilad, A.A.; Bulte, J.W.M. Supercharged green fluorescent proteins as bimodal reporter genes for CEST MRI and optical imaging. Chem. Commun. 2015, 51, 4869–4871. [Google Scholar] [CrossRef] [PubMed]
- Bar-Shir, A.; Liu, G.; Chan, K.W.; Oskolkov, N.; Song, X.; Yadav, N.N.; Walczak, P.; McMahon, M.T.; van Zijl, P.C.; Bulte, J.W.; et al. Human protamine-1 as an MRI reporter gene based on chemical exchange. ACS Chem. Biol. 2014, 9, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Gilad, A.A.; McMahon, M.T.; Walczak, P.; Winnard, P.T.; Raman, V.; van Laarhoven, H.W.M.; Skoglund, C.M.; Bulte, J.W.M.; van Zijl, P.C.M. Artificial reporter gene providing MRI contrast based on proton exchange. Nat. Biotechnol. 2007, 25, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Airan, R.D.; Bar-Shir, A.; Liu, G.; Pelled, G.; McMahon, M.T.; van Zijl, P.C.M.; Bulte, J.W.M.; Gilad, A.A. MRI biosensor for protein kinase A encoded by a single synthetic gene. Magn. Reson. Med. 2012, 68, 1919–1923. [Google Scholar] [CrossRef] [PubMed]
- Bar-Shir, A.; Liu, G.; Liang, Y.; Yadav, N.N.; McMahon, M.T.; Walczak, P.; Nimmagadda, S.; Pomper, M.G.; Tallman, K.A.; Greenberg, M.M.; et al. Transforming thymidine into a magnetic resonance imaging probe for monitoring gene expression. J. Am. Chem. Soc. 2013, 135, 1617–1624. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Liang, Y.; Bar-Shir, A.; Chan, K.W.Y.; Galpoththawela, C.S.; Bernard, S.M.; Tse, T.; Yadav, N.N.; Walczak, P.; McMahon, M.T.; et al. Monitoring enzyme activity using a diamagnetic chemical exchange saturation transfer magnetic resonance imaging contrast agent. J. Am. Chem. Soc. 2011, 133, 16326–16329. [Google Scholar] [CrossRef] [PubMed]
- Arifin, D.R.; Kedziorek, D.A.; Fu, Y.; Chan, K.W.Y.; McMahon, M.T.; Weiss, C.R.; Kraitchman, D.L.; Bulte, J.W.M. Microencapsulated cell tracking. NMR Biomed. 2013, 26, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Paredes-Juarez, G.A.; Barnett, B.P.; Bulte, J.W.M. Noninvasive tracking of alginate-microencapsulated cells. In Cell Microencapsulation: Methods and Protocols; Opara, E.C., Ed.; Springer: New York, NY, USA, 2017; pp. 143–155. [Google Scholar]
- Barnett, B.P.; Arepally, A.; Stuber, M.; Arifin, D.R.; Kraitchman, D.L.; Bulte, J.W.M. Synthesis of magnetic resonance–, X-ray– and ultrasound-visible alginate microcapsules for immunoisolation and noninvasive imaging of cellular therapeutics. Nat. Protoc. 2011, 6, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- Arifin, D.R.; Valdeig, S.; Anders, R.A.; Bulte, J.W.M.; Weiss, C.R. Magnetoencapsulated human islets xenotransplanted into swine: A comparison of different transplantation sites. Xenotransplantation 2016, 23, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Azene, N.; Ehtiati, T.; Flammang, A.; Gilson, W.D.; Gabrielson, K.; Weiss, C.R.; Bulte, J.W.M.; Solaiyappan, M.; Johnston, P.V.; et al. Fused, X-ray and MR Imaging guidance of intrapericardial delivery of microencapsulated human mesenchymal stem cells in immunocompetent swine. Radiology 2014, 272, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Bulte, J.W.M. In vivo MRI cell tracking: Clinical studies. Am. J. Roentgenol. 