
Neuroprotection by Caffeine in Hyperoxia-Induced Neonatal Brain Injury

 ,
,
Abstract
:1. Introduction
2. Results
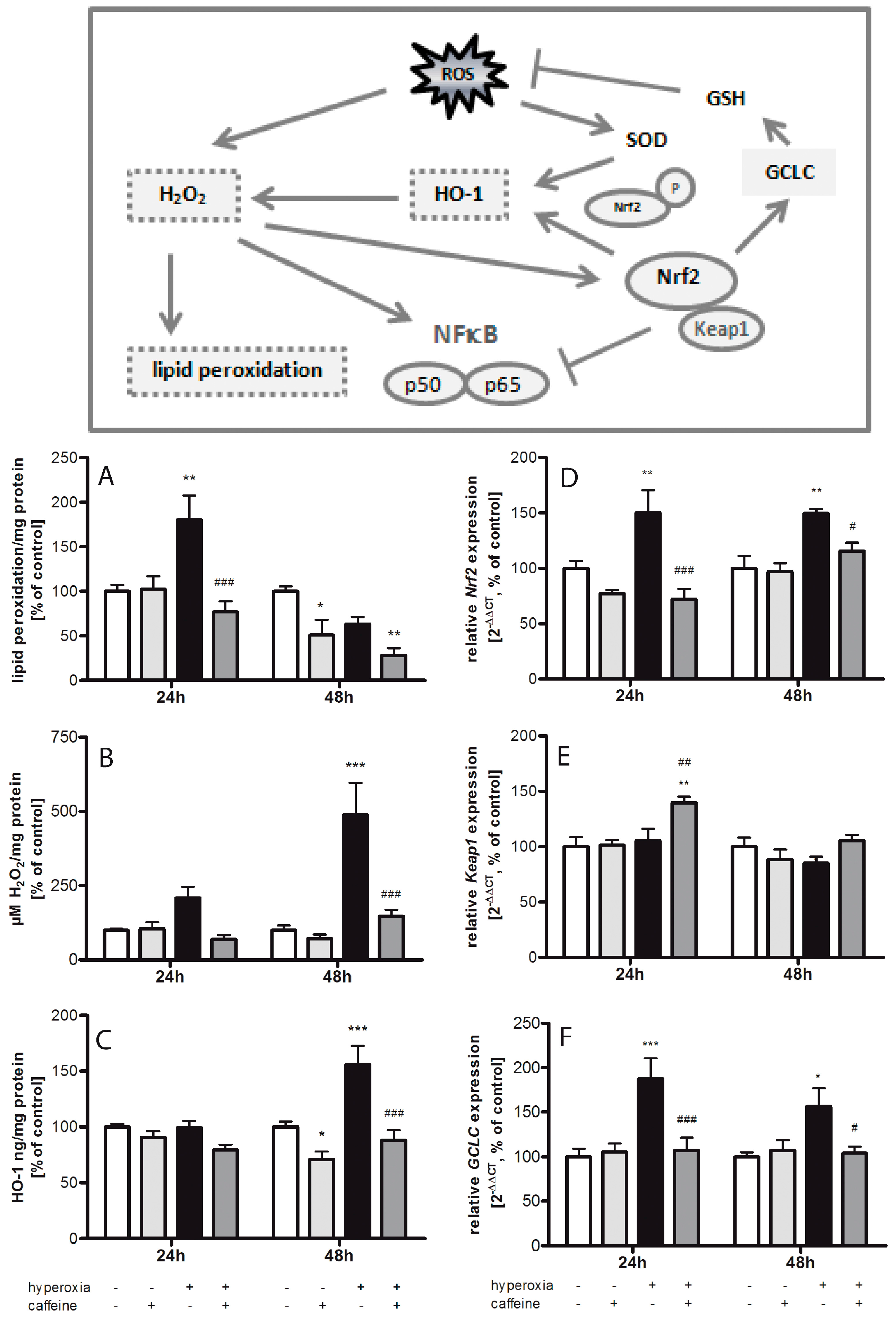
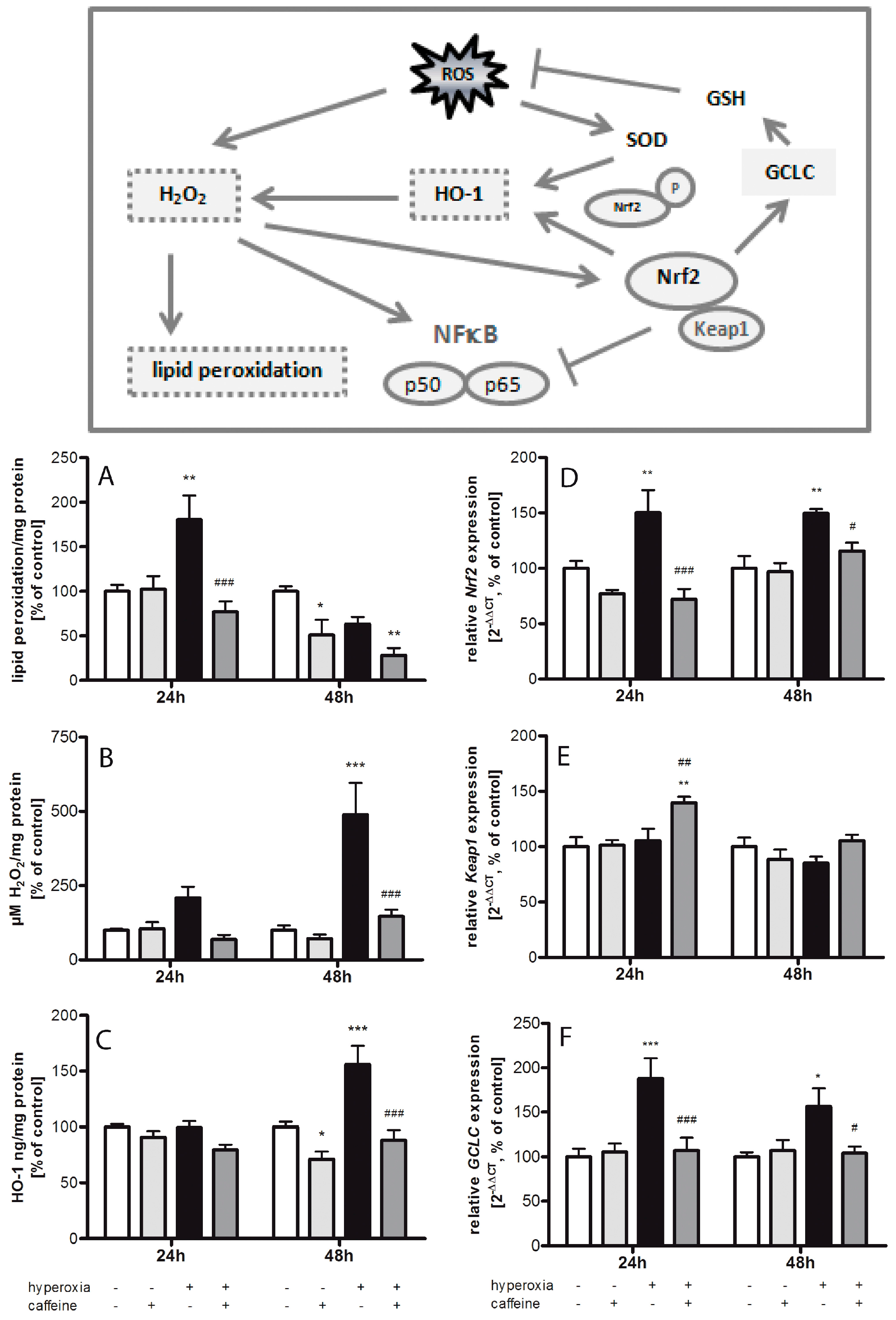
2.1. Hyperoxia Induces Oxidative Stress Which Is Counteracted by Caffeine
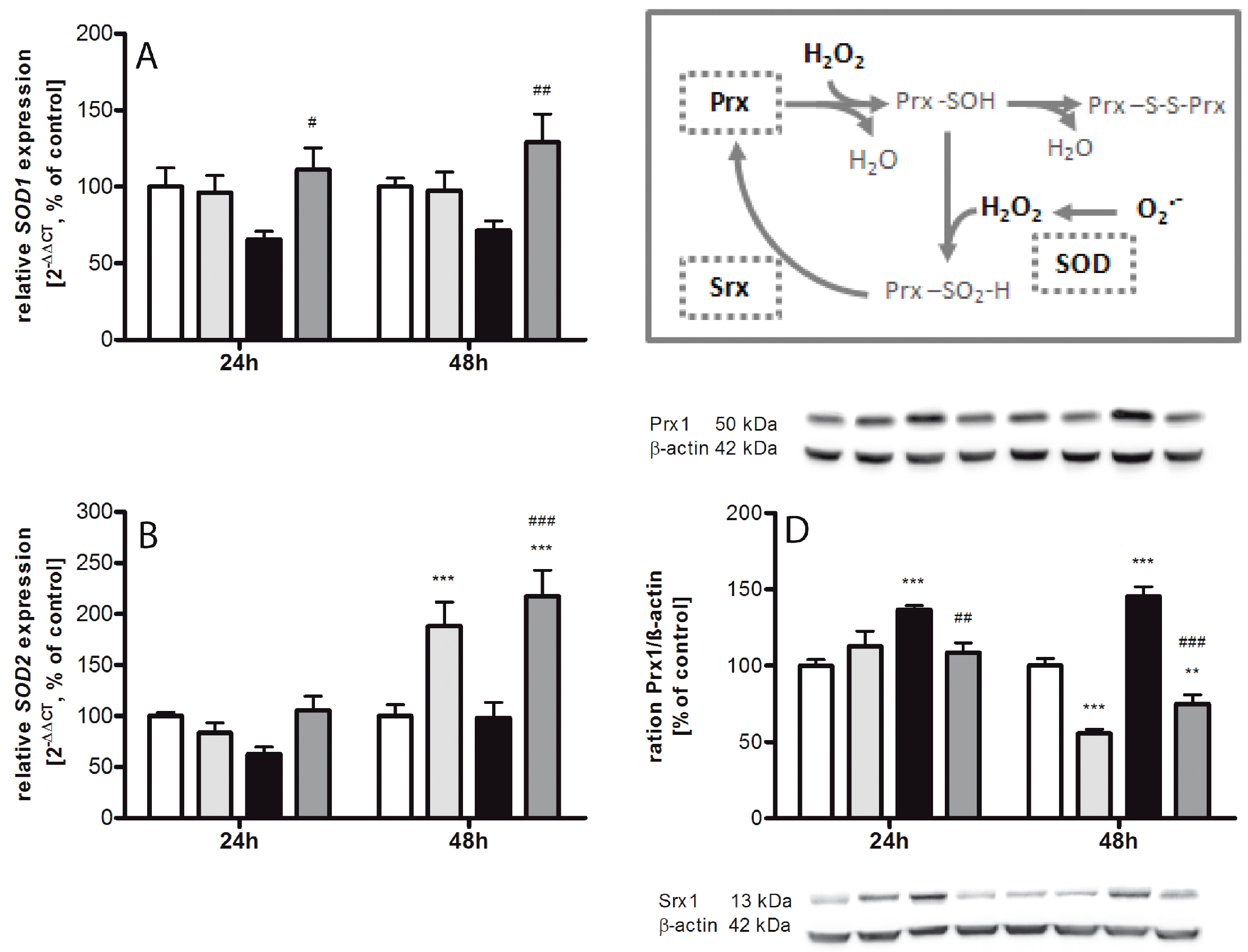
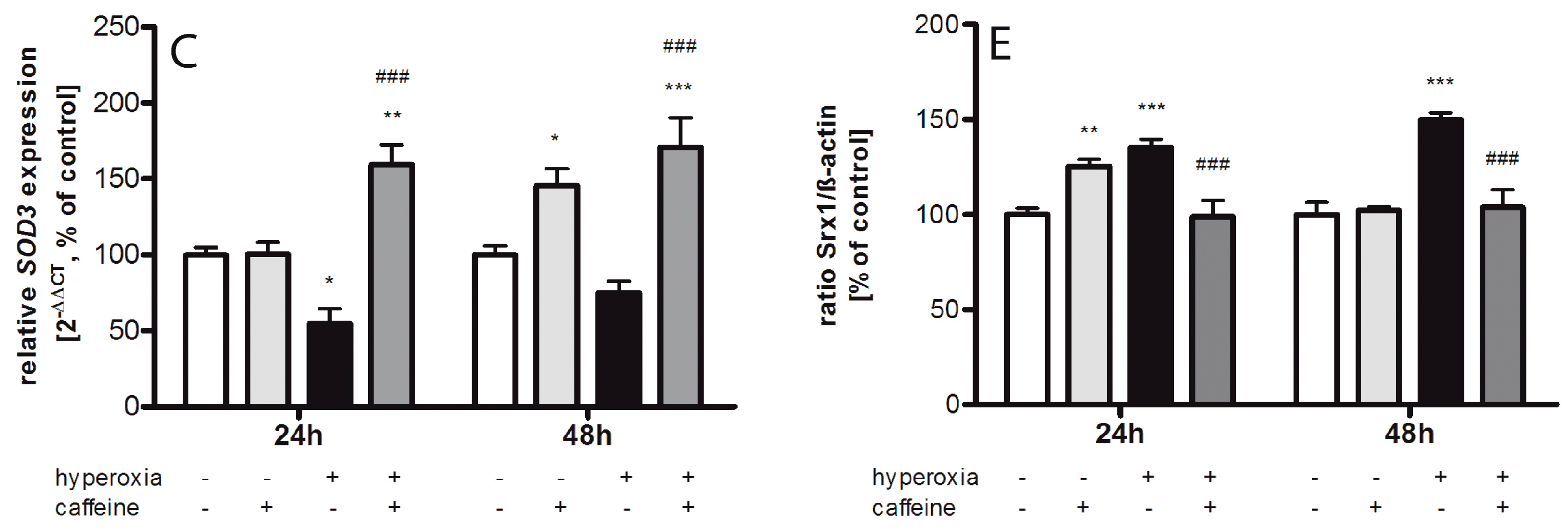
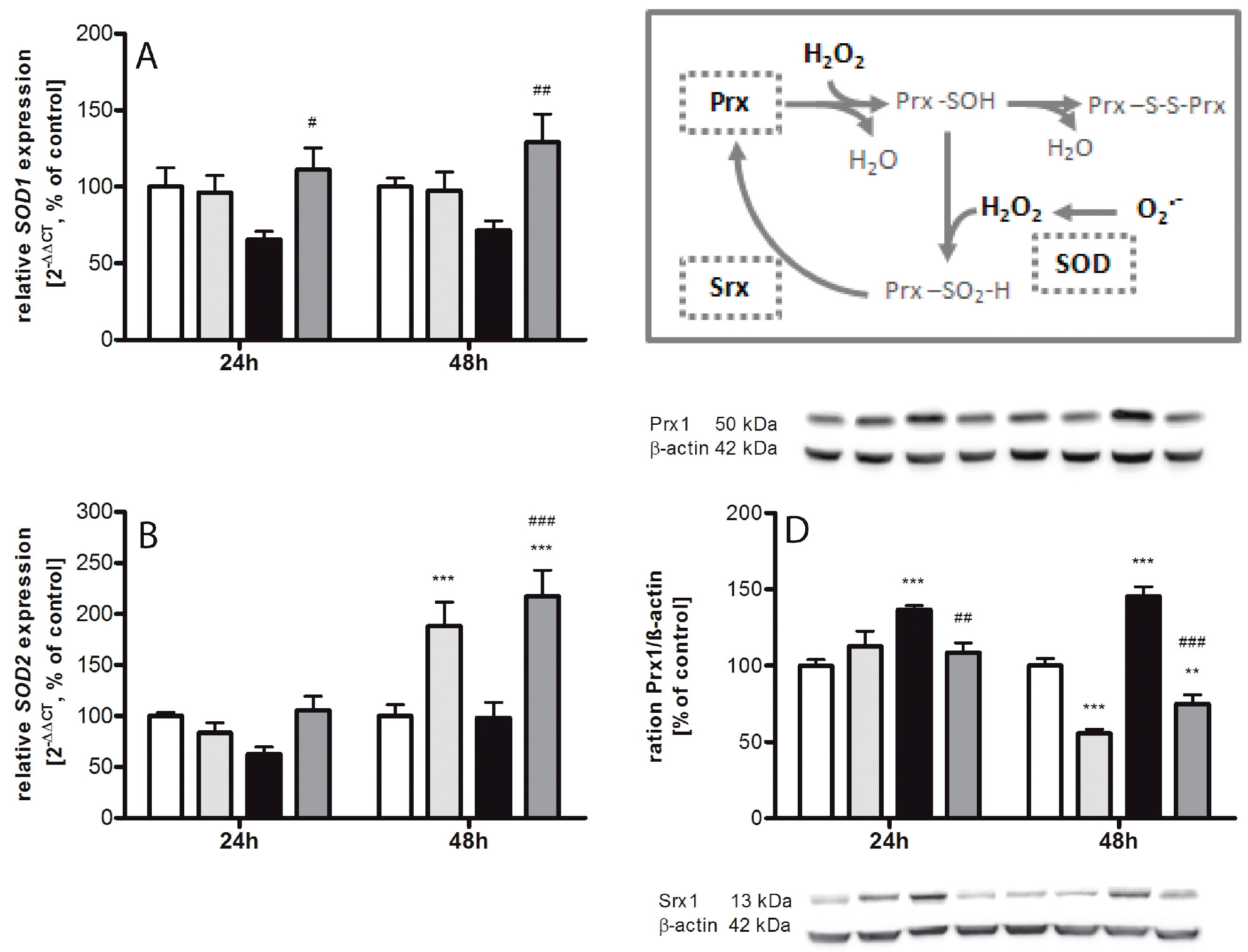
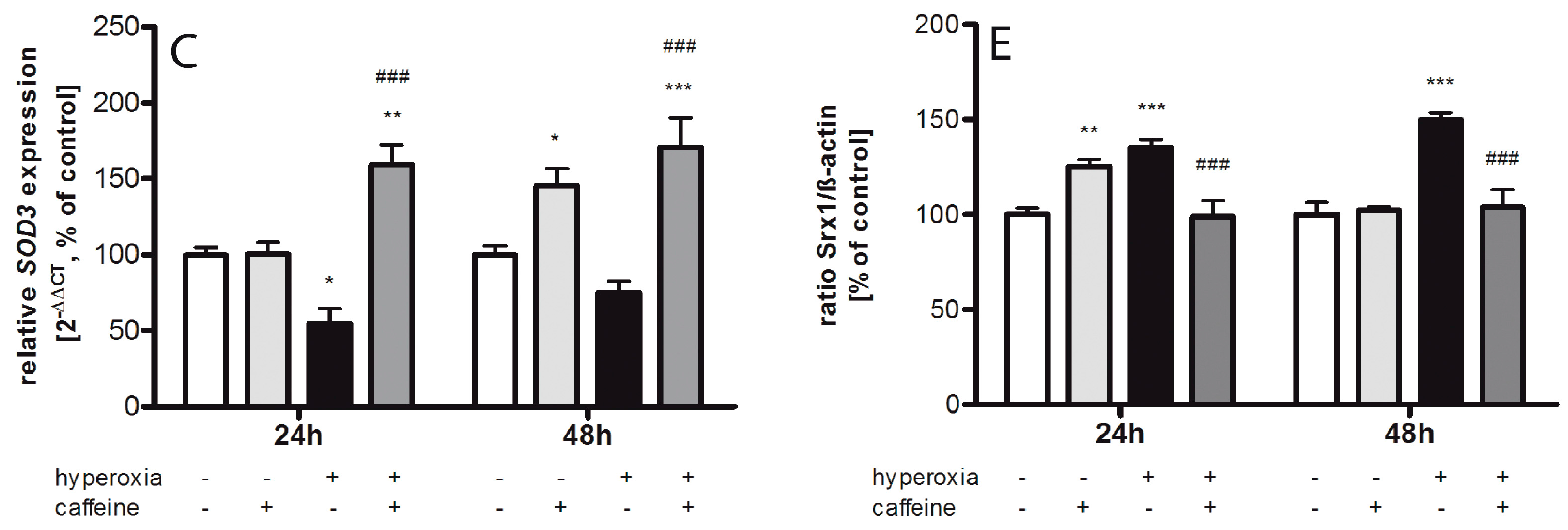
2.2. Regulating Effect of Caffeine on the Imbalance of the Sulfiredoxin/Peroxiredoxin System after Hyperoxia
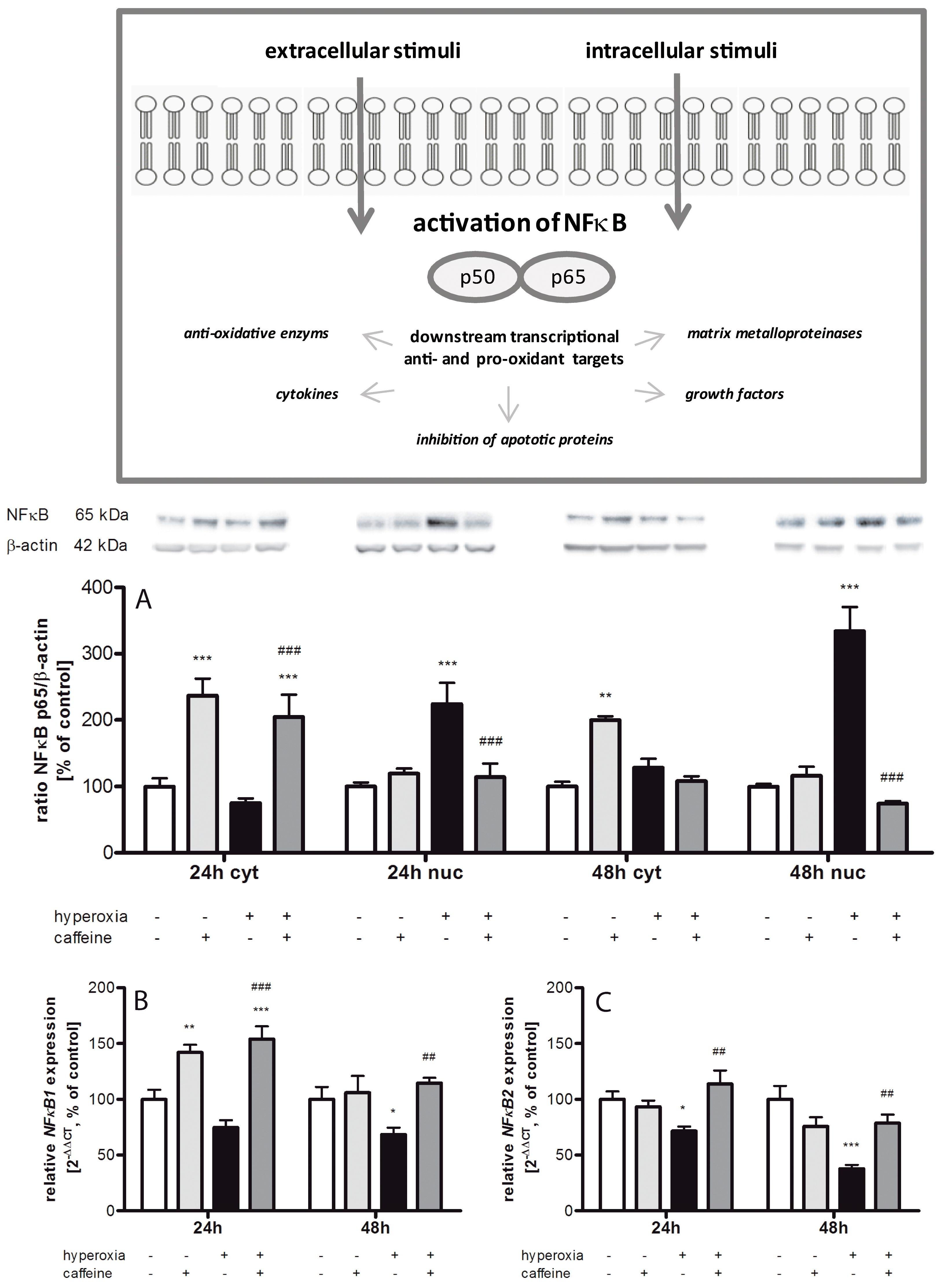
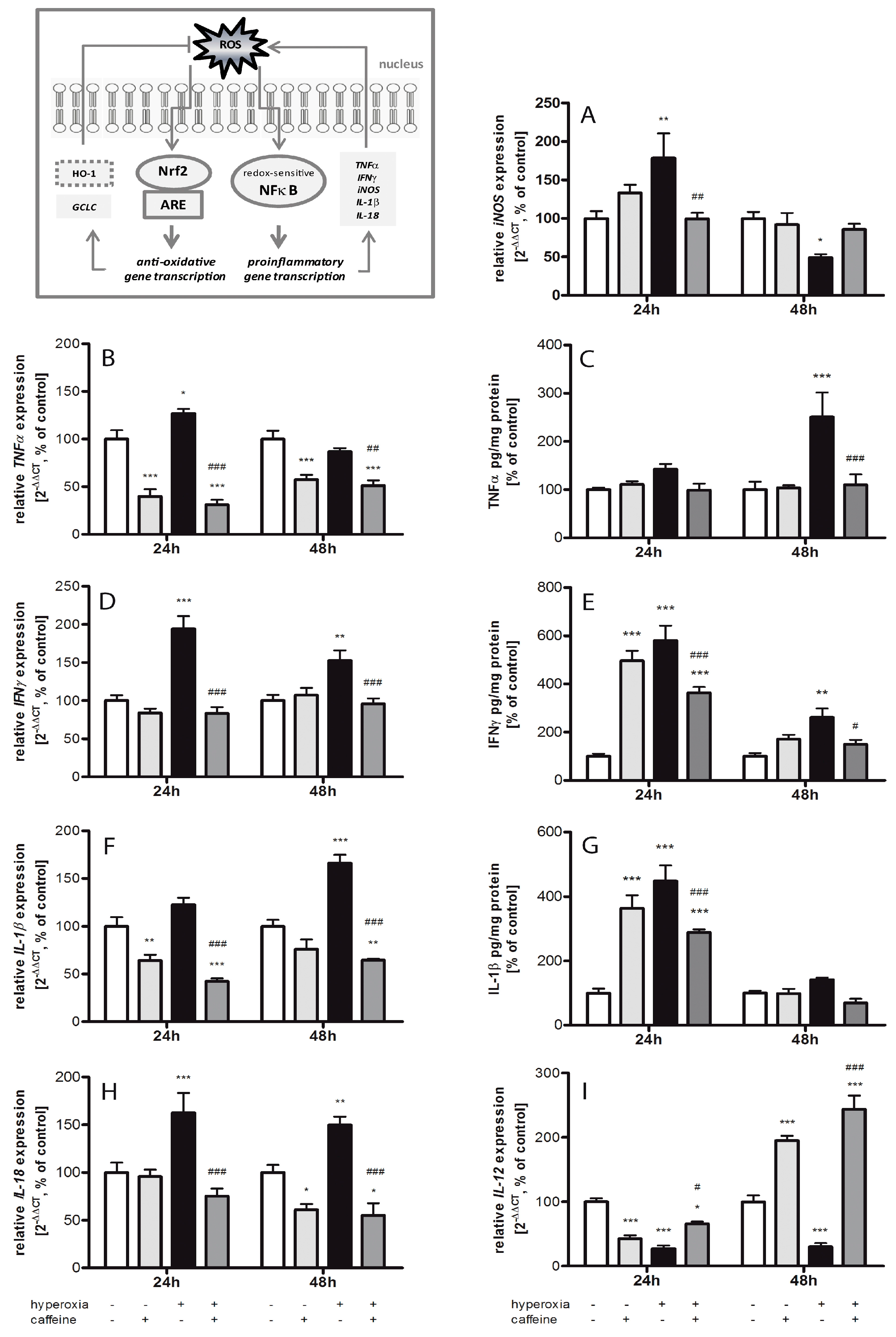
2.3. Effects of Caffeine on the Inflammatory Cytokine Expression
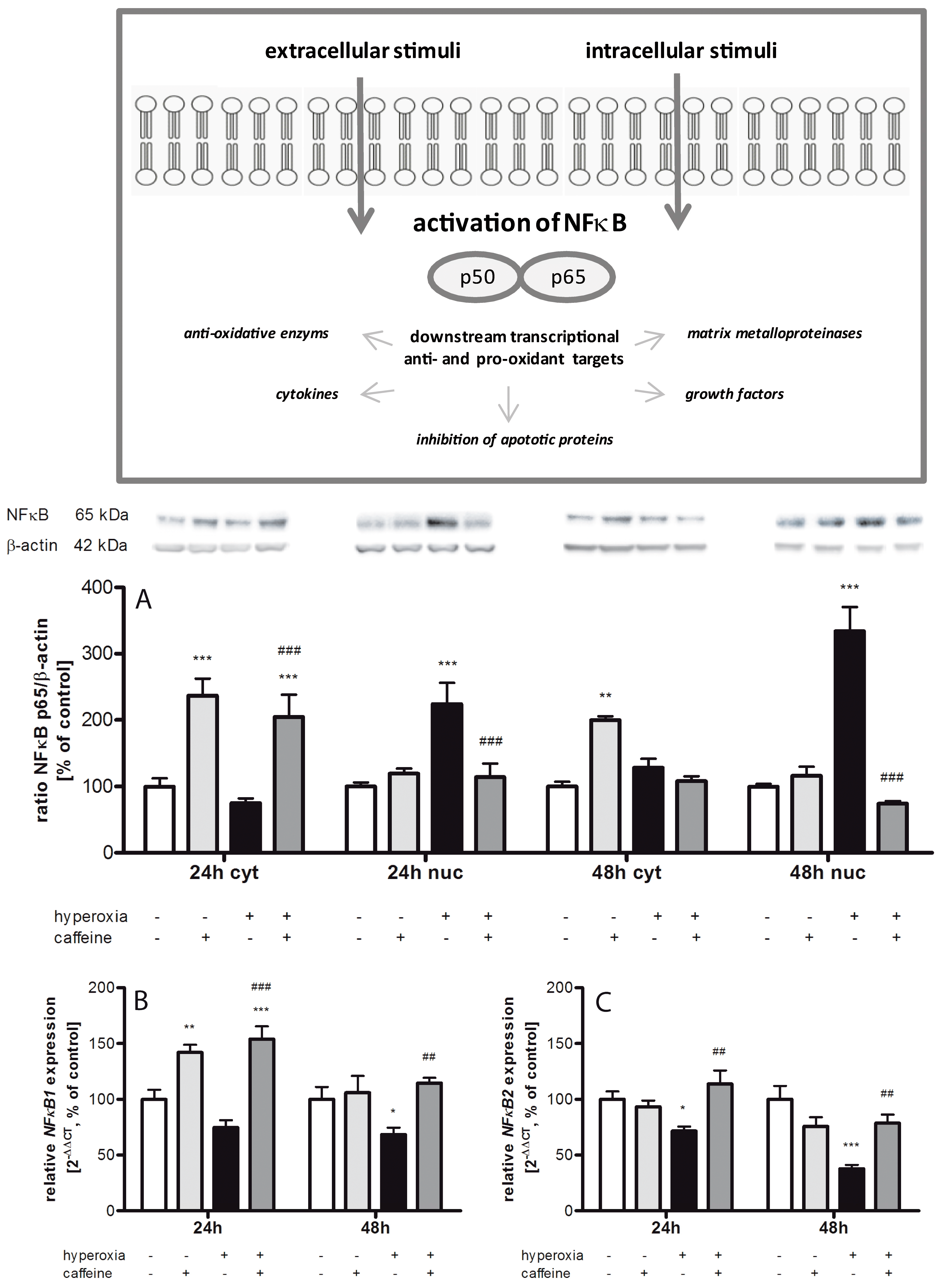
2.4. Hyperoxia Modulates Gene Expression of Transcription Factors and Caffeine Counteracts
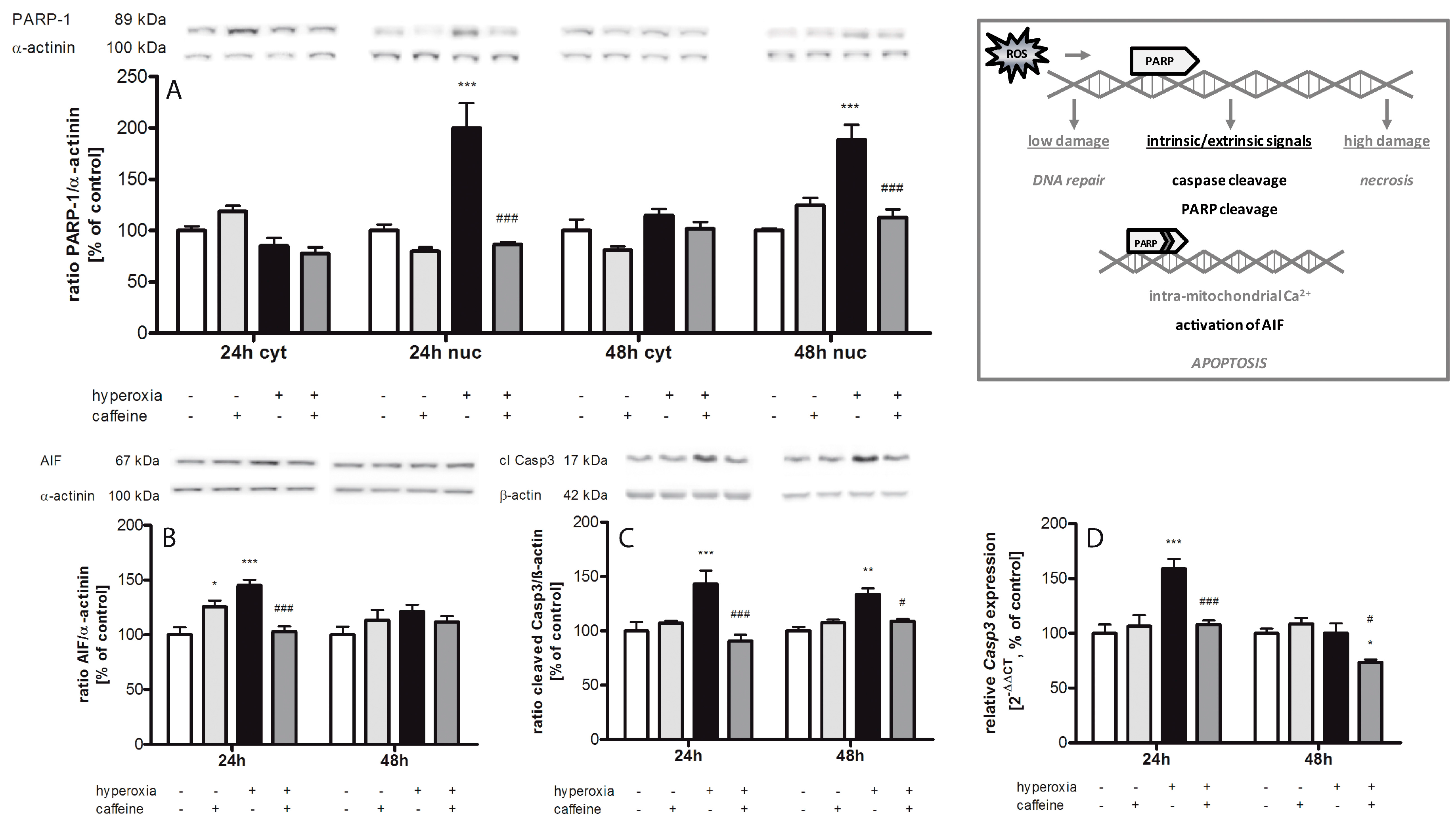
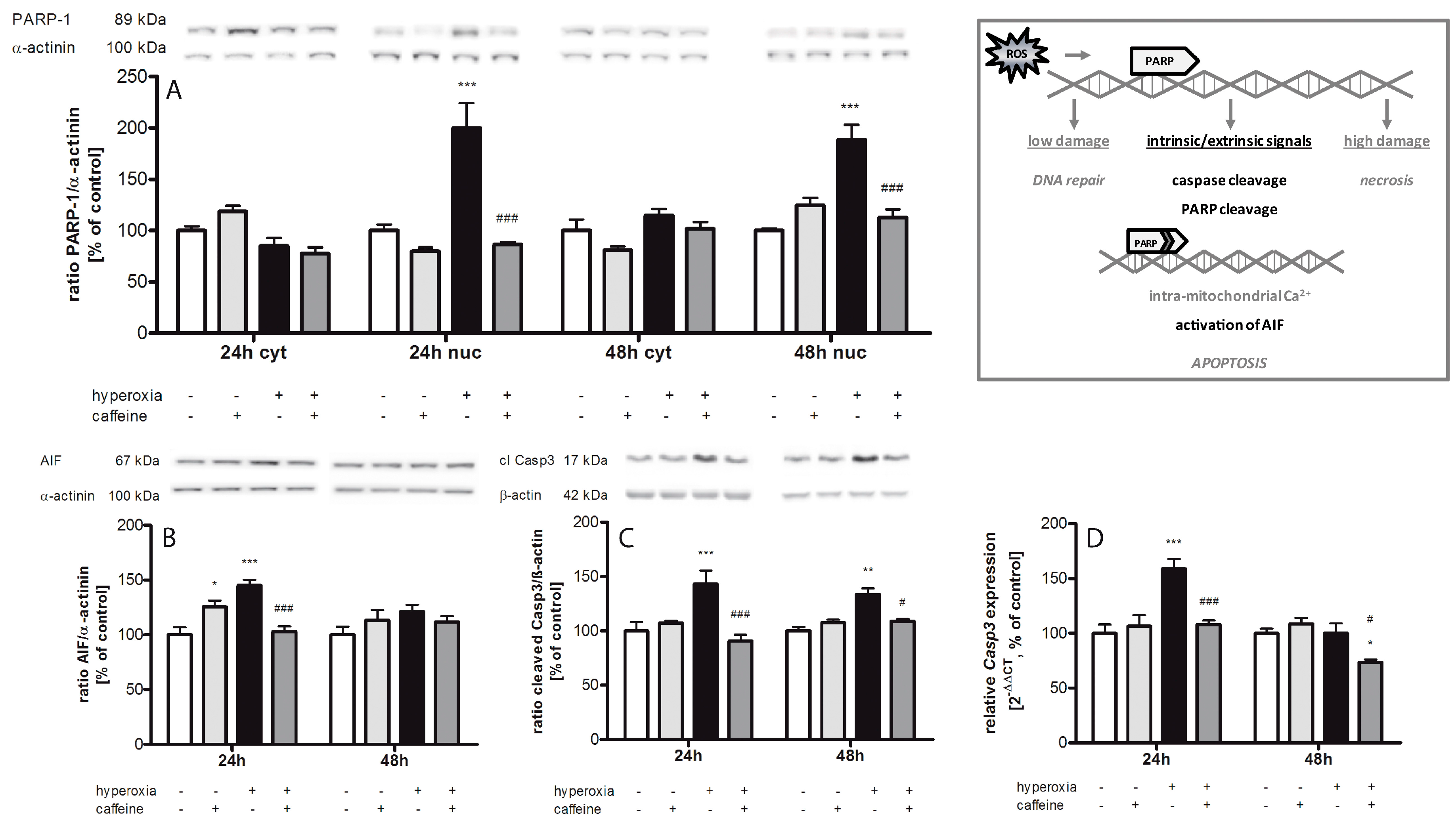
2.5. Caffeine Prevents Hyperoxia-Mediated Increase in Apoptotic Gene Expression
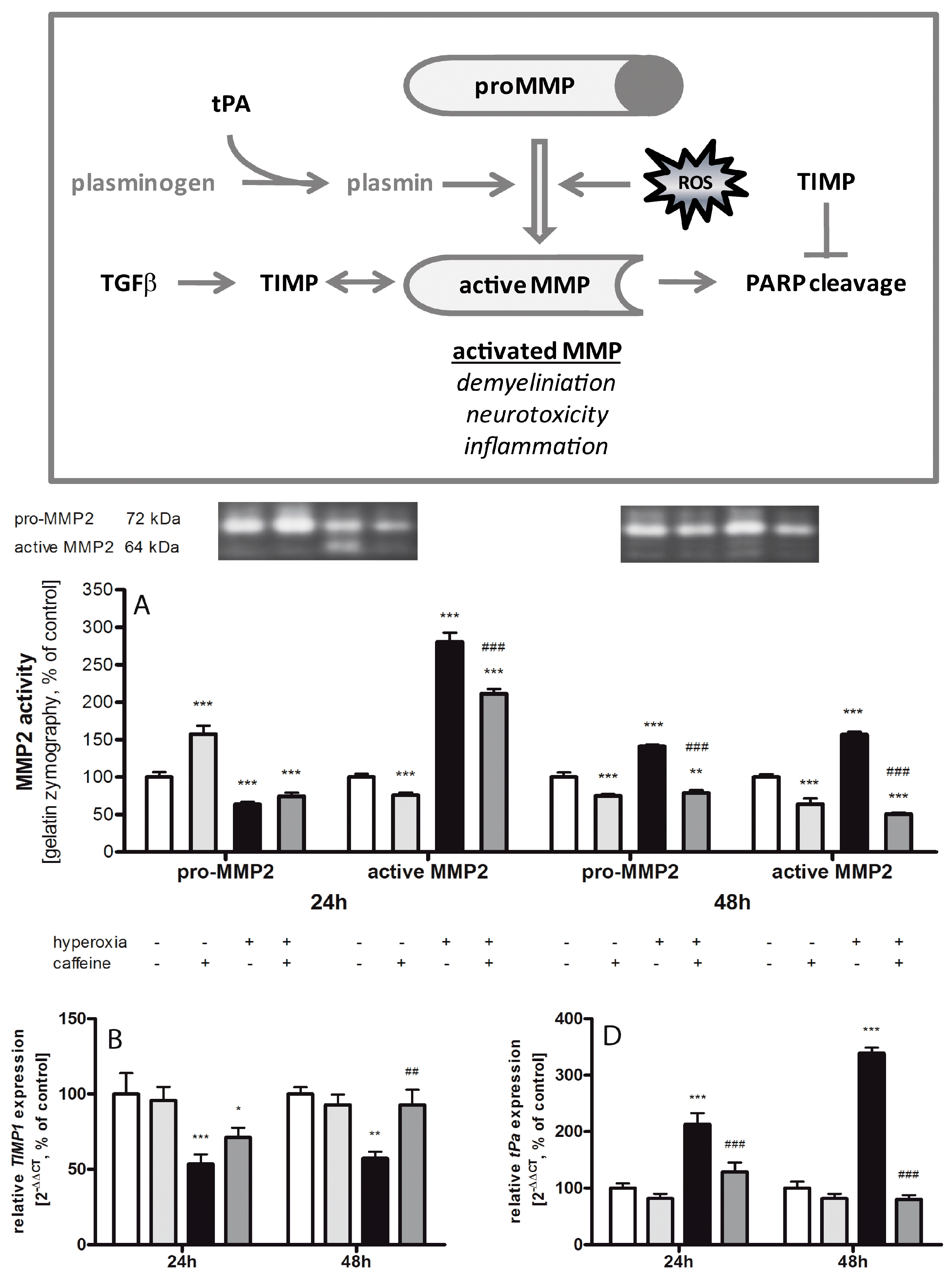
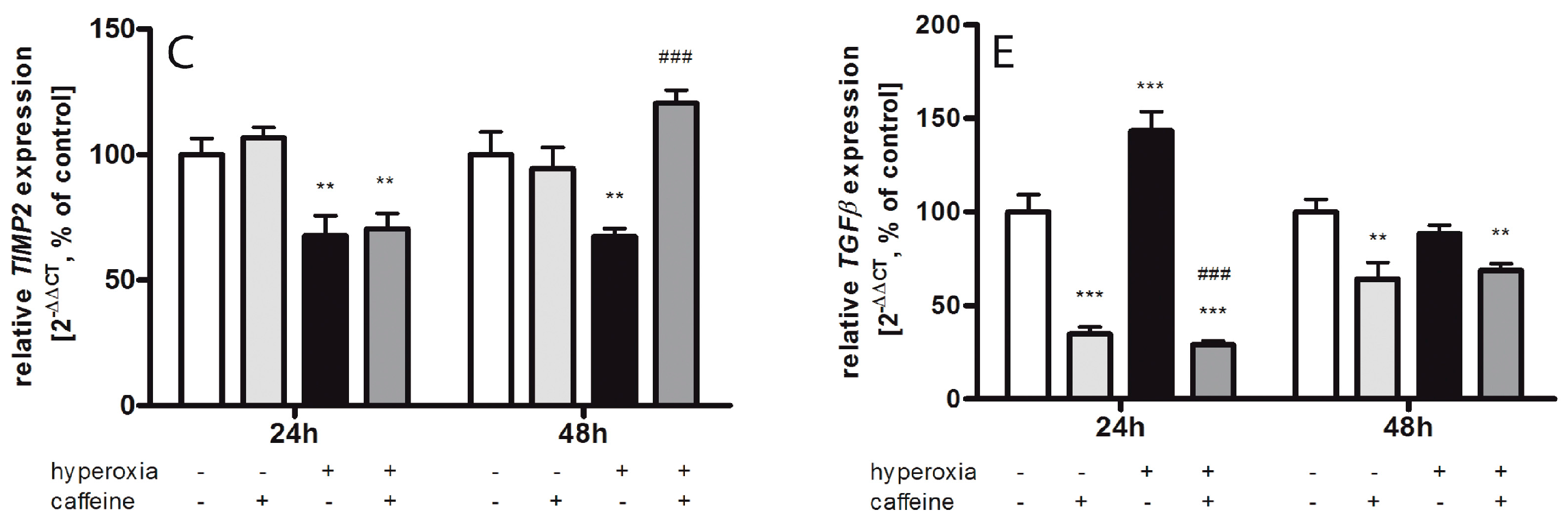
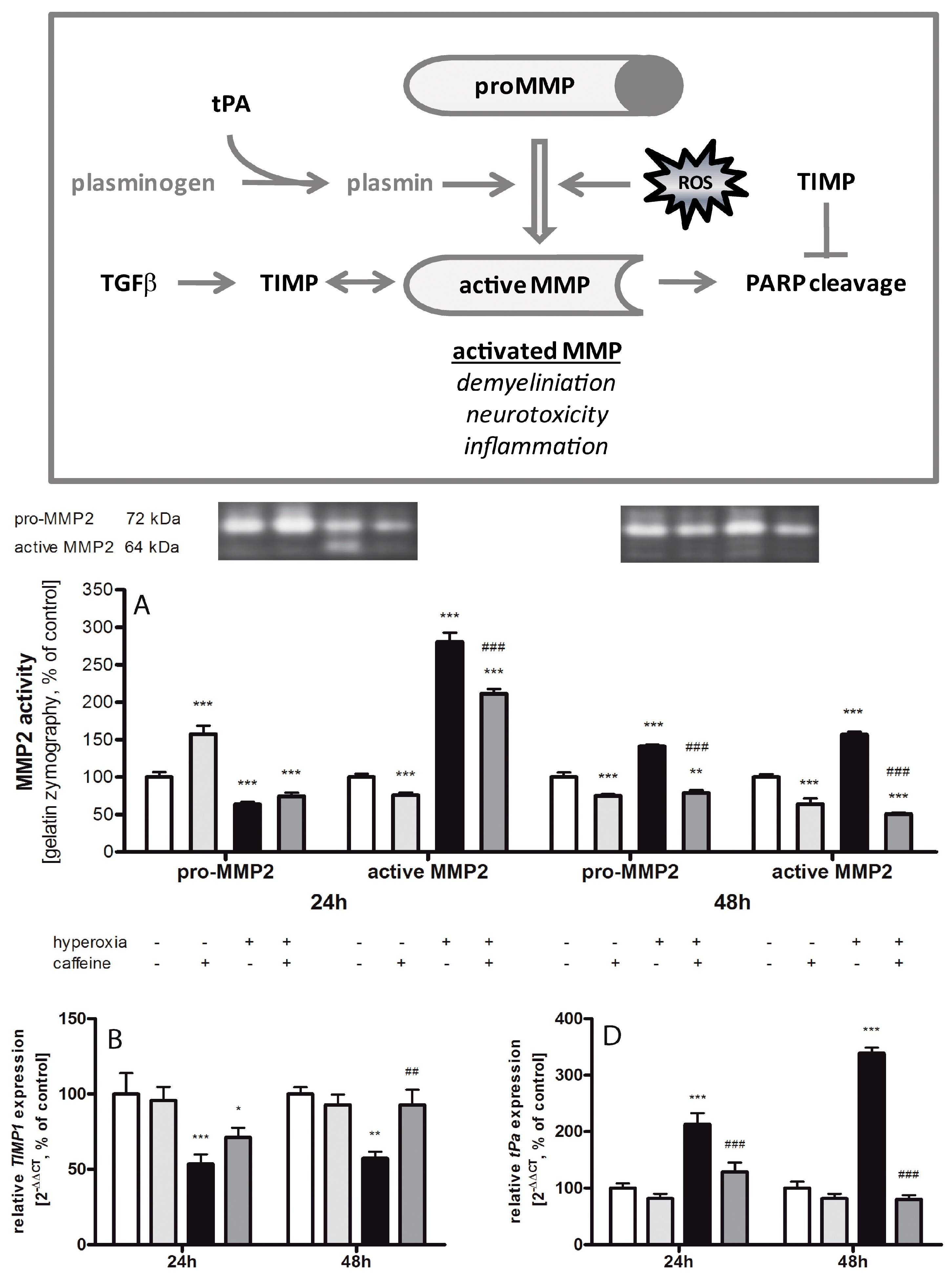
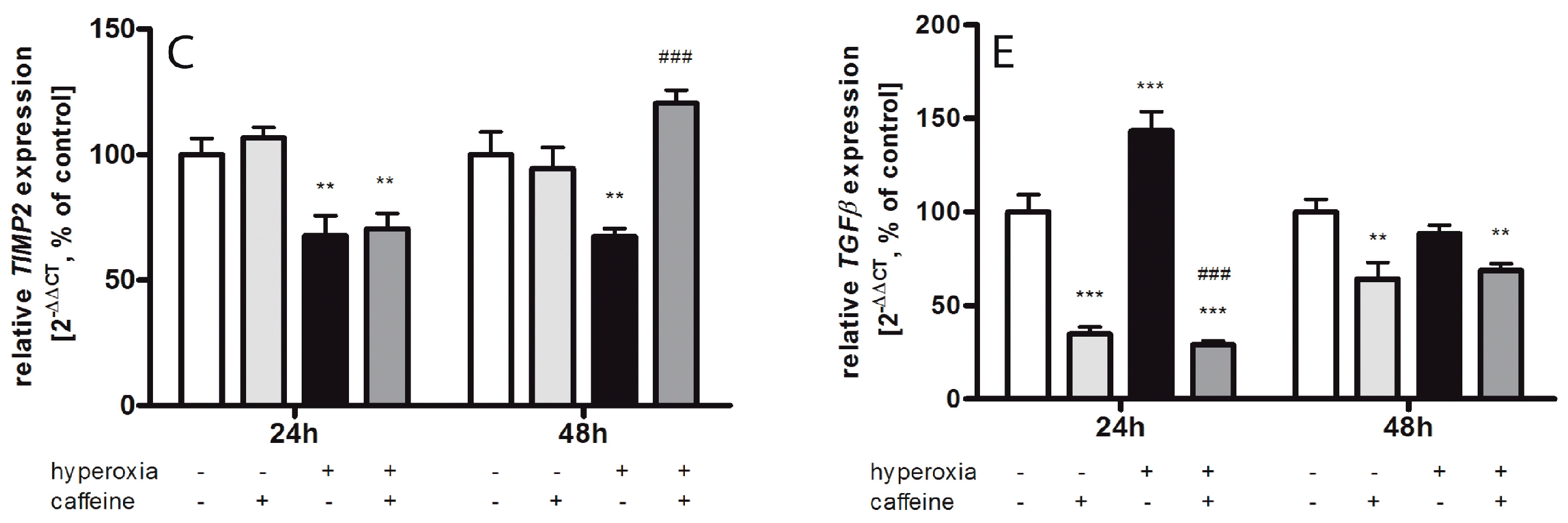
2.6. Caffeine Effects on Matrix Metalloproteinases and the tPa/Plasminogen System
3. Discussion
3.1. Oxidative Stress Response
3.2. Inflammatory Response via NFκB Pathway
3.3. Apoptosis
3.4. Fibrinolytic and Matrix Metalloproteinases System
3.5. Caffeine and Neuroprotection
4. Materials and Methods
4.1. Animals and Study Design
4.2. Tissue Preparation
4.3. Protein Extraction
4.4. Immunoblotting
4.5. Gelatin Zymography
4.6. Thiobarbituric Acid Reactive Substances (TBARS) Assay
4.7. Heme Oxygenase-1 (HO-1) Assay
4.8. Hydrogen Peroxide Assay
4.9. Enzyme-Linked Immunosorbent Assays (ELISAs)
4.10. RNA Extraction and Real-Time PCR
4.11. Statistical Analyses
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Marlow, N.; Wolke, D.; Bracewell, M.A.; Samara, M.; Group, E.S. Neurologic and developmental disability at six years of age after extremely preterm birth. N. Engl. J. Med. 2005, 352, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Wilson-Costello, D.; Friedman, H.; Minich, N.; Fanaroff, A.A.; Hack, M. Improved survival rates with increased neurodevelopmental disability for extremely low birth weight infants in the 1990s. Pediatrics 2005, 115, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Volpe, J.J. Perinatal brain injury: From pathogenesis to neuroprotection. Ment. Retard. Dev. Disabil. Res. Rev. 2001, 7, 56–64. [Google Scholar] [CrossRef]
- Perrone, S.; Tataranno, M.L.; Stazzoni, G.; Buonocore, G. Biomarkers of oxidative stress in fetal and neonatal diseases. J. Mater. Fetal Neonatal Med. 2012, 25, 2575–2578. [Google Scholar] [CrossRef] [PubMed]
- Saugstad, O.D. Update on oxygen radical disease in neonatology. Curr. Opin. Obstet. Gynecol. 2001, 13, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Stone, W.L.; Shah, D.; Hollinger, S.M. Retinopathy of prematurity: An oxidative stress neonatal disease. Front. Biosci. 2016, 21, 165–177. [Google Scholar] [CrossRef]
