
Neuroprotective and Anti-Apoptotic Effects of CSP-1103 in Primary Cortical Neurons Exposed to Oxygen and Glucose Deprivation

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
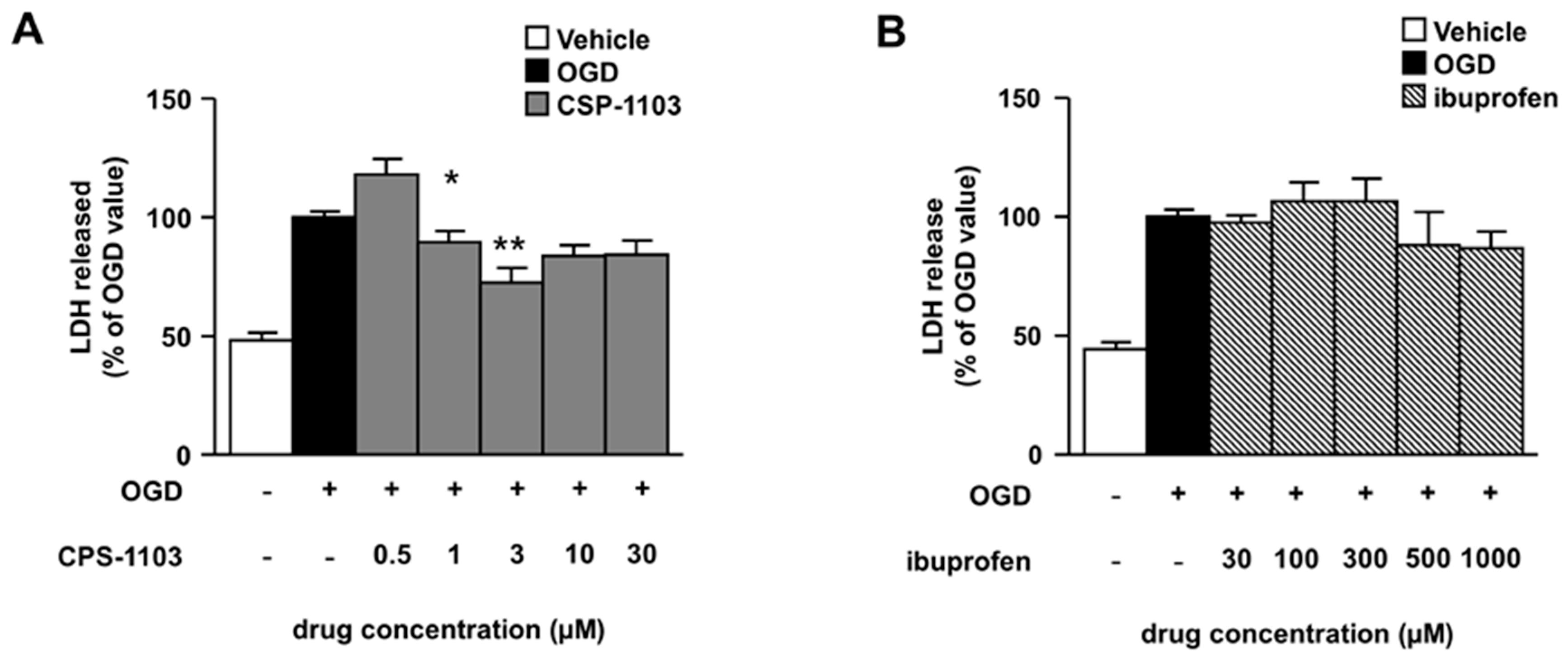
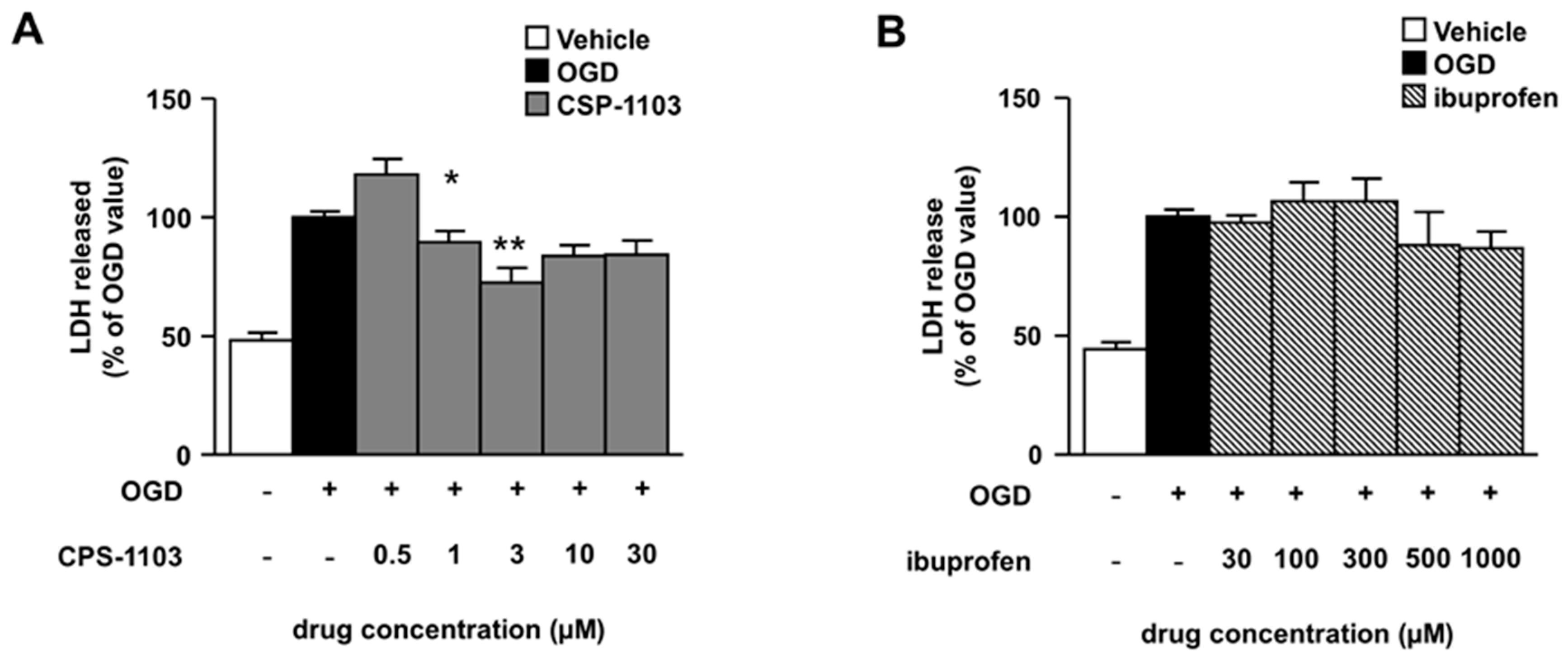
2.1. CSP-1103, but Not Ibuprofen, Reduces Necrosis Induced by OGD in Primary Cortical Neurons
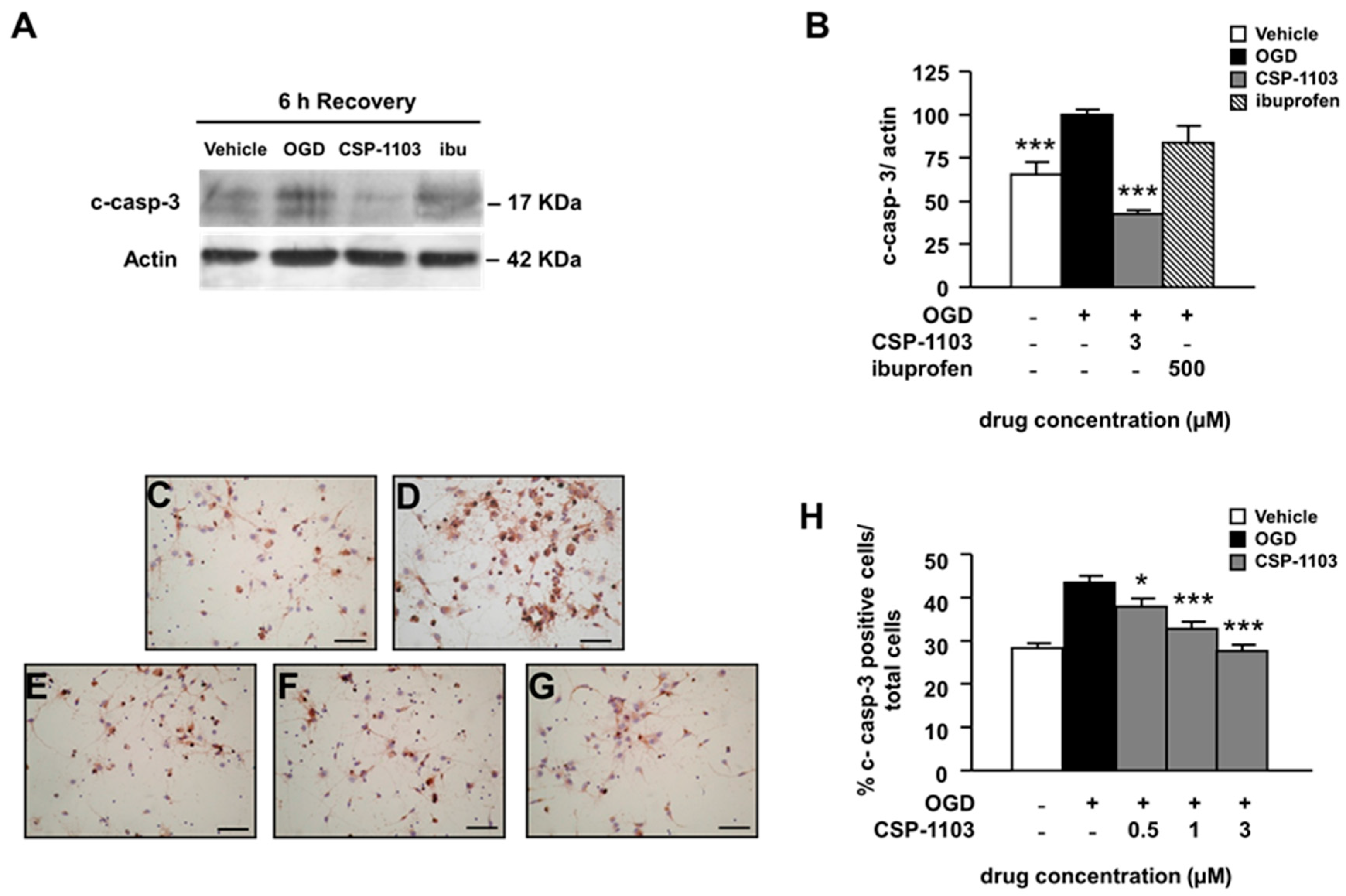
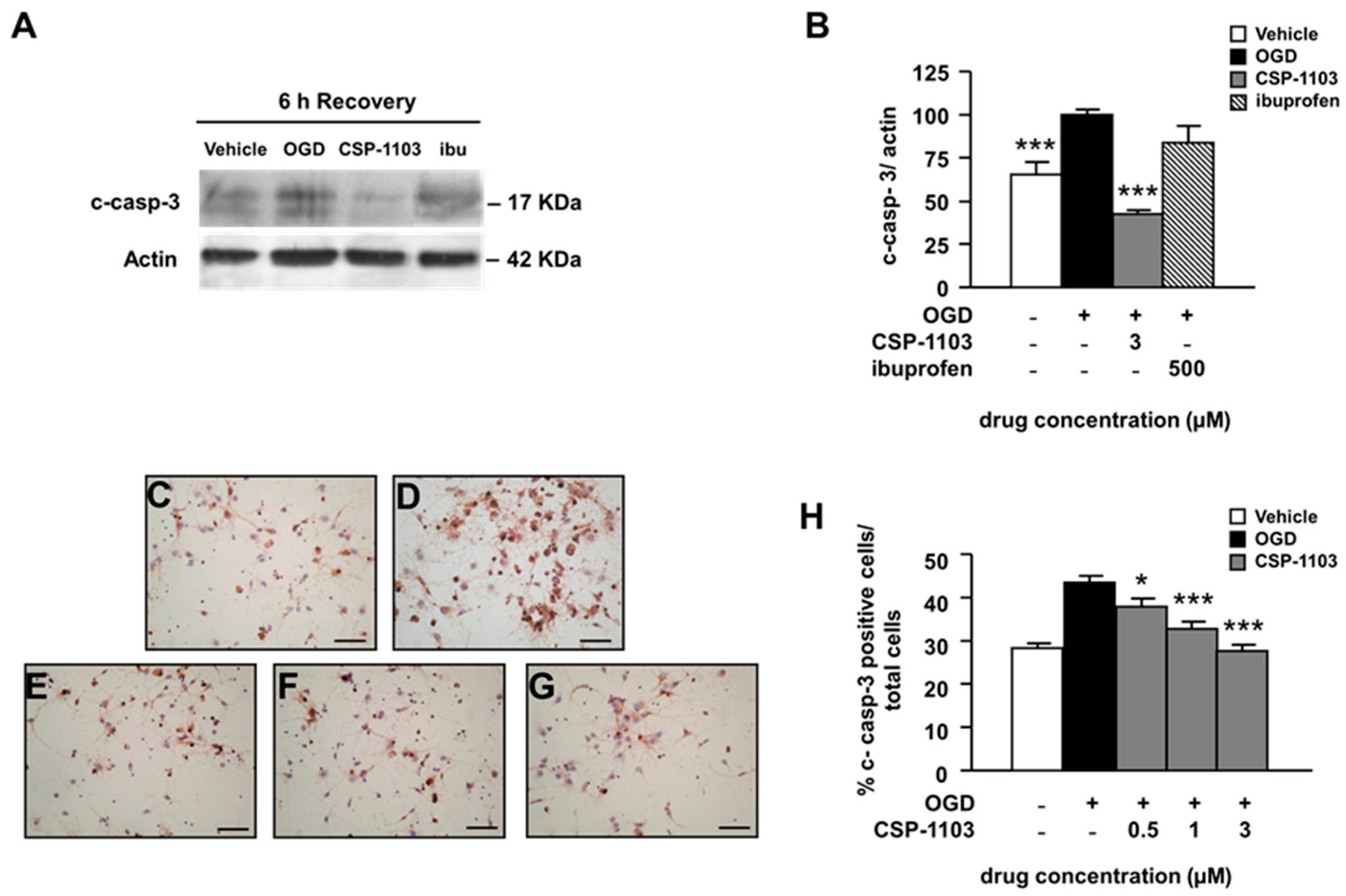
2.2. CSP-1103, but Not Ibuprofen, Prevents Caspase-3 Activation Induced by OGD in Primary Cortical Neurons
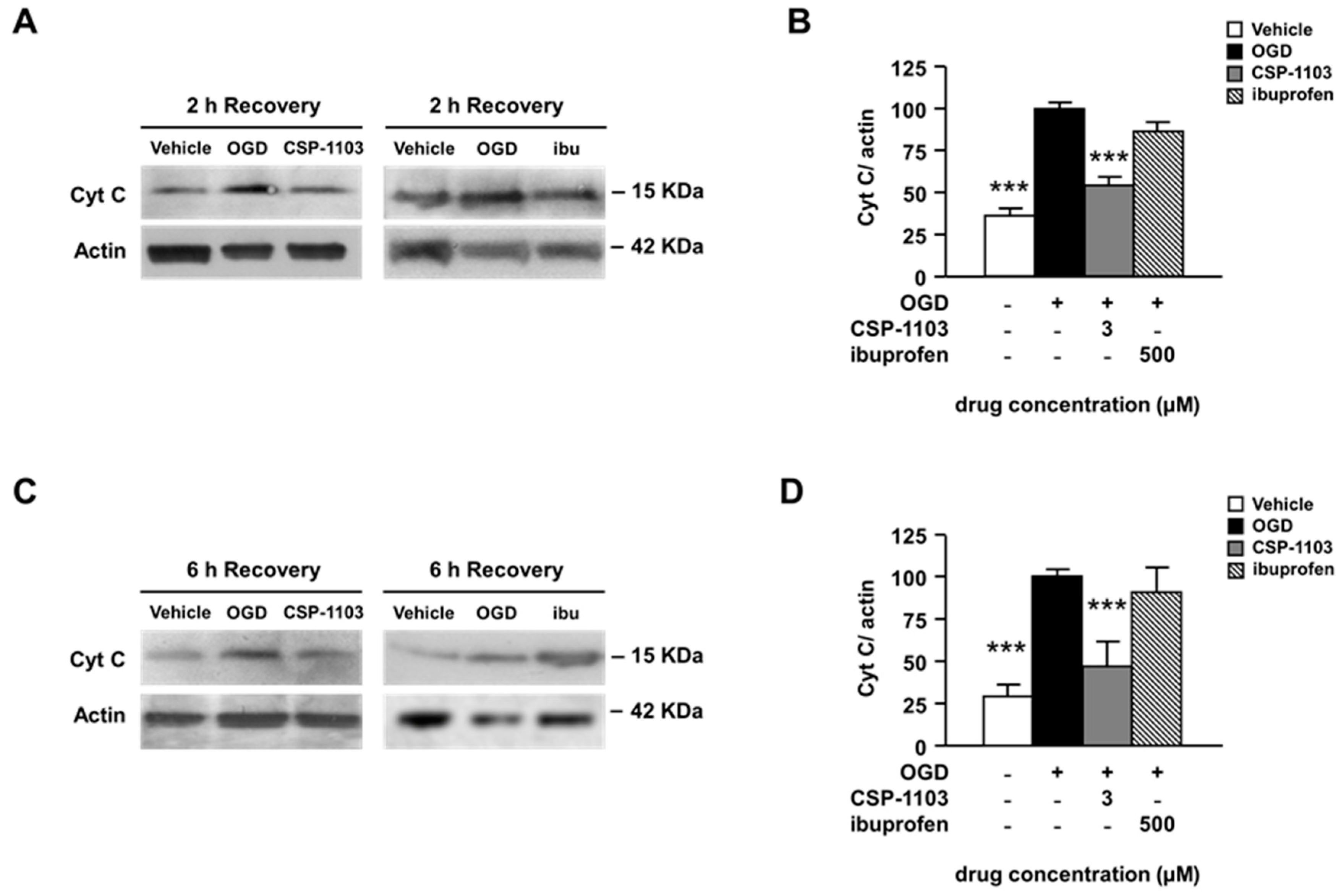
2.3. CSP-1103, but Not Ibuprofen, Prevents Cytochrome C Release Induced by OGD in Primary Cortical Neurons
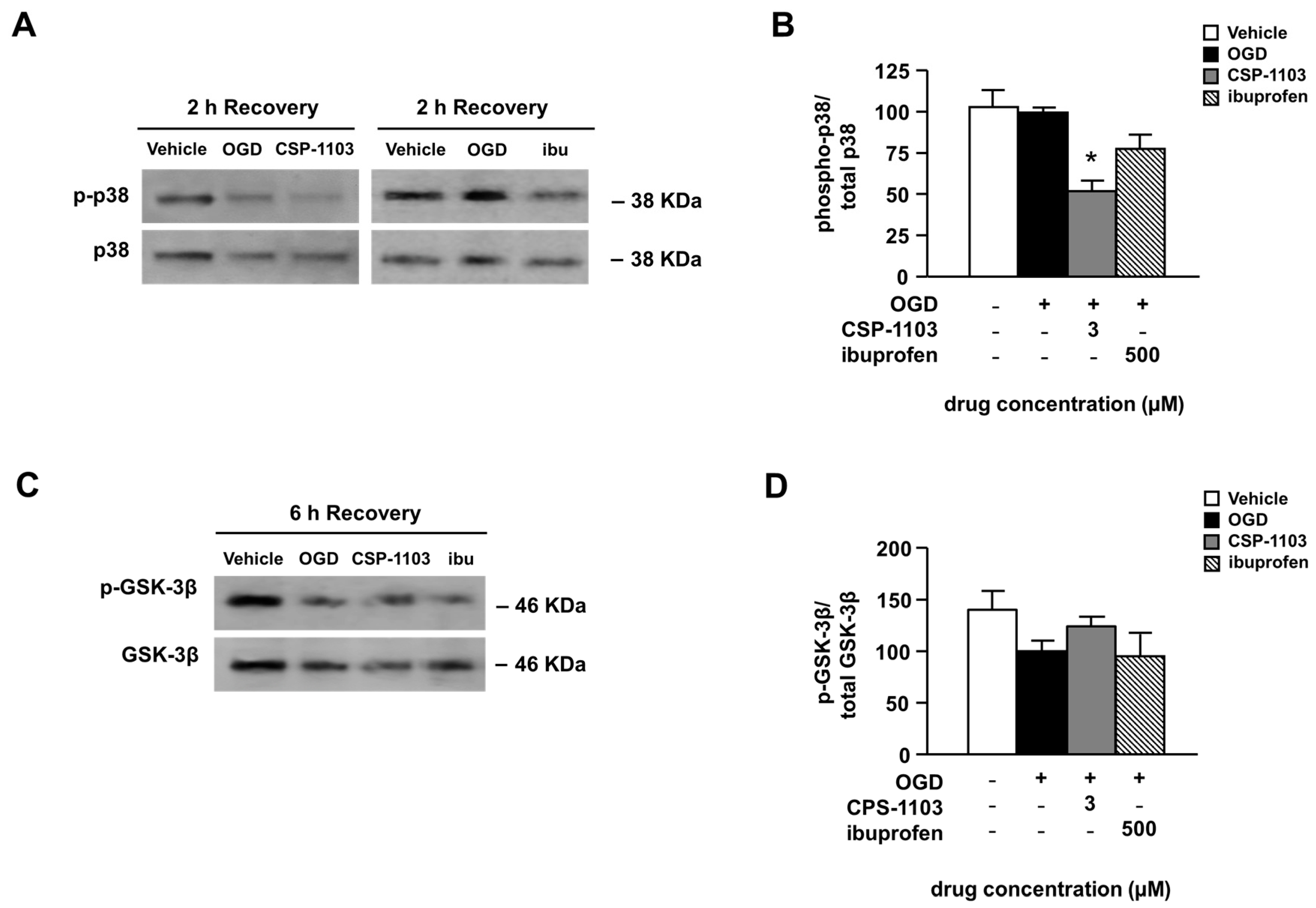
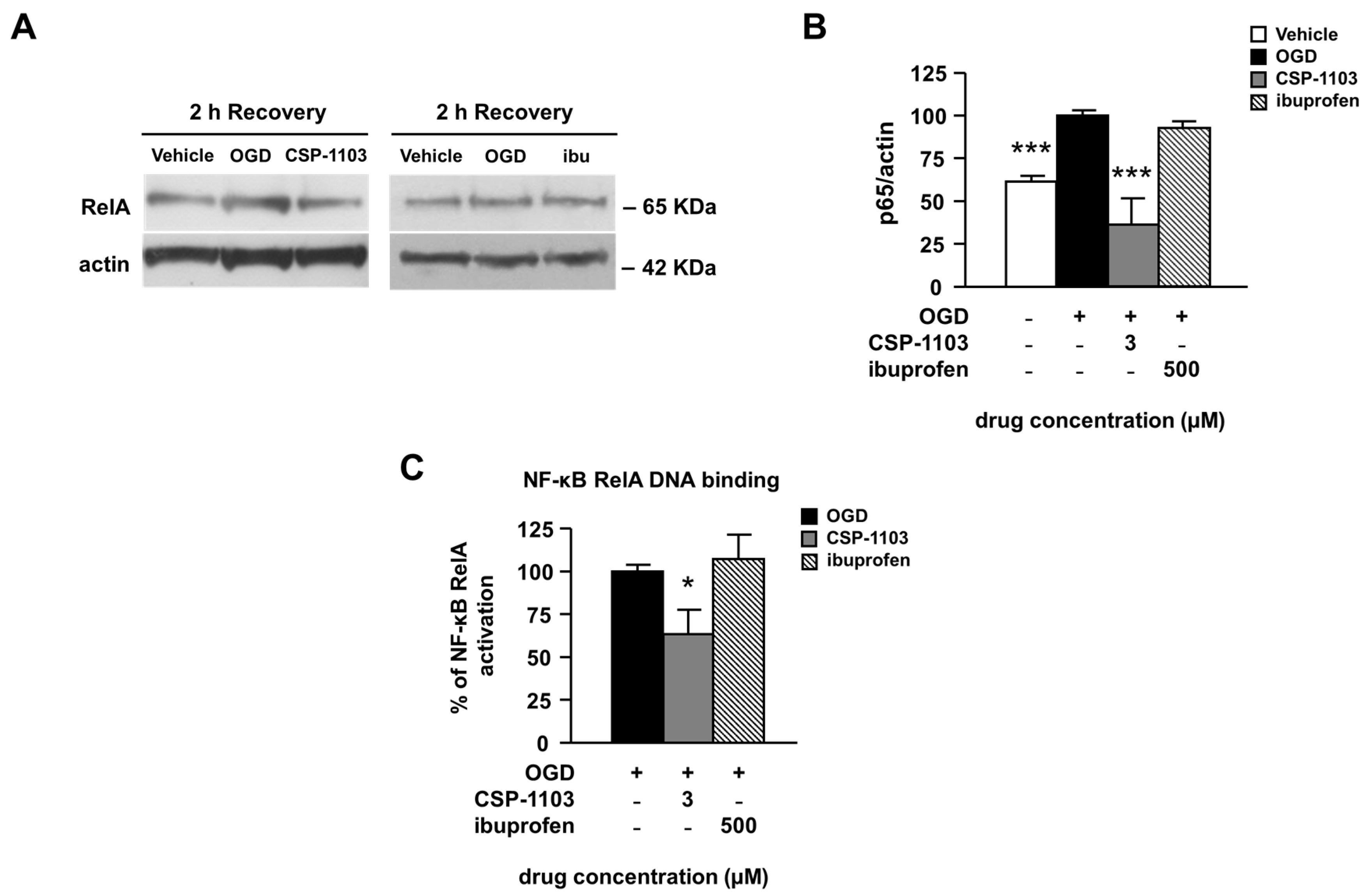
2.4. Effect of CSP-1103 and Ibuprofen, on p38 MAPK, GSK-3β, and NF-κB Activation in Primary Cortical Neurons Exposed to OGD
3. Discussion
4. Materials and Methods
4.1. Oxygen-Glucose Deprivation in Nearly Pure Primary Mouse Cortical Neurons
4.2. Immunocytochemistry
4.3. Western Blot Analysis
4.4. DNA-Based ELISA