2009, 193, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Evgenov, N.V.; Medarova, Z.; Dai, G.; Bonner-Weir, S.; Moore, A. In vivo imaging of islet transplantation. Nat. Med. 2006, 12, 144–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraitchman, D.L.; Kedziorek, D.A.; Bulte, J.W.M. MR Imaging of transplanted stem cells in myocardial infarction molecular imaging. In Molecular Imaging; Shah, K., Ed.; Humana Press: New York, NY, USA, 2011; pp. 141–152. [Google Scholar]
- Mao, X.; Xu, J.; Cui, H. Functional nanoparticles for magnetic resonance imaging. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2016. [Google Scholar] [CrossRef] [PubMed]
- De León-Rodríguez, L.M.; Martins, A.F.; Pinho, M.C.; Rofsky, N.M.; Sherry, A.D. Basic, MR relaxation mechanisms and contrast agent design. J. Magn. Reson. Imaging 2015, 42, 545–565. [Google Scholar] [CrossRef] [PubMed]
- Kraitchman, D.L.; Caravan, P. Magnetic resonance labeling of stem cells: Is positive tracking a plus or a minus? JACC Cardiovasc. Imaging 2009, 2, 1123–1125. [Google Scholar] [CrossRef] [PubMed]
- Cromer Berman, S.M.; Walczak, P.; Bulte, J.W.M. Tracking stem cells using magnetic nanoparticles. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2011, 3, 343–355. [Google Scholar]
- Qi, Y.; Feng, G.; Huang, Z.; Yan, W. The application of super paramagnetic iron oxide-labeled mesenchymal stem cells in cell-based therapy. Mol. Biol. Rep. 2013, 40, 2733–2740. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.K.; Kadayakkara, D.K.; Bar-Shir, A.; Gilad, A.A.; McMahon, M.T.; Bulte, J.W.M. Advances in using MRI probes and sensors for in vivo cell tracking as applied to regenerative medicine. Dis. Model. Mech. 2015, 8, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Reimer, P.; Balzer, T. Ferucarbotran (Resovist): A new clinically approved RES-specific contrast agent for contrast-enhanced MRI of the liver: Properties, clinical development, and applications. Eur. Radiol. 2003, 13, 1266–1276. [Google Scholar] [PubMed]
- Coyne, D.W. Ferumoxytol for treatment of iron deficiency anemia in patients with chronic kidney disease. Expert Opin. Pharmacother. 2009, 10, 2563–2568. [Google Scholar] [CrossRef] [PubMed]
- Castaneda, R.T.; Khurana, A.; Khan, R.; Daldrup-Link, H.E. Labeling stem cells with ferumoxytol, an FDA-approved iron oxide nanoparticle. J. Vis. Exp. 2011, 3482. [Google Scholar] [CrossRef] [PubMed]
- Granot, D.; Nkansah, M.K.; Bennewitz, M.F.; Tang, K.S.; Markakis, E.A.; Shapiro, E.M. Clinically viable magnetic poly(lactide-co-glycolide) (PLGA) particles for MRI-based cell tracking. Magn. Reson. Med. 2014, 71, 1238–1250. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, E.M. Biodegradable, polymer encapsulated, metal oxide particles for MRI-based cell tracking. Magn. Reson. Med. 2015, 73, 376–389. [Google Scholar] [CrossRef] [PubMed]