- Buonocore, G.; Perrone, S.; Longini, M.; Vezzosi, P.; Marzocchi, B.; Paffetti, P.; Bracci, R. Oxidative stress in preterm neonates at birth and on the seventh day of life. Pediatr. Res. 2002, 52, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Vento, M.; Escobar, J.; Cernada, M.; Escrig, R.; Aguar, M. The use and misuse of oxygen during the neonatal period. Clin. Perinatol. 2012, 39, 165–176. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, D.J.; Fernandes, C.J. Free radicals and diseases in premature infants. Antioxid. Redox Signal. 2004, 6, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Chou, Y.H. Antioxidant profiles in full term and preterm neonates. Chang Gung Med. J. 2005, 28, 846–851. [Google Scholar] [PubMed]
- Prasanthi, J.R.; Dasari, B.; Marwarha, G.; Larson, T.; Chen, X.; Geiger, J.D.; Ghribi, O. Caffeine protects against oxidative stress and Alzheimer’s disease-like pathology in rabbit hippocampus induced by cholesterol-enriched diet. Free Radic. Biol. Med. 2010, 49, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Ullah, F.; Ali, T.; Ullah, N.; Kim, M.O. Caffeine prevents d-galactose-induced cognitive deficits, oxidative stress, neuroinflammation and neurodegeneration in the adult rat brain. Neurochem. Int. 2015, 90, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Henderson-Smart, D.J.; de Paoli, A.G. Methylxanthine treatment for apnoea in preterm infants. Cochrane Database Syst. Rev. 2010, CD000140. [Google Scholar] [CrossRef]
- Schmidt, B.; Roberts, R.S.; Davis, P.; Doyle, L.W.; Barrington, K.J.; Ohlsson, A.; Solimano, A.; Tin, W. Caffeine therapy for apnea of prematurity. N. Engl. J. Med. 2006, 354, 2112–2121. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B.; Roberts, R.S.; Davis, P.; Doyle, L.W.; Barrington, K.J.; Ohlsson, A.; Solimano, A.; Tin, W. Long-term effects of caffeine therapy for apnea of prematurity. N. Engl. J. Med. 2007, 357, 1893–1902. [Google Scholar] [CrossRef] [PubMed]
- Endesfelder, S.; Zaak, I.; Weichelt, U.; Bührer, C.; Schmitz, T. Caffeine protects neuronal cells against injury caused by hyperoxia in the immature brain. Free Radic. Biol. Med. 2014, 67, 221–234. [Google Scholar] [CrossRef]
- Rivkees, S.A.; Wendler, C.C. Adverse and protective influences of adenosine on the newborn and embryo: Implications for preterm white matter injury and embryo protection. Pediatr. Res. 2011, 69, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Chavez Valdez, R.; Ahlawat, R.; Wills-Karp, M.; Nathan, A.; Ezell, T.; Gauda, E.B. Correlation between serum caffeine levels and changes in cytokine profile in a cohort of preterm infants. J. Pediatr. 2011, 158, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Weichelt, U.; Cay, R.; Schmitz, T.; Strauss, E.; Sifringer, M.; Buhrer, C.; Endesfelder, S. Prevention of hyperoxia-mediated pulmonary inflammation in neonatal rats by caffeine. Eur. Respir. J. 2013, 41, 966–973. [Google Scholar] [CrossRef] [PubMed]