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| TUNEL | terminal deoxynucleotidyl transferase dUTP nick end labeling |
| NSAID | non-steroidal anti-inflammatory drug |
| AD | Alzheimer’s disease |
| Aβ | β-amyloid |
| sCD40L | soluble CD40 ligand |
| TNF-α | tumor necrosis factor α |
| CSF | cerebrospinal fluid |
| AICD | amyloid precursor protein intracellular domain |
| TRAIL | tumor necrosis factor related apoptosis inducing ligand |
| OGD | oxygen-glucose deprivation |
| LDH | lactate dehydrogenase |
| c-casp-3 | cleaved caspase-3 protein |
| WB | western blot |
| MAPK | mitogen-activated protein kinase |
| GSK-3β | glycogen synthase kinase-3β |
| NF-κB | nuclear factor kappa B |
| DAB | 3,3′-diaminobenzidine |
| HRP | horseradish peroxidase |
References
- Donnan, G.A.; Fisher, M.; Macleod, M.; Davis, S.M. Stroke. Lancet 2008, 371, 1612–1623. [Google Scholar] [CrossRef]
- Broughton, B.R.; Reutens, D.C.; Sobey, C.G. Apoptotic mechanisms after cerebral ischemia. Stroke 2009, 40, e331–e339. [Google Scholar] [CrossRef] [PubMed]
- Benchoua, A.; Guégan, C.; Couriaud, C.; Hosseini, H.; Sampaïo, N.; Morin, D.; Onténiente, B. Specific caspase pathways are activated in the two stages of cerebral infarction. J. Neurosci. 2001, 21, 7127–7134. [Google Scholar] [PubMed]