- Lü, J.M.; Wang, X.; Marin-Muller, C.; Wang, H.; Lin, P.H.; Yao, Q.; Chen, C. Current advances in research and clinical applications of PLGA-based nanotechnology. Exp. Rev. Mol. Diagn. 2009, 9, 325–341. [Google Scholar] [CrossRef] [PubMed]
- Nkansah, M.K.; Thakral, D.; Shapiro, E.M. Magnetic poly(lactide-co-glycolide) and cellulose particles for MRI-based cell tracking. Magn. Reson. Med. 2011, 65, 1776–1785. [Google Scholar] [CrossRef] [PubMed]
- Louie, A. MRI biosensors: A short primer. J. Magn. Reson. Imaging 2013, 38, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, E.M.; Skrtic, S.; Koretsky, A.P. Sizing it up: Cellular, MRI using micron-sized iron oxide particles. Magn. Reson. Med. 2005, 53, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Leder, A.; Raschzok, N.; Schmidt, C.; Arabacioglu, D.; Butter, A.; Kolano, S.; de Sousa Lisboa, L.S.; Werner, W.; Polenz, D.; Reutzel-Selke, A.; et al. Micron-sized iron oxide-containing particles for microRNA-targeted manipulation and MRI-based tracking of transplanted cells. Biomaterials 2015, 51, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Iordanova, B.; Robison, C.; Goins, W.; Ahrens, E. Single chain ferritin chimera as an improved MRI gene reporter. Prilozi 2010, 31, 151–155. [Google Scholar] [PubMed]
- Cohen, B.; Ziv, K.; Plaks, V.; Harmelin, A.; Neeman, M. Ferritin nanoparticles as magnetic resonance reporter gene. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2009, 1, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Chen, R.; Anikeeva, P.; Jasanoff, A. Engineering intracellular biomineralization and biosensing by a magnetic protein. Nat. Commun. 2015, 6, 8721. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Woo, J.; Choi, Y.; Hwang, E.H.; Choi, S.K.; Cho, K.W.; Moon, W.K. Noninvasive, MRI and multilineage differentiation capability of ferritin-transduced human mesenchymal stem cells. NMR Biomed. 2015, 28, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Noad, J.; Gonzalez-Lara, L.E.; Broughton, H.C.; McFadden, C.; Chen, Y.; Hess, D.A.; Foster, P.J. MRI tracking of transplanted iron-labeled mesenchymal stromal cells in an immune-compromised mouse model of critical limb ischemia. NMR Biomed. 2013, 26, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Nedopil, A.; Klenk, C.; Kim, C.; Liu, S.; Wendland, M.; Golovko, D.; Schuster, T.; Sennino, B.; McDonald, D.M.; Daldrup-Link, H.E. MR signal characteristics of viable and apoptotic human mesenchymal stem cells in MASI for treatment of osteoarthritis. Invest. Radiol. 2010, 45, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Van Buul, G.M.; Kotek, G.; Wielopolski, P.A.; Farrell, E.; Bos, P.K.; Weinans, H.; Grohnert, A.U.; Jahr, H.; Verhaar, J.A.N.; Krestin, G.P.; et al. Clinically translatable cell tracking and quantification by MRI in cartilage repair using superparamagnetic iron oxides. PLoS ONE 2011, 6, e17001. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, D.V.; Bernstein, A.S.; Pagel, M.D. A review of responsive MRI contrast agents: 2005–2014. Contrast Media Mol. Imaging 2015, 10, 245–265. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.; Osborne, E.A.; Louie, A.Y. Activatable, T1 and T2 magnetic resonance imaging contrast agents. Ann. Biomed. Eng. 2011, 39, 1335–1348. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Artemov, D. Monitoring of release of cargo from nanocarriers by MRI/MR spectroscopy (MRS): Significance of T2/T2* effect of iron particles. Magn. Reson. Med. 2009, 61, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Onuki, Y.; Jacobs, I.; Artemov, D.; Kato, Y. Noninvasive visualization of in vivo release and intratumoral distribution of surrogate MR contrast agent using the dual MR contrast technique. Biomaterials 2010, 31, 7132–7138. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Momin, E.; Choi, J.; Yuan, K.; Zaidi, H.; Kim, J.; Park, M.; Lee, N.; McMahon, M.T.; Quinones-Hinojosa, A.; et al. Mesoporous silica-coated hollow manganese oxide nanoparticles as positive T1 contrast agents for labeling and MRI tracking of adipose-derived mesenchymal stem cells. J. Am. Chem. Soc. 2011, 133, 2955–2961. [Google Scholar] [CrossRef] [PubMed]
- Létourneau, M.; Tremblay, M.; Faucher, L.; Rojas, D.; Chevallier, P.; Gossuin, Y.; Lagueux, J.; Fortin, M.-A. MnO-labeled cells: Positive contrast enhancement in MRI. J. Phys. Chem. B 2012, 116, 13228–13238. [Google Scholar] [CrossRef] [PubMed]
- Guillet-Nicolas, R.; Laprise-Pelletier, M.; Nair, M.M.; Chevallier, P.; Lagueux, J.; Gossuin, Y.; Laurent, S.; Kleitz, F.; Fortin, M.A. Manganese-impregnated mesoporous silica nanoparticles for signal enhancement in MRI cell labelling studies. Nanoscale 2013, 5, 11499–11511. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Shao, Y.; He, H.; Tan, Y.; Tian, X.; Xie, F.; Li, L. Gadolinium3+-doped mesoporous silica nanoparticles as a potential magnetic resonance tracer for monitoring the migration of stem cells in vivo. Int. J. Nanomed. 2013, 8, 119–127. [Google Scholar]
- Hsiao, J.K.; Tsai, C.P.; Chung, T.H.; Hung, Y.; Yao, M.; Liu, H.M.; Mou, C.Y.; Yang, C.S.; Chen, Y.C.; Huang, D.M. Mesoporous silica nanoparticles as a delivery system of gadolinium for effective human stem cell tracking. Small 2008, 4, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Guenoun, J.; Koning, G.A.; Doeswijk, G.; Bosman, L.; Wielopolski, P.A.; Krestin, G.P.; Bernsen, M.R. Cationic, Gd-DTPA liposomes for highly efficient labeling of mesenchymal stem cells and cell tracking with MRI. Cell Transplant. 2012, 21, 191–205. [Google Scholar] [CrossRef]