- Taha, D.; Kirkby, S.; Nawab, U.; Dysart, K.C.; Genen, L.; Greenspan, J.S.; Aghai, Z.H. Early caffeine therapy for prevention of bronchopulmonary dysplasia in preterm infants. J. Mater. Fetal Neonatal Med. 2014, 27, 1698–1702. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.M.; Leong, T.; Carlton, D.P.; Vyas-Read, S. Early caffeine therapy and clinical outcomes in extremely preterm infants. J. Perinatol. 2013, 33, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.; Maier, K.; Linderkamp, O.; Hentschel, R. Effect of caffeine on oxygen consumption and metabolic rate in very low birth weight infants with idiopathic apnea. Pediatrics 2001, 107, 660–663. [Google Scholar] [CrossRef] [PubMed]
- Steer, P.; Flenady, V.; Shearman, A.; Charles, B.; Gray, P.H.; Henderson-Smart, D.; Bury, G.; Fraser, S.; Hegarty, J.; Rogers, Y.; et al. High dose caffeine citrate for extubation of preterm infants: A randomised controlled trial. Arch. Dis. Child. Fetal Neonatal Ed. 2004, 89, F499–F503. [Google Scholar] [CrossRef] [PubMed]
- Henderson-Smart, D.J.; Steer, P.A. Caffeine versus theophylline for apnea in preterm infants. Cochrane Database Syst. Rev. 2010, CD000273. [Google Scholar] [CrossRef]
- Shi, X.; Dalal, N.S.; Jain, A.C. Antioxidant behaviour of caffeine: Efficient scavenging of hydroxyl radicals. Food Chem. Toxicol. 1991, 29, 1–6. [Google Scholar] [CrossRef]
- Devasagayam, T.P.; Kamat, J.P.; Mohan, H.; Kesavan, P.C. Caffeine as an antioxidant: Inhibition of lipid peroxidation induced by reactive oxygen species. Biochim. Biophys. Acta 1996, 1282, 63–70. [Google Scholar] [CrossRef]
- Barcelos, R.P.; Souza, M.A.; Amaral, G.P.; Stefanello, S.T.; Bresciani, G.; Fighera, M.R.; Soares, F.A.; Barbosa, N.V. Caffeine supplementation modulates oxidative stress markers in the liver of trained rats. Life Sci. 2014, 96, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Tunc, T.; Aydemir, G.; Karaoglu, A.; Cekmez, F.; Kul, M.; Aydinoz, S.; Babacan, O.; Yaman, H.; Sarici, S.U. Toll-like receptor levels and caffeine responsiveness in rat pups during perinatal period. Regul. Pept. 2013, 182, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Valdez, R.; Wills-Karp, M.; Ahlawat, R.; Cristofalo, E.A.; Nathan, A.; Gauda, E.B. Caffeine modulates TNF-α production by cord blood monocytes: The role of adenosine receptors. Pediatr. Res. 2009, 65, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, G.; Hu, J.L.; Fu, X.H.; Zeng, Y.J.; Zhou, Y.G.; Xiong, G.; Yang, N.; Dai, S.S.; He, F.T. Chronic or high dose acute caffeine treatment protects mice against oleic acid-induced acute lung injury via an adenosine A2A receptor-independent mechanism. Eur. J. Pharmacol. 2011, 654, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, S.; Schnermann, J.; Noorbakhsh, F.; Henry, S.; Yong, V.W.; Winston, B.W.; Warren, K.; Power, C. A1 adenosine receptor upregulation and activation attenuates neuroinflammation and demyelination in a model of multiple sclerosis. J. Neurosci. 2004, 24, 1521–1529. [Google Scholar] [CrossRef] [PubMed]