- Ferrer, I.; Friguls, B.; Dalfó, E.; Justicia, C.; Planas, A.M. Caspase-dependent and caspase-independent signalling of apoptosis in the penumbra following middle cerebral artery occlusion in the adult rat. Neuropathol. Appl. Neurobiol. 2003, 29, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.R.; Wang, J.X.; Wang, J.; Xu, R.; Zhao, J.K.; Wang, D.S. Ischemia induces endoplasmic reticulum stress and cell apoptosis in human brain. Neurosci. Lett. 2010, 475, 132–135. [Google Scholar] [CrossRef] [PubMed]
- Mitsios, N.; Gaffney, J.; Krupinski, J.; Mathias, R.; Wang, Q.; Hayward, S.; Rubio, F.; Kumar, P.; Kumar, S.; Slevin, M. Expression of signaling molecules associated with apoptosis in human ischemic stroke tissue. Cell Biochem. Biophys. 2007, 47, 73–86. [Google Scholar] [CrossRef]
- Rami, A.; Sims, J.; Botez, G.; Winckler, J. Spatial resolution of phospholipid scramblase 1 (plscr1), caspase-3 activation and DNA-fragmentation in the human hippocampus after cerebral ischemia. Neurochem. Int. 2003, 43, 79–87. [Google Scholar] [CrossRef]