- Hedlund, A.; Ahrén, M.; Gustafsson, H.; Abrikossova, N.; Warntjes, M.; Jönsson, J.I.; Uvdal, K.; Engström, M. Gd2O3 nanoparticles in hematopoietic cells for MRI contrast enhancement. Int. J. Nanomed. 2011, 6, 3233–3240. [Google Scholar]
- Gianolio, E.; Arena, F.; Strijkers, G.J.; Nicolay, K.; Högset, A.; Aime, S. Photochemical activation of endosomal escape of MRI-Gd-agents in tumor cells. Magn. Reson. Med. 2011, 65, 212–219. [Google Scholar] [CrossRef]
- Terreno, E.; Geninatti Crich, S.; Belfiore, S.; Biancone, L.; Cabella, C.; Esposito, G.; Manazza, A.D.; Aime, S. Effect of the intracellular localization of a Gd-based imaging probe on the relaxation enhancement of water protons. Magn. Reson. Med. 2006, 55, 491–497. [Google Scholar] [CrossRef] [PubMed]
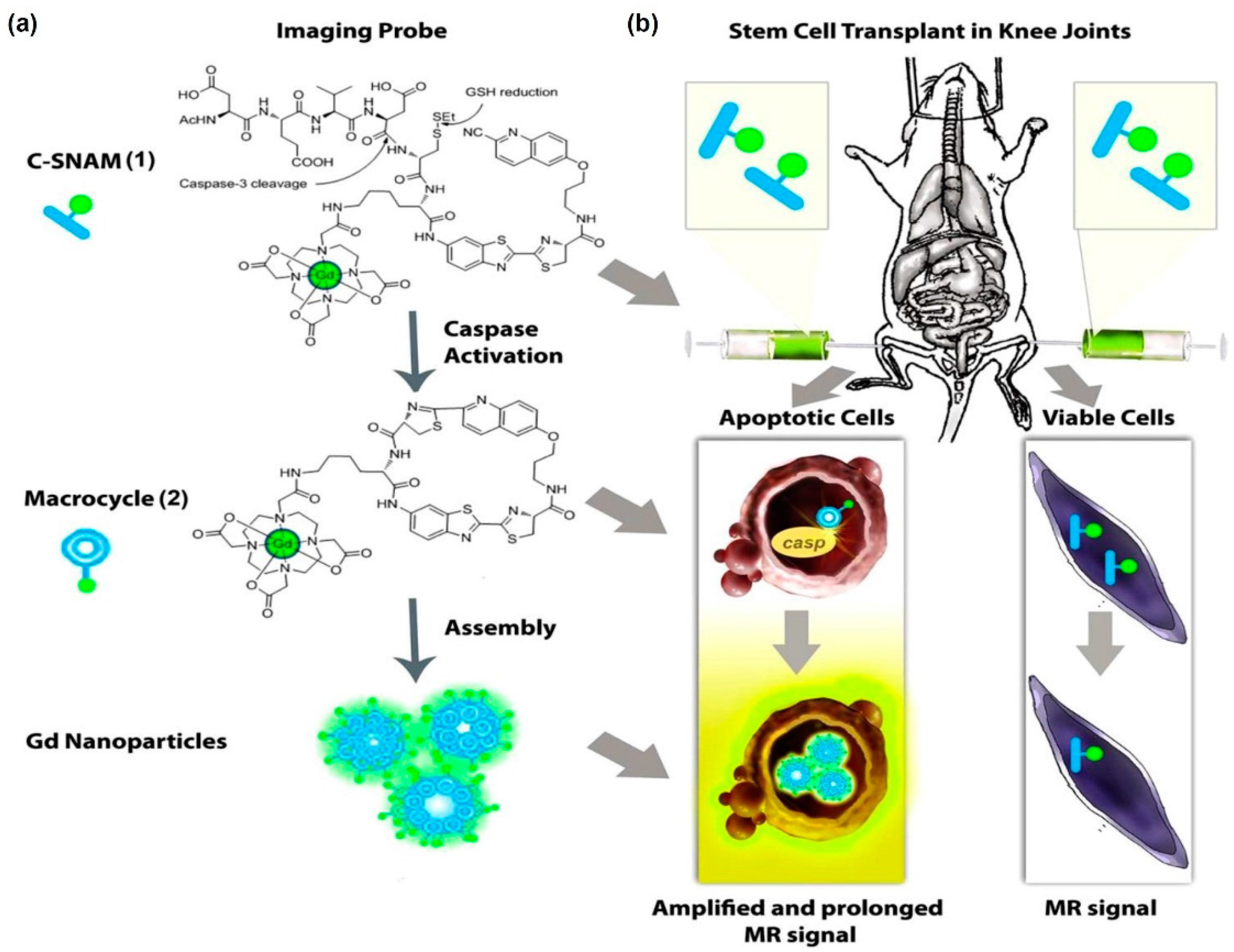
- Nejadnik, H.; Ye, D.; Lenkov, O.D.; Donig, J.S.; Martin, J.E.; Castillo, R.; Derugin, N.; Sennino, B.; Rao, J.; Daldrup-Link, H. Magnetic resonance imaging of stem cell apoptosis in arthritic joints with a caspase activatable contrast agent. ACS Nano 2015, 9, 1150–1160. [Google Scholar] [CrossRef] [PubMed]
- Regueiro-Figueroa, M.; Gündüz, S.; Patinec, V.; Logothetis, N.K.; Esteban-Gómez, D.; Tripier, R.; Angelovski, G.; Platas-Iglesias, C. Gd3+-Based magnetic resonance imaging contrast agent responsive to Zn2+. Inorg. Chem. 2015, 54, 10342–10350. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.; Louie, A.Y. Strategies for the development of Gd-based “q”-activatable MRI contrast agents. NMR Biomed. 2013, 26, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.T.; Cheng, C.M.; Su, Y.Z.; Lee, W.T.; Hsu, J.S.; Liu, G.C.; Cheng, T.L.; Wang, Y.M. Synthesis and characterization of a new bioactivated paramagnetic gadolinium(III) complex [Gd(DOTA-FPG)(H2O)] for tracing gene expression. Bioconjug. Chem. 2007, 18, 1716–1727. [Google Scholar] [CrossRef] [PubMed]
- Duimstra, J.A.; Femia, F.J.; Meade, T.J. A gadolinium chelate for detection of β-glucuronidase: A self-Immolative approach. J. Am. Chem. Soc. 2005, 127, 12847–12855. [Google Scholar] [CrossRef] [PubMed]
- Fujisaki, K.; Ono-Fujisaki, A.; Kura-Nakamura, N.; Komune, N.; Hirakawa, N.; Tsuruya, N.; Komune, S.; Lida, M. Rapid deterioration of renal insufficiency after magnetic resonance imaging with gadolinium-based contrast agent. Clin. Nephrol. 2011, 75, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Buhaescu, I.; Izzedine, H. Gadolinium-induced nephrotoxicity. Int. J. Clin. Pract. 2008, 62, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Bar-Shir, A.; Bulte, J.W.M.; Gilad, A.A. Molecular engineering of nonmetallic biosensors for CEST MRI. ACS Chem. Biol. 2015, 10, 1160–1170. [Google Scholar] [CrossRef] [PubMed]
- Sherry, A.D.; Woods, M. Chemical exchange saturation transfer contrast agents for magnetic resonance imaging. Ann. Rev. Biomed. Eng. 2008, 10, 391–411. [Google Scholar] [CrossRef] [PubMed]