- Ikonomidou, C.; Kaindl, A.M. Neuronal death and oxidative stress in the developing brain. Antioxid. Redox Signal. 2011, 14, 1535–1550. [Google Scholar] [CrossRef] [PubMed]
- Chua, C.O.; Vinukonda, G.; Hu, F.; Labinskyy, N.; Zia, M.T.; Pinto, J.; Csiszar, A.; Ungvari, Z.; Ballabh, P. Effect of hyperoxic resuscitation on propensity of germinal matrix haemorrhage and cerebral injury. Neuropathol. Appl. Neurobiol. 2010, 36, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Waldbaum, S.; Patel, M. Mitochondrial dysfunction and oxidative stress: A contributing link to acquired epilepsy? J. Bioenerg. Biomembr. 2010, 42, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef] [PubMed]
- Ho, E.; Karimi Galougahi, K.; Liu, C.C.; Bhindi, R.; Figtree, G.A. Biological markers of oxidative stress: Applications to cardiovascular research and practice. Redox Biol. 2013, 1, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Zaghloul, N.; Nasim, M.; Patel, H.; Codipilly, C.; Marambaud, P.; Dewey, S.; Schiffer, W.K.; Ahmed, M. Overexpression of extracellular superoxide dismutase has a protective role against hyperoxia-induced brain injury in neonatal mice. FEBS J. 2012, 279, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Sifringer, M.; Brait, D.; Weichelt, U.; Zimmerman, G.; Endesfelder, S.; Brehmer, F.; von Haefen, C.; Friedman, A.; Soreq, H.; Bendix, I.; et al. Erythropoietin attenuates hyperoxia-induced oxidative stress in the developing rat brain. Brain Behav. Immun. 2010, 24, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Morse, D.; Choi, A.M. Heme oxygenase-1: From bench to bedside. Am. J. Respir. Crit. Care Med. 2005, 172, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem. Pharmacol. 2013, 85, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Fang, Y.Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [PubMed]
- Yang, H.; Magilnick, N.; Lee, C.; Kalmaz, D.; Ou, X.; Chan, J.Y.; Lu, S.C. Nrf1 and Nrf2 regulate rat glutamate-cysteine ligase catalytic subunit transcription indirectly via NF-κB and AP-1. Mol. Cell. Biol. 2005, 25, 5933–5946. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Bendix, I.; Weichelt, U.; Strasser, K.; Serdar, M.; Endesfelder, S.; von Haefen, C.; Heumann, R.; Ehrkamp, A.; Felderhoff-Mueser, U.; Sifringer, M. Hyperoxia changes the balance of the thioredoxin/peroxiredoxin system in the neonatal rat brain. Brain Res. 2012, 1484, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.; Patil, J.; D’Angelo, B.; Weber, S.G.; Mallard, C. NRF2-regulation in brain health and disease: Implication of cerebral inflammation. Neuropharmacology 2014, 79, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Esterbauer, H.; Cheeseman, K.H. Determination of aldehydic lipid peroxidation products: Malonaldehyde and 4-hydroxynonenal. Methods Enzymol. 1990, 186, 407–421. [Google Scholar] [PubMed]