- Sairanen, T.; Szepesi, R.; Karjalainen-Lindsberg, M.L.; Saksi, J.; Paetau, A.; Lindsberg, P.J. Neuronal caspase-3 and parp-1 correlate differentially with apoptosis and necrosis in ischemic human stroke. Acta Neuropathol. 2009, 118, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Imbimbo, B.P.; Del Giudice, E.; Colavito, D.; D’Arrigo, A.; Dalle Carbonare, M.; Villetti, G.; Facchinetti, F.; Volta, R.; Pietrini, V.; Baroc, M.F.; et al. 1-(3′,4′-dichloro-2-fluoro[1,1′-biphenyl]-4-yl)-cyclopropanecarboxylic acid (CHF5074), a novel gamma-secretase modulator, reduces brain β-amyloid pathology in a transgenic mouse model of Alzheimer’s disease without causing peripheral toxicity. J. Pharmacol. Exp. Ther. 2007, 323, 822–830. [Google Scholar] [CrossRef] [PubMed]
- Balducci, C.; Mehdawy, B.; Mare, L.; Giuliani, A.; Lorenzini, L.; Sivilia, S.; Giardino, L.; Calzà, L.; Lanzillotta, A.; Sarnico, I.; et al. The γ-secretase modulator CHF5074 restores memory and hippocampal synaptic plasticity in plaque-free TG2576 mice. J. Alzheimers Dis. 2011, 24, 799–816. [Google Scholar] [PubMed]