- Hancu, I.; Dixon, W.T.; Woods, M.; Vinogradov, E.; Sherry, A.D.; Lenkinski, R.E. CEST and PARACEST MR contrast agents. Acta Radiol. 2010, 51, 910–923. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, F.J.; Ling, W.; Ferrauto, G.; Aime, S.; Modo, M. Simultaneous, MR imaging for tissue engineering in a rat model of stroke. Sci. Rep. 2015, 5, 14597. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Roose, B.W.; Palovcak, E.J.; Carnevale, V.; Dmochowski, I.J. A genetically encoded β-lactamase reporter for ultrasensitive 129Xe, NMR in mammalian cells. Angew. Chem. 2016, 55, 8984–8987. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, M.G.; Ramirez, R.M.; Sperling, L.J.; Sun, G.; Sun, J.; Pines, A.; Schaffer, D.V.; Bajaj, V.S. Genetically encoded reporters for hyperpolarized xenon magnetic resonance imaging. Nat. Chem. 2014, 6, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Hill, P.A.; Dmochowski, I.J. Utilizing a water-soluble cryptophane with fast xenon exchange rates for picomolar sensitivity NMR measurements. Anal. Chem. 2012, 84, 9935–9941. [Google Scholar] [CrossRef] [PubMed]
- Stevens, T.K.; Palaniappan, K.K.; Ramirez, R.M.; Francis, M.B.; Wemmer, D.E.; Pines, A. HyperCEST detection of a 129Xe-based contrast agent composed of cryptophane—A molecular cages on a bacteriophage scaffold. Magn. Reson. Med. 2013, 69, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Taratula, O.; Dmochowski, I.J. Functionalized 129Xe contrast agents for magnetic resonance imaging. Curr. Opin. Chem. Biol. 2010, 14, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Stevens, T.K.; Ramirez, R.M.; Pines, A. Nanoemulsion contrast agents with sub-picomolar sensitivity for xenon NMR. J. Am. Chem. Soc. 2013, 135, 9576–9579. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, E.T.; Bulte, J.W.M. Tracking immune cells in vivo using magnetic resonance imaging. Nat. Rev. Immunol. 2013, 13, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, M.; Boehm-Sturm, P.; Figdor, C.G.; de Vries, I.J.; Hoehn, M. Labeling cells for in vivo tracking using 19F MRI. Biomaterials 2012, 33, 8830–8840. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, M.; Heerschap, A.; Ahrens, E.T.; Figdor, C.G.; de Vries, I.J.M. 19F MRI for quantitative in vivo cell tracking. Trends Biotechnol. 2010, 28, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, E.T.; Helfer, B.M.; O’Hanlon, C.F.; Schirda, C. Clinical cell therapy imaging using a perfluorocarbon tracer and fluorine-19 MRI. Magn. Reson. Med. 2014, 72, 1696–1701. [Google Scholar] [CrossRef] [PubMed]
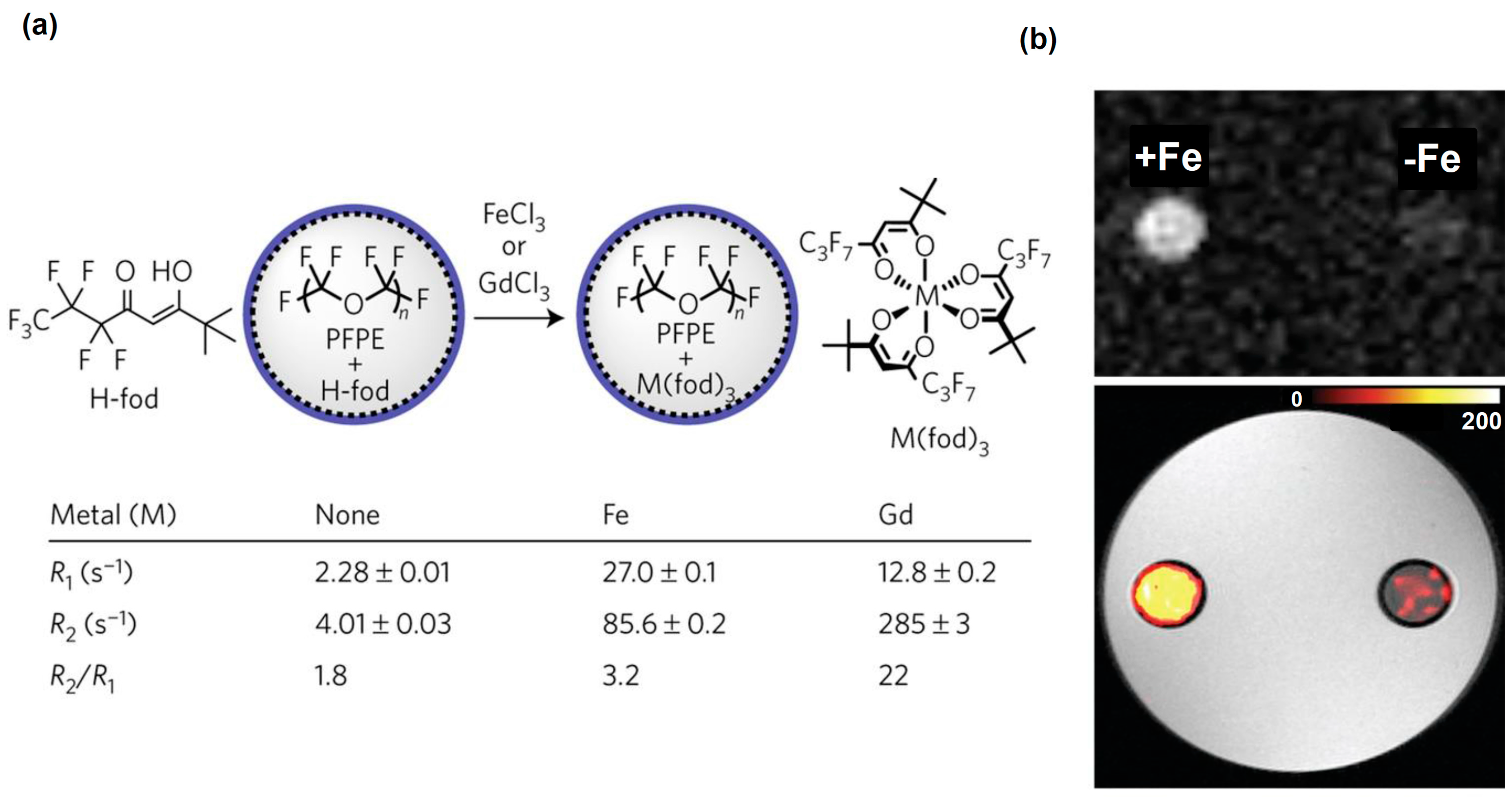