- Ahotupa, M.; Mantyla, E.; Peltola, V.; Puntala, A.; Toivonen, H. Pro-oxidant effects of normobaric hyperoxia in rat tissues. Acta Physiol. Scand. 1992, 145, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 5th ed.; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Gupta, S.P.; Srivastava, G.; Srivastava, P.K.; Singh, M.P. Role of secondary mediators in caffeine-mediated neuroprotection in maneb- and paraquat-induced Parkinson's disease phenotype in the mouse. Neurochem. Res. 2012, 37, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Sifringer, M.; von Haefen, C.; Krain, M.; Paeschke, N.; Bendix, I.; Buhrer, C.; Spies, C.D.; Endesfelder, S. Neuroprotective effect of dexmedetomidine on hyperoxia-induced toxicity in the neonatal rat brain. Oxidative Med. Cell. Longev. 2015, 2015, 530371. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Kubo, H.; Kobayashi, S.; Ishizawa, K.; Sasaki, H. Interferon-gamma: A key contributor to hyperoxia-induced lung injury in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 287, L1042–L1047. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and ugly. Am. J. Physiol. 1996, 271, C1424–C1437. [Google Scholar] [PubMed]
- Griscavage, J.M.; Wilk, S.; Ignarro, L.J. Inhibitors of the proteasome pathway interfere with induction of nitric oxide synthase in macrophages by blocking activation of transcription factor NF-κB. Proc. Natl. Acad. Sci. USA 1996, 93, 3308–3312. [Google Scholar] [CrossRef] [PubMed]
- Hoehn, T.; Felderhoff-Mueser, U.; Maschewski, K.; Stadelmann, C.; Sifringer, M.; Bittigau, P.; Koehne, P.; Hoppenz, M.; Obladen, M.; Buhrer, C. Hyperoxia causes inducible nitric oxide synthase-mediated cellular damage to the immature rat brain. Pediatr. Res. 2003, 54, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Ikeno, S.; Nagata, N.; Yoshida, S.; Takahashi, H.; Kigawa, J.; Terakawa, N. Immature brain injury via peroxynitrite production induced by inducible nitric oxide synthase after hypoxia-ischemia in rats. J. Obstet. Gynaecol. Res. 2000, 26, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Taoufik, Y.; de Goer de Herve, M.G.; Giron-Michel, J.; Durali, D.; Cazes, E.; Tardieu, M.; Azzarone, B.; Delfraissy, J.F. Human microglial cells express a functional IL-12 receptor and produce IL-12 following IL-12 stimulation. Eur. J. Immunol. 2001, 31, 3228–3239. [Google Scholar] [CrossRef]
- Chang, H.D.; Radbruch, A. The pro- and anti-inflammatory potential of interleukin-12. Ann. N. Y. Acad. Sci. 2007, 1109, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.A.; Langrish, C.L.; Chen, Y.; Blumenschein, W.; McClanahan, T.; Kastelein, R.A.; Sedgwick, J.D.; Cua, D.J. Divergent pro- and anti-inflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J. Exp. Med. 2003, 198, 1951–1957. [Google Scholar] [CrossRef] [PubMed]