- Imbimbo, B.P.; Hutter-Paier, B.; Villetti, G.; Facchinetti, F.; Cenacchi, V.; Volta, R.; Lanzillotta, A.; Pizzi, M.; Windisch, M. Chf5074, a novel gamma-secretase modulator, attenuates brain β-amyloid pathology and learning deficit in a mouse model of Alzheimer’s disease. Br. J. Pharmacol. 2009, 156, 982–993. [Google Scholar] [CrossRef] [PubMed]
- Sivilia, S.; Lorenzini, L.; Giuliani, A.; Gusciglio, M.; Fernandez, M.; Baldassarro, V.A.; Mangano, C.; Ferraro, L.; Pietrini, V.; Baroc, M.F.; et al. Multi-target action of the novel anti-Alzheimer compound chf5074: In vivo study of long term treatment in TG2576 mice. BMC Neurosci. 2013, 14, 44. [Google Scholar] [CrossRef] [PubMed]
- Imbimbo, B.P.; Frigerio, E.; Breda, M.; Fiorentini, F.; Fernandez, M.; Sivilia, S.; Giardino, L.; Calzà, L.; Norris, D.; Casula, D.; et al. Pharmacokinetics and pharmacodynamics of CHF5074 after short-term administration in healthy subjects. Alzheimer Dis. Assoc. Disord. 2013, 27, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Porrini, V.; Lanzillotta, A.; Branca, C.; Benarese, M.; Parrella, E.; Lorenzini, L.; Calzà, L.; Flaibani, R.; Spano, P.F.; Imbimbo, B.P.; et al. CHF5074 (CSP-1103) induces microglia alternative activation in plaque-free Tg2576 mice and primary glial cultures exposed to β-amyloid. Neuroscience 2014, 302, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.; Sharma, S.; Winston, J.; Nunez, M.; Bottini, G.; Franceschi, M.; Scarpini, E.; Frigerio, E.; Fiorentini, F.; Fernandez, M.; et al. CHF5074 reduces biomarkers of neuroinflammation in patients with mild cognitive impairment: A 12-week, double-blind, placebo-controlled study. Curr Alzheimer Res 2013, 10, 742–753. [Google Scholar] [CrossRef] [PubMed]
- Town, T.; Nikolic, V.; Tan, J. The microglial “Activation” Continuum: From innate to adaptive responses. J. Neuroinflamm. 2005, 2, 24. [Google Scholar] [CrossRef] [PubMed]
- Imbimbo, B.P.; Fernandez, M.; Giardino, L.; Calzà, L.; Chain, D.; Margolin, R. Relationship between cerebrospinal fluid (CSF) biomarkers and cognitive performance of patients with mild cognitive impairment (MCI) after long-term treatment with chf5074. In Proceedings of the 17th Alzheimer’s Association International Conference, Alzheimer's & Dementia 2014, Copenhagen, Denmark, 12–17 July 2014; Volume 10, p. 273.