- Kislukhin, A.A.; Xu, H.; Adams, S.R.; Narsinh, K.H.; Tsien, R.Y.; Ahrens, E.T. Paramagnetic fluorinated nanoemulsions for sensitive cellular fluorine-19 magnetic resonance imaging. Nat. Mater. 2016, 15, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Hockett, F.D.; Wallace, K.D.; Schmieder, A.H.; Caruthers, S.D.; Pham, C.T.N.; Wickline, S.A.; Lanza, G.M. Simultaneous, dual frequency 1H and 19F open, coil imaging of arthritic, rabbit knee at 3 T. IEEE Trans. Med. Imaging 2011, 30, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Hockett, F.D.; Chen, J.; Zhang, L.; Caruthers, S.D.; Lanza, G.M.; Wickline, S.A. A generalized strategy for designing 19F/1H dual-frequency MRI coil for small animal imaging at 4.7 Tesla. J. Magn. Reson. Imaging 2011, 34, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Schanne, F.A.; Dowd, T.L.; Gupta, R.K.; Rosen, J.F. Development of 19F NMR for measurement of [Ca2+]i and [Pb2+]i in cultured osteoblastic bone cells. Environ. Health Perspect. 1990, 84, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Bar-Shir, A.; Gilad, A.A.; Chan, K.W.Y.; Liu, G.; van Zijl, P.C.M.; Bulte, J.W.M.; McMahon, M.T. Metal ion sensing using ion chemical exchange saturation transfer 19F MRI. J. Am. Chem. Soc. 2013, 135, 12164–12167. [Google Scholar] [CrossRef] [PubMed]
- Bar-Shir, A.; Yadav, N.N.; Gilad, A.A.; van Zijl, P.C.M.; McMahon, M.T.; Bulte, J.W.M. Single 19F probe for simultaneous detection of multiple metal ions using miCEST MRI. J. Am. Chem. Soc. 2015, 137, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Jianxin, Y.; Zhenyi, M.; Yingming, L.; Kenneth, S.K.; Li, L.; Ralph, P.M. Synthesis and evaluation of a novel gene reporter molecule: Detection of b-galactosidase activity using 19F NMR of a fluorinated vitamin B6 conjugate+. Med. Chem. 2005, 1, 255–262. [Google Scholar]
- Kodibagkar, V.D.; Yu, J.; Liu, L.; Hetherington, H.P.; Mason, R.P. Imaging β-galactosidase activity using 19F chemical shift imaging of LacZ gene-reporter molecule 2-fluoro-4-nitrophenol-β-d-galactopyranoside. Magn. Reson. Imaging 2006, 24, 959–962. [Google Scholar] [CrossRef]
- Yu, J.; Liu, L.; Kodibagkar, V.D.; Cui, W.; Mason, R.P. Synthesis and evaluation of novel enhanced gene reporter molecules: Detection of β-galactosidase activity using 19F NMR of trifluoromethylated aryl β-d-galactopyranosides. Bioorg. Med. Chem. 2006, 14, 326–333. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
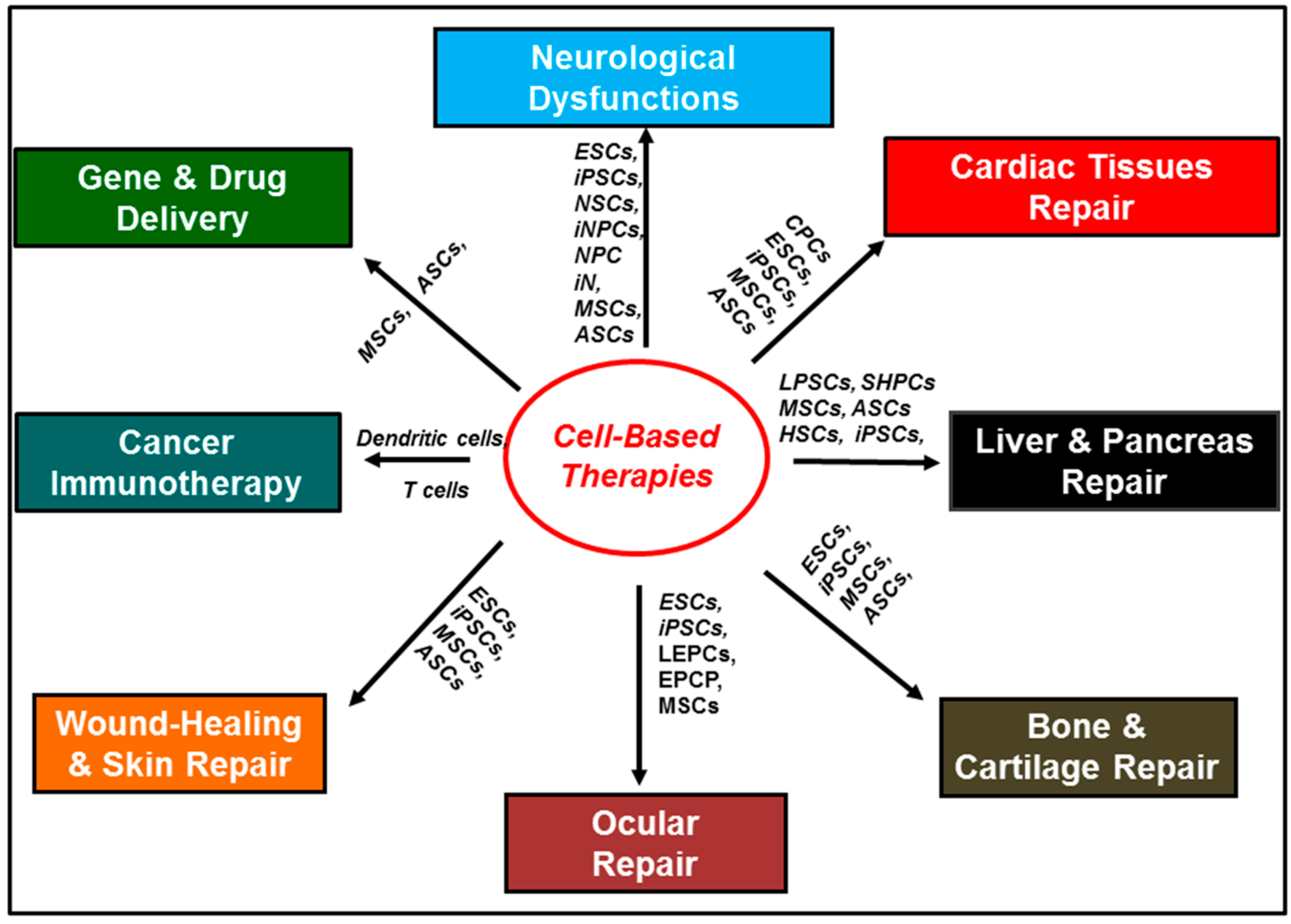
| Disease Type | Examples of Cells Tested | Cell Therapy Rationale | References |
|---|---|---|---|
| Neurological Dysfunctions | |||
| Parkinson’s disease | Embryonic stem cells (ESCs); neural stem cells (NSCs); neural progenitor cells (NPCs); mesenchymal stem cells (MSCs); induced pluripotent stem cells (iPSC); induced neuronal cells (iN); induced neuronal progenitor cells (iNPCs). | Cell replacement therapy; immunomodulatory and neuroprotective properties | [11,12,13,14,15,16,17] |
| Alzheimer’s disease | ESCs; NSCs; NPCs; MSCs; iPSCs; iN; iNPCs | Cell replacement therapy; immunomodulatory and neuroprotective properties | [12,18,19,20] |
| Huntington’s disease | ESCs; NSC; NPC; MSC; adipose-derived stem cells (ADSCs). | Cell replacement therapy; immunomodulatory and neuroprotective properties | [12,13,14] |