- Zeidan-Chulia, F.; Gelain, D.P.; Kolling, E.A.; Rybarczyk-Filho, J.L.; Ambrosi, P.; Terra, S.R.; Pires, A.S.; da Rocha, J.B.; Behr, G.A.; Moreira, J.C. Major components of energy drinks (caffeine, taurine, and guarana) exert cytotoxic effects on human neuronal SH-SY5Y cells by decreasing reactive oxygen species production. Oxidative Med. Cell. Longev. 2013, 2013, 791795. [Google Scholar] [CrossRef] [PubMed]
- Leon-Carmona, J.R.; Galano, A. Is caffeine a good scavenger of oxygenated free radicals? J. Phys. Chem. B 2011, 115, 4538–4546. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, K.K.; Chu, C.; Couroucli, X.; Moorthy, B.; Lingappan, K. Differential concentration-specific effects of caffeine on cell viability, oxidative stress, and cell cycle in pulmonary oxygen toxicity in vitro. Biochem. Biophys. Res. Commun. 2014, 450, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Pohanka, M. Caffeine alters oxidative homeostasis in the body of BALB/c mice. Bratislavske Lekarske Listy 2014, 115, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Motterlini, R.; Foresti, R.; Bassi, R.; Green, C.J. Curcumin, an antioxidant and anti-inflammatory agent, induces heme oxygenase-1 and protects endothelial cells against oxidative stress. Free Radic. Biol. Med. 2000, 28, 1303–1312. [Google Scholar] [CrossRef]
- Ran, Q.; Liang, H.; Gu, M.; Qi, W.; Walter, C.A.; Roberts, L.J., 2nd; Herman, B.; Richardson, A.; van Remmen, H. Transgenic mice overexpressing glutathione peroxidase 4 are protected against oxidative stress-induced apoptosis. J. Biol. Chem. 2004, 279, 55137–55146. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, T.; Krabbe, G.; Weikert, G.; Scheuer, T.; Matheus, F.; Wang, Y.; Mueller, S.; Kettenmann, H.; Matyash, V.; Buhrer, C.; et al. Minocycline protects the immature white matter against hyperoxia. Exp. Neurol. 2014, 254, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Kilicdag, H.; Daglioglu, Y.K.; Erdogan, S.; Zorludemir, S. Effects of caffeine on neuronal apoptosis in neonatal hypoxic-ischemic brain injury. J. Mater. Fetal Neonatal Med. 2014, 27, 1470–1475. [Google Scholar] [CrossRef] [PubMed]
- Geraets, L.; Moonen, H.J.; Wouters, E.F.; Bast, A.; Hageman, G.J. Caffeine metabolites are inhibitors of the nuclear enzyme poly(ADP-ribose)polymerase-1 at physiological concentrations. Biochem. Pharmacol. 2006, 72, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Barkett, M.; Gilmore, T.D. Control of apoptosis by Rel/NF-κB transcription factors. Oncogene 1999, 18, 6910–6924. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Dee, C.M.; Shen, J. Interaction of free radicals, matrix metalloproteinases and caveolin-1 impacts blood-brain barrier permeability. Front. Biosci. 2011, 3, 1216–1231. [Google Scholar] [CrossRef]