- Branca, C.; Sarnico, I.; Ruotolo, R.; Lanzillotta, A.; Viscomi, A.R.; Benarese, M.; Porrini, V.; Lorenzini, L.; Calzà, L.; Imbimbo, B.P.; et al. Pharmacological targeting of the β-amyloid precursor protein intracellular domain. Sci. Rep. 2014, 4, 4618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardossi-Piquard, R.; Checler, F. The physiology of the β-amyloid precursor protein intracellular domain aicd. J. Neurochem. 2012, 120, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Ronsisvalle, N.; di Benedetto, G.; Parenti, C.; Amoroso, S.; Bernardini, R.; Cantarella, G. CHF5074 protects sh-sy5y human neuronal-like cells from amyloidβ 25–35 and tumor necrosis factor related apoptosis inducing ligand toxicity in vitro. Curr. Alzheimer Res. 2014, 11, 714–724. [Google Scholar] [CrossRef] [PubMed]
- Mango, D.; Barbato, G.; Piccirilli, S.; Panico, M.B.; Feligioni, M.; Schepisi, C.; Graziani, M.; Porrini, V.; Benarese, M.; Lanzillotta, A.; et al. Electrophysiological and metabolic effects of CHF5074 in the hippocampus: Protection against in vitro ischemia. Pharmacol. Res. 2014, 81, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Sarnico, I.; Lanzillotta, A.; Boroni, F.; Benarese, M.; Alghisi, M.; Schwaninger, M.; Inta, I.; Battistin, L.; Spano, P.; Pizzi, M. NF-κB p50/rela and c-rel-containing dimers: Opposite regulators of neuron vulnerability to ischaemia. J. Neurochem. 2009, 108, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Valerio, A.; Dossena, M.; Bertolotti, P.; Boroni, F.; Sarnico, I.; Faraco, G.; Chiarugi, A.; Frontini, A.; Giordano, A.; Liou, H.C.; et al. Leptin is induced in the ischemic cerebral cortex and exerts neuroprotection through NF-κB/C-Rel-dependent transcription. Stroke 2009, 40, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Cory, S. Apoptosomes: Engines for caspase activation. Curr. Opin. Cell Biol. 2002, 14, 715–720. [Google Scholar] [CrossRef]
- Ghavami, S.; Hashemi, M.; Ande, S.R.; Yeganeh, B.; Xiao, W.; Eshraghi, M.; Bus, C.J.; Kadkhoda, K.; Wiechec, E.; Halayko, A.J.; et al. Apoptosis and cancer: Mutations within caspase genes. J. Med. Genet. 2009, 46, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, Y.; Steller, H. Programmed cell death in animal development and disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef] [PubMed]
- Iwata, Y.; Nicole, O.; Zurakowski, D.; Okamura, T.; Jonas, R.A. Ibuprofen for neuroprotection after cerebral ischemia. J. Thorac. Cardiovasc. Surg. 2010, 139, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Park, E.M.; Cho, B.P.; Volpe, B.T.; Cruz, M.O.; Joh, T.H.; Cho, S. Ibuprofen protects ischemia-induced neuronal injury via up-regulating interleukin-1 receptor antagonist expression. Neuroscience 2005, 132, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Sugino, T.; Nozaki, K.; Takagi, Y.; Hattori, I.; Hashimoto, N.; Moriguchi, T.; Nishida, E. Activation of mitogen-activated protein kinases after transient forebrain ischemia in gerbil hippocampus. J. Neurosci. 2000, 20, 4506–4514. [Google Scholar] [PubMed]
- Bhuiyan, M.I.; Jung, S.Y.; Kim, H.J.; Lee, Y.S.; Jin, C. Major role of the PI3K/Akt pathway in ischemic tolerance induced by sublethal oxygen-glucose deprivation in cortical neurons in vitro. Arch. Pharm. Res. 2011, 34, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, C.; Li, Y.; Dong, L.; Cui, L.; Wang, L.; Liu, Z.; Qiao, H.; Zhu, C.; Xing, Y.; et al. Neuroprotection of early and short-time applying berberine in the acute phase of cerebral ischemia: Up-regulated PAKT, PGSK and PCREB, down-regulated NF-κB expression, ameliorated BBB permeability. Brain Res. 2012, 1459, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.; Zhao, H.; Hua Sun, G.; Cheng, D.; Qiao, Y.; Luo, J.; Martin, K.; Steinberg, G.K.; Harrison, S.D.; Yenari, M.A. Glycogen synthase kinase 3β inhibitor Chir025 reduces neuronal death resulting from oxygen-glucose deprivation, glutamate excitotoxicity, and cerebral ischemia. Exp. Neurol. 2004, 188, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Lanzillotta, A.; Sarnico, I.; Benarese, M.; Branca, C.; Baiguera, C.; Hutter-Paier, B.; Windisch, M.; Spano, P.; Imbimbo, B.P.; Pizzi, M. The γ-secretase modulator CHF5074 reduces the accumulation of native hyperphosphorylated tau in a transgenic mouse model of Alzheimer’s disease. J. Mol. Neurosci. 2011, 45, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Takahashi-Yanaga, F. Activator or inhibitor? GSK-3 as a new drug target. Biochem. Pharmacol. 2013, 86, 191–199. [Google Scholar] [PubMed]
- Lanzillotta, A.; Porrini, V.; Bellucci, A.; Benarese, M.; Branca, C.; Parrella, E.; Spano, P.F.; Pizzi, M. NF-κB in innate neuroprotection and age-related neurodegenerative diseases. Front. Neurol. 2015, 6, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, F.C.; Irving, E.A.; Ray, A.M.; Lee, J.C.; Kassis, S.; Kumar, S.; Badger, A.M.; Legos, J.J.; Erhardt, J.A.; Ohlstein, E.H.; et al. Inhibition of p38 mitogen-activated protein kinase provides neuroprotection in cerebral focal ischemia. Med. Res. Rev. 2001, 21, 129–145. [Google Scholar] [CrossRef]