| Amyotrophic lateral sclerosis | ESCs; NSCs; iPSCs; embryonic germinal stem cells (EGC) | Cell replacement therapy; immunomodulatory and neuroprotective properties | [12] |
| Multiple sclerosis | ESCs; iPSCs; MSCs; ADSCs; | Cell replacement therapy; immunomodulatory and neuroprotective properties | [20,21] |
| Central and Peripheral Nervous System (CNS and PNS) Injuries | |||
| Spinal cord injuries | ESCs; MSCs; adipose-derived mesenchymal stem cells | Cell replacement therapy; neuroprotective properties. | [22,23,24] |
| Stroke | MSCs; ESCs; NSCs; iPSCs | Cell replacement therapy; immunomodulatory and neuroprotective properties. | [25,26,27] |
| Traumatic brain injuries | MSCs; iPSCs; bone-marrow-derived multipotent adult progenitor cells (MAPCs) | Cell replacement therapy; immunomodulatory and neuroprotective properties. | [28,29,30] |
| Radiotherapy-induced brain injuries | NSCs; ESCs; MSCs | Cell replacement therapy; immunomodulatory and neuroprotective properties. | [31,32,33,34,35] |
| Tissue Repair | |||
| Skin (wound healing) | MSCs; ASCs; iPSCs; hematopoietic stem cells (HSCs); endothelial progenitor cells (EPCs) | Cell replacement therapy; paracrine action; modulation of physiological responses. | [36,37] |
| Heart | Cardiac progenitor cells (CPCs); MSCs; ASCs; iPSCs | Cell replacement therapy; paracrine action; modulation of physiological responses. | [38,39,40] |
| Eyes | Lens epithelial progenitor cells (LEPCs); epithelial progenitor cells (EPCP); inducible progenitor cells (iPSCs); MSCs. | Cell replacement therapy; paracrine action; modulation of physiological responses. | [41,42,43,44] |
| Liver | Small hepatocytes-like progenitor cells (SHPCs); Liver stem cells/progenitor cells LPSCs; Sinosoidal endothelial progenitor cells (SEPCs); Hematopoeitic Stem cells (HSCs); MSCs. | Cell replacement therapy; paracrine action; modulation of physiological responses. | [45,46] |
| Bone and cartilage | MSCs; ASCs. | Cell replacement therapy; paracrine action; modulation of physiological responses. | [47,48,49,50,51] |
| Cancer Immunotherapy | |||
| Cancer | Dendritic cells; T cells | Stimulate immune response. | [52,53,54,55,56,57] |
| Drug and Gene Delivery | |||
| Cancer | MSCs; ASCs. | Migratory properties. | [58,59,60] |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ngen, E.J.; Artemov, D. Advances in Monitoring Cell-Based Therapies with Magnetic Resonance Imaging: Future Perspectives. Int. J. Mol. Sci. 2017, 18, 198. https://doi.org/10.3390/ijms18010198
Ngen EJ, Artemov D. Advances in Monitoring Cell-Based Therapies with Magnetic Resonance Imaging: Future Perspectives. International Journal of Molecular Sciences. 2017; 18(1):198. https://doi.org/10.3390/ijms18010198
Chicago/Turabian StyleNgen, Ethel J., and Dmitri Artemov. 2017. "Advances in Monitoring Cell-Based Therapies with Magnetic Resonance Imaging: Future Perspectives" International Journal of Molecular Sciences 18, no. 1: 198. https://doi.org/10.3390/ijms18010198