- Coutts, A.; Chen, G.; Stephens, N.; Hirst, S.; Douglas, D.; Eichholtz, T.; Khalil, N. Release of biologically active TGF-β from airway smooth muscle cells induces autocrine synthesis of collagen. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 280, L999–L1008. [Google Scholar] [PubMed]
- Millis, A.J.; Hoyle, M.; McCue, H.M.; Martini, H. Differential expression of metalloproteinase and tissue inhibitor of metalloproteinase genes in aged human fibroblasts. Exp. Cell Res. 1992, 201, 373–379. [Google Scholar] [CrossRef]
- Sifringer, M.; Genz, K.; Brait, D.; Brehmer, F.; Lober, R.; Weichelt, U.; Kaindl, A.M.; Gerstner, B.; Felderhoff-Mueser, U. Erythropoietin attenuates hyperoxia-induced cell death by modulation of inflammatory mediators and matrix metalloproteinases. Dev. Neurosci. 2009, 31, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Okazaki, R.; Morioka, K.; Nakamura, K.; Tanaka, S.; Ogata, T. Lipopolysaccharide preconditioning facilitates M2 activation of resident microglia after spinal cord injury. J. Neurosci. Res. 2014, 92, 1647–1658. [Google Scholar] [CrossRef] [PubMed]
- Saad, M.A.; Abdelsalam, R.M.; Kenawy, S.A.; Attia, A.S. Ischemic preconditioning and postconditioning alleviates hippocampal tissue damage through abrogation of apoptosis modulated by oxidative stress and inflammation during transient global cerebral ischemia-reperfusion in rats. Chem. Biol. Interact. 2015, 232, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Parmar, J.; Jones, N.M. Hypoxic preconditioning can reduce injury-induced inflammatory processes in the neonatal rat brain. Int. J. Dev. Neurosci. 2015, 43, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G. Cardioprotection: Chances and challenges of its translation to the clinic. Lancet 2013, 381, 166–175. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Yellon, D.M. Ischaemic conditioning and reperfusion injury. Nat. Rev. Cardiol. 2016, 13, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Robertson, N.J.; Tan, S.; Groenendaal, F.; van Bel, F.; Juul, S.E.; Bennet, L.; Derrick, M.; Back, S.A.; Valdez, R.C.; Northington, F.; et al. Which neuroprotective agents are ready for bench to bedside translation in the newborn infant? J. Pediatr. 2012, 160, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| cDNA | Oligonucleotide Sequence 5′–3′ | Accession No. |
|---|---|---|
| HPRT | ||
| forward | GGAAAGAACGTCTTGATTGTTGAA | NM_012583.2 |
| reverse | CCAACACTTCGAGAGGTCCTTTT | |
| probe | CTTTCCTTGGTCAAGCAGTACAGCCCC | |
| Casp3 | ||
| forward | ACAGTGGAACTGACGATGATATGG | NM_012922.2 |
| reverse | AATAGTAACCGGGTGCGGTAGA | |
| probe | ATGCCAGAAGATACCAGTGG | |
| GCLC | ||
| forward | GGAGGACAACATGAGGAAACG | NM_012815.2 |
| reverse | GCTCTGGCAGTGTGAATCCA | |
| probe | GAGGCTACTTCTGTATTAGG | |
| IFN-γ | ||
| forward | GCAAAAGGACGGTAACACGAA | NM_138880.2 |
| reverse | ATGGCCTGGTTGTCTTTCAAGA | |
| probe | TCTCTTTCTACCTCAGACTC | |
| IL-1β | ||
| forward | CTCCACCTCAATGGACAGAACA | NM_031512.2 |
| reverse | CACAGGGATTTTGTCGTTGCT | |
| probe | CTCCATGAGCTTTGTACAAG | |
| IL-12b | ||