- Guo, R.B.; Wang, G.F.; Zhao, A.P.; Gu, J.; Sun, X.L.; Hu, G. Paeoniflorin protects against ischemia-induced brain damages in rats via inhibiting MAPKS/NF-κB-mediated inflammatory responses. PLoS ONE 2012, 7, e49701. [Google Scholar] [CrossRef] [PubMed]
- Harari, O.A.; Liao, J.K. Nf-κb and innate immunity in ischemic stroke. Ann. N. Y. Acad. Sci. 2010, 1207, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Grice, S.C.; Chappell, E.T.; Prough, D.S.; Whitley, J.M.; Su, M.; Watkins, W.D. Ibuprofen improves cerebral blood flow after global cerebral ischemia in dogs. Stroke 1987, 18, 787–791. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, J.E.; Steimle, C.N.; Zelenock, G.B.; d’Alecy, L.G. Ibuprofen improves survival and neurologic outcome after resuscitation from cardiac arrest. Resuscitation 1986, 14, 199–212. [Google Scholar] [CrossRef]
- Patel, P.M.; Drummond, J.C.; Sano, T.; Cole, D.J.; Kalkman, C.J.; Yaksh, T.L. Effect of ibuprofen on regional eicosanoid production and neuronal injury after forebrain ischemia in rats. Brain Res. 1993, 614, 315–324. [Google Scholar] [CrossRef]
- Antezana, D.F.; Clatterbuck, R.E.; Alkayed, N.J.; Murphy, S.J.; Anderson, L.G.; Frazier, J.; Hurn, P.D.; Traystman, R.J.; Tamargo, R.J. High-dose ibuprofen for reduction of striatal infarcts during middle cerebral artery occlusion in rats. J. Neurosurg. 2003, 98, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Cole, D.J.; Patel, P.M.; Reynolds, L.; Drummond, J.C.; Marcantonio, S. Temporary focal cerebral ischemia in spontaneously hypertensive rats: The effect of ibuprofen on infarct volume. J. Pharmacol. Exp. Ther. 1993, 266, 1713–1717. [Google Scholar] [PubMed]
- López-Villodres, J.A.; De La Cruz, J.P.; Muñoz-Marin, J.; Guerrero, A.; Reyes, J.J.; González-Correa, J.A. Cytoprotective effect of nonsteroidal antiinflammatory drugs in rat brain slices subjected to reoxygenation after oxygen-glucose deprivation. Eur. J. Pharm. Sci. 2012, 45, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Hypoxia/ischemia activate processing of amyloid precursor protein: Impact of vascular dysfunction in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2016. [Google Scholar] [CrossRef] [PubMed]
- Ułamek-Kozioł, M.; Pluta, R.; Bogucka-Kocka, A.; Januszewski, S.; Kocki, J.; Czuczwar, S.J. Brain ischemia with alzheimer phenotype dysregulates alzheimer’s disease-related proteins. Pharmacol. Rep. 2016, 68, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.A.; Suh, Y.H. Possible roles of amyloid intracellular domain of amyloid precursor protein. BMB Rep. 2010, 43, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Trazzi, S.; Fuchs, C.; de Franceschi, M.; Mitrugno, V.M.; Bartesaghi, R.; Ciani, E. App-dependent alteration of GSK3β activity impairs neurogenesis in the ts65dn mouse model of down syndrome. Neurobiol. Dis. 2014, 67, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Gong, K.; Song, B.; Ma, T.; van Laar, T.; Gong, Y.; Zhang, L. The app intracellular domain (aicd) inhibits wnt signalling and promotes neurite outgrowth. Biochim. Biophys. Acta 2012, 1823, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Lanzillotta, A.; Pignataro, G.; Branca, C.; Cuomo, O.; Sarnico, I.; Benarese, M.; Annunziato, L.; Spano, P.; Pizzi, M. Targeted acetylation of NF-κB/Rela and histones by epigenetic drugs reduces post-ischemic brain injury in mice with an extended therapeutic window. Neurobiol. Dis. 2013, 49, 177–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Movsesyan, V.A.; Stoica, B.A.; Faden, A.I. Mglur5 activation reduces β-amyloid-induced cell death in primary neuronal cultures and attenuates translocation of cytochrome c and apoptosis-inducing factor. J. Neurochem. 2004, 89, 1528–1536. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.C.; Faller, D.V. A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic. Acids Res. 1991, 19, 2499. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porrini, V.; Sarnico, I.; Benarese, M.; Branca, C.; Mota, M.; Lanzillotta, A.; Bellucci, A.; Parrella, E.; Faggi, L.; Spano, P.; et al. Neuroprotective and Anti-Apoptotic Effects of CSP-1103 in Primary Cortical Neurons Exposed to Oxygen and Glucose Deprivation. Int. J. Mol. Sci. 2017, 18, 184. https://doi.org/10.3390/ijms18010184
Porrini V, Sarnico I, Benarese M, Branca C, Mota M, Lanzillotta A, Bellucci A, Parrella E, Faggi L, Spano P, et al. Neuroprotective and Anti-Apoptotic Effects of CSP-1103 in Primary Cortical Neurons Exposed to Oxygen and Glucose Deprivation. International Journal of Molecular Sciences. 2017; 18(1):184. https://doi.org/10.3390/ijms18010184
Chicago/Turabian StylePorrini, Vanessa, Ilenia Sarnico, Marina Benarese, Caterina Branca, Mariana Mota, Annamaria Lanzillotta, Arianna Bellucci, Edoardo Parrella, Lara Faggi, Pierfranco Spano, and et al. 2017. "Neuroprotective and Anti-Apoptotic Effects of CSP-1103 in Primary Cortical Neurons Exposed to Oxygen and Glucose Deprivation" International Journal of Molecular Sciences 18, no. 1: 184. https://doi.org/10.3390/ijms18010184