| forward | TGCTGCTCCACAAGAAGGAA | NM_022611.1 |
| reverse | TTGGTGCTTCACACTTCAGGAA | |
| probe | ATGGAATTTGGTCCACCGAG | |
| IL-18 | ||
| forward | CGGAGCATAAATGACCAAGTTCTC | NM_019165.1 |
| reverse | TGGGATTCGTTGGCTGTTC | |
| probe | TTGACAAAAGAAACCCGCCTG | |
| iNOS | ||
| forward | AGCTGTAGCACTGCATCAGAAATG | NM_012611.3 |
| reverse | CAGTAATGGCCGACCTGATGT | |
| probe | CAGACACATACTTTACGCCAC | |
| Keap1 | ||
| forward | GATCGGCTGCACGGAACT | NM_057152.2 |
| reverse | GCAGTGTGACAGGTTGAAGAACTC | |
| probe | CTCGGGAGTATATCTACATGC | |
| NFκB1 | ||
| forward | GACCCAAGGACATGGTGGTT | NM_001276711.1 |
| reverse | TCATCCGTGCTTCCAGTGTTT | |
| probe | CTGGGAATACTTCACGTGAC | |
| NFκB2 | ||
| forward | GCCTAAACAGCGAGGCTTCA | NM_001008349.1 |
| reverse | TCTTCCGGCCCTTCTCACT | |
| probe | TTTCGATATGGCTGTGAAGG | |
| Nrf2 | ||
| forward | ACTCCCAGGTTGCCCACAT | NM_031789.2 |
| reverse | GCGACTCATGGTCATCTACAAATG | |
| probe | CTTTGAAGACTGTATGCAGC | |
| SOD1 | ||
| forward | CAGAAGGCAAGCGGTGAAC | NM_017050.1 |
| reverse | CCCCATATTGATGGACATGGA | |
| probe | TACAGGATTAACTGAAGGCG | |
| SOD2 | ||
| forward | GACCTACGTGAACAATCTGAACGT | NM_017051.2 |
| reverse | AGGCTGAAGAGCAACCTGAGTT | |
| probe | ACCGAGGAGAAGTACCACGA | |
| SOD3 | ||
| forward | GGAGAGTCCGGTGTCGACTTAG | NM_012880.1 |
| reverse | CTCCATCCAGATCTCCAGGTCTT | |
| probe | CTGGTTGAGAAGATAGGCGA | |
| TGF-β | ||
| forward | CCTGCAGAGATTCAAGTCAACTGT | NM_021578.2 |
| reverse | GTCAGCAGCCGGTTACCAA | |
| probe | CAACAATTCCTGGCGTT | |
| TIMP1 | ||
| forward | CGGACCTGGTTATAAGGGCTAA | NM_053819.1 |
| reverse | CGTCGAATCCTTTGAGCATCT | |
| probe | AGAAATCATCGAGACCACCT | |
| TIMP2 | ||
| forward | GGCAACCCCATCAAGAGGAT | NM_021989.2 |
| reverse | GGGCCGTGTAGATAAATTCGAT | |
| probe | AGATGTTCAAAGGACCTGAC | |
| tPa | ||
| forward | TCAGAAGAGGAGCTCGGTCCTA | NM_013151.2 |
| reverse | TGGGACGTAGCCATGACTGAT | |
| probe | CAGAGATGAACAGACTCAGA | |
| TNFα | ||
| forward | CCCCCAATCTGTGTCCTTCTAAC | NM_012675.2 |
| reverse | CGTCTCGTGTGTTTCTGAGCAT | |
| probe | TAGAAAGGGAATTGTGGCTC | |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Endesfelder, S.; Weichelt, U.; Strauß, E.; Schlör, A.; Sifringer, M.; Scheuer, T.; Bührer, C.; Schmitz, T. Neuroprotection by Caffeine in Hyperoxia-Induced Neonatal Brain Injury. Int. J. Mol. Sci. 2017, 18, 187. https://doi.org/10.3390/ijms18010187
Endesfelder S, Weichelt U, Strauß E, Schlör A, Sifringer M, Scheuer T, Bührer C, Schmitz T. Neuroprotection by Caffeine in Hyperoxia-Induced Neonatal Brain Injury. International Journal of Molecular Sciences. 2017; 18(1):187. https://doi.org/10.3390/ijms18010187
Chicago/Turabian StyleEndesfelder, Stefanie, Ulrike Weichelt, Evelyn Strauß, Anja Schlör, Marco Sifringer, Till Scheuer, Christoph Bührer, and Thomas Schmitz. 2017. "Neuroprotection by Caffeine in Hyperoxia-Induced Neonatal Brain Injury" International Journal of Molecular Sciences 18, no. 1: 187. https://doi.org/10.3390/ijms18010187